DOI:
10.1039/C9RA08525D
(Paper)
RSC Adv., 2020,
10, 1711-1723
Retracted Article: Circular RNA PTK2 modifies the progression and radiosensitivity in gastric cancer via miR-369-3p/ZEB1 axis†
Received
18th October 2019
, Accepted 3rd December 2019
First published on 9th January 2020
Abstract
Gastric cancer (GC) is one of the common cancers worldwide. Emerging findings imply that aberrant expression of circular RNA_0003221 (circPTK2) is involved in GC. Nevertheless, the function of circPTK2 in GC needs more explanation. Profiles of circPTK2, microRNA (miR)-369-3p, and zinc finger E-box binding homeobox 1 (ZEB1) were determined by quantitative real-time polymerase chain reaction assay. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, flow cytometry, and transwell assays were employed to estimate cell proliferation, apoptosis, and mobility in GC cells, respectively. Meanwhile, levels of ZEB1 and other indicated proteins were tested using western blot. Survival fraction was assessed utilizing clonogenic assay. Additionally, the role of circPTK2 in tumorigenesis was investigated via a xenograft tumor model. Dual-luciferase reporter, RNA immunoprecipitation, and RNA pull-down assays were conducted to confirm the interrelation between miR-369-3p and circPTK2 or ZEB1. Levels of circPTK2 and ZEB1 were markedly augmented, but miR-369-3p was downregulated in GC tissues and cells. CircPTK2 depletion restrained cell growth, metastasis, and epithelial–mesenchymal transition, and promoted radiosensitivity in GC cells. And circPTK2 depletion reduced tumor growth and metastasis in vivo. Moreover, the effect of circPTK2 silencing on cellular phenotypes and radiosensitivity was regained by miR-369-3p inhibitor. Furthermore, upregulation of ZEB1 could overturn miR-369-3p mimic-induced effect on cell metastasis and radiosensitivity of GC cells. Mechanically, circPTK2 was a sponge of miR-369-3p to separate ZEB1. CircPTK2/miR-369-3p/ZEB1 axis modulated cell behaviors and radiosensitivity in GC; thus circPTK2 might serve as a promising target for GC therapy.
Introduction
Gastric cancer (GC) is one of the common diseases and is characterized as an aggressive malignancy.1 Most patients with GC are usually in advanced stages when they are diagnosed and accompanied by several unique features, such as lymph node and distant metastasis.2 Up to now, surgical resection and chemotherapy have been the major therapeutic approaches for GC, whereas recurrence and metastasis are the barriers for survival times and prognosis.2 Although remarkable researches have led to partial understanding of the molecular mechanisms of cancer,3 the therapeutic strategies and efficiencies are limited. Hence, it is necessary to discover several biomarkers associated with GC pathogenesis.
Recently, a series of transcriptomes have been defined as non-coding RNAs (ncRNAs).4 Emerging evidence has disclosed that ncRNAs, which were deemed as transcriptional “noise” at first, play critical roles in gene regulation.5 Circular RNAs (circRNAs) belong to ncRNAs with covalently closed-loop structures.6,7 Growing numbers of ectopic circRNAs are observed in diverse cancers.8 Apart from that, circRNAs possess various biological characteristics, such as being not easily degraded by exonuclease RNase R and acting as complementary endogenous RNA (ceRNA) to regulate gene expression.9 Moreover, the relation between circRNAs and tumor growth or radiosensitivity has been studied.10 For example, circRNA-0000285 is involved in radiosensitivity and regarded as a prognostic biomarker for nasopharyngeal carcinoma.11 Circ_0003221 (circPTK2) is implicated in tumor growth and metastasis of bladder cancer.12 Thus, a reasonable assumption is that circRNAs, including circPTK2, are tightly related to cellular processes.
Over the past few years, more and more studies have been devoted to shedding light on the spongy role of circRNAs for microRNAs.6 MiRNAs are small ncRNAs with 18–24 nucleotides (nts), involved in diverse cancer progression.13 MiRNAs have been identified as involved in the radioresistance of esophageal cancer cells.14 For example, miRNA-223 expedites GC metastasis by targeting tumor suppressor genes.15 MiR-200c is strongly associated with radiosensitivity in breast cancer.16 Besides, miR-369-3p works as a tumor suppressor to modify the onset and progress of GC.17 However, the function of miR-369-3p in radiosensitivity is unclear. Zinc finger E-box binding homeobox 1 (ZEB1) is a master monitor of the epithelial–mesenchymal transition (EMT), thereby participating in tumorigenesis in human cancer.18,19 And the aberrant expression of ZEB1 is involved in the outcome of radioresistance.19 Synthetically, we hypothesized that ZEB1 might be a requisite for miR-369-3p-mediated cell phenotypes.
In this paper, we identified the expression patterns of circPTK2, miR-369-3p, and ZEB1 in GC specimens and cell lines. The working pathway of them in cell pathogenesis and radiosensitivity was screened in subsequent assays.
Materials and methods
Clinical specimens and cell culture
Primer GC tissues (n = 48) and para-noncancerous specimens (n = 48) were obtained from GC patients who underwent surgical resection in Gansu Provincial Hospital. These clinical samples were immediately stored in nitrogen until use, and the specific characteristics of the samples are introduced in Table 1. On this basis, the GC tissues were classified, including lymph node metastasis (n = 30) or not (n = 18). Besides, we also grouped the collected specimens according to distance metastasis (n = 28) or non-metastasis (n = 20). Above all, written informed consents were acquired from all participators, and the study was authorized by the Ethics Committee of Gansu Provincial Hospital. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of National Tissue Engineering Center (China). GC cell lines (AGS, SNU-638, and HGC-27) and gastric mucosa cell line (GES-1) were purchased from CoBioer (Nanjing, China) and kept in Roswell Park Memorial Institute (RPMI)-1640 medium (Hyclone, Logan, UT, USA). In addition, the RPMI-1640 medium was supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 1× antibiotic (Gibco; 100 U mL−1 penicillin and 100 mg mL−1 streptomycin). All the cells were grown in an incubator with 5% CO2 at 37 °C.
Table 1 Association of circPTK2 expression with clinicopathologic characteristics of gastric cancer
| Parameter |
Case |
CircPTK2 expression |
P value |
| High (n = 24) |
Low (n = 24) |
| Gender |
| Female |
23 |
13 |
10 |
0.314 |
| Male |
25 |
11 |
14 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Age (years) |
| ≤60 |
28 |
16 |
12 |
0.202 |
| >60 |
20 |
8 |
12 |
|
| T stage |
| T1–T2 |
27 |
16 |
11 |
0.168 |
| T3–T4 |
21 |
8 |
13 |
|
| Lymph node metastasis |
| No |
30 |
18 |
12 |
0.011 |
| Yes |
18 |
6 |
12 |
|
| Distance metastasis |
| No |
28 |
18 |
10 |
0.008 |
| Yes |
20 |
6 |
14 |
Cell transfection
The full-length sequence of circPTK2 was amplified and used to construct the overexpression vector of circPTK2. What is more, the overexpression of ZEB1 was created, and the control of the overexpression vector was generated in Ribobio (Guangzhou, China). Short hairpin RNA (shRNA) targeting circPTK2 (sh-circPTK2) and its negative scramble (sh-NC) were designed and synthesized in GenePharma (Shanghai, China). After that, miR-369-3p mimic (miR-369-3p) and inhibitor (anti-miR-369-3p) and their control (miR-NC and anti-miR-NC) were simultaneously generated in Ribobio. Cell transfection was carried out by Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) as per the producer's introductions. Also, sh-circPTK2 and sh-NC were used for packaging lentivirus.
Quantitative real-time polymerase chain reaction (qRT-PCR) assay
RNA separation was performed with Trizol (Invitrogen) in accordance with the manufacturer's manuals. The extracted RNA acted as the template to form complementary DNA (cDNA) with the help of M-MLV reverse transcriptase (Promega, Madison, WI, USA). Expression levels of circPTK2, miR-369-3p, and ZEB1 were evaluated using the SYBR Green assay (Takara, Dalian, China) according to the producer's descriptions. RNase R digestion was performed following previously reported procedures.20 Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; for circPTK2 and ZEB1) and U6 (for miR-369-3p) served as the internal standards, and the relative level was calculated using the 2−ΔΔCt method. Special primer sequences were as follows: circPTK2 (forward: 5′-ATCATACTGGGAGATGCGGG-3′, reverse: 5′-AGTTGGGGTCAAGGTAAGCA-3′); miR-369-3p (forward: 5′-TCTGGACGACGAATGTTGC-3′, reverse: 5′-GATACGGACGGCTAGTGGA-3′); ZEB1 (forward: 5′-AGCGAGGTAAAGTTGCGTCT-3′, reverse: 5′-AGGTTTTCTGGGCCATACCG-3′); GAPDH (forward: 5′-ACTCCTCCACCTTTGACGC-3′, reverse: 5′-GCTGTAGCCAAATTCGTTGTC-3′); U6 (forward: 5′-CTCGCTTCGGCAGCACA-3′, reverse: 5′-AACGCTTCACGAATTTGCGT-3′).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
AGS and SNU-638 cells (5 × 103) were seeded into a 96-well plate with 200 μL complement medium. After incubation for the indicated time, the medium was replaced with fresh medium containing 0.5 mg mL−1 MTT (Sigma, St. Louis, MO, USA) for 4 h. Subsequently, the formed precipitate was dissolved by 200 μL dimethyl sulfoxide (Sigma), and the optical density values were examined using a microplate reader at 470 nm.
Flow cytometry assay
The cells were collected with ice-cold PBS (Gibco) and stained with the reagents of the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (Sigma) in accordance with the manufacturer's protocols. After incubation for 25 min in the dark, the apoptotic signals were captured using flow cytometry (BD Biosciences, San Jose, CA, USA) and analyzed with the use of BD FACS Diva software V6.1.3.
Western blot assay
The protocol was operated as per the introduction in a previous paper.21 Briefly, the protein lysates were segregated by 12% SDS-PAGE and then blotted on polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The immunoblots were performed by incubation with unique antibodies for B-cell lymphoma-2 (Bcl-2; 1:2000, ab182858), cleaved-caspase 3 (c-caspase 3; 1:500, ab13847), E-cadherin (1:1000, ab76055), N-cadherin (1:500, ab18203), poly(ADP-ribose) polymerase (PARP; 1:1000, ab191217), proliferating cell nuclear antigen (PCNA; 1:6000, ab92552), vimentin (1:1000, ab20346), ZEB1 (1:500, ab203829), and GAPDH (1:5000, ab8245). All antibodies were obtained from Abcam (Cambridge, MA, USA).
Transwell assay
24-well chambers (8 μm pore size; Corning, Corning, NY, USA) were employed for cell migration and invasion assay in vitro. Firstly, transfected AGS and SNU-638 cells (1 × 105 per mL) were re-suspended in 100 μL serum-free media and then added in the upper chamber with (for cell invasion) or without (for cell migration) pre-coated Matrigel (Corning). Next, the medium with 10% FBS was supplemented into the lower chamber. After culture for 48 h at 37 °C, the non-migrated cells were physically abandoned, but the migrated or invaded cells in the lower side of the membranes were stained with 0.5% crystal violet (Sigma) after methanol treatment. Five randomly fields were observed and chosen to count the rates of migrated or invaded cells.
Clonogenic assay
The AGS and SNU-638 cells transfected accordingly were treated with different doses (0, 2, 4, 6, or 8 Gy) of irradiation. After replacement with fresh medium, the cells were cultured for two weeks. The colonies were stained using 0.5% crystal violet (Sigma) for 20 min. Colonies with at least 50 cells were counted under a light microscope.
Xenograft tumor model
Four-to six-week-old athymic nude mice were purchased from Vital River Laboratory Animal Technology (Beijing, China). Briefly, a total of 12 nude mice were divided into two groups randomly, and AGS cells (2 × 106) with non-depleted or depleted circPTK2 under 200 μL PBS were subcutaneously injected into the left flank of the mice (n = 6/group). Beginning five days post-injection, tumor growth was monitored every five days, and the tumor length (L) and width (W) were measured for tumor volume calculation, following the formula: volume = L × W2 × 0.5. After injection for 30 days, xenograft tumors were taken out and weighed. Above all, animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Gansu Provincial Hospital and approved by the Animal Ethics Committee of Gansu Provincial Hospital.
Cell cytoplasm/nucleus fraction isolation
The extraction of cytoplasm and nucleus (AGS and SUN-638 cells) was performed using nuclear and cytoplasmic extraction reagents (Thermo Scientific, Waltham, MA, USA) in accordance with user guidebook. Then, the levels of circPTK2 and U6 in extracted sections were determined by means of qRT-PCR assay.
Dual-luciferase reporter assay
The putative miR-369-3p targeted common sequences in wildtype circPTK2 and the relative mutant were subcloned into the downstream of the luciferase gene in basic vector (pmirGLO; Promega), thereby constructing circPTK2-WT and circPTK2-MUT. Similarly, the common fragments between miR-369-3p and the 3′ untranslated region (3′UTR) of ZEB1 were also amplified and formed luciferase reporters in the same way, named as ZEB1 3′UTR-WT and ZEB1 3′UTR-MUT. Subsequently, AGS and SNU-638 cells were co-transfected with the created luciferase reporters and miR-369-3p or miR-NC with the use of Lipofectamine 2000 (Invitrogen) as per manuals. After culture for 48 h, a dual-luciferase assay system (Promega) was employed to measure the luciferase activities of firefly and renilla. Renilla luciferase activity acted as the normalization of firefly luciferase activity in this assay.
RNA immunoprecipitation (RIP) assay
RIP assay was implemented by means of an EZ-Magna RIP kit (Millipore). AGS and SNU-638 cells were harvested in NP-40 RIP lysis buffer (Sigma) at 80% confluence. Next, 100 μL of lysates was incubated with RIP buffer containing magnetic beads combined with Argonaute2 (Ago2; Millipore) or immunoglobulin G (IgG; Millipore) antibody. Then, the RNA combined with Ago2 antibody was separated by Trizol (Invitrogen), and the presence of the direct target between miR-369-3p and circPTK2 was demonstrated by adopting qRT-PCR analysis.
RNA pull-down assay
Biotin-labeled circPTK2 (Bio-circPTK2) and ZEB1 (Bio-ZEB1) and their control (Bio-NC) were synthesized in Ribobio (Guangzhou, China). The RIP lysates from AGS and SNU-638 cells were incubated with Bio-circPTK2 or Bio-ZEB1 for 2 h at 25 °C, and the complexes were captured by streptavidin-coupled Dynabeads (Invitrogen). After washing with RIP buffer, the level of miR-369-3p in the pull-down was measured utilizing qRT-PCR.
Statistical analysis
The data from at least three independent assays were exhibited as mean ± standard deviation (SD). Group difference was assessed utilizing Student's t-test (two groups) and one-way analysis of variance with Tukey's test (three or more groups). Kaplan–Meier analysis was performed for the overall survival of GC patients. P < 0.05 was regarded as an indicator of statistical significance.
Results
CircPTK2 at high level was closely associated with GC development
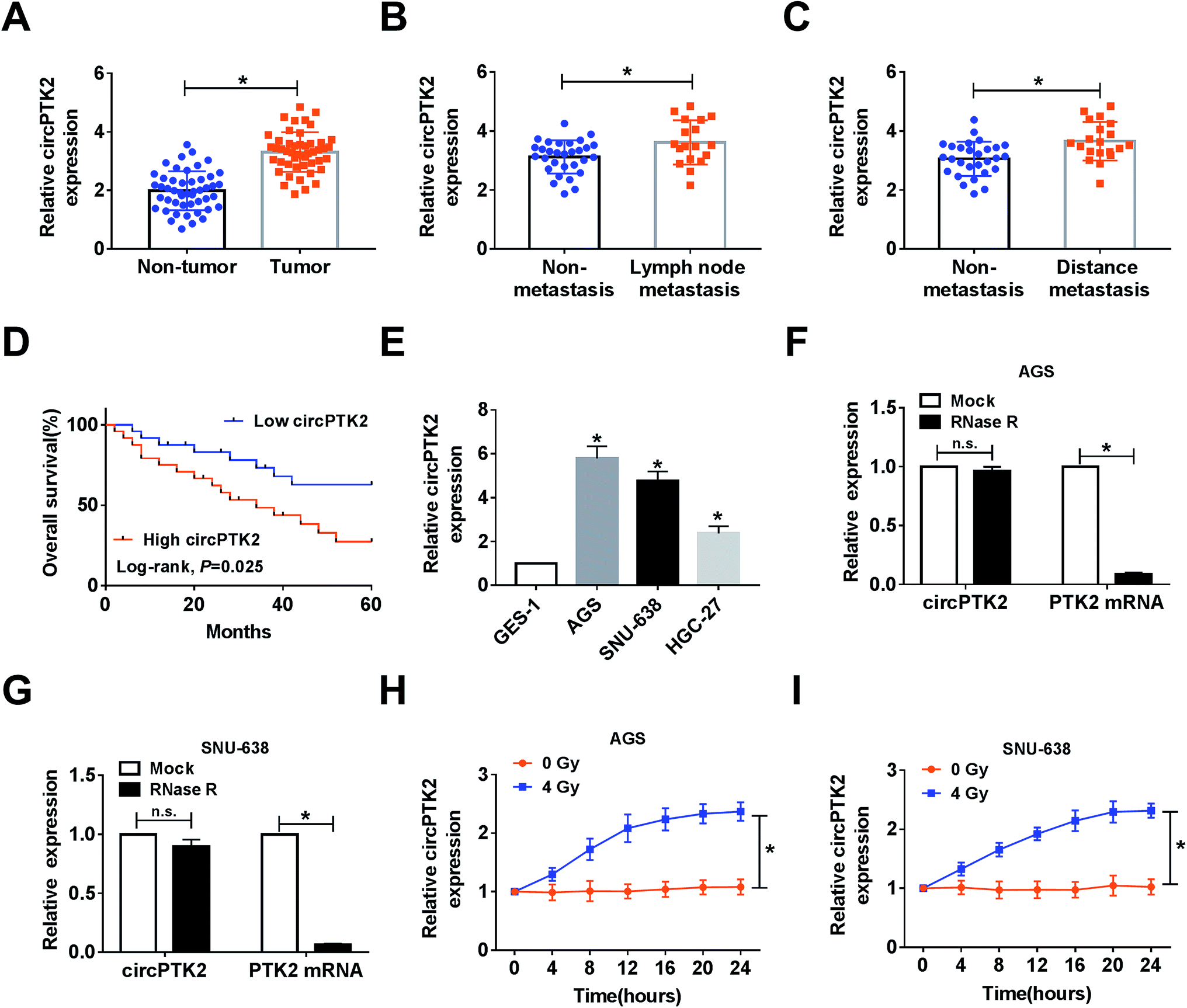
In the assay, we found that circPTK2 was upregulated in GC tissues compared with adjacent samples (Fig. 1A). Furthermore, the GC tissues were classified according to the clinical characteristics, and qRT-PCR analysis demonstrated that the level of circPTK2 was continuously augmented in lymph node metastasis and distance metastasis with respect to related controls (Fig. 1B and C). More importantly, we discovered that the high level of circPTK2 was tightly associated with poor prognosis of GC patients, exhibited as Kaplan–Meier survival curves in Fig. 1D. Subsequently, we selected three GC cell lines (AGS, SNU-638, and HGC-27), and an evident high level of circPTK2 was observed in GC cells. Then, AGS and SNU-638 cells were chosen for subsequent assays (Fig. 1E). What is more, RNase R was recruited to treat the extracted RNA, and qRT-PCR analysis showed that circPTK2 could resist RNase R exonuclease in AGS and SNU-638 cells (Fig. 1F and G). Apart from that, the role of irradiation in altering the level of circPTK2 was investigated. Results from qRT-PCR assay indicated that 4 Gy exposure could sharply improve the level of circPTK2 in both AGS and SNU-638 cells (Fig. 1H and I). All the data suggested that circPTK2 was closely related to tumor growth, metastasis, and radiosensitivity in GC.
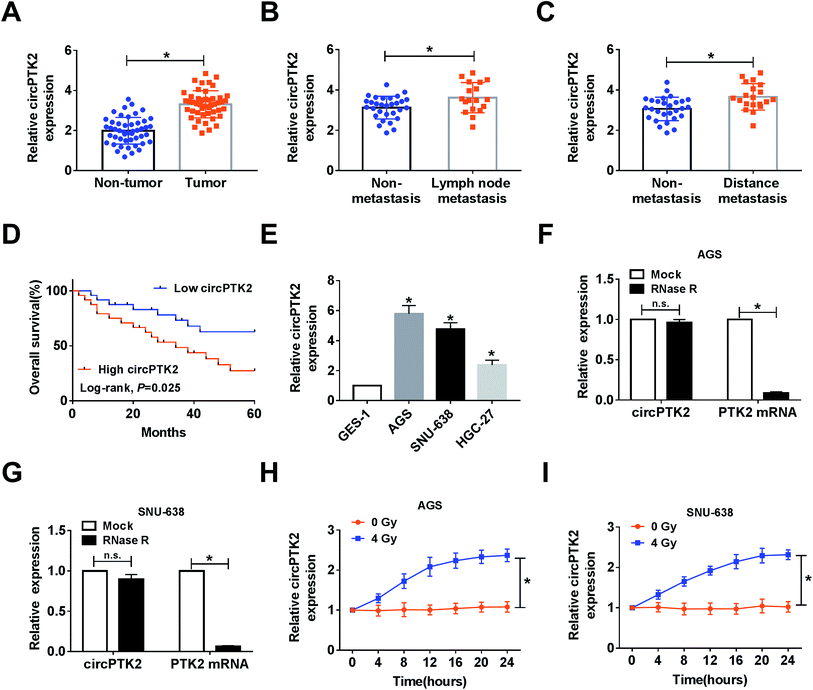 |
| | Fig. 1 CircPTK2 with high level was closely associated with GC development. (A–C) qRT-PCR analysis for circPTK2 level in tumor samples, lymph node metastasis, and distance metastasis in comparison with related controls. (D) Kaplan–Meier analysis for the overall survival of the GC patients with high or low level of circPTK2. (E) qRT-PCR analysis for the level of circPTK2 in GC cell lines. (F and G) The levels of circPTK2 and PTK2 RNA after treatment with RNase R in AGS and SNU-638 cells. (H and I) CircPTK2 expression alteration in AGS and SNU-638 cells after 0 or 4 Gy of irradiation treatment for the indicated time. *P < 0.05. | |
Knockdown of circPTK2 reduced cell metastasis and increased radiosensitivity in GC cells
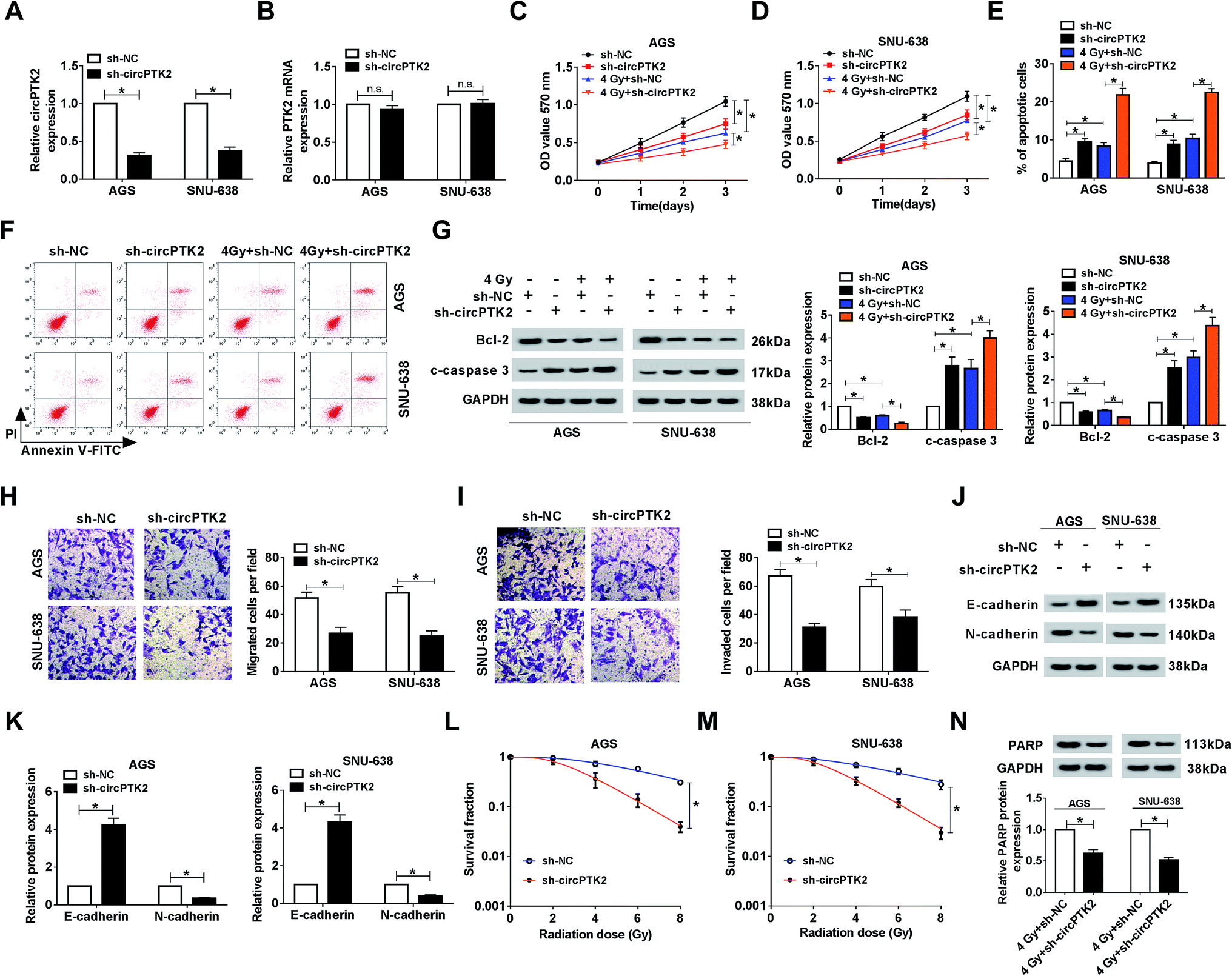
The ectopic expression of circPTK2 in GC tissues and cells prompted us to investigate its potential role in GC. As depicted in Fig. 2A, the level of circPTK2 was clearly decreased in AGS and SNU-638 cells with sh-circPTK2 transfection. However, the absence of circPTK2 did not affect the expression of linear PTK2 (Fig. 2B). Then, cell proliferation was assessed using MTT assay. The results indicated that circPTK2 deficiency could suppress cell proliferation, and simultaneous irradiation exposure aggravated this repressive effect in circPTK2-silenced GC cells (Fig. 2C and D). Conversely, cell apoptosis was boosted as a result of circPTK2 reduction, and such hindered effect was enhanced via 4 Gy treatment in AGS and SNU-638 cells (Fig. 2E and F). Subsequently, we also identified the alterations of apoptosis-related proteins (Bcl-2 and c-caspase 3) in vitro. The low level of Bcl-2 and the high level of c-caspase 3 verified the synergistic effects of circPTK2 silencing and irradiation on cell apoptosis in both AGS and SNU-638 cells (Fig. 2G). In addition, transwell analysis revealed that cell migration and invasion were both retarded in circPTK2-silenced GC cells (Fig. 2H and I). For the analysis of western blot, E-cadherin was triggered but N-cadherin was downregulated in the two GC cell lines with circPTK2 deficiency. Namely, circPTK2 knockdown resulted in the repression of EMT in vitro (Fig. 2J and K). Moreover, we discovered that the survival fraction was significantly decreased in the sh-circPTK2 transfected group under the indicated irradiation exposure (Fig. 2L and M). Besides, PARP is a DNA repair-associated protein; the alteration of PARP level was detected in the assay. As shown in Fig. 2N, circPTK2 silencing caused a curb of the level of mature PARP in vitro. These findings proved that circPTK2 reduction could block cell growth and metastasis and enhance radiosensitivity in the two GC cell lines.
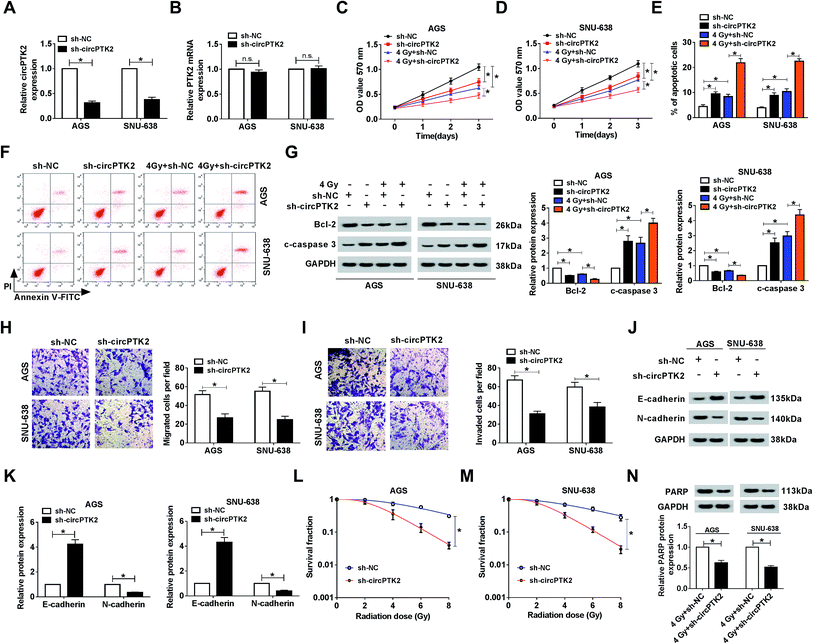 |
| | Fig. 2 Knockdown of circPTK2 diminished cell growth and metastasis and increased radiosensitivity in GC cells. (A) The level of circPTK2 in the two GC cell types after sh-circPTK2 transfection. (B–G) AGS and SNU-638 cells were introduced with sh-circ-PTK2 or sh-NC under 4 Gy irradiation exposure or not. (B and C) The effects of circPTK2 depletion and 4 Gy treatment on the proliferation in AGS and SNU-638 cells. (D and E) Flow cytometry analysis for cell apoptosis in vitro. (F and G) Levels of Bcl-2 and c-caspase 3 in the two GC cell lines. (H and I) Transwell assay for the capacities of cell migration and invasion in AGS and SNU-638 cells with circPTK2 silencing. (J and K) The impact of circPTK2 silencing on the levels of E-cadherin and N-cadherin in vitro. (L and M) Clonogenic assay for the survival fraction in sh-circPTK2- or sh-NC-treated AGS and SNU-638 cells. (N) The alteration of PARP level in circPTK2-deficient GC cells under 4 Gy irradiation exposure. *P < 0.05. | |
CircPTK2 depletion caused a decrease of tumor growth in vivo
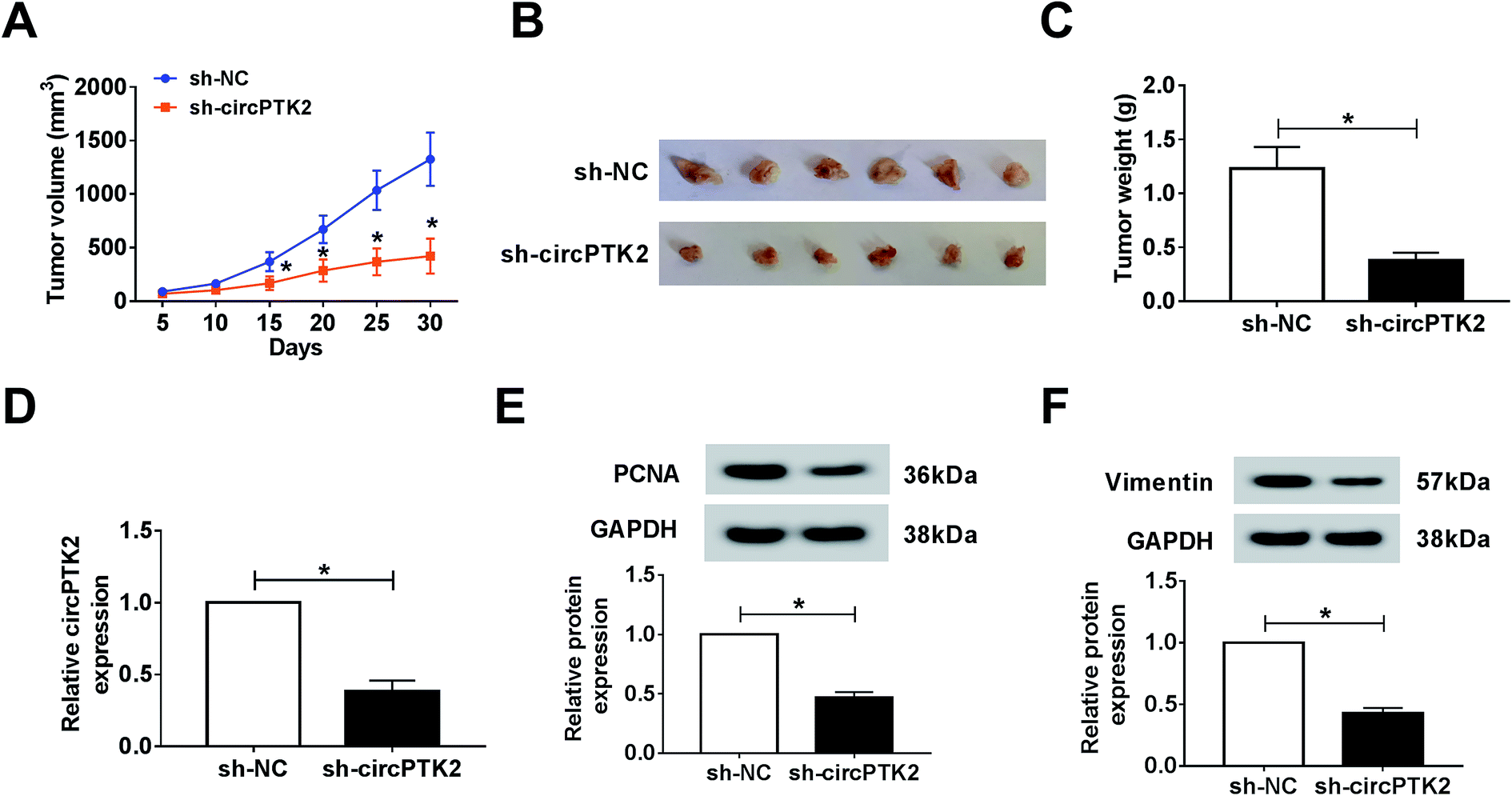
Owing to the oncogenic role of circPTK2 in vitro, we further explored its role via a xenograft tumor model. Firstly, lentivirus-mediated AGS cells (sh-circPTK2 or sh-NC) were injected into nude mice. After 30 days, the volume and weight of xenograft tumors were strongly diminished in circPTK2-deficient samples (Fig. 3A–C). Next, an effective decrease of circPTK2 level was observed in sh-circPTK2-mediated tissues (Fig. 3D). PCNA and vimentin served as indicators of cell proliferation and EMT, respectively. The low levels of them elucidated that both cell proliferation and metastasis might be reduced in the sh-circPTK2 transfected group (Fig. 3E and F). In brief, circPTK2 knockdown led to the suppression of tumor growth and metastasis in vivo.
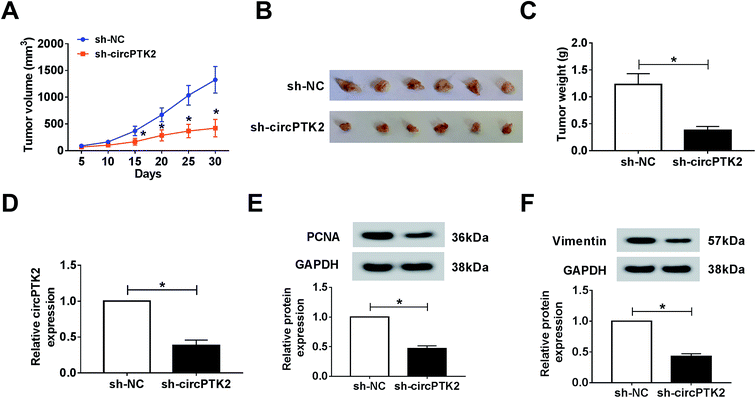 |
| | Fig. 3 CircPTK2 depletion caused a decrease of tumor growth in vivo. (A and B) The data of tumor volume and weight after injection with sh-circPTK2 or sh-NC. (C and D) qRT-PCR analysis for the level of circPTK2 in xenograft tumors. (E and F) Relative levels of PCNA and vimentin in excised tumor specimens. *P < 0.05. | |
CircPTK2 was a sponge of miR-369-3p
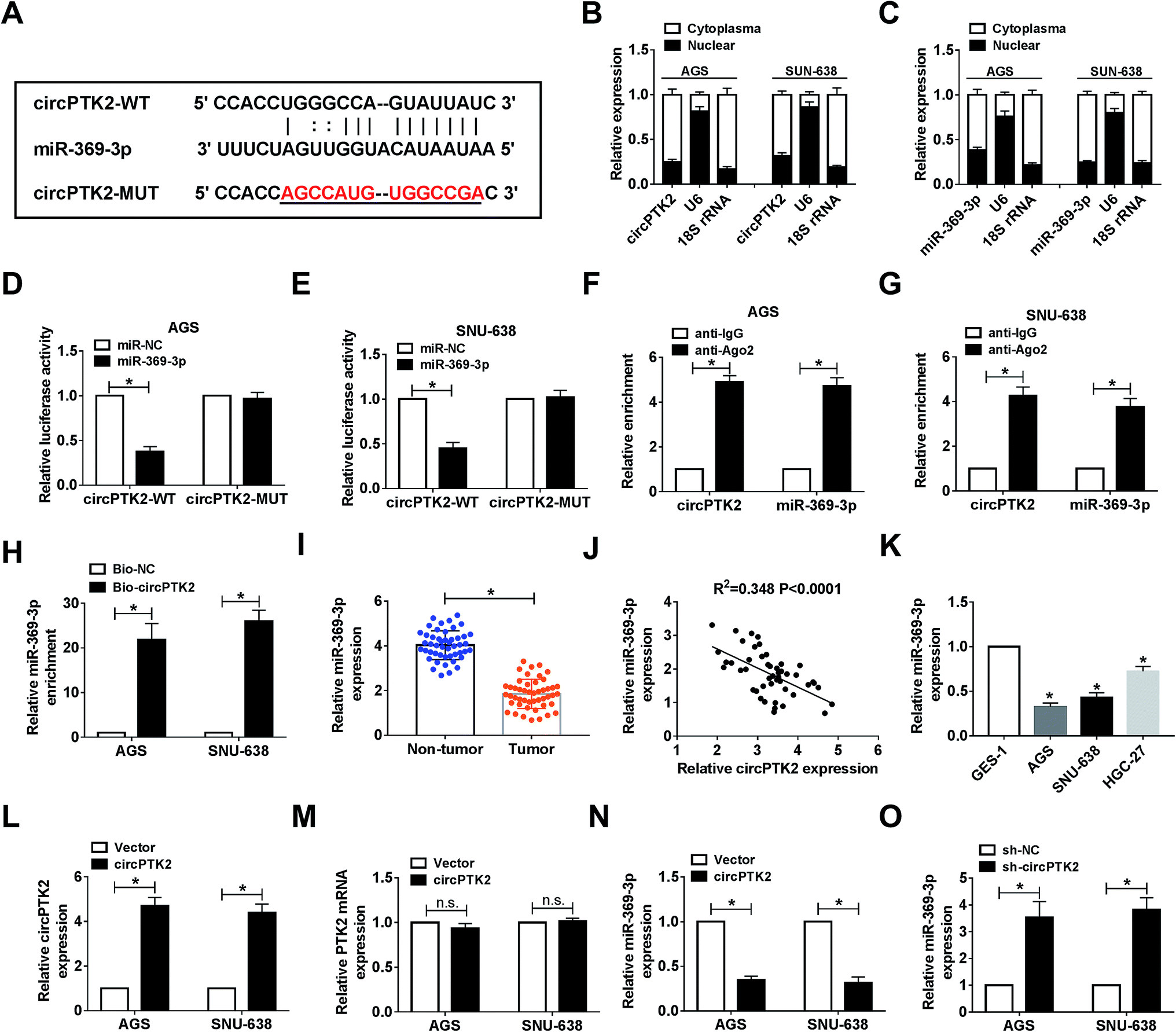
Given the vital role of circPTK2 in the cell behaviors and tumor growth of GC, we subsequently sought the targets of circPTK2. As predicted by starBase, there were complementary sequences between circPTK2 and miR-369-3p (Fig. 4A). Next, we found that both circPTK2 and miR-369-3p were enriched in cytoplasm in AGS and SNU-638 cells (Fig. 4B and C). To confirm the relationship between circPTK2 and miR-369-3p, dual-luciferase reporter assay was carried out. In the assay, miR-369-3p reduced the luciferase activity of circPTK2-WT reporter, whereas it had no apparent effect on luciferase activity of circPTK2-MUT reporter system (Fig. 4D and E). Other assays, including RIP and RNA pull-down, were performed to verify the interaction between miR-369-3p and circPTK2, the enriched levels of miR-369-3p and circPTK2 in the Ago2 group, as well as the abundance of miR-369-3p in Bio-circPTK2-introduced GC cells validating that miR-369-3p was a target of circPTK2 (Fig. 4F–H).
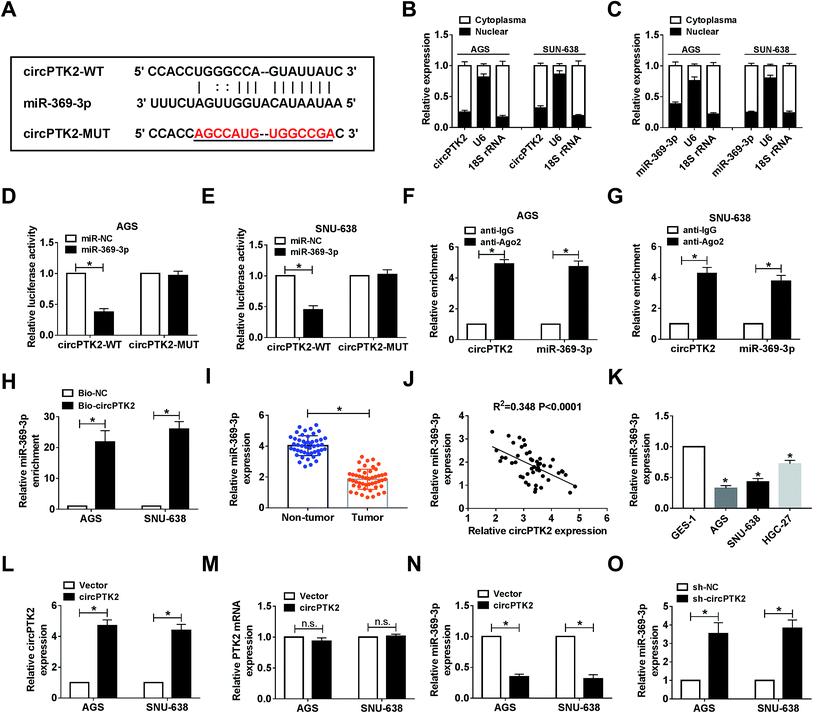 |
| | Fig. 4 CircPTK2 was a sponge of miR-369-3p. (A) The complementary binding sites between miR-369-3p and circPTK2. (B–E) Dual-luciferase reporter analysis for the interaction between miR-369-3p and circPTK2 in AGS and SNU-638 cells. (F–H) RIP and RNA pull-down analyses for the clarification of the relationship between miR-369-3p and circPTK2. (I) Relative level of miR-369-3p in GC tumors. (J) Correlation between miR-369-3p level and circPTK2 level in GC tumors. (K) qRT-PCR analysis for the level of miR-369-3p in the three GC cell types. (L and M) Relative level of circPTK2 in AGS and SNU-638 cells with circPTK2 transfection. (N and O) The influence of circPTK2 overexpression or deficiency on the level of miR-369-3p in vitro. *P < 0.05. | |
Then, an evident curb of miR-369-3p level was observed in GC tissues in comparison with non-cancerous samples (Fig. 4I). Also, circPTK2 was passively correlated with miR-369-3p in clinical GC cells (Fig. 4J). On this basis, we evaluated the level of miR-369-3p in GC cells. qRT-PCR analysis indicated that miR-369-3p was expressed at a low level in selected GC cells (Fig. 4K). To further clarify the regulatory mechanism between circPTK2 and miR-369-3p, we firstly investigated the efficiency of circPTK2 vector in GC cells. Results from qRT-PCR assay corroborated that transfection with circPTK2 could markedly facilitate the level of circPTK2 in vitro (Fig. 4L). Besides, the transfection of circPTK2 had no effect on altering the level of the linear PTK2 in the two GC cell lines (Fig. 4M). Subsequently, circPTK2 or sh-circPTK2 was introduced into AGS and SNU-638 cells, separately, and circPTK2 inversely regulated miR-369-3p in the two GC cell lines (Fig. 4N and O). Collectively, circPTK2 was a ceRNA of miR-369-3p.
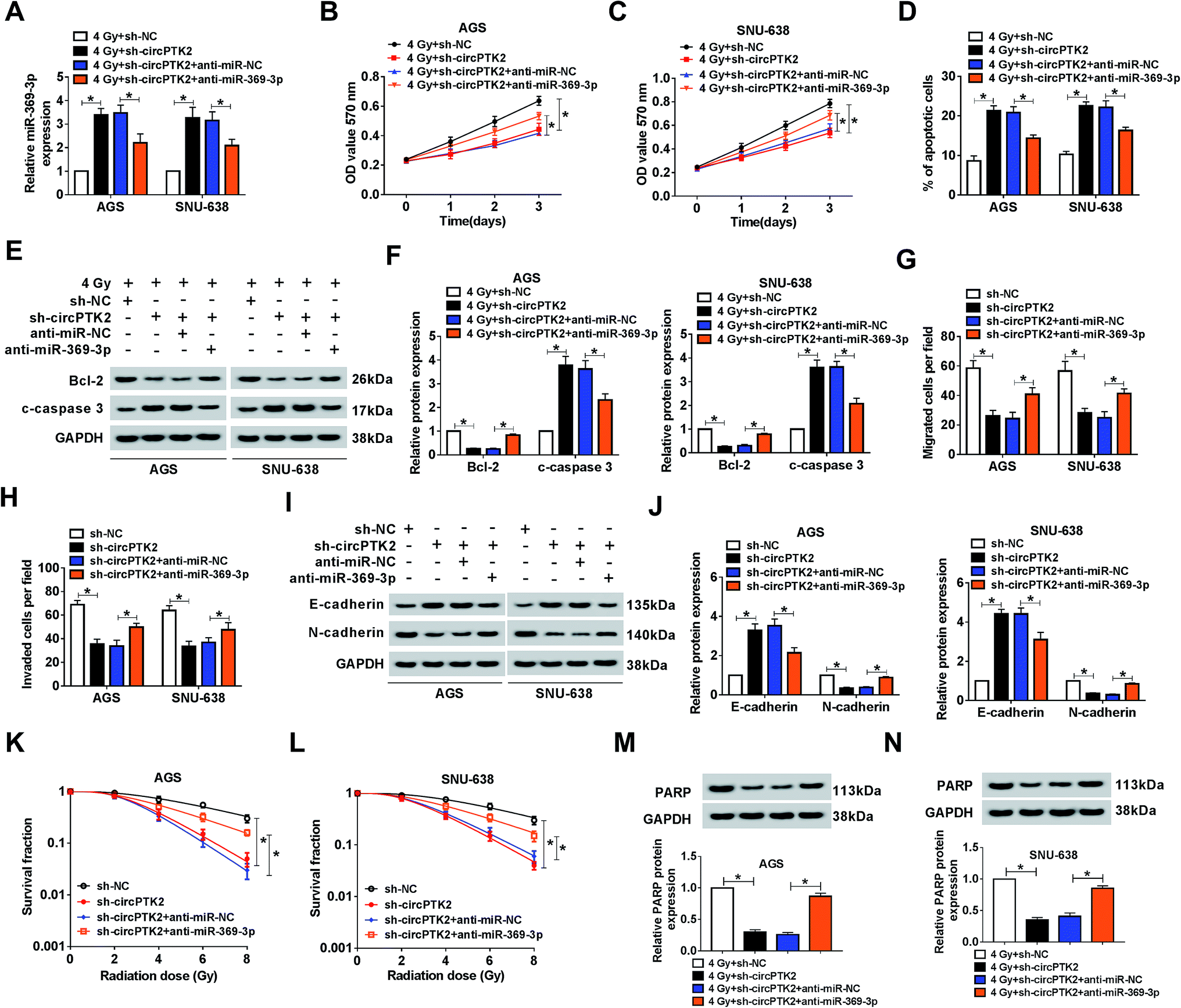
The impact of circPTK2 silencing on radiosensitivity and cell mobility was abrogated by miR-369-3p inhibition
Considering that circPTK2 was a sponge of miR-369-3p, we guessed that circPTK2 regulated cell phenotypes and radiosensitivity via targeting miR-369-3p in GC. In the assay, sh-NC, sh-circPTK2, sh-circPTK2 + anti-miR-NC, or sh-circPTK2 + anti-miR-369-3p was transfected into AGS and SNU-638 cells with 4 Gy exposure. qRT-PCR analysis manifested that miR-369-3p inhibitor could reverse the strong influence of circPTK2 repression on the level of miR-369-3p in the two GC cell lines under 4 Gy treatment (Fig. 5A). Afterwards, reintroduction of miR-369-3p inhibitor particularly restored the reductive effect of sh-circPTK2 transfection in 4 Gy exposed AGS and SNU-638 cells (Fig. 5B and C). Additionally, the apoptotic rate of the two GC cell lines increased as a result of circPTK2 silencing, but co-transfection with anti-miR-369-3p could overturn this effect in GC cells with irradiation exposure (Fig. 5D). Interestingly, the effect of miR-369-3p or circPTK2 deficiency on cell apoptosis was further illuminated via measuring the levels of Bcl-2 and c-caspase 3 in irradiated GC cells (Fig. 5E and F). Synchronously, the impacts of circPTK2 and miR-369-3p on cell mobility were uncovered. As introduced in Fig. 5G and H, the decreases of cell migration and invasion caused by circPTK2 downregulation were partially strengthened via reintroduction with miR-369-3p inhibitor in vitro. Not only that, the level of EMT was impeded in circPTK2-silenced GC cells, while this effect was recovered after simultaneous inhibition of miR-369-3p (Fig. 5I and J). After which, knockdown of miR-369-3p could remarkably revert the inhibiting effect of circPTK2 depletion on survival fractions of GC cells with indicated irradiation exposure (Fig. 5K and L). Furthermore, DNA repair-related protein was apparently decreased by circPTK2 silencing, while this repressive impact was notably rescued by miR-369-3p inhibition in radiation-treated AGS and SNU-638 cells (Fig. 5M and N). That is to say, circPTK2 exerted its roles via sponging miR-369-3p in GC cells.
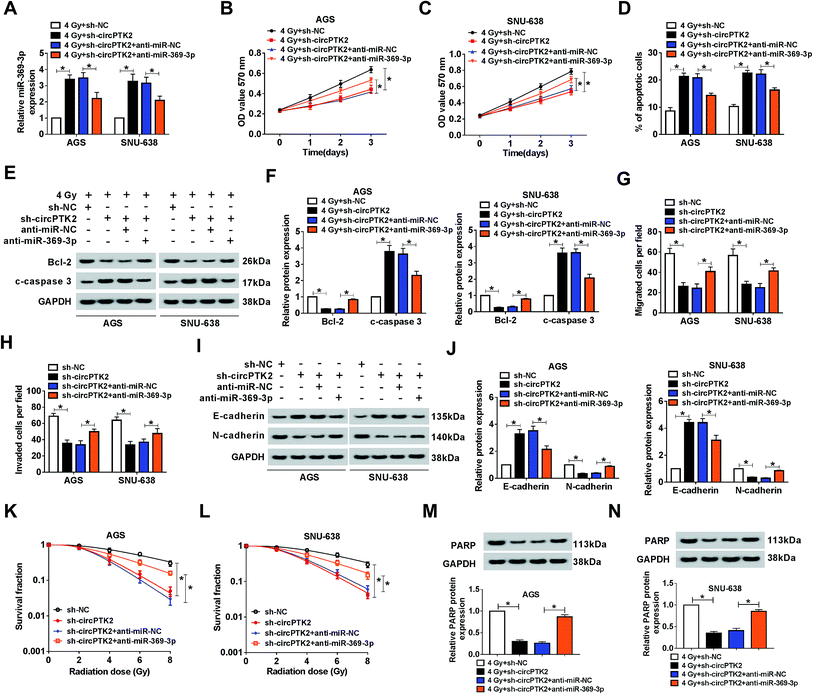 |
| | Fig. 5 The impact of circPTK2 silencing on cell behaviors was abrogated by miR-369-3p inhibition. AGS and SNU-638 cells were each transfected with sh-NC, sh-circPTK2, sh-circPTK2 + anti-miR-NC, or sh-circPTK2 + anti-miR-369-3p. (A) Relative level of miR-369-3p in vitro. (B and C) MTT analysis for the level of cell proliferation in GC cells. (D) The impact of circPTK2 or miR-369-3p deficiency on cell apoptosis in irradiation-exposed AGS and SNU-638 cells. (E and F) western blot analysis for the level of apoptosis-related proteins in vitro. (G and H) The alterations of cell migration and invasion in AGS and SNU-638 cells. (I and J) Relative levels of E-cadherin and N-cadherin in the two GC cell types. (K and L) The role of circPTK2 depletion or miR-369-3p inhibition in colony formation in GC cells with indicated treatment. (M and N) Relative level of PARP in AGS and SNU-638 cells under 4 Gy exposure. *P < 0.05. | |
ZEB1 was a target of miR-369-3p
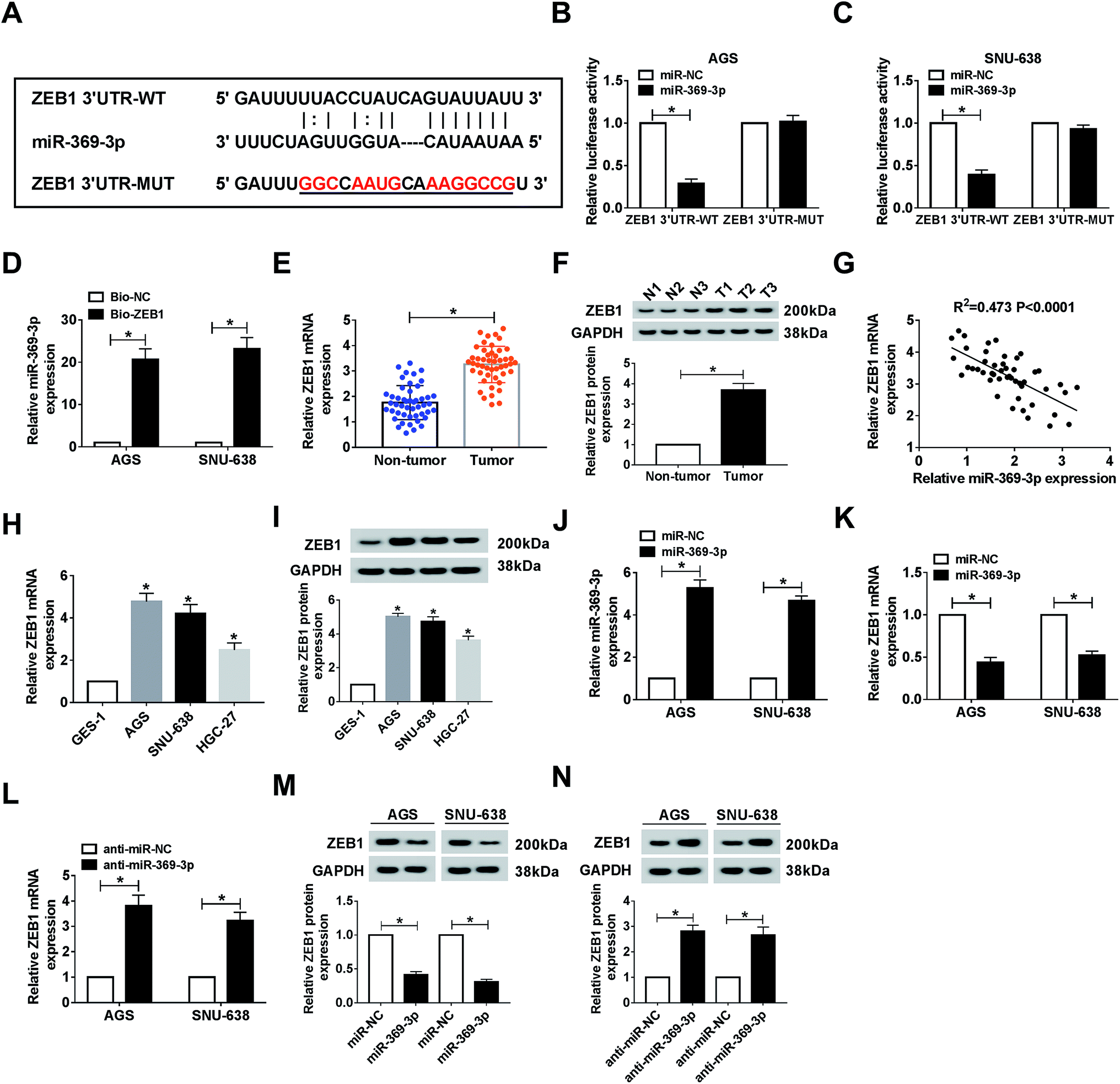
As mentioned above, we attempted to search for the probable targets of miR-369-3p. As a result of starBase prediction, ZEB1 was found to possess binding sites with miR-369-3p (Fig. 6A). To shed light on the interaction between miR-369-3p and ZEB1, dual-luciferase reporter and RNA pull-down assays were conducted. MiR-369-3p mimic could reduce nearly 70% of the luciferase activity in the wildtype group, while it had no distinct effect on altering the fluorescence intensity of ZEB1 3′UTR-MUT in both AGS and SNU-638 cells (Fig. 6B and C). The improved enrichment of miR-369-3p in Bio-ZEB1-introduced GC cells further confirmed that ZEB1 was a target of miR-369-3p (Fig. 6D). Next, a significantly high level of ZEB1 was found in GC tissues compared with matched controls (Fig. 6E and F). Synchronously, we determined that miR-369-3p was inversely correlated with ZEB1 in clinical GC tissues (Fig. 6G). At the same time, similar tendencies of the mRNA and protein levels were observed in GC cells (Fig. 6H and I). Besides, miR-369-3p transfection could substantially enhance the level of miR-369-3p in vitro (Fig. 6J). Expectably, transfection with miR-369-3p resulted in an apparent decline, but silence of miR-369-3p elicited a forceful enhancement of ZEB1 level in terms of mRNA and protein expression in vitro (Fig. 6K–N). In a word, ZEB1 was directly targeted by miR-369-3p.
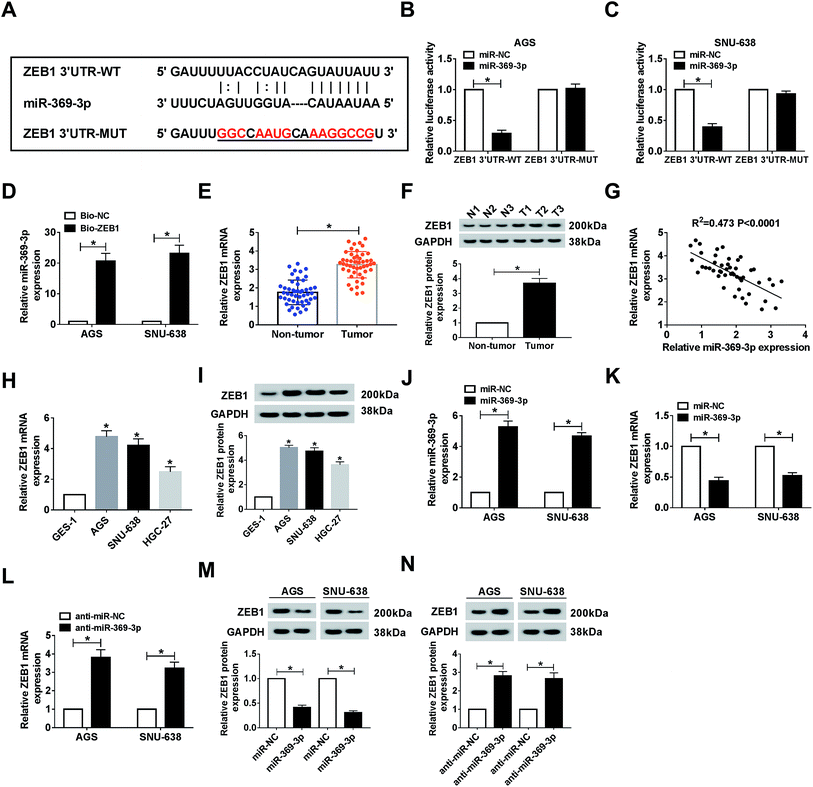 |
| | Fig. 6 ZEB1 was a target of miR-369-3p. (A) Common binding sites between miR-369-3p and ZEB1. (B–D) Dual luciferase reporter and RNA pull-down analyses for the interaction between miR-369-3p and ZEB1 in AGS and SNU-638 cells. (E and F) Relative mRNA and protein levels of ZEB1 in GC tumors. (G) The correlation between miR-369-3p and ZEB1 in clinical tumors. (H and I) qRT-PCR and western blot analysis for the level of ZEB1 in AGS and SNU-638 cells in terms of mRNA and protein expression. (J) The level of miR-369-3p in the two GC cell types after miR-369-3p transfection. (K–N) Relative mRNA and protein levels of ZEB1 in AGS and SNU-638 cells with miR-369-3p upregulation or downregulation. *P < 0.05. | |
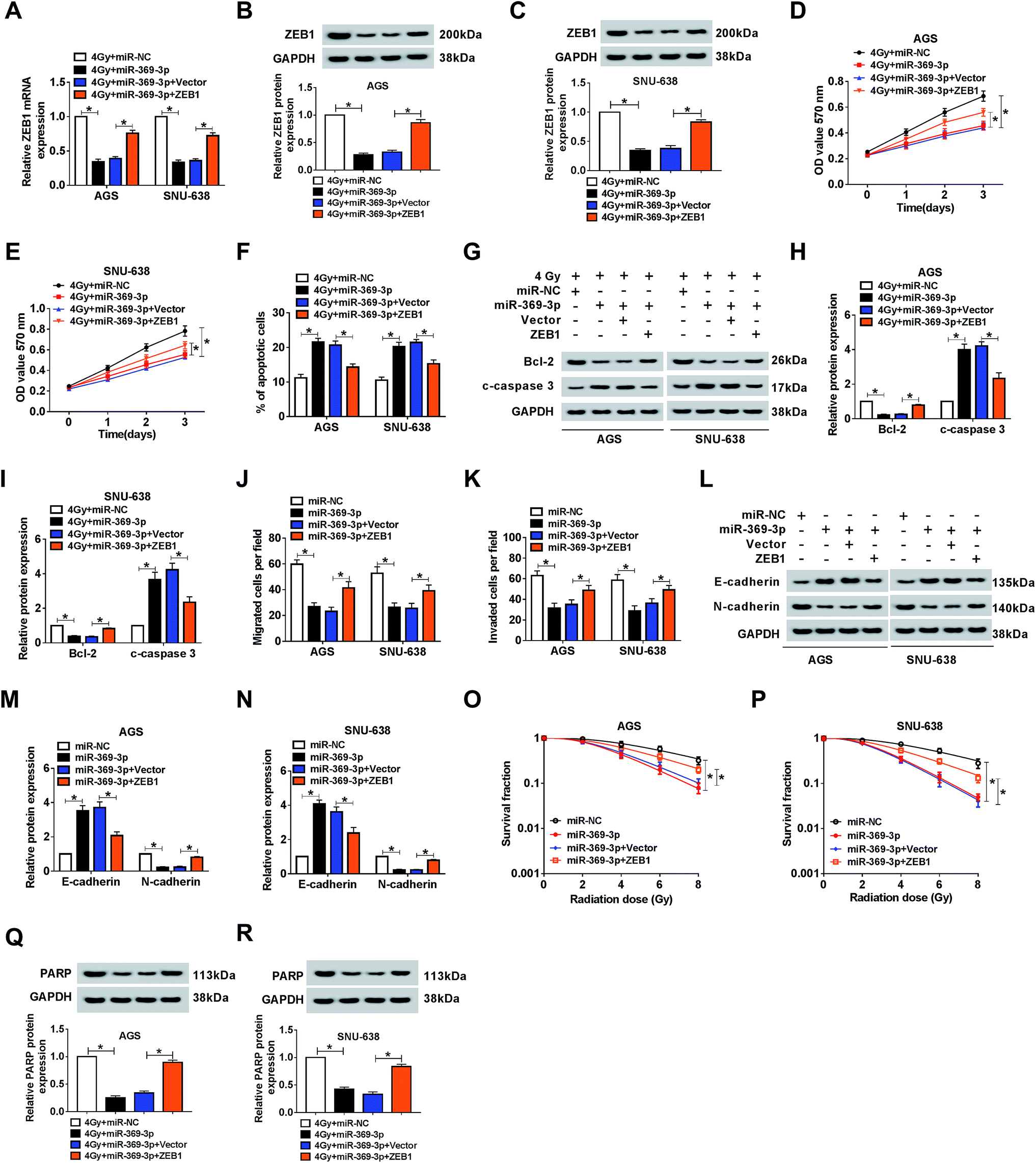
Effect of miR-369-3p mimic on cell phenotypes and radiosensitivity was abolished by ZEB1 upregulation in AGS and SNU-638 cells
Because of the relationship between miR-369-3p and ZEB1, the functional mechanism needed to be explained. Firstly, miR-369-3p alone or along with ZEB1 was transfected into AGS and SNU-638 cells with 4 Gy exposure. As shown in Fig. 7A–C, miR-369-3p-mediated curb in ZEB1 level was sharply reduced after ZEB1 upregulation. In addition, miR-369-3p led to the decline of cell proliferation, and such repressive effect was obviously abolished via overexpression of ZEB1 in the two GC cell lines under irradiation exposure (Fig. 7D and E). Meanwhile, flow cytometry and western blot analyses revealed that reintroduction of ZEB1 abrogated the intensive influence of miR-369-3p on cell apoptosis in radiation-exposed AGS and SNU-638 cells (Fig. 7F–I). Interestingly, cell migration and invasion were diminished as a result of miR-369-3p introduction, and such inhibitory effect was clearly eliminated via co-transfection with ZEB1 in both AGS and SNU-638 cells (Fig. 7J and K). At the same time, the hindered level of EMT resulting from miR-369-3p mimic was significantly promoted after overexpression of ZEB1 in vitro (Fig. 7L–N). Subsequently, we also determined the effects of miR-369-3p and ZEB1 in colony formation in GC cells under indicated treatment. As shown in Fig. 7O and P, miR-369-3p-mediated repression of survival fractions was evidently improved in vitro. Lastly, miR-369-3p upregulation caused a virtual weakened in DNA repair in 4 Gy-exposed GC cells, whereas this effect was strongly eliminated by regaining of ZEB1 (Fig. 7Q and R). The evidence pointed to miR-369-3p modulating cell metastasis and radiosensitivity via reducing ZEB1 in GC cells.
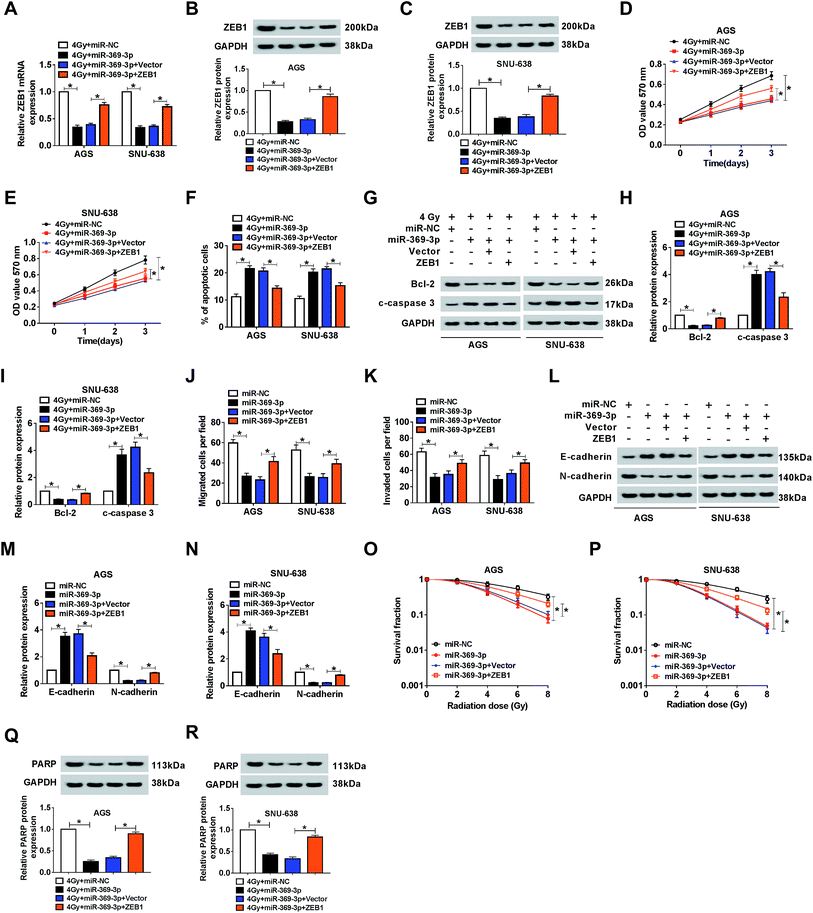 |
| | Fig. 7 Effect of miR-369-3p mimic on cell phenotypes and radiosensitivity was abolished by ZEB1 upregulation in AGS and SNU-638 cells. MiR-NC, miR-369-3p, miR-369-3p + vector, or miR-369-3p + ZEB1 was introduced into each of AGS and SNU-638 cells under irradiation exposure. (A–C) Relative level of ZEB1 in the two GC cell lines. (D and E) MTT analysis for the ability of cell proliferation in AGS and SNU-638 cells with miR-369-3p or ZEB1 transfection. (F) The apoptotic rate of GC cells under 4 Gy exposure. (G–I) western blot analysis for the expression alterations of apoptosis-related proteins in vitro. (J and K) The effect of miR-369-3p or ZEB1 transfection on cell migration and invasion in AGS and SNU-638 cells. (L–N) Relative levels of EMT-related proteins. (O and P) Clonogenic assay for the survival fraction in AGS and SNU-638 cells with related treatment. (Q and R) Level of PARP in 4 Gy-exposed AGS and SNU-638 cells with miR-369-3p or ZEB1 introduction. *P < 0.05. | |
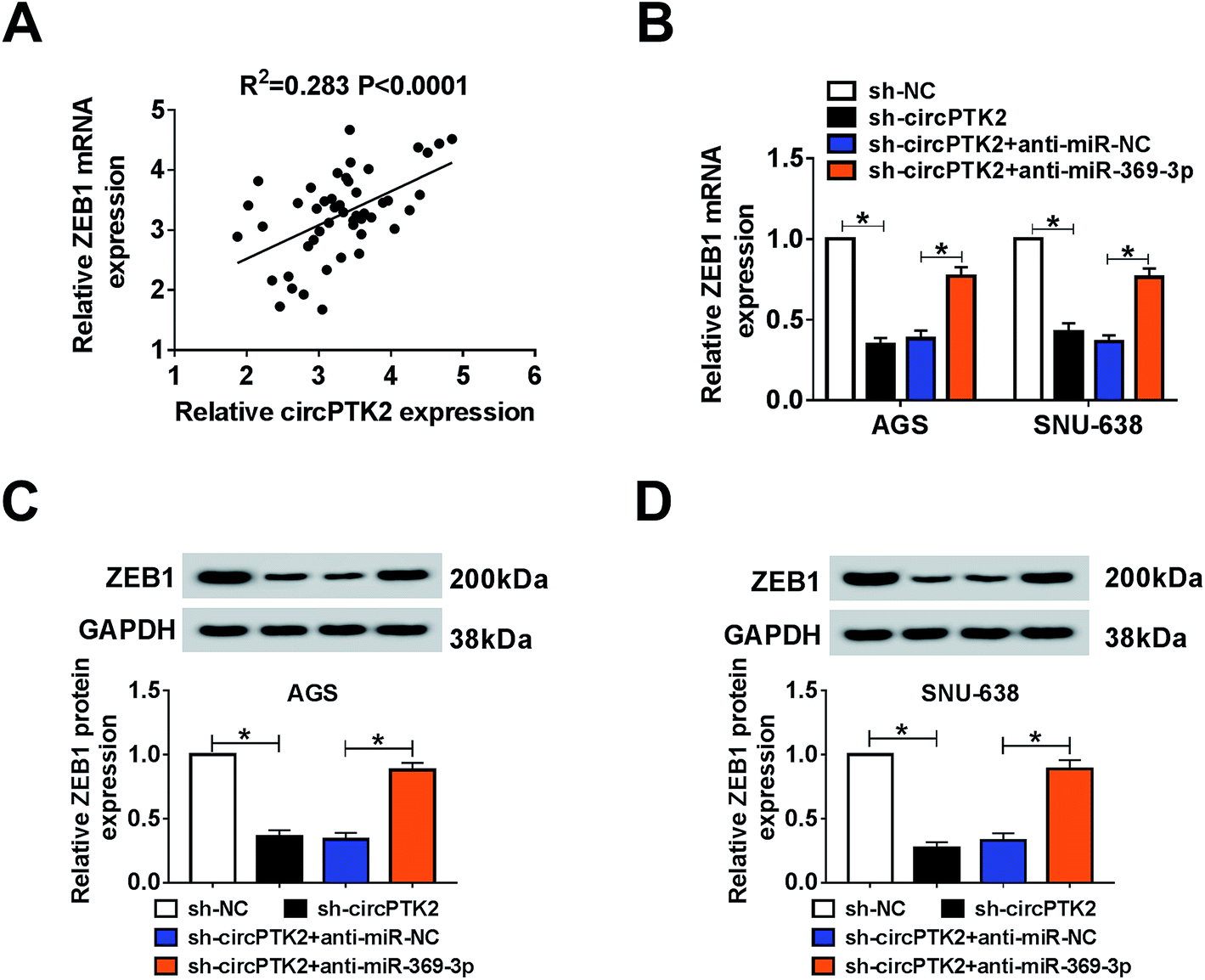
ZEB1 was co-regulated by circPTK2 and miR-369-3p
As described previously, we systemically uncovered the molecular mechanism between ZEB1 and circPTK2 or miR-369-3p. Firstly, a positive association between circPTK2 and ZEB1 was determined in GC specimens (Fig. 8A). Subsequently, sh-circPTK2 alone or combined with anti-miR-369-3p was introduced into AGS and SNU-638 cells, and sh-circPTK2-mediated decrease of the ZEB1 level was partially regained via miR-369-3p inhibition (Fig. 8B). Also, a similar alteration of the protein level of ZEB1 was observed in the two GC cell lines (Fig. 8C and D). In sum, circPTK2 exerted its oncogenic role via miR-369-3p/ZEB1 axis in GC cells.
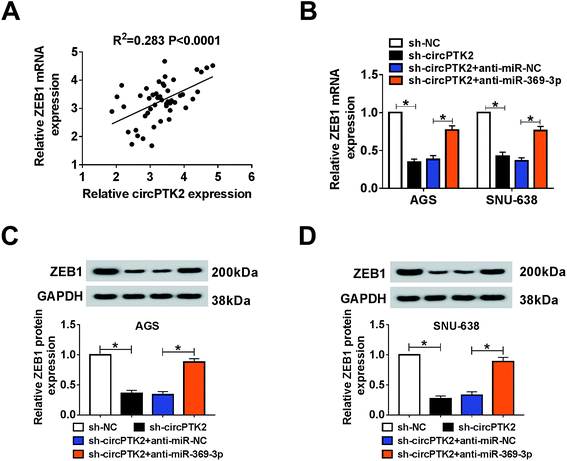 |
| | Fig. 8 ZEB1 was co-regulated by circPTK2 and miR-369-3p. (A) Spearman rank correlation for the relationship between circPTK2 and ZEB1 levels in GC tissues. (B–D) qRT-PCR and western blot analyses for the level of ZEB1 in terms of mRNA and protein expression in AGS and SNU-638 cells under circPTK2 silencing or miR-369-3p inhibition. *P < 0.05. | |
Discussion
Until now, circRNAs have been identified to be a group of ncRNAs with many potential functions, and the abundant and conserved RNA features have been expounded.22 Accruing reports demonstrate that circRNAs act with important functions in transcriptional regulation and tumorigenesis.23 Because of the structure of circRNAs, they are very stable in resisting the degradation by RNase R. On this basis, circRNAs might serve as a class of promising biomarkers for cancer treatments. Among them, hsa_circ_0001564, a sponge of mRNAs, functioned as an oncogene in osteosarcoma, showing as the repression of cell proliferation, along with promotions of cell cycle arrest and apoptosis under hsa_circ_0001564 deficiency.24 CircPTK2 could restrain EMT stimulated by transforming growth factor-β in non-small cell lung cancer.25 Despite the fact that the oncogenic function of circPTK2 has been investigated in several cancers, the underlying mechanism of it in GC needed to be highlighted. In our study, a high level of circPTK2 was observed in GC tissues and cell lines, and its knockdown apparently retarded cell growth and metastasis in vitro. Interestingly, irradiation exposure could strongly associate with circPTK2 depletion to modulate cell proliferation and apoptosis in the selected GC cells. The above evidence meant that circPTK2 also improved the radiosensitivity according to the alterations of cellular behaviors and survival fraction in vitro. In addition, the oncogenic role of circPTK2 was further clarified by assessing DNA repair-related protein (PARP). In general, we validated that circPTK2 had an oncogenic role in the progression of GC in vitro and in vivo. When circPTK2 was silenced, the tumor growth (in vivo) and aggressive cell behaviors (in vitro), was decreased significantly.
Mechanically, accumulating evidence has demonstrated that circRNAs take charge of ceRNAs of miRNAs to regulate downstream targets.26 Recent findings reveal that circRNAs possess at least a complementary sequence of miRNA.27 In this present research, the possible target miRNAs of circPTK2 were predicted by starBase software. As shown in Fig. S2,† several downregulated miRNAs in GC were chosen, and the depletion of circPTK2 could upregulate the levels of the miRNAs (miR-369-3p, miR-367-3p, miR-134-5p, miR-200b-3p, and miR-137), of which miR-369-3p was the most increased in AGS cells. Subsequent dual-luciferase reporter, RIP, and RNA pull-down assays were administrated to elucidate the interaction between miR-369-3p and circPTK2. In regard to miR-369-3p, it has attracted plenty of attention previously. For instance, a notable low expression of miR-369-3p was found, and the upregulation of miR-369-3p could efficiently impede cell proliferation and migration in glioblastoma cells.28 Consistently, miR-369-3p with a low level participated in the regulation of malignant cellular phenotypes in colorectal cancer.29 Currently, miR-369-3p was consistently downregulated in GC tissues and cells, and the level of miR-369-3p was negatively correlated with circPTK2 in GC tissues. However, the knowledge of miR-369-3p-mediated radiosensitivity in GC was limited. Based on this ectopic expression of miR-369-3p, we further found that the supplement of miR-369-3p could diminish aggressive behaviors and strengthen radiosensitivity in AGS and SNU-638 cells. Expectantly, miR-369-3p worked as a target of circPTK2 to mediate cellular phenotypes in GC cells.
Subsequently, the targets of miR-369-3p were predicted by starBase software, and some targets were found which acted with oncogenic function in the development of GC. Also, the supplement of miR-369-3p could decrease the expression of some genes (Fig. S3†). On this basis, ZEB1 was chosen as the research objective. Firstly, we proved the upregulation of ZEB1 in GC tissues and cells, which was in keeping with earlier findings.30 In addition, available evidence has manifested that ZEB1 is connected with aggressive phenotypes, such as proliferation, mobility, and EMT in the progression of GC.31 Besides, ZEB1, inhibited by miR-205, could affect radiosensitivity in breast cancer.32 In the current research, we first validated that ZEB1 was a direct target of miR-369-3p. Functional assays indicated that ZEB1 depletion especially suppressed aggressive behaviors, and facilitated radiosensitivity in GC cells. Simultaneously, ZEB1 could eliminate the role of miR-369-3p mimic in cellular regulation and radiosensitivity. Overall, ZEB1, a target of miR-369-3p, was deemed as an oncogenic factor in the GC process.
Conclusion
In summary, the present study found that circPTK2 and ZEB1 with high levels and miR-369-3p with low expression took part in pathogenesis and tumorigenesis in GC. Deficiency of circPTK2 reduced cell proliferation, migration, and invasion, and contributed to radiosensitivity in AGS and SNU-638 cells. Also, circPTK2 exerted its effect via ZEB1 by sponging miR-369-3p in GC cells. Namely, circPTK2 might be a novel proliferative regulator and prognostic mediator in GC (Fig. S1†).
Conflicts of interest
The authors declare that they have no financial conflicts of interest.
References
- A. Ferro, B. Peleteiro, M. Malvezzi, C. Bosetti, P. Bertuccio, F. Levi, E. Negri, C. La Vecchia and N. Lunet, Eur. J. Cancer, 2014, 50, 1330–1344 CrossRef PubMed.
- P. Karimi, F. Islami, S. Anandasabapathy, N. D. Freedman and F. Kamangar, Cancer Epidemiol., Biomarkers Prev., 2014, 23, 700–713 CrossRef PubMed.
- N. Nagarajan, D. Bertrand, A. M. Hillmer, Z. J. Zang, F. Yao, P. E. Jacques, A. S. Teo, I. Cutcutache, Z. Zhang, W. H. Lee, Y. Y. Sia, S. Gao, P. N. Ariyaratne, A. Ho, X. Y. Woo, L. Veeravali, C. K. Ong, N. Deng, K. V. Desai, C. C. Khor, M. L. Hibberd, A. Shahab, J. Rao, M. Wu, M. Teh, F. Zhu, S. Y. Chin, B. Pang, J. B. So, G. Bourque, R. Soong, W. K. Sung, B. Tean Teh, S. Rozen, X. Ruan, K. G. Yeoh, P. B. Tan and Y. Ruan, Genome Biol., 2012, 13, R115 CrossRef PubMed.
- E. Birney, J. A. Stamatoyannopoulos, A. Dutta, R. Guigo, T. R. Gingeras, E. H. Margulies, Z. Weng, M. Snyder, E. T. Dermitzakis, R. E. Thurman, M. S. Kuehn, C. M. Taylor, S. Neph, C. M. Koch, S. Asthana, A. Malhotra, I. Adzhubei, J. A. Greenbaum, R. M. Andrews, P. Flicek, P. J. Boyle, H. Cao, N. P. Carter, G. K. Clelland, S. Davis, N. Day, P. Dhami, S. C. Dillon, M. O. Dorschner, H. Fiegler, P. G. Giresi, J. Goldy, M. Hawrylycz, A. Haydock, R. Humbert, K. D. James, B. E. Johnson, E. M. Johnson, T. T. Frum, E. R. Rosenzweig, N. Karnani, K. Lee, G. C. Lefebvre, P. A. Navas, F. Neri, S. C. Parker, P. J. Sabo, R. Sandstrom, A. Shafer, D. Vetrie, M. Weaver, S. Wilcox, M. Yu, F. S. Collins, J. Dekker, J. D. Lieb, T. D. Tullius, G. E. Crawford, S. Sunyaev, W. S. Noble, I. Dunham, F. Denoeud, A. Reymond, P. Kapranov, J. Rozowsky, D. Zheng, R. Castelo, A. Frankish, J. Harrow, S. Ghosh, A. Sandelin, I. L. Hofacker, R. Baertsch, D. Keefe, S. Dike, J. Cheng, H. A. Hirsch, E. A. Sekinger, J. Lagarde, J. F. Abril, A. Shahab, C. Flamm, C. Fried, J. Hackermuller, J. Hertel, M. Lindemeyer, K. Missal, A. Tanzer, S. Washietl, J. Korbel, O. Emanuelsson, J. S. Pedersen, N. Holroyd, R. Taylor, D. Swarbreck, N. Matthews, M. C. Dickson, D. J. Thomas, M. T. Weirauch, J. Gilbert, J. Drenkow, I. Bell, X. Zhao, K. G. Srinivasan, W. K. Sung, H. S. Ooi, K. P. Chiu, S. Foissac, T. Alioto, M. Brent, L. Pachter, M. L. Tress, A. Valencia, S. W. Choo, C. Y. Choo, C. Ucla, C. Manzano, C. Wyss, E. Cheung, T. G. Clark, J. B. Brown, M. Ganesh, S. Patel, H. Tammana, J. Chrast, C. N. Henrichsen, C. Kai, J. Kawai, U. Nagalakshmi, J. Wu, Z. Lian, J. Lian, P. Newburger, X. Zhang, P. Bickel, J. S. Mattick, P. Carninci, Y. Hayashizaki, S. Weissman, T. Hubbard, R. M. Myers, J. Rogers, P. F. Stadler, T. M. Lowe, C. L. Wei, Y. Ruan, K. Struhl, M. Gerstein, S. E. Antonarakis, Y. Fu, E. D. Green, U. Karaoz, A. Siepel, J. Taylor, L. A. Liefer, K. A. Wetterstrand, P. J. Good, E. A. Feingold, M. S. Guyer, G. M. Cooper, G. Asimenos, C. N. Dewey, M. Hou, S. Nikolaev, J. I. Montoya-Burgos, A. Loytynoja, S. Whelan, F. Pardi, T. Massingham, H. Huang, N. R. Zhang, I. Holmes, J. C. Mullikin, A. Ureta-Vidal, B. Paten, M. Seringhaus, D. Church, K. Rosenbloom, W. J. Kent, E. A. Stone, S. Batzoglou, N. Goldman, R. C. Hardison, D. Haussler, W. Miller, A. Sidow, N. D. Trinklein, Z. D. Zhang, L. Barrera, R. Stuart, D. C. King, A. Ameur, S. Enroth, M. C. Bieda, J. Kim, A. A. Bhinge, N. Jiang, J. Liu, F. Yao, V. B. Vega, C. W. Lee, P. Ng, A. Shahab, A. Yang, Z. Moqtaderi, Z. Zhu, X. Xu, S. Squazzo, M. J. Oberley, D. Inman, M. A. Singer, T. A. Richmond, K. J. Munn, A. Rada-Iglesias, O. Wallerman, J. Komorowski, J. C. Fowler, P. Couttet, A. W. Bruce, O. M. Dovey, P. D. Ellis, C. F. Langford, D. A. Nix, G. Euskirchen, S. Hartman, A. E. Urban, P. Kraus, S. Van Calcar, N. Heintzman, T. H. Kim, K. Wang, C. Qu, G. Hon, R. Luna, C. K. Glass, M. G. Rosenfeld, S. F. Aldred, S. J. Cooper, A. Halees, J. M. Lin, H. P. Shulha, X. Zhang, M. Xu, J. N. Haidar, Y. Yu, Y. Ruan, V. R. Iyer, R. D. Green, C. Wadelius, P. J. Farnham, B. Ren, R. A. Harte, A. S. Hinrichs, H. Trumbower, H. Clawson, J. Hillman-Jackson, A. S. Zweig, K. Smith, A. Thakkapallayil, G. Barber, R. M. Kuhn, D. Karolchik, L. Armengol, C. P. Bird, P. I. de Bakker, A. D. Kern, N. Lopez-Bigas, J. D. Martin, B. E. Stranger, A. Woodroffe, E. Davydov, A. Dimas, E. Eyras, I. B. Hallgrimsdottir, J. Huppert, M. C. Zody, G. R. Abecasis, X. Estivill, G. G. Bouffard, X. Guan, N. F. Hansen, J. R. Idol, V. V. Maduro, B. Maskeri, J. C. McDowell, M. Park, P. J. Thomas, A. C. Young, R. W. Blakesley, D. M. Muzny, E. Sodergren, D. A. Wheeler, K. C. Worley, H. Jiang, G. M. Weinstock, R. A. Gibbs, T. Graves, R. Fulton, E. R. Mardis, R. K. Wilson, M. Clamp, J. Cuff, S. Gnerre, D. B. Jaffe, J. L. Chang, K. Lindblad-Toh, E. S. Lander, M. Koriabine, M. Nefedov, K. Osoegawa, Y. Yoshinaga, B. Zhu and P. J. de Jong, Nature, 2007, 447, 799–816 CrossRef CAS PubMed.
- T. Nagano and P. Fraser, Cell, 2011, 145, 178–181 CrossRef CAS PubMed.
- Q. Zheng, C. Bao, W. Guo, S. Li, J. Chen, B. Chen, Y. Luo, D. Lyu, Y. Li, G. Shi, L. Liang, J. Gu, X. He and S. Huang, Nat. Commun., 2016, 7, 11215 CrossRef CAS PubMed.
- J. Liu, T. Liu, X. Wang and A. He, Mol. Cancer, 2017, 16, 58 CrossRef CAS PubMed.
- R. Zhou, Y. Wu, W. Wang, W. Su, Y. Liu, Y. Wang, C. Fan, X. Li, G. Li, Y. Li, W. Xiong and Z. Zeng, Cancer Lett., 2018, 425, 134–142 CrossRef CAS PubMed.
- L. D. Hou and J. Zhang, Int. J. Immunopathol. Pharmacol., 2017, 30, 1–6 CrossRef CAS PubMed.
- H. Su, F. Lin, X. Deng, L. Shen, Y. Fang, Z. Fei, L. Zhao, X. Zhang, H. Pan, D. Xie, X. Jin and C. Xie, J. Transl. Med., 2016, 14, 225 CrossRef PubMed.
- M. Shuai, J. Hong, D. Huang, X. Zhang and Y. Tian, Oncol. Lett., 2018, 16, 6495–6501 CAS.
- Z. Q. Xu, M. G. Yang, H. J. Liu and C. Q. Su, J. Cell. Biochem., 2018, 119, 3317–3325 CrossRef CAS PubMed.
- R. Rupaimoole and F. J. Slack, Nat. Rev. Drug Discovery, 2017, 16, 203–222 CrossRef CAS PubMed.
- H. Su, X. Jin, X. Zhang, S. Xue, X. Deng, L. Shen, Y. Fang and C. Xie, Cell Biol. Int., 2014, 38, 318–325 CrossRef CAS PubMed.
- X. Li, Y. Zhang, H. Zhang, X. Liu, T. Gong, M. Li, L. Sun, G. Ji, Y. Shi, Z. Han, S. Han, Y. Nie, X. Chen, Q. Zhao, J. Ding, K. Wu and F. Daiming, Mol. Cancer Res., 2011, 9, 824–833 CrossRef CAS PubMed.
- J. Lin, C. Liu, F. Gao, R. E. Mitchel, L. Zhao, Y. Yang, J. Lei and J. Cai, J. Cell. Biochem., 2013, 114, 606–615 CrossRef CAS PubMed.
- L. Dong, Z. Zhang, J. Xu, F. Wang, Y. Ma, F. Li, C. Shen, Z. Liu, J. Zhang, C. Liu, P. Yi and J. Yu, Mol. Oncol., 2019, 13, 1605–1620 CAS.
- J. Caramel, M. Ligier and A. Puisieux, Cancer Res., 2018, 78, 30–35 CrossRef CAS PubMed.
- P. Zhang, Y. Sun and L. Ma, Cell Cycle, 2015, 14, 481–487 CrossRef CAS PubMed.
- M. Danan, S. Schwartz, S. Edelheit and R. Sorek, Nucleic Acids Res., 2012, 40, 3131–3142 CrossRef CAS PubMed.
- J. Zheng, X. Liu, P. Wang, Y. Xue, J. Ma, C. Qu and Y. Liu, Mol. Ther., 2016, 24, 1199–1215 CrossRef CAS PubMed.
- N. R. Pamudurti, O. Bartok, M. Jens, R. Ashwal-Fluss, C. Stottmeister, L. Ruhe, M. Hanan, E. Wyler, D. Perez-Hernandez, E. Ramberger, S. Shenzis, M. Samson, G. Dittmar, M. Landthaler, M. Chekulaeva, N. Rajewsky and S. Kadener, Mol. Cell, 2017, 66, 9–21 CrossRef CAS PubMed.
- S. Haque and L. W. Harries, Genes, 2017, 8, 353 CrossRef PubMed.
- Y. Z. Song and J. F. Li, Biochem. Biophys. Res. Commun., 2018, 495, 2369–2375 CrossRef CAS PubMed.
- L. Wang, X. Tong, Z. Zhou, S. Wang, Z. Lei, T. Zhang, Z. Liu, Y. Zeng, C. Li, J. Zhao, Z. Su, C. Zhang, X. Liu, G. Xu and H. T. Zhang, Mol. Cancer Res., 2018, 17, 140 CrossRef PubMed.
- G. Militello, T. Weirick, D. John, C. Doring, S. Dimmeler and S. Uchida, Briefings Bioinf., 2017, 18, 780–788 CAS.
- L. F. Thomas and P. Saetrom, Bioinformatics, 2014, 30, 2243–2246 CrossRef CAS PubMed.
- T. Shahar, A. Granit, D. Zrihan, T. Canello, H. Charbit, O. Einstein, U. Rozovski, S. Elgavish, Z. Ram, T. Siegal and I. Lavon, J. Neuro-Oncol., 2016, 130, 413–422 CrossRef CAS PubMed.
- J. Wang, H. Wang, A. Liu, C. Fang, J. Hao and Z. Wang, Oncotarget, 2015, 6, 19456–19468 Search PubMed.
- X. Zhou, Y. Wang, B. Shan, J. Han, H. Zhu, Y. Lv, X. Fan, M. Sang, X. D. Liu and W. Liu, Med. Oncol., 2015, 32, 428 CrossRef PubMed.
- C. Xu, M. Li, L. Zhang, Y. Bi, P. Wang, J. Li and X. Jiang, Mol. Med. Rep., 2016, 13, 4767–4773 CrossRef CAS PubMed.
- P. Zhang, L. Wang, C. Rodriguez-Aguayo, Y. Yuan, B. G. Debeb, D. Chen, Y. Sun, M. J. You, Y. Liu, D. C. Dean, W. A. Woodward, H. Liang, X. Yang, G. Lopez-Berestein, A. K. Sood, Y. Hu, K. K. Ang, J. Chen and L. Ma, Nat. Commun., 2014, 5, 5671 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08525d |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Ke Zhanga,
Jianping Wanga,
Qiang Daic and
Rong Wangd
*b,
Ke Zhanga,
Jianping Wanga,
Qiang Daic and
Rong Wangd