
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: LncRNA SNHG5 regulates the cell viability and apoptosis of glioma cells by the miR-1297/KPNA2 axis
Xueyuan Li†
a,
Qiankun Liu†a,
Kang Wanga,
Wenzheng Luoa,
Tiansong Liangb,
Shanpeng Yuana,
Yingwei Zhena and
Dongming Yan *a
*a
aDepartment of Neurosurgery, The First Affiliated Hospital of Zhengzhou University, No. 1, Longhu Middle Ring Road, Jinshui District, Zhengzhou City 450000, Henan Province, China. E-mail: mrdmyan@163.com; Tel: +86-371-66278032
bDepartment of Radiotherapy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
First published on 8th January 2020
Abstract
Long non-coding RNA small nucleolar RNA host gene 5 (lncRNA SNHG5) has been reported to participate in the occurrence and development of glioma. However, the function and underlying molecular mechanisms of SNHG5 in glioma remain largely unknown. The expressions of SNHG5, microRNA-1297 (miR-1297) and karyopherin subunit alpha 2 (KPNA2) in glioma tissues and cells were evaluated by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) or western blot. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay and flow cytometry were used to detect cell viability and apoptosis, respectively. Western blot was also performed to detect the expressions of autophagy-associated proteins. The relationship among lncRNA SNHG5, miR-1297 and KPNA2 was verified by luciferase reporter assay and RNA immunoprecipitation (RIP) assay. SNHG5 and KPNA2 were over expressed, and the level of miR-1297 was down-regulated in glioma tissues and cell lines. Knockdown of SHNG5 promoted apoptosis, while suppressing cell viability and autophagy of A172 and LN340 cells. Meanwhile, SHNG5 harbored the binding sites with miR-1297, and a negative correlation between the expression of SNHG5 and miR-1297 in glioma tissues was also observed. Interestingly, silencing of miR-1297 undermined the SHNG5 depletion-mediated effect on cell viability, apoptosis, and autophagy. KPNA2 was a direct target of miR-1297, and negatively regulated by miR-1297. More importantly, gain of KPNA2 mitigated the effect of SHNG5l knockdown on glioma cells. Silencing of SNHG5 had an implication in inhibiting apoptosis and stimulating cell viability and autophagy by the miR-1297/KPNA2 axis in glioma.
Introduction
Glioma is the most common primary brain tumors with a 5 year overall survival rate less than 35%.1 Surgical resection combined with chemoradiotherapy is thought to be the best therapeutic method for these patients.2 But the survival rate of glioma patients has not significantly improved.3 Therefore, it is very necessary to clarify the underlying mechanisms of glioma and seek novel targets for early diagnosis and therapy of glioma.Long non-coding RNAs (lncRNAs) are primarily 200 nucleotides (nts) to a 100 kb polyadenylation transcript which are normally transcribed by RNA polymerase II and controlled by various types of transcription factors.4 It has been reported that lncRNAs participate in many biological functions, including chromosome remodeling, intracellular, and trafficking transcription, mainly involved in cell processes.5 Jin et al. found that down-regulated of lncRNA cancer susceptibility candidate 11 inhibits the proliferation, migration and metastatic growth of glioma cells.6 In another report, Li et al. revealed that lncRNA growth arrest-specific transcript 5 effectively suppresses the proliferation, migration and invasion of glioma cells and promoted cell apoptosis by regulating GSTM3 expression.7 Those data suggested the involvement of lncRNAs in the development of cancers. Small nucleolar RNA Host Gene 5 (SNHG5) is a newly discovered lncRNA. At present, a lot of researchers have clarified the relation between SNHG5 and malignant tumors. Of note, Zhao et al. demonstrated that SNHG5 is dysregulated in gastric cancer (GC) and first discovered that SNHG5/miR-32/KLF4 axis is potentially to the benefit of GC diagnosis and therapy.8 Recently, with the development of research, lncRNA SNHG5 has been reported to affect glioma, as well. Li et al. illustrated that SNHG5 regulates the tumorigenesis of glioma in vivo, and the fundamental mechanism in this process may be the SNHG5/miR-205/E2F3 axis.9 Nevertheless, the function and mechanisms of lncRNA SNHG5 in glioma is still relatively little known.
MicroRNAs (miRNAs) are known as non-coding RNAs with 22–24 nts in length. Through inducing mRNA decay and translation repression, miRNAs are able to regulate complementary mRNAs.10 Many miRNAs are dysregulated in glioma and affects the biological process of tumor cells, including cell cycle, apoptosis, proliferation and migration, such as miR-182, miR-132 and miR-218-5p.11–13 MiR-1297 is considered to be a tumor suppressor in kinds of human cancers, including prostate cancer,14 colorectal cancer,15 pancreatic cancer,16 and gastric cancer.17 Nevertheless, its role in glioma remains elusive. In glioblastomas, low expression of karyopherin subunit alpha 2 (KPNA2) has been found to significantly inhibit glioma cell growth and invasion.18 However, whether KPNA2 participates in the SNHG5-mediated function has not been reported.
In this study, we found that SNHG5 was significantly up-regulated in A172 and LN340 cells than other glioma cell lines (U118MG, LN464, SNB19, LN18, T98G, and U251MG cell lines). Therefore, we selected A172 and LN340 cells acted as our objects. Then, we aimed to investigate whether miR-1297 and KPNA2 are involved in SNHG5-mediated function, including viability, apoptosis, and autophagy, in glioma cell lines A172 and LN340.
Materials and methods
Clinical samples and cell lines
39 pairs of clinical samples consisting of cancerous tissues and adjacent normal tissues were received from patients with glioma in The First Affiliated Hospital of Zhengzhou University. These patients were confirmed by MRI imaging and pathology. They did not suffer from any other type of cancer, infectious diseases, autoimmune diseases, etc. All experiments were performed in accordance with the Declaration of Helsink. Experiments were approved by the ethics committee at The First Affiliated Hospital of Zhengzhou University. Informed consents were obtained from human participants of this study. The glioma specimens were labelled and put into liquid nitrogen quickly, and then stored at −80 °C.Normal human astrocytes (NHAs) were purchased from JENNIO Biological Technology (Guangzhou, China). The glioma A172 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), and glioma LN340 cells were purchased from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in Dulbecco's modified eagle's medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA). All cells were maintained in a humidified incubator at 37 °C with 5% CO2.
Cell transfection
Lentiviral small hairpin RNA (shRNA) targetingSNHG5 (sh-SNHG5) and its negative control sh-NC, SHNG5 overexpression plasmid (SNHG5), KPNA2 overexpression plasmid (KPNA2) and the negative controlpcDNA 3.0 empty vector (vector), miR-1297 inhibitor (anti-miR-1297) and its negative control (anti-miR-NC), miR-1297 mimic (miR-1297) and its negative control (miR-NC) were constructed by Ribo Bio Corporation (Guangzhou, China). These vectors were transfected into glioma cells by Liposome 3000 (Thermo Fisher Scientific) according to experimental needs. The transfection efficiency was tested by quantitative reverse transcription-polymerase chain reaction (qRT-PCR).qRT-PCR
Total RNA of glioma tissues or cultured cells was isolated with Trizol reagent (Invitrogen, Carlsbad, CA, USA). Then the total RNA was reverse transcribed into cDNA using the M-MLV reverse transcription kit (Takara, Tokyo, Japan). QPCR was conducted using ABI 7300 Fast Real-time PCR System (Biosystems, Foster city, CA, USA) with a SYBR Premix Ex Taq kit (TaKaRa). The primer sequences were as follows:SNHG5, 5′-CGAGTAGCCAGTGAAGATAATG-3′ (forward) and 5′-CACACAACA GTCAAGTAAACC-3′ (reverse); miR-1297, 5′-ACACTCCAGCTGGGTCCTTCATTCCA-3’ (forward) and 5′-GTGCAGGGTCCGAGGT-3′ (reverse); U6, 5′-CTCGCTTCGGCAGCACA-3’ (forward) and 5′-AACGCTTCACGAATTTGCGT-3’ (reverse); KPNA2, 5′-ATTGCAGGTGATGGCTCAGT-3′ (forward) and 5′-CTGCTCAACAGCATCTATCG-3′ (reverse); glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-GCACCGTCAAGGCTGAGAAC-3’ (forward) and 5′-TGGTGAAGACGCCAGTGGA-3’ (reverse). GAPDH and U6 were employed as internal controls.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay
All cell were seeded in a 96-well plate for incubation of 24 h, 48 h, and 72 h. Then MTT was added to each well with incubation of 4 h. After the supernatant was removed, DMSO was added and the light absorption (490 nm) value was measured.Flow cytometry
The cells (5 × 105) were dissociated with 0.25% trypsin without ethylene diamine tetraacetic acid, then washed with PBS, and resuspended in 500 μL of binding buffer. Next, 5 μL of Annexin V-FITC/PI and PI were added to the binding buffer. After incubation for 15 min at room temperature in the darkness, flow cytometry analysis was performed on the BD FACS Calibur (BD Biosciences, Franklin Lakes, NJ, USA).Caspase-3 activity assay
The activity of caspase-3 was evaluated by the caspase activity kit referring to the user manual. Briefly, at 24 h post incubation, cells were collected and treated with ice-cold radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China). Then, cell suspension was centrifuged and washed. The concentration of extracted protein was determined by bicinchoninic acid kit. Finally, the activity of caspase-3 was measured by microplate reader.Western blot
Proteins were extracted from cultured cells by lysis buffer containing 1% Triton X-100, 10 mmol L−1 Tris–HCl (pH 7.4) and proteases/phosphates inhibitors (Roche Diagnostics, Indianapolis, IN, USA). The protein concentration was calculated by the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Then the proteins were electrophoresed in SDS-PAGE gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (Life Technologies, Gaithersburg, MD, USA). After blockage with 5% skim milk, the membrane was probed with primary antibodies. Next, the membrane was probed with horseradish peroxidase conjugated secondary antibodies. Finally, the visualization was enhanced by a chemiluminescence detection system (Pierce, Rockford, IL, USA). The primary antibodies were: anti-caspase-3 (ab197202), anti-LC3 (ab225383), anti-Beclin-1 (ab207612), anti-p62 (ab109012), anti-KPNA2 (ab84440), and anti-GAPDH (ab181602) (Abcam, Cambridge, MA, USA).Luciferase reporter assay
A wild-type (WT) fragment of SNHG5 or KPNA2 harboring the miR-1297 binding site and their mutated (MUT) seed sequences were assembled into pMIR-REPORT luciferase reporter vectors. Then A172 and LN340 cells were co-transfected with miR-1297 mimic or miR-NC and WT-SNHG5, MUT-SNHG5, WT-KPNA2, MUT-KPNA2. Subsequently, the luciferase activity was evaluated by a Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA).RNA immunoprecipitation (RIP) assay
RIP assay was performed using the Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA) following the manufacturer's protocols. Cells were collected and lysed in complete RIP lysis buffer, and then 100 μL of cell extracts were incubated with RIP buffer containing magnetic beads conjugated with human anti-Ago2 antibody (Abcam), normal mouse IgG (Abcam) was used as negative control. Samples were treated with Proteinase K and then immunoprecipitated RNAs were isolated. The co-precipitated RNAs were subjected to qRT-PCR analysis.Statistical analysis
In this study, we have repeated three times for each experiment. The form of mean ± standard deviation (SD) was used to convey the data. All statistical analysis was assessed by Student's t test or One-Way ANOVA. P < 0.05 indicated the statistical significance. The data analysis software was GraphPad Prism 6.0 (GraphPad Software, San Diego, CA).Results
Loss of SNHG5 inhibits cell viability and autophagy and promotes cell apoptosis of glioma cells
Firstly, to verify the expression of SNHG5 in glioma tissues, we performed qRT-PCR analysis in 39 pairs of cancerous tissues and adjacent normal tissues. The results showed that the relative expression of SNHG5 in tumor tissues was higher than that in normal tissues (Fig. 1A). Meanwhile, lncRNA SNHG5 was also over expressed in A172 and LN340 cells compared that of NHAs (Fig. 1B). Then, we performed loss-of function experiment to determine the function of SNHG5 in glioma cells. The transfection efficiency demonstrated that SNHG5 was decreased in sh-SNHG5-transfected cells, indicating that the results in the following experiments were significant (Fig. 2A). Subsequently, siRNA-mediated knockdown of SNHG5 impaired cell viability (Fig. 2B and C). Flow cytometry assay further displayed that the apoptotic rate was increased after transfection with sh-SNHG5 (Fig. 2D). As shown in Fig. 2E, the activity of apoptotic protein caspase-3 was also significantly increased. Further analysis of autophagy protein expression indicated that knockdown of SNHG5 decreased the ratio of the LC3-I to LC3-II proteins and the expression of Beclin-1, while increased the expression of p62 (Fig. 2F and G). Those data implicated that SNHG5 may play oncogene role in the development of glioma.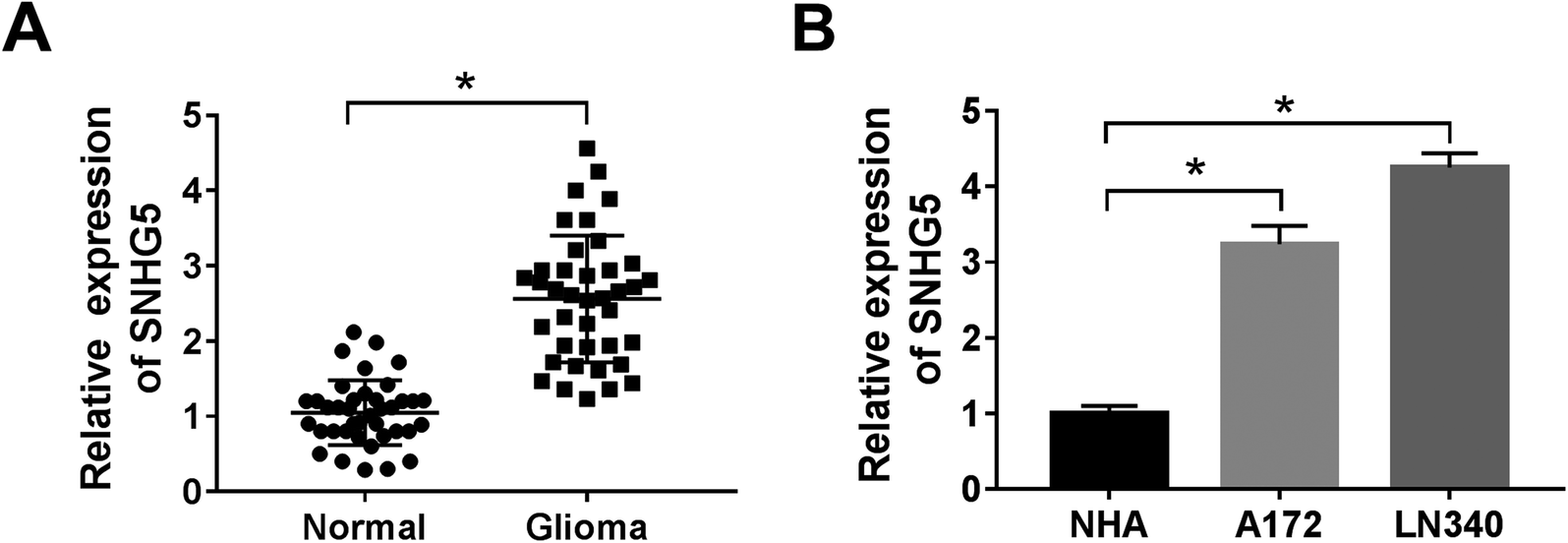 | ||
| Fig. 1 The expression of lncRNA SNHG5 in glioma tissues and cells. (A) QRT-PCR was conducted to monitor the expression of lncRNA SNHG5 in glioma tissues, normal tissues. (B) The expression level of SNHG5 in glioma cell lines (A172 and LN340) and normal human astrocytes (NHAs). *P < 0.05. | ||
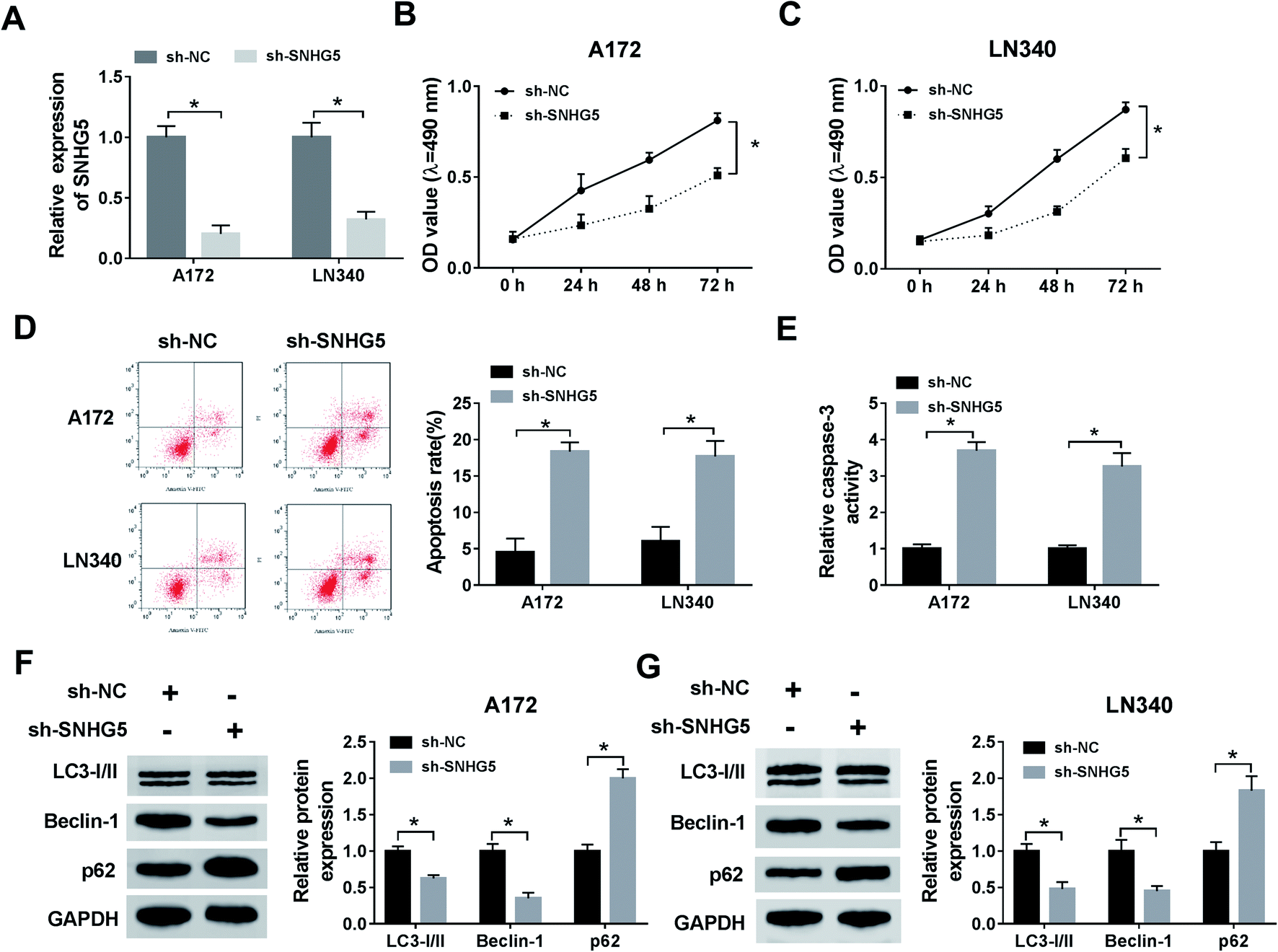 | ||
| Fig. 2 The effect of SNHG5 knockdown on proliferation, apoptosis, and autophagy in glioma cells. (A) QRT-PCR was used to examine the level of SNHG5 in A172 or LN340 cells transfected with sh-SNHG5 and sh-NC. (B and C) MTT assay was conducted to test cell proliferation. (D and E) Flow cytometry assay was used to determine cell apoptosis. (F and G) The expression of autophagy associated proteins including LC3I/II, Beclin-1 and p62 in cells transfected with sh-SNHG5 or sh-NC was determined. *P < 0.05. | ||
miR-1297 is sponged by SNHG5
In order to explore the underlying mechanism of lncRNA SNHG5 in glioma cells, we used starBase to predict the possible target miRNAs. As delineated in Fig. 3A, miR-1297 may be a target of SNHG5, and the potential binding sites were showed. Then, the luciferase reporter plasmids of WT and MUT of SNHG5 were constructed. Results showed that luciferase activity was significantly decreased in miR-1297 overexpression-mediated cells of WT-SNHG5 group. However, there was no obvious change in MUT-SNHG5 group (Fig. 3B and C). The RIP assay showed that a significant amount of miR-1297 or SNHG5 was detected in anti-Ago2 group, further clarifying the direct interaction between SNHG5 and miR-1297 (Fig. 3D and E). Next, we examined the level of miR-1297 in glioma tissues and cell lines. As demonstrated in Fig. 3F and G, miR-1297 was remarkably lower in glioma tissues than that of normal tissues. Meanwhile, a down-regulation of miR-1297 was also detected in glioma cells. Furthermore, we investigated the correlation between the expression of LncRNA SNHG5 and miR-1297 in glioma tissues, a strong negative correlation was observed (Fig. 3H). It also indicated that the overexpression of SHNG5 resulted in the down-regulation of miR-1297 and low-expression of SNHG5 led to the up-regulation of miR-1297 (Fig. 3I).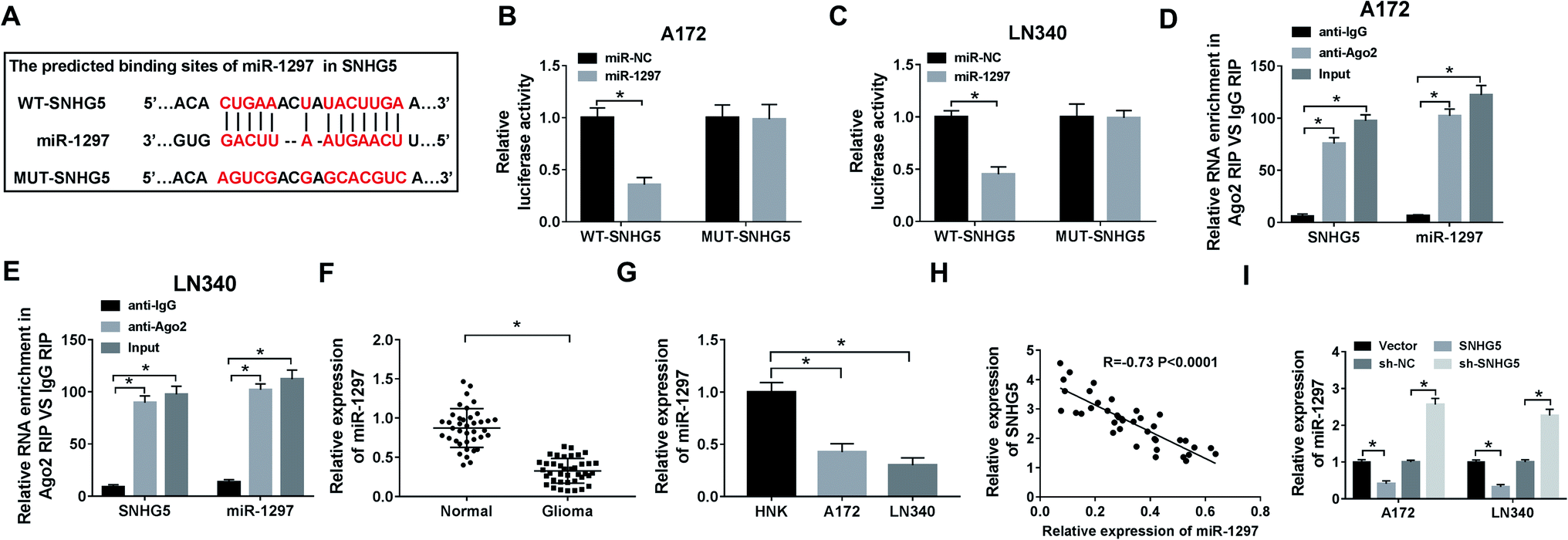 | ||
| Fig. 3 LncRNA SNHG5 is interacted with miR-1297. (A) The binding sites between SNHG5 and miR-1297 were predicted by starBase. (B and C) The luciferase activity was measured in A172 or LN340 cells co-transfected with WT-SNHG5 or MUT-SNHG5 luciferase reporter and miR-1297 mimic or miR-NC. (D and E) The RIP assay was performed and expression of SNHG5 ormiR-1297 was detected in the samples bound to the Ago2 antibody or IgG. (F and G) The level of miR-1297 in glioma tissues and cell lines was detected by qRT-PCR. (H) The correlation between the expression of SNHG5 and miR-1297 in glioma tissues was determined. (I) The level of miR-1297 in cells introduced with vector, SHNG5, sh-SNHG5 and sh-NC was tested. *P < 0.05. | ||
SNHG5 promotes cell viability and autophagy and suppresses apoptosis in glioma cells by regulating miR-1297
We further evaluated whether miR-1297 is associated with SNHG5-mediated function in glioma cells. The results showed that down-regulation of SNHG5 elevated the level of miR-1297, after co-transfection of anti-miR-1297, the miR-1297 returned to near original level (Fig. 4A). Meanwhile, the MTT assay and flow cytometry analysis indicated that knockdown of SNHG5 inhibited viability and promoted apoptosis and this effect was relieved after the anti-miR-1297 transfection (Fig. 4B, C and D). Also, silence of SNHG5 promoted the activity of caspase-3, which was abated by low expression of miR-1297 (Fig. 4E). Moreover, decreased expression of miR-1297 abolished the SNHG5 depletion on autophagy (Fig. 4F and G).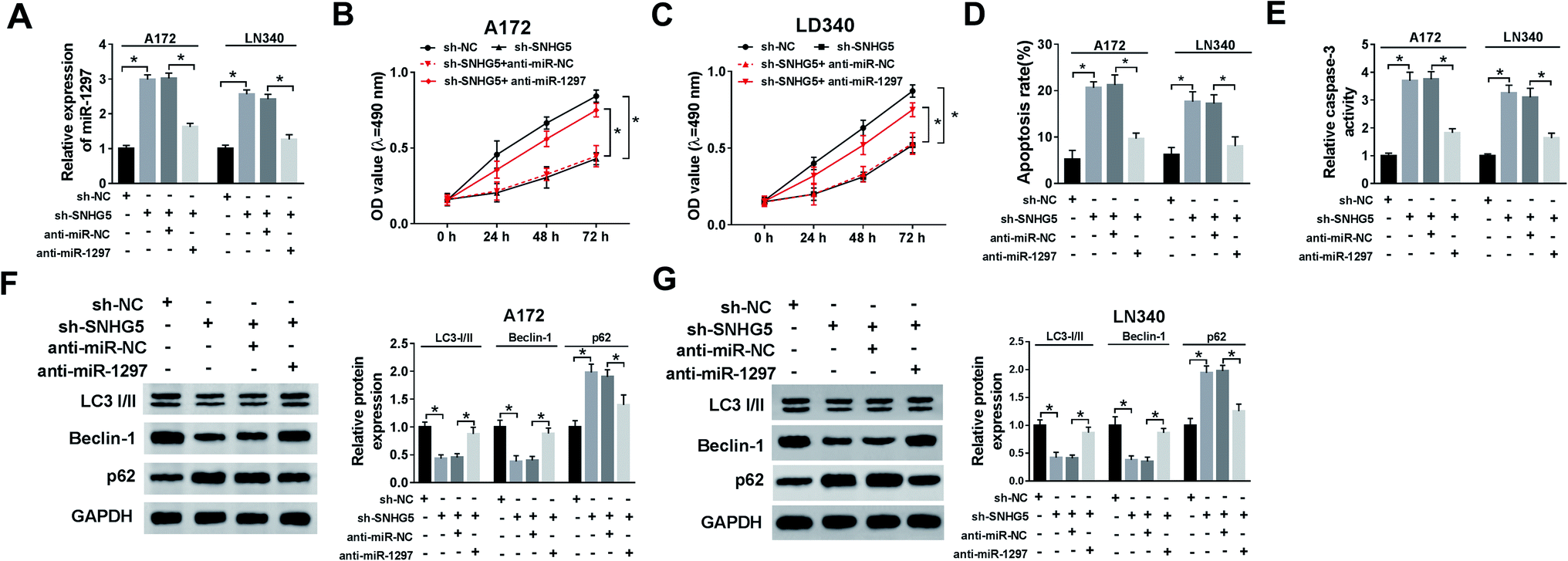 | ||
| Fig. 4 The effect of miR-1297 down-regulation on loss of SNHG5 on glioma cells. (A) The level of miR-1297 in cells transfected with sh-SNHG5, anti-miR-1297 or their negative controls. (B and C) Cell proliferation was determined in cells after transfection. (D and E) Cell apoptosis was examined in cells of each group. (F and G) The protein levels of LC3I/II, Beclin-1 and p62 in cells transfected was detected. *P < 0.05. | ||
KPNA2 is a target of miR-1297
To explore the targeted genes of miR-1297, the analysis of starBase was performed. According to the result, several target genes of miR-1297 were predicted. Among these genes, KPNA2 as a potential target associated with glioma was selected and the predicted binding sites were displayed in Fig. 5A. According to luciferase reporter assay, miR-1297 significantly decreased the luciferase activity of miR-1297 up-regulation cells of WT-KPNA2 group (Fig. 5B and C). The results indicated that miR-1297 directly bound to KPNA2. The relative higher level of KPNA2 was detected in glioma tissues (Fig. 5D). The correlation analysis demonstrated that KPNA2 expression had a negative correlation with miR-1297 expression (Fig. 5E). Likewise, the mRNA and protein expression of KPNA2 in cancer cells were significantly higher than that in normal cells (Fig. 5F and G). Moreover, the introduction of miR-1297 inhibitor led to the increase of KPNA2 at mRNA level, while overexpression of miR-1297 inhibited the expression of KPNA2 (Fig. 5H and I). Similarly, the protein level of KPNA2 could be up-regulated by miR-1297 inhibitor and down-regulated by miR-1297 mimic in glioma cells (Fig. 5J and K), clarifying the negative regulatory effect of miR-1297 on KPNA2.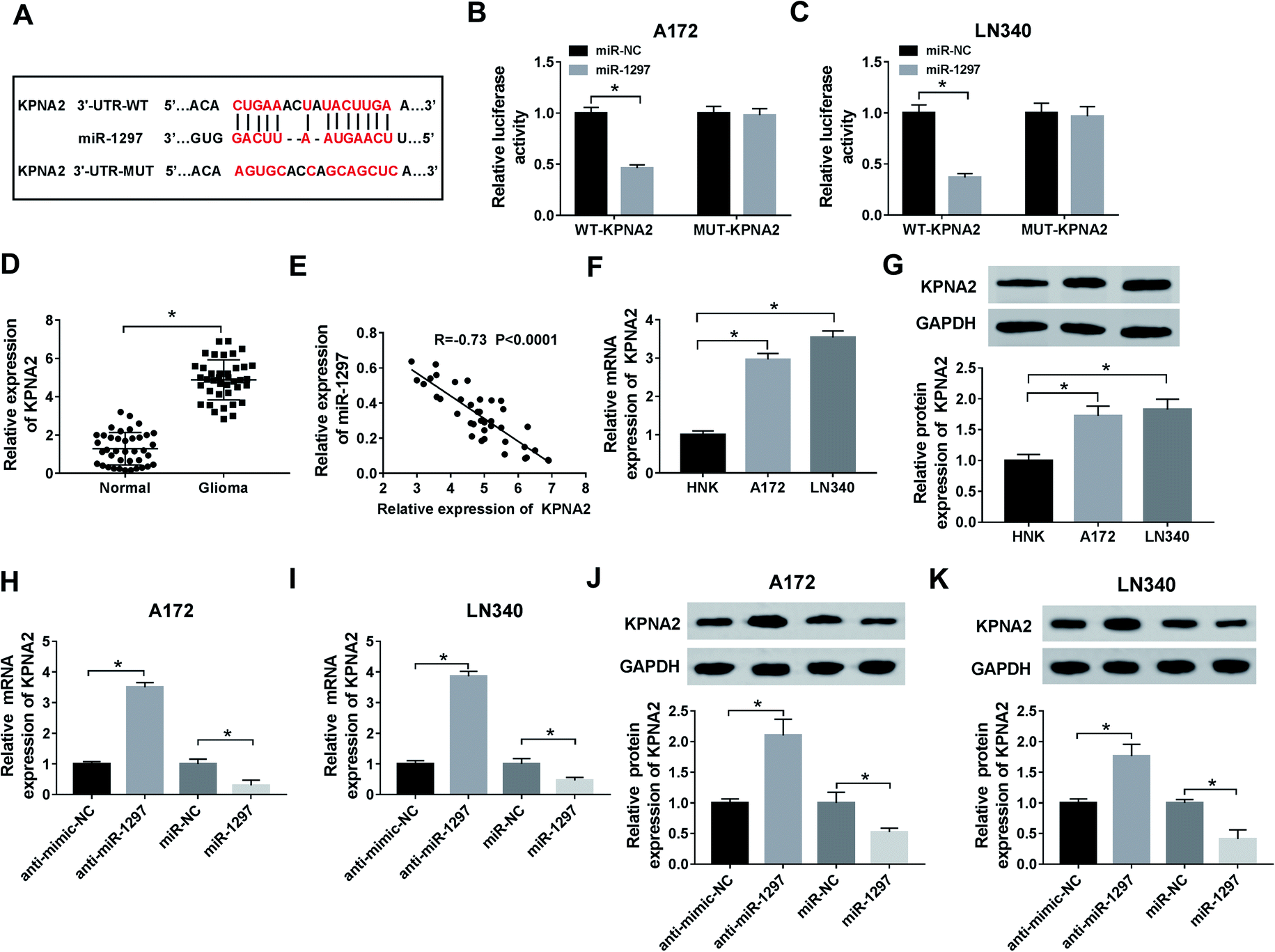 | ||
| Fig. 5 KPNA2 is a direct target of miR-1297. (A) The predicted binding sites between miR-1297 and KPNA2 were shown. (B and C) Luciferase reporter assay was conducted to verify the targeted binding between miR-1297 and KPNA2. (D) The enrichment of KPNA2 in glioma and normal tissues was measured. (E) The correlation between the expression of miR-1297 and KPNA2 was analyzed. (F and G) The mRNA and protein expression of KPNA2 in NHA, A172 and LN340 cell lines was detected. (H–K) QRT-PCR and western blot analyzed the expression of KPNA2 in glioma cell lines transfected with miR-1297 mimic, inhibitor or corresponding negative control. *P < 0.05. | ||
SNHG5 governs the expression of KPNA2 by targeting miR-1297 to regulate cell viability, apoptosis, and autophagy
Finally, we further confirmed the whether miR-1297/KPNA2 axis is related the SNHG5-induced function in glioma cells. The transfection of miR-1297 mimic resulted in down-regulated KPNA2, while the simultaneous up-regulation of miR-1297 and SNHG5 returned KPNA2 expression to original level at both mRNA and protein levels (Fig. 6A–D). This suggested that SNHG5 regulated KPNA2 expression by targeting miR-1297. It was discovered that both the mRNA and protein levels of KPNA2 were reduced after SNHG5 was impaired, which then were rescued by the attenuation of KPNA2 (Fig. 7A, B and C). Down-regulation of SNHG5 suppressed cell viability and promoted apoptosis, and simultaneously up-regulation of KPNA5 undermined those changes (Fig. 7D–G). In addition, the changes in the expression of autophagy associated proteins induced by sh-SNHG5 transfection were also rescued by the co-transfection of sh-SNHG5 and KPNA2 (Fig. 7H and I). These findings indicated that KPNA2 might be required for SNHG5 in the regulation of cell viability, apoptosis and autophagy in glioma.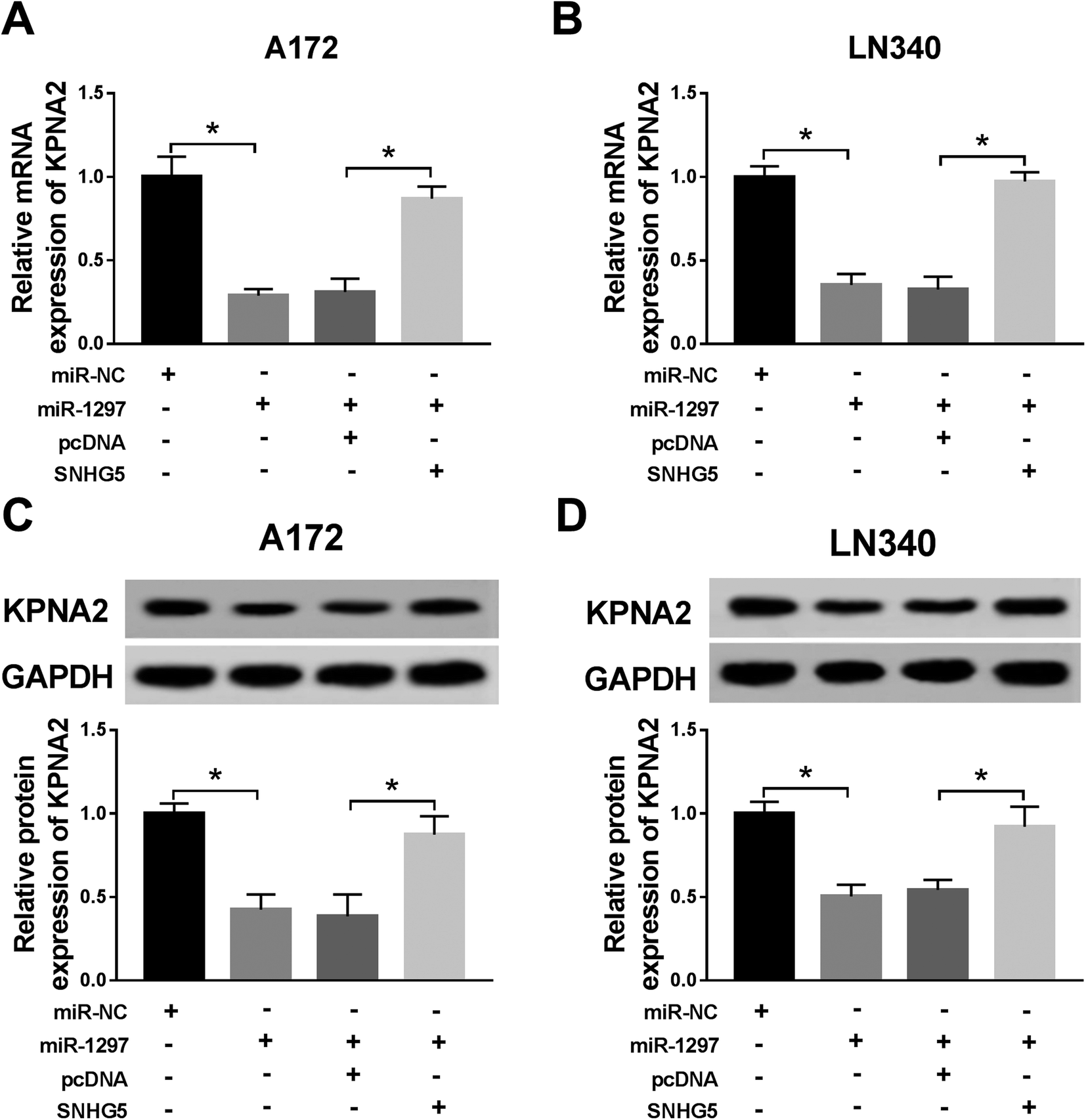 | ||
| Fig. 6 SNHG5 regulates KPNA2 via targeting miR-1297. (A and B) The mRNA level of KPNA2 in glioma cell lines transfected with miR-1297 mimic, SNHG5 overexpression plasmid or combination was analyzed. (C and D) Western blot analysis of KPNA2 in A172 and LN340 cells in different treatment group. *P < 0.05. | ||
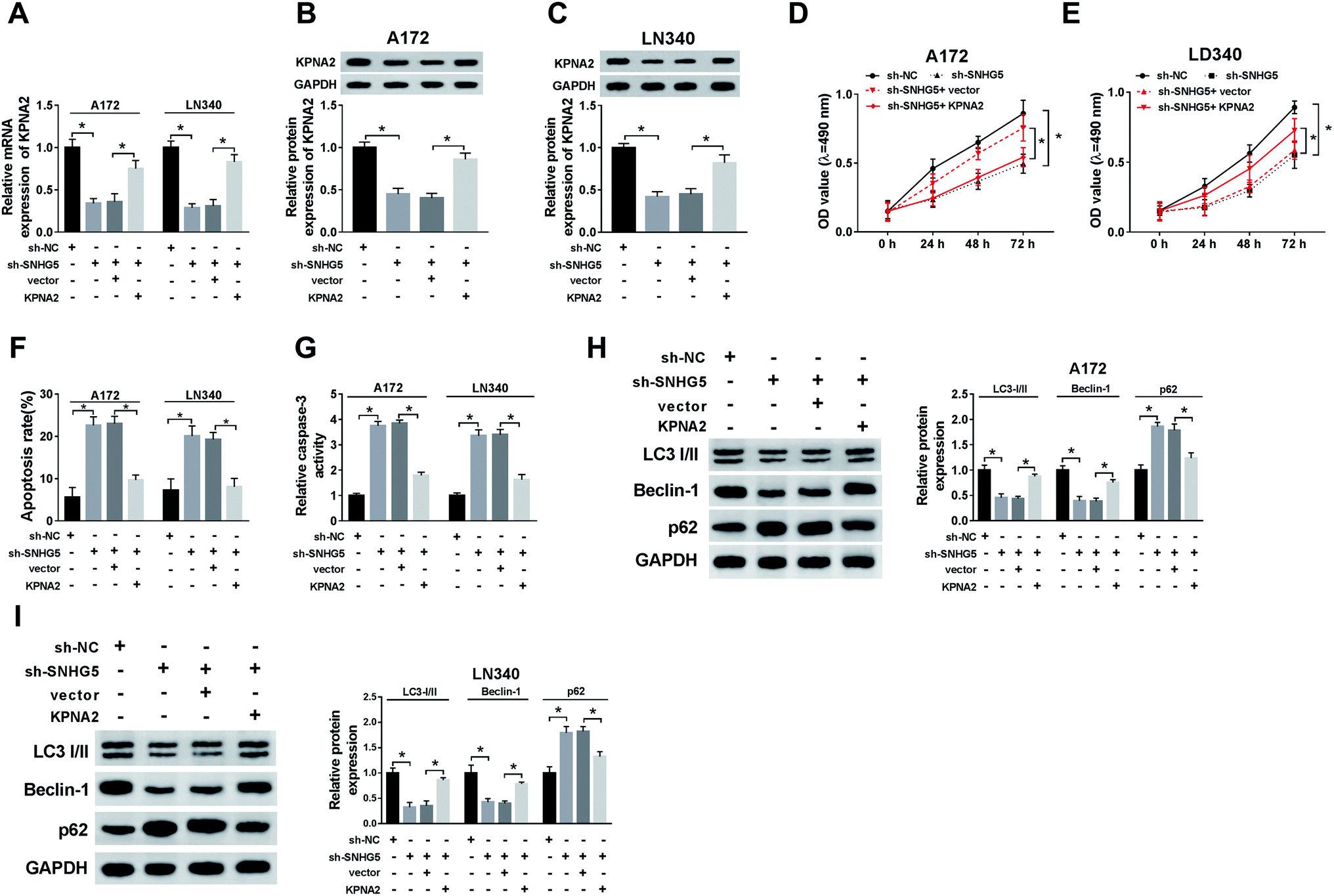 | ||
| Fig. 7 The effect of KPNA2 up-regulation on SNHG5 depletion-mediated cell function. (A, B and C) The mRNA and protein expression of KPNA2 in cells transfected with sh-NC, sh-SNHG5, sh-SNHG5 + vector, and sh-SNHG5 + KPNA2 overexpression plasmid was monitored. (D and E) The cell proliferation and apoptosis of glioma cells after indicated transfection were tested. (F–I) The apoptosis rate, caspase-3 activity and autophagy associated proteins expression in glioma cells after indicated transfection were analyzed. *P < 0.05. | ||
Discussion
Although the conventional treatments including surgical resection, radiotherapy and chemotherapy have been improved, the prognosis of patients with malignant glioma has remained unsatisfactory in the past few decades.19,20 Hence, there is a necessity to increase the treatment efficiency and deepen the understanding of glioma pathogenic mechanisms at the molecular level.So far, many lncRNAs have been reported to be strong candidates for cancer biomarkers with the potential to be applied in clinical practice. The brain is the organ with highest number of lncRNAs that are expressed, and these lncRNAs are found to be related to development and function of the brain.21 The roles of SNHG5 in various tumors have been investigated. For example, high expression of SNGH5 promotes cell proliferation and is corresponded with a bad prognosis of bladder cancer.22 Li et al. found that SNHG5 facilitates human hepatocellular carcinoma (HCC) development by adjusting miR-26a-5p/GSK3β.23 Then, Zhao et al. elucidated that SNHG5 inhibits GC cell proliferation and metastasis in vitro and in vivo.8 SNHG5 is also up-regulated in colorectal cancer, the low-expression of which similarly promotes cell cycle arrest and apoptosis in vitro and limits tumor genesis in vivo.4 Taken together, lncRNA SNHG5 indicates a poor prognosis of different tumors, with the promotion of viability and apoptosis of cancer cells. More importantly, it has been reported that SNHG5 regulates the proliferation and metastasis of osteosarcoma cells.24 Meanwhile, high expression of SNHG5 in glioma has been reported. Besides, on the basis of online database analysis and a series of experiments, Hu et al. further indicated that SNHG5 could affect the proliferation, apoptosis and autophagy of U251 and U87 cells.25 In the present study, we also found that SNHG5 was up-regulated in glioma tissues and cells. Moreover, low-expressed SNHG5 also led to cell proliferation and autophagy inhibition, and apoptosis promotion, which is consistent with previous studies.25
Studies have shown that lncRNAs can affect miRNA pathways by the ceRNA mechanism. That is, for achieving the purpose of regulating gene expression, ceRNAs competitively bind miRNA to inhibit the binding of its targets.26,27 For example, Zhang et al. indicated that lncRNA HCG11 interacts with miR-4425 to release the expression of MTA3 so as to mediate glioma growth.28 Zhou et al. found that lncRNA NEAT1 could facilitate glioma development by increasing the expression of SOX2 through suppressing miR-132 (ref. 29). Here, we discovered that the expression of miR-1297 was negatively related to the expression of SNHG5. We assumed that SNHG5 also targeted miRNAs and further identified the binding site between miR-1297 and SNHG5. Meanwhile, luciferase reporter assays and RIP assay also confirmed the relationship between them. Previous study has demonstrated that miR-1297 inhibits prostate cancer cell proliferation and invasion by targeting the AEG-1/Wnt signaling pathway.14 Interestingly, many studies also found that miR-1297 plays a tumor promoting factor in many cancers such as liver cancer,30 human breast cancer,31 non-small cell lung cancer32 and human cervical cancer.33,34 Wang et al. explained that miR-1297 is a tumor-inhibiting factor in glioma.35 In this report, we also disclosed that loss of miR-1297 recuperates the SNHG5 knockdown-mediated inhibitory effect on cell proliferation. Those data also indicated that SNHG5 may exhibit a suppressor role in glioma by sponging miR-1297. KPNA2 could act as a potential diagnostic in various cancers. KPNA2 is up-regulated in many human cancers, such as colorectal cancer,36 ovarian cancer37 and HCC.38 For example, over-expression of KPNA2 increases migration and proliferation in HCC.38 Moreover, Zhang et al. also reported that knockdown of KPNA2 significantly inhibits the growth and invasion of glioma cells.18 In our research, we found that KPNA2 was a target of miR-1297. More importantly, SNHG5 regains the expression of KPNA2 by down regulating miR-1297. Those data suggested that KPNA2 may be involved in the SNHG5-mediated cell regulation. We further manifested that gain of KPNA2 reversed the SNHG5 depletion-induced function on glioma cells. These lines of evidence suggested that SNHG5 may be responsible for glioma proliferation, apoptosis, and autophagy by miR-1297/KPNA2 axis.
However, our study also have some limitations. The role of SNHG5/miR-1297/KPNA2 pathway was only investigated in vitro. Their function should be further verified in vivo.
In summary, we uncovered that lncRNA SNHG5 was up-regulated in glioma. It was firstly reported that a novel lncRNA-miRNA-mRNA regulatory network SNHG5/miR-1297/KPNA2 in glioma. Knockdown of SNHG5 could inhibit cell viability and autophagy while promote apoptosis of glioma cell lines through miR-1297/KPNA2 axis. This may provide fundamental basis for the novel biomarkers and therapeutic strategies for glioma.
Conflicts of interest
The authors declare that they have no financial conflicts of interest.Funding
This work was supported by Henan Science and Technology Research Project (No. 172102410015), Henan Province Higher Education Research Project (No. 18A320077) and Medical Science and Technology Project of Henan Province (No. 2018020043).References
- S. Lapointe, A. Perry and N. A. Butowski, Lancet, 2018, 392, 432–446 CrossRef.
- P. D. Delgado-Lopez and E. M. Corrales-Garcia, Clin. Transl. Oncol., 2016, 18, 1062–1071 CrossRef CAS PubMed.
- R. M. Young, A. Jamshidi, G. Davis and J. H. Sherman, Ann. Transl. Med., 2015, 3, 121 Search PubMed.
- N. D. Damas, M. Marcatti, C. Come, L. L. Christensen, M. M. Nielsen, R. Baumgartner, H. M. Gylling, G. Maglieri, C. F. Rundsten, S. E. Seemann, N. Rapin, S. Thezenas, S. Vang, T. Orntoft, C. L. Andersen, J. S. Pedersen and A. H. Lund, Nat. Commun., 2016, 7, 13875 CrossRef CAS PubMed.
- L. Bolha, M. Ravnik-Glavac and D. Glavac, Dis. Markers, 2017, 2017, 7243968 Search PubMed.
- J. Jin, S. Zhang, Y. Hu, Y. Zhang, C. Guo and F. Feng, Biomed. Pharmacother., 2019, 116, 108968 CrossRef CAS PubMed.
- G. Li, Y. Cai, C. Wang, M. Huang and J. Chen, J. Neuro-Oncol., 2019, 143, 525–536 CrossRef CAS PubMed.
- L. Zhao, T. Han, Y. Li, J. Sun, S. Zhang, Y. Liu, B. Shan, D. Zheng and J. Shi, FASEB J., 2017, 31, 893–903 CrossRef CAS PubMed.
- X. Li, L. Liu, Y. Luo, S. Cui, W. Chen, A. Zeng, Y. Shi and L. Luo, Biosci. Rep., 2019, 39 DOI:10.1042/BSR20190668.
- H. O. Iwakawa and Y. Tomari, Trends Cell Biol., 2015, 25, 651–665 CrossRef CAS PubMed.
- F. M. Kouri, C. Ritner and A. H. Stegh, Cell Cycle, 2015, 14, 3794–3800 CrossRef CAS PubMed.
- H. Wang, X. T. Li, C. Wu, Z. W. Wu, Y. Y. Li, T. Q. Yang, G. L. Chen, X. S. Xie, Y. L. Huang, Z. W. Du and Y. X. Zhou, OncoTargets Ther., 2015, 8, 3211–3218 CAS.
- Z. Wu, Y. Han, Y. Li, X. Li, T. Sun, G. Chen, Y. Huang, Y. Zhou and Z. Du, Oncol. Rep., 2016, 35, 869–877 CrossRef CAS PubMed.
- X. Liang, H. Li, D. Fu, T. Chong, Z. Wang and Z. Li, Biochem. Biophys. Res. Commun., 2016, 480, 208–214 CrossRef CAS PubMed.
- Y. Wang, J. Xue, H. Kuang, X. Zhou, L. Liao and F. Yin, DNA Cell Biol., 2017, 36, 991–999 CrossRef CAS PubMed.
- Z. Chen, Y. Ma, Y. Pan, H. Zhu, C. Yu and C. Sun, Mol. Cell. Probes, 2018, 40, 19–26 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, Q. Guo, X. Hu, Z. Zhao, L. Ni, L. Liu, X. Wang, Z. Wang, D. Tong, S. Chang, Y. Cao and C. Huang, Anticancer Drugs, 2019, 30, 803–811 CrossRef CAS PubMed.
- Z. Zhang, X. Huang, J. Li, H. Fan, F. Yang, R. Zhang, Y. Yang, S. Feng, D. He, W. Sun and T. Xin, J. Cancer Res. Ther., 2019, 15, 927–932 CrossRef PubMed.
- L. P. Taylor, Neurology, 2010, 75, S28–S32 CrossRef PubMed.
- A. Omuro and L. M. DeAngelis, JAMA, 2013, 310, 1842–1850 CrossRef CAS PubMed.
- J. Aprea and F. Calegari, EMBO J., 2015, 34, 2865–2884 CrossRef CAS PubMed.
- Z. Ma, S. Xue, B. Zeng and D. Qiu, Oncol. Lett., 2018, 15, 1924–1930 Search PubMed.
- Y. Li, D. Guo, Y. Zhao, M. Ren, G. Lu, Y. Wang, J. Zhang, C. Mi, S. He and X. Lu, Cell Death Dis., 2018, 9, 888 CrossRef PubMed.
- C. Ju, R. Zhou, J. Sun, F. Zhang, X. Tang, K. K. Chen, J. Zhao, X. Lan, S. Lin, Z. Zhang and X. B. Lv, Cancer Cell Int., 2018, 18, 141 CrossRef PubMed.
- X. Hu, Y. Hong and C. Shang, J. Cancer, 2019, 10, 1333–1340 CrossRef CAS PubMed.
- Y. Lian, F. Xiong, L. Yang, H. Bo, Z. Gong, Y. Wang, F. Wei, Y. Tang, X. Li, Q. Liao, H. Wang, M. Zhou, B. Xiang, X. Wu, Y. Li, X. Li, X. Chen, G. Li, C. Guo, Z. Zeng and W. Xiong, J. Exp. Clin. Cancer Res., 2018, 37, 253 CrossRef CAS PubMed.
- L. Liu, S. Cui, T. Wan, X. Li, W. Tian, R. Zhang, L. Luo and Y. Shi, J. Cell. Physiol., 2018, 233, 6822–6831 CrossRef CAS PubMed.
- L. Zhang, Y. Cao, X. Kou, L. Che, X. Zhou, G. Chen and J. Zhao, J. Gene Med., 2019, 21, e3074 CrossRef CAS PubMed.
- K. Zhou, C. Zhang, H. Yao, X. Zhang, Y. Zhou, Y. Che and Y. Huang, Mol. Cancer, 2018, 17, 105 CrossRef PubMed.
- C. Liu, C. Wang, J. Wang and H. Huang, Oncol. Lett., 2016, 12, 5177–5182 CrossRef CAS PubMed.
- C. Liu, Z. Liu, X. Li, X. Tang, J. He and S. Lu, Oncol. Rep., 2017, 38, 2435–2443 CrossRef CAS PubMed.
- W. Bu and T. Luo, DNA Cell Biol., 2017, 36, 976–982 CrossRef CAS PubMed.
- Z. Wang, S. He, P. Guo, X. Guo and J. Zheng, Oncol. Rep., 2017, 38, 3121–3129 CrossRef CAS PubMed.
- Z. Chen, M. Zhang, Y. Qiao, J. Yang and Q. Yin, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1120–1126 CrossRef CAS PubMed.
- J. Wang, X. Xu, S. Mo, Y. Tian, J. Wu, J. Zhang and J. Zhao, Am. J. Transl. Res., 2016, 8, 2149–2158 CAS.
- T. Takada, S. Tsutsumi, R. Takahashi, K. Ohsone, H. Tatsuki, T. Suto, T. Kato, T. Fujii, T. Yokobori and H. Kuwano, J. Surg. Oncol., 2016, 113, 213–217 CrossRef CAS PubMed.
- L. Huang, Y. Zhou, X. P. Cao, J. X. Lin, L. Zhang, S. T. Huang and M. Zheng, Tumor Biol., 2017, 39, 1010428317706289 Search PubMed.
- X. Guo, Z. Wang, J. Zhang, Q. Xu, G. Hou, Y. Yang, C. Dong, G. Liu, C. Liang, L. Liu, W. Zhou and H. Liu, Acta Biochim. Biophys. Sin., 2019, 51, 285–292 CrossRef CAS PubMed.
Footnote |
| † Xueyuan Li and Qiankun Liu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |