DOI:
10.1039/C9RA09136J
(Paper)
RSC Adv., 2020,
10, 1361-1370
Excellent performance of aromatic polyguanamines induced by multiple hydrogen bondable tetraazacalix[2]arene[2]-triazine ring in their main chain†
Received
5th November 2019
, Accepted 19th December 2019
First published on 8th January 2020
Abstract
A series of poly(guanamine) (c-PG)s containing tetraazacalix[2]arene[2]-triazine (mPDA2CyC2) were successfully prepared by solution polycondensation of mPDA2CyC2 with various aromatic diamines in an aprotic organic solvent with a lithium chloride additive (5 wt%) at 150 °C for 6 hours. The number-average molecular weights (Mn)s of these c-PG polymers reached up to 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, with a relatively broad molecular weight distribution (Mw/Mn) of 5.3. They showed good solubility in aprotic organic solvents, such as N-methylpyrrolidone and N,N-dimethylacetamide at a concentration of 2 mg mL−1. The glass transition temperatures (Tg) of the c-PG polymers were in the range 359 °C–392 °C, approximately 160 °C higher than those of counterpart polymers (i.e., with no aza-calixarene-based PG (l-PG)). The coefficients of thermal expansion (CTEs) of the c-PG polymers were 29.7–48.1 ppm K−1 (at 100 °C–150 °C), much lower than those of l-PG samples, i.e., 59.1–85.1 ppm K−1. Transparent and almost colorless c-PG films were successfully prepared by a solution casting method, showing maximum tensile strength (σS), modulus (Eγ), and elongation at break (Eb) values of 151 MPa, 6.3 GPa, and 4.4%, respectively, for the c-PG polymer from mPDA2CyC2 and 4,4′-oxydianiline monomers. The corresponding l-PG film exhibited σS, Eγ, and Eb values of just 76 MPa, 5.4 GPa, and 1.6%, respectively. These outstanding thermal and mechanical properties of the c-PG polymers can be attributed to their multiple hydrogen bonding interaction between mPDA2CyC2 residues in the polymer backbone. This interaction was identified by infrared spectroscopy measurements at the broad absorption band around 3000–3400 cm−1.
500, with a relatively broad molecular weight distribution (Mw/Mn) of 5.3. They showed good solubility in aprotic organic solvents, such as N-methylpyrrolidone and N,N-dimethylacetamide at a concentration of 2 mg mL−1. The glass transition temperatures (Tg) of the c-PG polymers were in the range 359 °C–392 °C, approximately 160 °C higher than those of counterpart polymers (i.e., with no aza-calixarene-based PG (l-PG)). The coefficients of thermal expansion (CTEs) of the c-PG polymers were 29.7–48.1 ppm K−1 (at 100 °C–150 °C), much lower than those of l-PG samples, i.e., 59.1–85.1 ppm K−1. Transparent and almost colorless c-PG films were successfully prepared by a solution casting method, showing maximum tensile strength (σS), modulus (Eγ), and elongation at break (Eb) values of 151 MPa, 6.3 GPa, and 4.4%, respectively, for the c-PG polymer from mPDA2CyC2 and 4,4′-oxydianiline monomers. The corresponding l-PG film exhibited σS, Eγ, and Eb values of just 76 MPa, 5.4 GPa, and 1.6%, respectively. These outstanding thermal and mechanical properties of the c-PG polymers can be attributed to their multiple hydrogen bonding interaction between mPDA2CyC2 residues in the polymer backbone. This interaction was identified by infrared spectroscopy measurements at the broad absorption band around 3000–3400 cm−1.
Introduction
For decades, attention has been paid to the synthesis of polymers having a cyclic skeleton, such as crown ethers,1–6 cyclodextrins,7 and calixarenes,8 for application to ion recognition, separation, batteries, and supramolecular chemistry. Among them, calixarenes have been attracted great attention because of their rich functionalization chemistry and the well-defined molecular recognition capabilities.9,10 There are a number of reports on the synthesis of polymers having calix[4]arenes as a bulky pendant group in the polymer backbone to study their metal coordination abilities.11,12 Conjugated polymers are one of the hot research topics both in academic and industry, and calix[4]arenes were incorporated as a pendant side group, which enhanced the polymer solubility and was utilized as sensors toward various classes of molecules.13–19 On the other hands, calixarenes are known to have a rigid structure, making their incorporation into a polymer backbone relatively difficult unless flexible solubilizing groups are introduced at the same time. Dondoni et al. synthesized a bisphenol A-type polymer having a calix[4]arene moiety in its main chain and studied Ag ion recognition.20 Kim et al. reported the synthesis of rigid polyimides containing 25,26,27,28-tetrapropoxycalix[4]arenes in the backbone; however, the effect of the incorporation of the calix[4]arene moiety was not adequately revealed probably because of the tightly packed nature of the parent polyimide backbone.21 Swager et al. reported the synthesis of polythiophene having 25,27-dimethoxy and 25,57-dipropoxycalix[4]arene and studied the spatial relationship between the electroactive polythiophene segments in terms of the electronic, optical, and optoelectronic properties.22 They also reported the synthesis of main chain p-tert-butylcalix[4]arene base polymers utilizing acetylene coupling polymerization.23 This p-tert-butylcalix[4]arene component was utilized for ring-opening metathesis copolymerization with cyclic alkenes, producing novel elastomers in which the calixarene and polyalkene components were functioned as hard and soft segments, respectively.24 Recently, novel aza- and/or oxo-bridged calix[2]arene[2]triazine derivatives with 1,3-alternative conformation were developed by Wang et al., and the structures were found to be the roughly coplanar two-triazine rings with almost perpendicular (to the planes) benzene rings defined by four bridged heteroatoms.25 Tuning of the cavity made by the calixarene moiety enabled them to work as fluorine sensors.26
Cyanuryl polyimines, known as poly(guanamine)s (PG)s, are condensation polymers. They are usually prepared from triazine dichloride and diamine monomers in solution, with a variety of derivatives having been synthesized to date.27–34 The guanamine functional group was also found in Wang's aza-bridged calix[2]arene[2]triazine (mPDA2CyC2), which has two active C–Cl bonds; therefore, we expected that a mPDA2CyC2 residue would exhibit a unique effect on PG structure. In particular, if rigid aromatic diamines were combined with mPDA2CyC2 during polycondensation, then a new series of condensation polymers having a tightly packed structure should be obtained. Our motivation in this research was in uncovering the unique and original nature of mPDA2CyC2-based PG polymers (c-PG), particularly in terms of thermal and mechanical properties, which can be significantly affected by the multiple hydrogen bonding abilities of mPDA2CyC2. As counterpart polymers, we also prepared non-calixarene-type PG polymers (l-PG), for which we compared the physical properties with those of the c-PG polymers. Herein, we report the synthesis and properties of novel azacalixarene containing c-PGs, utilizing mPDA2CyC2 as a cyclic guanamine monomer.
Results and discussion
Monomer and polymer synthesis
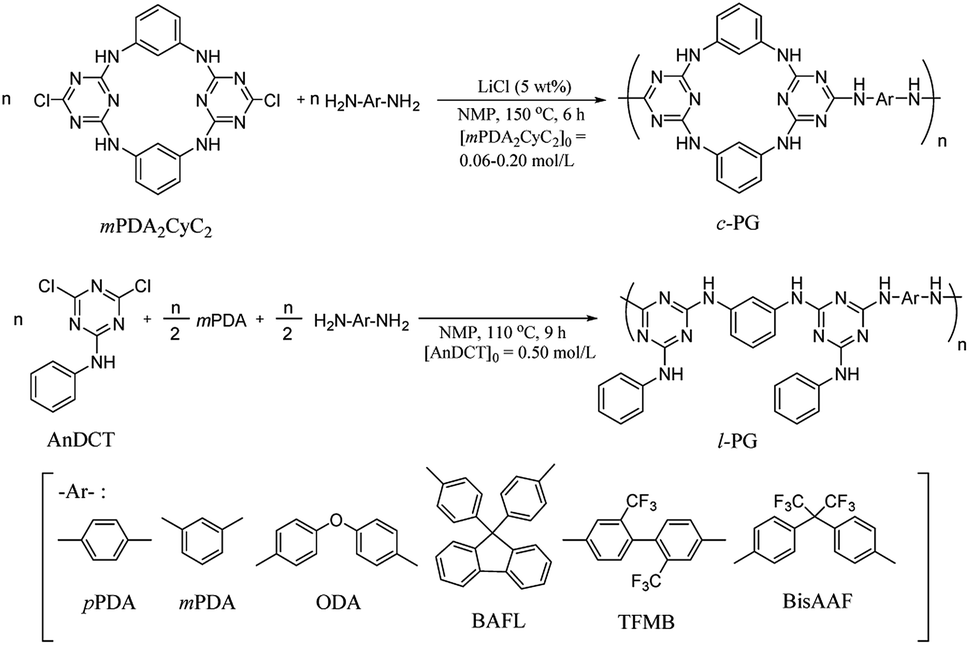
Cyclic guanamine calixarene (mPDA2CyC2) was prepared in accordance with Wang's method;25 the structure was confirmed by NMR and FAB MS spectroscopy (Scheme S1, Fig. S1–S3†). First, l-PG was prepared as a non-azacalixarene-type polymer in accordance with the literature.32 For instance, poly(AnDCT-mPDA) [l-PG(mPDA)] was prepared by solution polycondensation of AnDCT with mPDA in NMP (total monomer concentration was 1.0 mol L−1); 65% yield, Mn = 5000, Mw/Mn = 3.6 (Run 7, Table 1). Relatively low Mn of the polymer is attributable to the low solubility of l-PG(mPDA) with accompanying the formation of cyclic oligomers due to the meta phenylene linkage. By spontaneous polycondensation of AnDCT with mPDA and the corresponding diamine comonomers, the other l-PG polymers were prepared with reasonable Mn (Runs 8 and 9). In accordance with a similar procedure, the synthesis of c-PGs was then attempted using mPDA2CyC2 with various aromatic diamine monomers; however, the polymerization solution instantly became a gel within the reaction time. Therefore, polymerization was conducted in more dilute conditions (0.060 to 0.40 mol L−1 for mPDA2CyC2 depending on the diamine monomers used) with 5 wt% LiCl as a solubilizer at 150 °C for 6 hours (Scheme 1). To avoid gelation, the synthesis of c-PG polymers consisting of rigid diamine monomers, such as pPDA, required highly dilute conditions of below 0.10 mol L−1 for mPDA2CyC2. Thus, the final obtained Mn was only 6300, with a low polymer yield of 55% (Run 1, Table 1). However, diamine monomers having a bent structure or a bulky pendant group resulted in the formation of high-Mn polymers with good yields (Runs 2–5, Table 1). These higher Mn values of c-PGs compared with those of l-PGs can be explained by avoiding cyclic oligomer formation. For l-PG synthesis, although the addition of LiCl did not significantly improve the Mn values of the polymers, the corresponding films could be prepared (Runs 10–12, Table 1).
Table 1 Synthesis of c- and l-PG polymersa
| Run |
Polym. |
Add. |
[mPDA2CyC2]0 (mol L−1) |
Y (%) |
Mnb (kDa) |
Mw/Mnb |
Filmc |
| Conditions; in NMP at 150 °C for 6 h for c-PG and at 110 °C for 9 h for l-PG. Determined by GPC (NMP, PS standards). X: free-standing film cannot be prepared, B: brittle film, F: flexible film. |
| 1 |
c-PG (pPDA) |
LiCl |
0.060 |
55 |
6.3 |
5.0 |
X |
| 2 |
c-PG (mPDA) |
LiCl |
0.20 |
85 |
26.0 |
3.2 |
F |
| 3 |
c-PG (ODA) |
LiCl |
0.20 |
91 |
24.3 |
7.1 |
F |
| 4 |
c-PG (BAFL) |
LiCl |
0.25 |
92 |
24.9 |
7.3 |
F |
| 5 |
c-PG (TFMB) |
LiCl |
0.22 |
83 |
30.5 |
4.4 |
F |
| 6 |
c-PG (BisAAF) |
LiCl |
0.40 |
84 |
31.5 |
5.3 |
B |
| 7 |
l-PG (mPDA) |
— |
1.0 |
65 |
5.0 |
3.6 |
X |
| 8 |
l-PG (BAFL) |
— |
1.0 |
81 |
10.4 |
3.7 |
B |
| 9 |
l-PG (pPDA) |
— |
1.0 |
80 |
11.6 |
3.5 |
B |
| 10 |
l-PG (mPDA) |
LiCl |
1.0 |
66 |
7.3 |
3.8 |
B |
| 11 |
l-PG (BAFL) |
LiCl |
1.0 |
76 |
13.4 |
4.2 |
B |
| 12 |
l-PG (ODA) |
LiCl |
1.0 |
79 |
9.3 |
6.4 |
B |
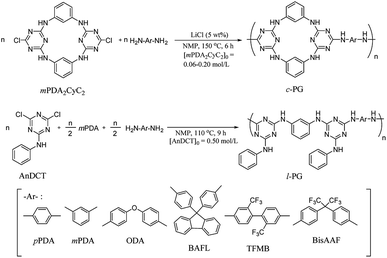 |
| | Scheme 1 Synthesis of c- and l-PG polymers. | |
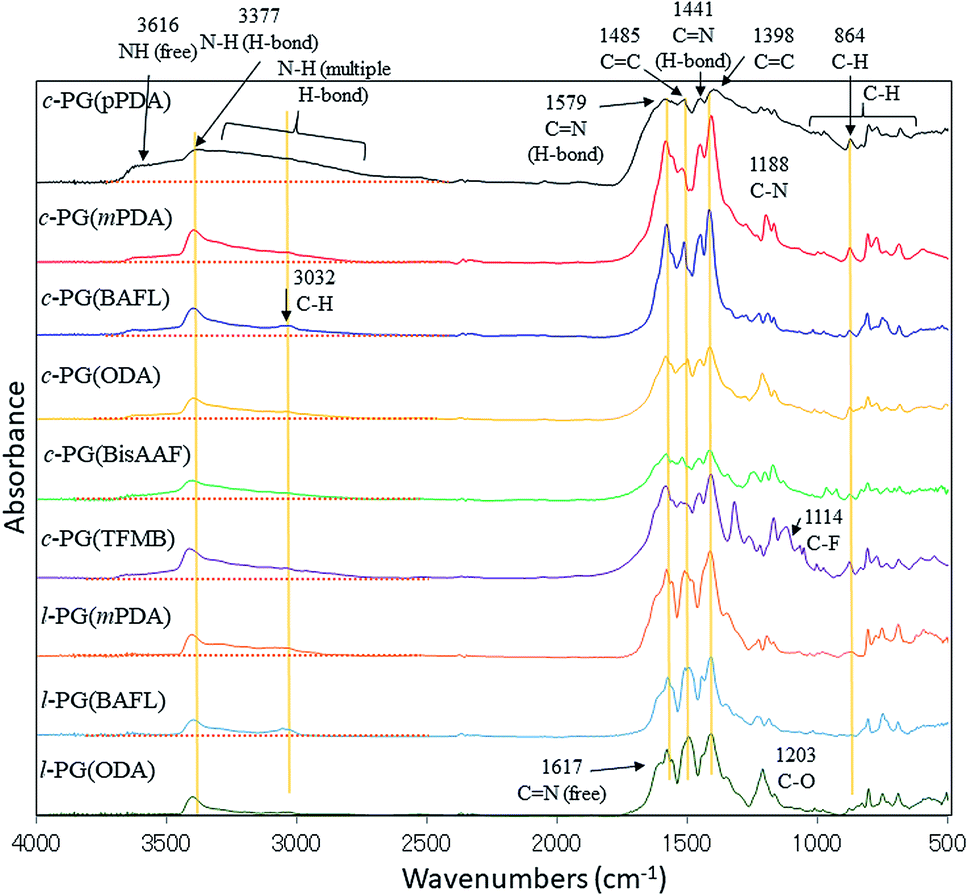
Fig. 1 shows the FT-IR spectra of the prepared polymers. Owing to the comparable structures of c-PG and l-PG (as illustrated in Fig. 2), their IR spectra appear almost identical, except for the shoulder peak at 3370–2800 cm−1, which results from the multiple hydrogen bonding absorption at the guanamine moiety, as reported in the literature.35 The absorption at 3616 cm−1 can be assigned to free guanamine NH stretching vibration and was only observed for c-PG polymers. The absorptions at 3377 and 3370–2800 cm−1 can be assigned to hydrogen-bonded NH stretching and multiple hydrogen-bonded NH stretching, respectively. The absorption at 1441 cm−1 can be assigned to a multiple hydrogen-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group, which is clearly observed, especially for c-PG samples. To study more about multiple hydrogen bond interactions between polymer chains, the FT-IR spectra of the PG polymer powder mixed with KBr were recorded at temperatures ranging from 20 to 200 °C (Fig. 3). As the temperature increased, the absorption intensities all decreased. These changes were not reversible even when the heated sample was cool down to room temperature. The most relevant changes were observed at 3000 to 3500 cm−1, which could be attributed to both the moisture desorption within polymer chains and the development of hydrogen bonding network.
N group, which is clearly observed, especially for c-PG samples. To study more about multiple hydrogen bond interactions between polymer chains, the FT-IR spectra of the PG polymer powder mixed with KBr were recorded at temperatures ranging from 20 to 200 °C (Fig. 3). As the temperature increased, the absorption intensities all decreased. These changes were not reversible even when the heated sample was cool down to room temperature. The most relevant changes were observed at 3000 to 3500 cm−1, which could be attributed to both the moisture desorption within polymer chains and the development of hydrogen bonding network.
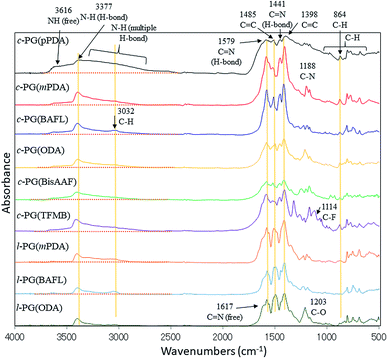 |
| | Fig. 1 FT-IR spectra of the prepared polymers (KBr method). | |
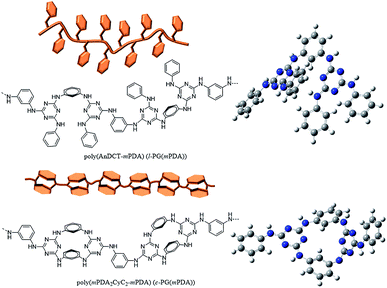 |
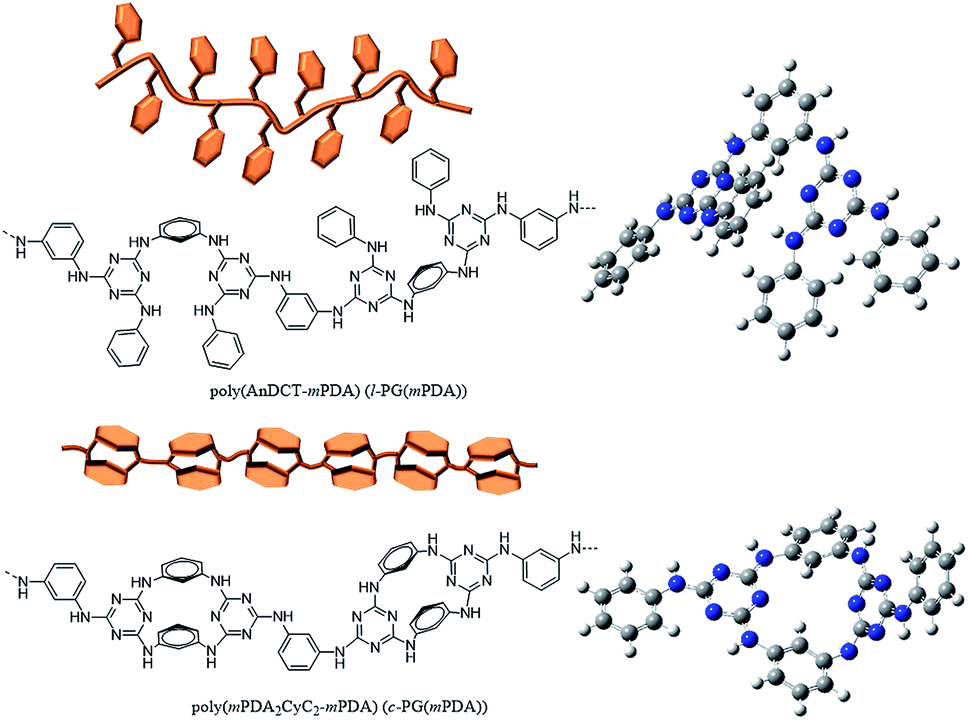
| | Fig. 2 Chemical structures, illustration, and optimized geometries of poly(AnDCT-mPDA) (l-PGA(mPDA)) (upper) and poly(mPDA2CyC2-mPDA) (c-PGA(mPDA)) (lower) as representatives of l- and c-PG polymers. The calculation was performed with the Gaussian 03W program and the Hartree–Fock method using the basis set of the 3-21G function. | |
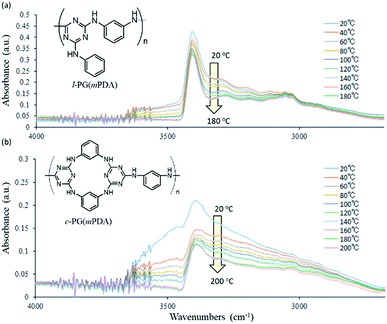 |
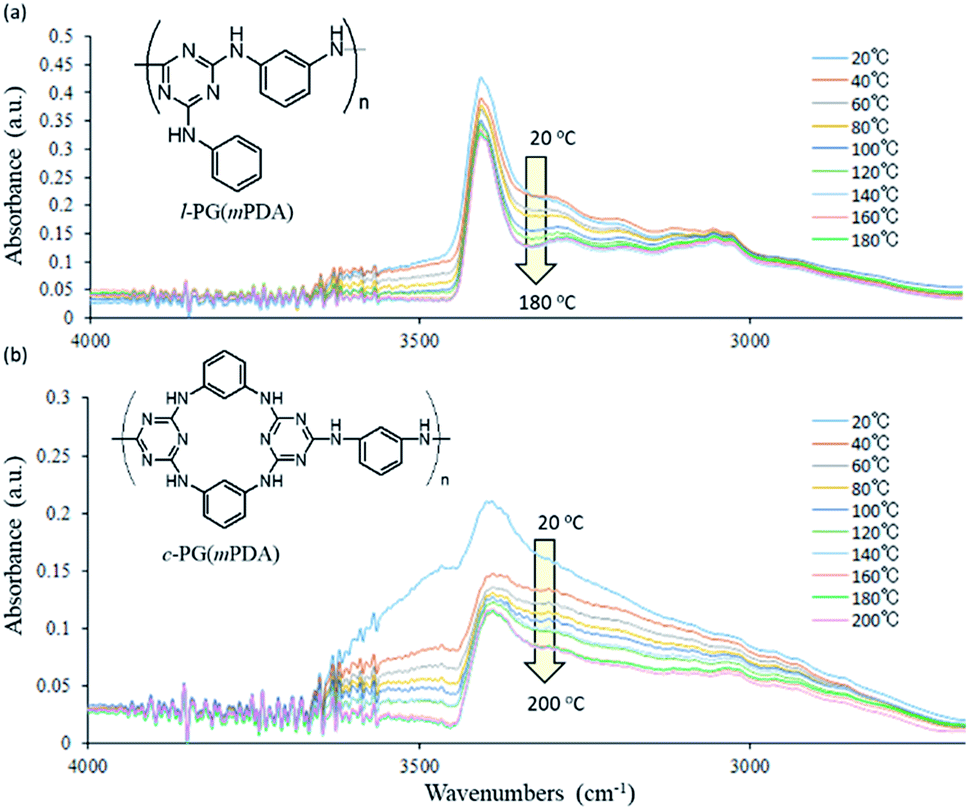
| | Fig. 3 Expanded FT-IR spectra changes of (a) l-PG(mPDA) and (b) c-PG(mPDA) heated from 20 to 200 °C. | |
Wang et al. performed temperature variable FT-IR study of diaminotriazine derivatives, in which clear decrease of the absorption at 3273 cm−1 (hydrogen bonded NH) was observed against that at 3379 cm−1 (free NH) especially above 54 °C that is the Tg of the samples.35 And these absorption changes were reversible with temperature. In contrast, our PG polymer samples only showed the absorption decrease, and the intensities were not recovered with temperature. These indicates that our l- and c-PG polymer samples should have high Tg above the FT-IR spectra measurement conditions of 200 °C. On the other hands, there are clear differences between the spectra from 3000 to 3500 cm−1, between l- and c-PG samples. The initially observed absorption at 3500 cm−1 of c-PG sample at 20 °C is much larger than that of l-PG, and in the spectrum of c-PG the shoulder peak at 3000 to 3300 cm−1 are more clearly observed even after heated to 200 °C. These suggests that c-PG polymers initially have some weak hydrogen bonding interaction between polymer chains, and these be developable with heating. In our current method to study FT-IR spectra, the attainable measurable temperature using heated sample was up to 200 °C that is far below the Tg of PG samples (220 °C for l-PG(mPDA) and 370 °C for c-PG(mPDA)), and thus the reversible FT-IR spectra changes were not observed.
The optimized geometry of the c-PG and l-PG models was determined with the Gaussian 03W program and the Hartree–Fock method, using the basis set of the 3-21G function (Fig. 2, right). The meta-linked bent structure of the l-PG polymer backbone based on mPDA2CyC2 and mPDA, together with the pendant aniline substituent, make the polymer structure cramped. On the contrary, the mPDA unit is covalently bonded at both sides in the c-PG polymer backbone; therefore, the c-PG polymer is free from the steric repulsion of the pendant aniline group. According to Wang's X-ray crystallographic study, mPDA2CyC2 has a boat-like geometry with two mPDA units and two-triazine rings.25 As a result, the mutually connected mPDA2CyC2 and mPDA monomer molecules make the c-PG polymer structure more extendable and flat, which significantly increase the hydrogen bonding opportunities between the polymer chains. These spectroscopic data and calculation results indicate that owing to multiple hydrogen bondable sites, the azacalixarene guanamine moieties are more tightly packed in the c-PG structure than they are in the linear structure of l-PG polymers.
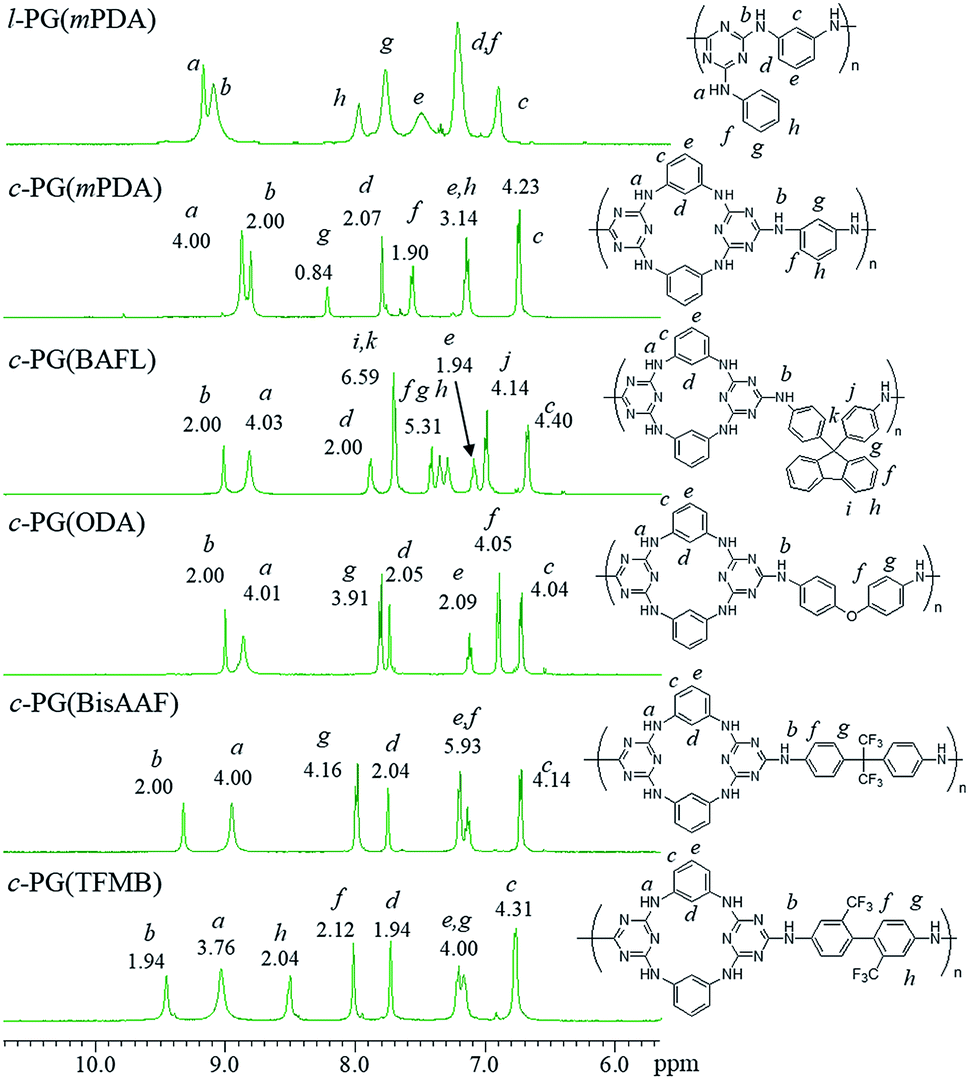
Fig. 4 shows the expanded 1H NMR spectra (from 6 to 11 ppm) of the prepared polymers (full spectra are given in the ESI as Fig. 4s†). The l-PG polymer (l-PGA(mPDA)) shows a typically broad signal observed at 7.50 (e) ppm, assignable to the mPDA unit. This is a result of the rotation limitation of the phenyl ring, which is caused by the weak hydrogen bonding between aromatic C–H (d) and triazine nitrogen. The guanamine protons are observed at 9.18 (a, 1H) and 9.10 (b, 2H) ppm. For the corresponding c-PG sample (c-PG(mPDA)), the proton labelled as h, assignable to the phenyl ring moiety of mPDA, is observed at 7.20 ppm, with proton e presenting as a sharper signal. The m-phenylene linkage in c-PG has more freedom in its rotation in solution. In addition, the electron density of the triazine moiety is higher than that in l-PG because of the additional introduction of the m-phenylenediamine group.
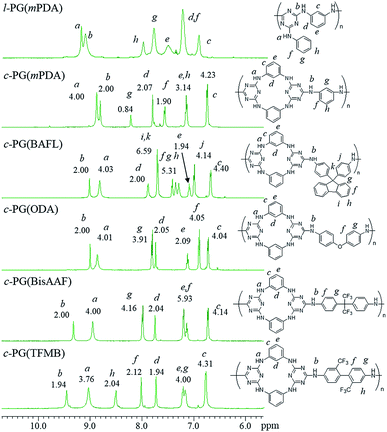 |
| | Fig. 4 1H NMR spectra of c- and l-PG samples in DMSO-d6. | |
Molecular orbital calculation study
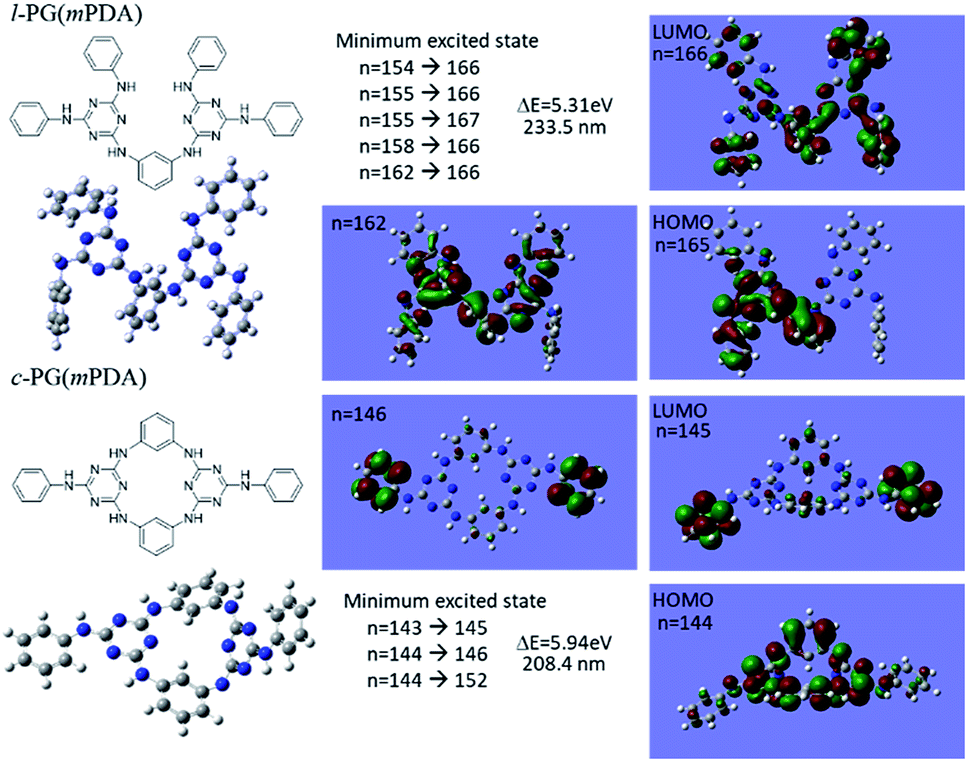
Although the structural similarity of the l- and c-PG polymers as explained in Fig. 2, both of the IR and NMR measurements gave the slightly different spectra. To study the relationship between the structure and the properties, the time dependent density functional theory (TD-DFT) was applied for the model compounds of the prepared polymers. Fig. 5 depicts the optimized geometry of the model compounds for l-PG(mPDA) and c-PG(mPDA) with the illustrations of the molecular orbitals. For the l-PG(mPDA) model, there were several minimum excited states such as from n = 154 to 166, and the band gap energy was calculated to be 5.31 eV (λ = 233.5 nm). On the other hands, the c-PG(mPDA) model showed larger band gap energy of 5.94 eV (λ = 208.4 nm). The corresponding molecular orbitals of the two model compounds indicate that the HOMO of the c-PG(mPDA) model is uniformly more delocalized at the azacalixarene moiety thanks to the coplanarity of the azacalixarene skeleton, which might enlarge the band gap energy of the c-PG polymer. From these calculation results, we expected that the c-PG polymer should have higher thermal, mechanical, and optical properties due to the accessible azacalixarene flat rings within the polymer chains.
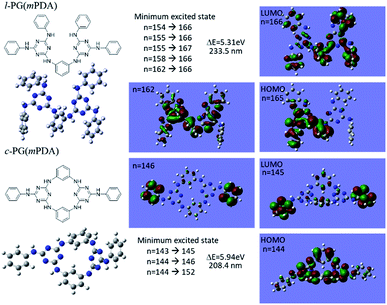 |
| | Fig. 5 Optimized geometry of model compounds for l-PG(mPDA) and c-PG(mPDA) with the molecular orbital illustrations as obtained from DFT/Hartree–Fock 3-21G level of calculation. | |
Polymer properties
The solubility values of the prepared c-PG polymers are summarized in Table 1s.† These polymers are soluble only in aprotic polar solvents, such as DMAc and NMP, at 25 °C and at 2 mg mL−1 concentration; they are insoluble in THF, CHCl3, γ-butyrolactone, propylene glycol monomethyl ether, and cyclohexanone. The rigid-rod polymer c-PG(pPDA) is only soluble in DMAc and NMP and is partially soluble in DMF and DMSO upon heating, which agrees with the observation that the polymer was precipitated even during polymerization.
The thermal properties of the c- and l-PG polymers were evaluated by TG/DTA, DSC, TMA, and DMA. Table 2 summarizes the glass transition temperatures (Tgs) measured by DMA, the 5 and 10 wt% weight loss temperatures (Td5 and Td10), and the char yield at 800 °C measured by TG. The Tg values of the c-PG samples range from 359 °C to 392 °C, approximately 160 °C higher than those of the reference l-PG polymers. The Td5 and Td10 data also illustrate the higher thermostability of the c-PG samples. These physical and chemical thermostabilities can be attributed to the azacalixarene moieties.
Table 2 Thermal properties of c- and l-PG polymersa
| Polymer |
Tg (°C) |
Td5a (°C) |
Td10a (°C) |
Td5a (°C) |
Td10a (°C) |
Charb (%) |
CTEc (ppm K−1) |
| DSC |
DMA |
TMA |
N2 |
Air |
| Determined by TGA (10 °C min). Char yield at 800 °C. Determined by TMA from 100–150 °C. Not detected. |
| c-PG (mPDA) |
—d |
368 |
372 |
506 |
542 |
444 |
448 |
61.8 |
48.10 |
| c-PG (ODA) |
—d |
359 |
357 |
513 |
539 |
448 |
451 |
59.7 |
38.14 |
| c-PG (BAFL) |
—d |
386 |
— |
522 |
542 |
476 |
480 |
61.1 |
29.68 |
| c-PG (BisAAF) |
—d |
—d |
392 |
513 |
534 |
479 |
498 |
55.0 |
44.35 |
| c-PG (TFMA) |
—d |
—d |
374 |
496 |
523 |
471 |
489 |
54.4 |
39.37 |
| l-PG (mPDA) |
218 |
—d |
—d |
416 |
436 |
404 |
432 |
50.2 |
—d |
| l-PG (BAFL) |
255 |
223 |
248 |
427 |
444 |
415 |
442 |
51.9 |
59.07 |
| l-PG (ODA) |
206 |
154 |
163 |
432 |
452 |
452 |
469 |
56.3 |
85.09 |
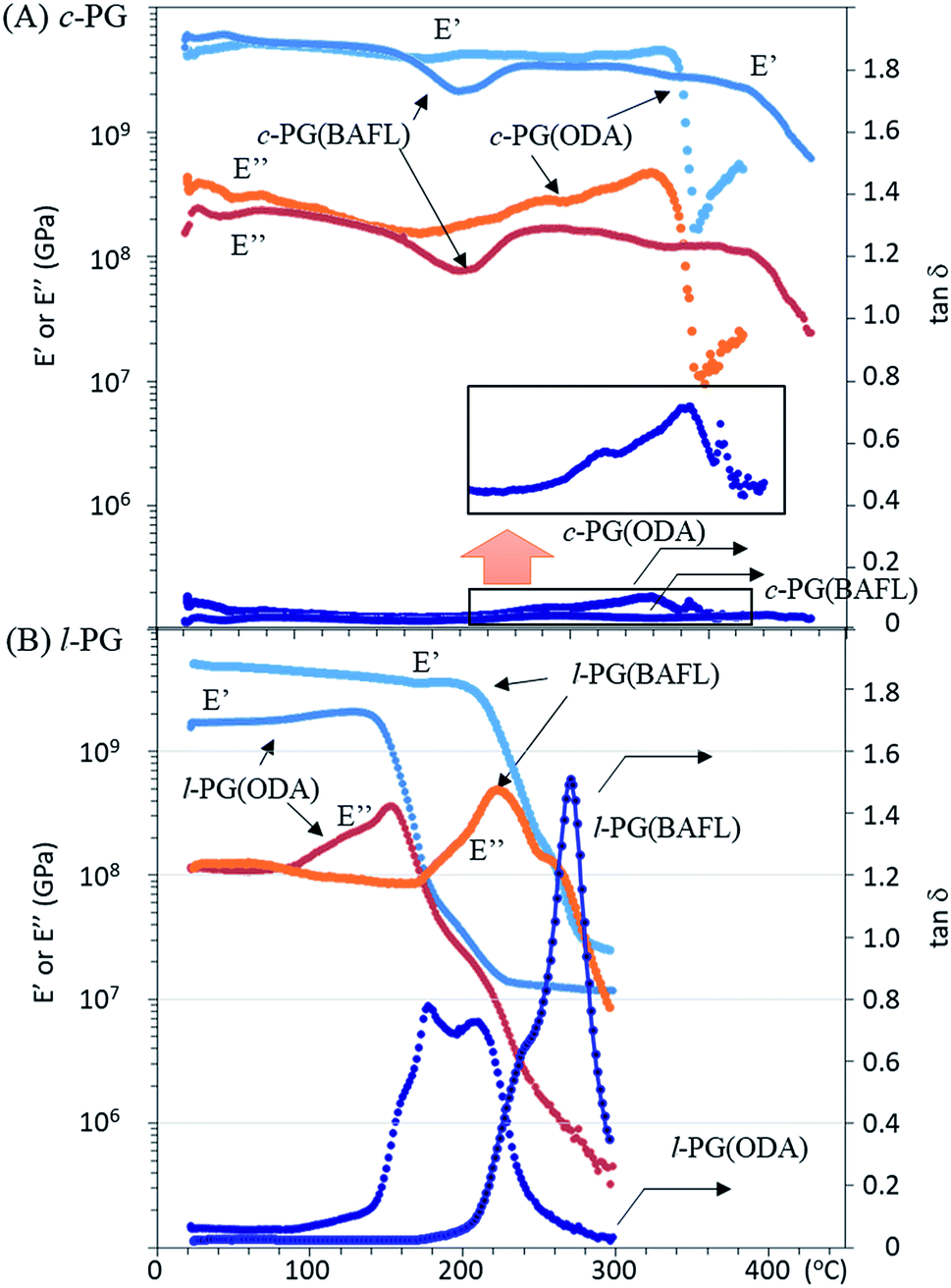
Fig. 6 depicts the DMA profiles of the c- and l-PG films dried at 150 °C for 12 hours. The l-PG(ODA) film initially showed a storage modulus E′ (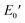 ) of 1.72 GPa. This value started to sharply decrease from 137 °C (Ts), reaching a rubbery plateau from 228 °C (Tr) at an E′ value (
) of 1.72 GPa. This value started to sharply decrease from 137 °C (Ts), reaching a rubbery plateau from 228 °C (Tr) at an E′ value (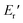 ) of 0.0135 GPa. For the l-PG(BAFL) film, the DMA curve showed better thermal stability owing to its rigid and bulky fluorene pendant effect, i.e., the values of Ts, Tr,
) of 0.0135 GPa. For the l-PG(BAFL) film, the DMA curve showed better thermal stability owing to its rigid and bulky fluorene pendant effect, i.e., the values of Ts, Tr, 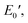 and
and 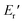 were 199.7 °C, 288.8 °C, 4.79 GPa, and 0.0257 GPa, respectively. For the c-PG films, the values of Ts, Tr,
were 199.7 °C, 288.8 °C, 4.79 GPa, and 0.0257 GPa, respectively. For the c-PG films, the values of Ts, Tr, 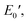 and
and 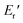 were significantly improved to 372.4 °C, 423.6 °C, 5.18 GPa, and 0.506 GPa for c-PG(ODA) and 387.1 °C, > 425.5 °C, 5.98 GPa, and > 0.615 GPa for c-PG(BAFL), respectively. The plot of tanδ and measured temperature for the l-PG(ODA) film shows two peaks at 177.1 °C and 204.5 °C, indicating that the polymer main chain produced a more stable packed structure with thermal agitation. The peaks show the Tg values of the polymers, reaching 0.8 and 1.6 for l-PG(ODA) and l-PG(BAFL), respectively. For the c-PG films, the tanδ profiles also show two peaks at 286.4 °C and 360.6 °C for c-PG(ODA) and at 259.4 °C and >424.0 °C for c-PG(BAFL). However, these peaks show maximum values of only 0.1059 for c-PG(ODA) and 0.0510 for c-PG(BAFL). These very small tanδ values indicate the absence of an E′′ contribution, which clearly suggests the dominant elastic nature of the c-PG polymers, with multiple densely bound hydrogen bonds at the azacalixarene moiety.
were significantly improved to 372.4 °C, 423.6 °C, 5.18 GPa, and 0.506 GPa for c-PG(ODA) and 387.1 °C, > 425.5 °C, 5.98 GPa, and > 0.615 GPa for c-PG(BAFL), respectively. The plot of tanδ and measured temperature for the l-PG(ODA) film shows two peaks at 177.1 °C and 204.5 °C, indicating that the polymer main chain produced a more stable packed structure with thermal agitation. The peaks show the Tg values of the polymers, reaching 0.8 and 1.6 for l-PG(ODA) and l-PG(BAFL), respectively. For the c-PG films, the tanδ profiles also show two peaks at 286.4 °C and 360.6 °C for c-PG(ODA) and at 259.4 °C and >424.0 °C for c-PG(BAFL). However, these peaks show maximum values of only 0.1059 for c-PG(ODA) and 0.0510 for c-PG(BAFL). These very small tanδ values indicate the absence of an E′′ contribution, which clearly suggests the dominant elastic nature of the c-PG polymers, with multiple densely bound hydrogen bonds at the azacalixarene moiety.
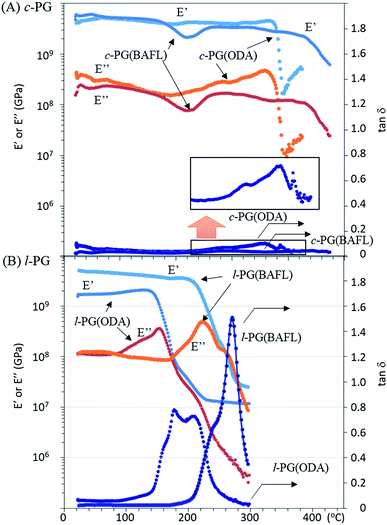 |
| | Fig. 6 DMA profiles of (A) c-PG and (B) l-PG films. | |
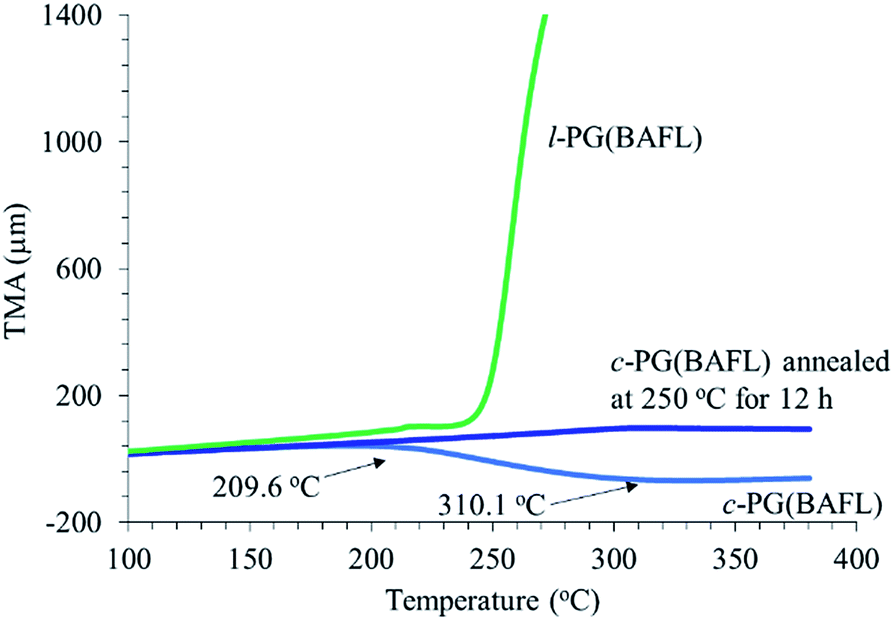
Fig. 7 depicts the TMA profiles of the c- and l-PG(BAFL) films. The shape of the l-PG(BAFL) film gradually changed and started to markedly expand at 238.9 °C. By contrast, the c-PG(BAFL) film mostly retained its shape within the measured temperature range up to 380 °C, indicating the effectiveness of azacalixarene packing. However, during TMA measurement, the c-PG(BAFL) film shrank from 209.6 °C to 310.1 °C, which was also observed for DMA (Fig. 6(A)). This may be caused by the reorientation of the polymer chains in the film upon thermal treatment. Pre-annealing of the film at 250 °C for 12 hours prevented such shrinkage, with the film retaining its shape above 380 °C. The CTE values were measured as 59.07 ppm K−1 for l-PB(BAFL) and 29.68 ppm K−1 for c-PB(BAFL) in the temperature range of 100 °C–150 °C. As illustrated in Fig. 2, as l-PG polymers have pendant aniline side groups that can freely rotate along with the polymer main chain, the film tended to become brittle even at a high molecular weight. By contrast, the c-PG polymers afford a flexible film and high thermal stability. This can be explained on the basis that the structure of the c-PG polymer is more extended and flat than that of the l-PG polymer. Moreover, the polymer main chains are functionalized with azacalixarene moieties, which can be further tightly packed with the multiple hydrogen bondable sites.
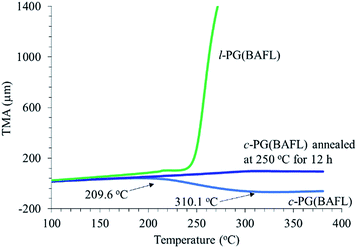 |
| | Fig. 7 TMA profiles of c- and l-PG films. | |
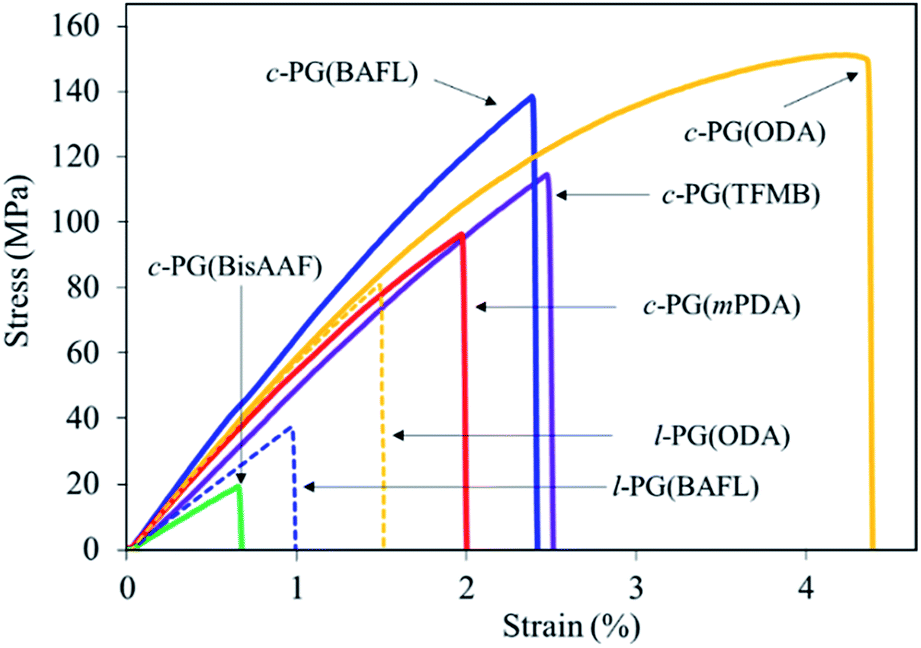
Table 3 and Fig. 8 summarize the mechanical properties of the polymer films. We first assumed that the c-PG films should have a higher modulus and thus be brittle in nature owing to the existence of azacalixarene moieties. However, the films we prepared were stronger than their linear counterparts (i.e., the l-PG films), whose tensile strength (σTS), elongation at break (Eb), and tensile modulus (Eγ) values were 37.0–76.0 MPa, 1.0–1.6%, and 4.2–5.4 GPa, respectively. As observed in Fig. 8, the l-PG films broke only below a strain of 1.5%. This behaviour is generally observed for PG polymers having a bulky pendant group like an aniline function, which restricts the effective entanglement between polymer main chains (Fig. 2). Surprisingly, the c-PG films (except for c-PG(BisAAF)) show much better tensile properties (σTS = 96–151 MPa, Eb = 2.0–4.4%, Eγ = 5.2–7.0 GPa), probably owing to the more extended and flat polymer backbone without freely rotatable aniline substituents, and stronger interaction between azacalixarene moieties. As observed in DMA and TMA analyses, the polymer main chains can be rearranged with outer stimuli. Therefore, assisted by azacalixarene interaction, the c-PG films are able to change their polymer chains in a manner suitable for enduring the outer load.
Table 3 Tensile properties of PG polymers
| Run |
Polym. |
Thickness (μm) |
σTSa (MPa) |
Ebb (%) |
Eγc (GPa) |
| Tensile strength. Elongation at break. Tensile modulus. |
| 1 |
c-PG(mPDA) |
15.0 |
96.0 |
2.0 |
5.9 |
| 2 |
c-PG(ODA) |
10.3 |
151 |
4.4 |
6.3 |
| 3 |
c-PG(BAFL) |
16.2 |
138 |
2.4 |
7.0 |
| 4 |
c-PG(TFMB) |
8.2 |
114 |
2.5 |
5.2 |
| 5 |
c-PG(BisAAF) |
21.0 |
19.0 |
0.7 |
3.1 |
| 6 |
l-PG(BAFL) |
70.4 |
37.0 |
1.0 |
4.2 |
| 7 |
l-PG(ODA) |
47.2 |
76.0 |
1.6 |
5.4 |
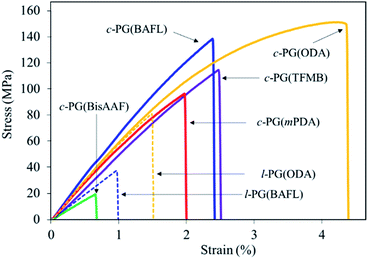 |
| | Fig. 8 Tensile properties of PG films. | |
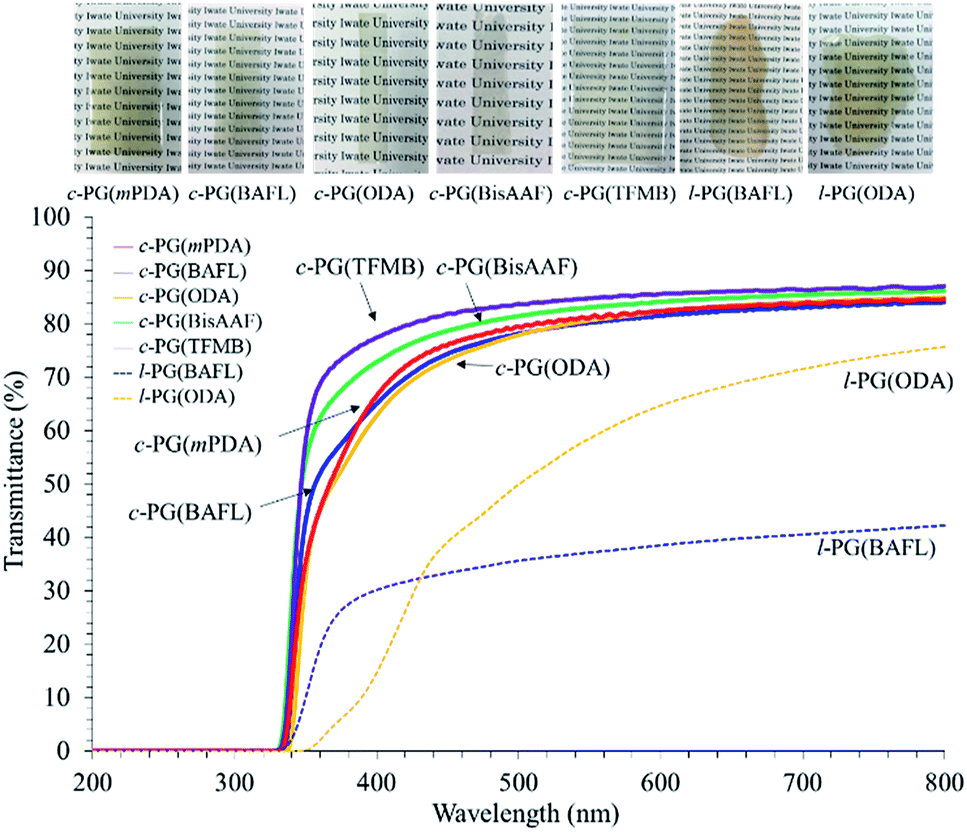
The optical properties of the prepared polymer films were analyzed using a UV-vis spectrophotometer and prism coupler, as summarized in Table 4 and Fig. 9.
Table 4 Optical properties of PG polymers
| Polymer |
Thickness (μm) |
|
na |
νb |
Δnc |
naved |
| 473 nm |
594 nm |
657 nm |
| TE, in-plane; TM, out-of-plane. Abbe's number. Birefringence at 594 nm. nave =((2 × nTE2 + nTM2)/3)0.5: nTE and nTM measured at 594 nm were used. |
| c-PG (mPDA) |
13.0 |
TE |
1.8251 |
1.7819 |
1.7679 |
15 |
0.0754 |
1.7571 |
| TM |
1.7357 |
1.7065 |
1.6957 |
20 |
| c-PG (BAFL) |
12.0 |
TE |
1.7839 |
1.7476 |
1.7352 |
17 |
0.0421 |
1.7337 |
| TM |
1.7347 |
1.7055 |
1.6953 |
20 |
| c-PG (ODA) |
9.0 |
TE |
1.8078 |
1.7677 |
1.7538 |
16 |
0.0793 |
1.7417 |
| TM |
1.7154 |
1.6884 |
1.6786 |
21 |
| c-PG (BisAAF) |
11.0 |
TE |
1.7237 |
1.6916 |
1.6817 |
19 |
0.0572 |
1.6728 |
| TM |
1.6560 |
1.6344 |
1.6255 |
23 |
| c-PG (TFMA) |
11.0 |
TE |
1.7398 |
1.7050 |
1.6938 |
17 |
0.0844 |
1.6773 |
| TM |
1.6413 |
1.6206 |
1.6130 |
24 |
| l-PG (BAFL) |
71.0 |
TE |
1.7666 |
1.7339 |
1.7218 |
18 |
0.0090 |
1.7309 |
| TM |
1.7498 |
1.7249 |
1.7129 |
20 |
| l-PG (ODA) |
47.0 |
TE |
1.7429 |
1.7133 |
1.7007 |
19 |
0.0058 |
1.7114 |
| TM |
1.7349 |
1.7075 |
1.6959 |
20 |
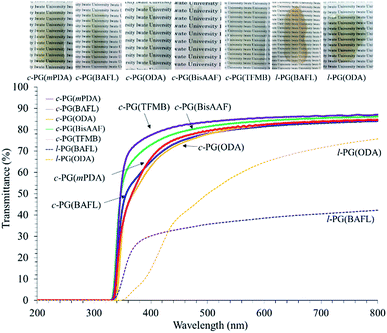 |
| | Fig. 9 UV-vis spectra of PG films. | |
The c-PG films were slightly yellow in colour but were more colourless and transparent compared with the l-PG films. The Mn values of the l-PG polymers were 7300–13400 and showed slightly better solubility in the aprotic solvent. However, the tensile and optical properties of l-PG polymers were obviously inferior to those of the c-PG films.
The refractive index (n) was measured by a prism coupler, and the results are summarized in Table 4. The PG films generally show a high refractive index (n594) of approximately 1.7 and low Abbe number (ν) owing to their tight packing and the high molecular refraction of the triazine component. l-PG(ODA) and l-PG(BAFL) showed average n594 values of 1.7114 and 1.7309, respectively, whereas the c-PG counterpart films showed values of 1.7417 and 1.7337, respectively. The slightly higher n594 values can be attributed to the azacalixarene moieties. The ν values of the c-PG films were larger than those of the l-PG films. These results can be explained by the strong interaction with azacalixarene, which somehow regulates the chain motion of the polymers in the film.
Table 5 summarizes the major properties of PG polymers categorized in their skeleton. As a solubilizing group, an alicyclic pendant such as adamantly group (AdDCT) is somewhat effective with keeping the thermal stability, but the n594 of the brittle thin film is only 1.672 (for BAFL).32 Simple anilino pendant in AnDCT is more effective to suppress over-aggregation, so the soluble transparent high Mn polymers are successfully obtained when BAFL is used to avoid cyclization.32 Tg of the polymer is 287 °C, and its n594 is improved up to 1.709. Semiaromatic-type PG polymers using α,ω-alkylene diamines as monomers generally show poor solubility in organic solvents with the strong aggregation and the packing caused by crystalline nature, which cause low transparency in the state of film.34 In order to secure the solubility, longer alkylene diamine segment such as DDDA is required, but the prepared polymer shows poor thermal stability (low Tg of 76 °C, low Td5 of 438 °C, and low char at 800 °C of only 7.0%). This can be caused by the weak packing of alkylene chain with the low weight fraction of triazine moiety. The film is relatively low transparency with low n594 of 1.616. In contrast with the above mentioned PG polymers, the c-PG polymers developed in this study showed good solubility in aprotic polar organic solvents, and the formation of the cyclic oligomer during the polymerization process be effectively avoidable. Extraordinary high thermal stabilities (Tg > 380 °C, Td5 > 520 °C, and char at 800 °C > 61%) with excellent optical properties (λcut off 332 nm, n594 > 1.75) should be the gifts from the cyclic skeleton of mPDA2CyC2 group with its dense packing utilizing its multiple hydrogen bondable ability.
Table 5 Properties of PG polymers categorized in the skeleton
| PG type |
Dichloridea |
Diamineb |
Mnc (kDa) |
Tg (°C) |
Td5(N2) (°C) |
Char (%) |
n594f |
| AdDCT: adamantylamino-s-triazine dichloride. HMDA: hexamethylene diamine, DDDA: dodecylene diamine. Determined by GPC (NMP, PS standards). Determined by DSC. Determined by DMA. Refractive index at 594 nm. |
| Alkyl side pendant |
AdDCT |
mPDA |
3.6 |
271d |
430 |
47.0 |
— |
| BAFL |
13.5 |
347d |
468 |
52.0 |
1.672 |
| Semiaromatic |
AnDCT |
HMDA |
(Insoluble) |
161d |
405 |
20.7 |
— |
| DDDA |
68.0 |
76d |
438 |
7.0 |
1.616 |
| Wholly aromatic |
AnDCT |
mPDA |
3.1 |
209d |
418 |
58.0 |
1.783 |
| BAFL |
36.1 |
287d |
448 |
54.5 |
1.709 |
| c-PG |
mPDA2CyC2 |
mPDA |
26.0 |
368e |
506 |
61.8 |
1.757 |
| BAFL |
24.9 |
386e |
522 |
61.1 |
1.734 |
Conclusions
In this study, we successfully synthesized a series of high molecular weight poly(guanamine) (c-PG)s containing an azacalixarene moiety. We revealed their outstanding thermal, mechanical, and optical properties and compared them with their linear counterparts (l-PG). Introducing multiple hydrogen bondable units of the azacalixarene moiety into the polymer skeleton provided elastomeric exclusively to the polymers, affording these unusual properties. We believe that our findings from this research will be applicable to the design and synthesis of a novel series of ring-containing polymers and materials.
Experimental
Materials
Cyanuric chloride (CyC) (TCI Chemical), N,N-diisopropylethylamine (DIPA) (Kanto Chemical), and ammonium hydroxide solution in water (Wako Pure Chemical) were purchased and used as received. m-Phenylene diamine (mPDA), p-phenylene diamine (pPDA), and 4,4′-oxydianiline (ODA) were purchased from TCI Chemical and purified via sublimation. 9,9-Bis(4-aminophenyl)fluorene (BAFL) was purified via recrystallization from ethanol before use. 2,2′-Bis(trifluoromethyl)benzidine (TFMB) and 2,2-bis(4-aminophenyl)hexafluoropropane (BisAAF) were purchased from TCI Chemical and used as received. 2-Anilino-4,6-dichloro-1,3,5-triazine (AnDCT) was prepared by the method given in our previous report.32 N-Methylpyrrolidone (NMP) was dried over calcium hydride (CaH2) and distilled under reduced pressure prior to use. Anhydrous tetrahydrofuran (THF) was purchased from Wako Pure Chemical and used as received. All other reagents and solvents were used as received.
Synthesis of mPDA2CyC2
The compound mPDA2CyC2 was prepared by Wang's method.25 Briefly, two equivalents of CyC were reacted with mPDA in the presence of DIPA in THF at −5 °C for 18 hours to give N,N′-bis(dichloro-s-triazinyl)-m-phenylenediamine (71% yield), which was further reacted with mPDA in acetone at 20 °C for 48 hours. The final product was purified by recrystallization with hexane–dichloromethane mixed solvent (45% yield). Mp > 300 °C (lit. 25, mp > 300 °C). FT-IR [KBr (cm−1)]: 3246 (N–H), 1596 (CC), 1576 (CN), 1558 (CC), 1388 (CC). 1H-NMR (400 MHz, DMSO-d6, ppm) δ 6.80 (dd, 4H, Ar-H), 7.23 (t, 2H, Ar-H), 7.76 (s, 2H, Ar-H), 10.01 (s, 4H, N–H). 13C-NMR (100 MHz, DMSO-d6, ppm) δ 118.2, 118.8, 129.0, 137.9, 164.3, 168.0. FAB-mass found: m/z = 439.0702 (C18H13Cl2N10) Err [ppm/mmu] = +3.7/+1.6.
Synthesis of linear-type PG (l-PG)
The typical procedure used for poly(AnDCT-mPDA) (l-PG(mPDA)) was as follows:32 all of the glass vessels were heated under a vacuum before use and were filled with and handled in a stream of dry nitrogen. In a two-neck flask equipped with a three-way stopcock, reflux condenser, and magnetic stirrer, mPDA (0.216 g, 2.00 mmol) was dissolved in dry NMP (4 mL) containing lithium chloride (5 wt%) at room temperature. To this solution, AnDCT (0.482 g, 2.00 mmol) was added in one portion. The mixture was stirred for 10 min, followed by stirring at 110 °C for 9 hours. The polymerization solution was then cooled to room temperature and poured into an excess amount of methanol containing NH3 (aq.) to remove HCl and inorganic salts produced during the reaction. The precipitate was dried at 60 °C for 12 hours, washed with hot methanol, and dried at 140 °C for 12 hours under reduced pressure. This procedure afforded the title polymer in 0.364 g (66%) yield. FT-IR [KBr (cm−1)]: 3385 (N–H), 3335–32887 (N–H, H-bonded), 1625 (CC), 1576 (CN), 1500 (CC), 1475 (CC), 1441, 1406, 1365 (CN), 1219, 1188, 1162 (C–N), 801, 767, 745, 686 (C–H). 1H NMR (400 MHz, DMSO-d6, ppm) δ 6.91 (aromatic CH, 1H), 7.22 (aromatic CH, 3H), 7.50 (aromatic CH, 1H), 7.78 (aromatic CH, 2H), 7.98 (aromatic CH, 1H), 9.10 (guanamine NH, 2H), 9.18 (aniline NH, 1H).
Synthesis of mPDA2CyC2 containing PG (c-PG)
The typical procedure used for poly(mPDA2CyC2-mPDA) (c-PG(mPDA)) was as follows: in a two-neck flask equipped with a three-way stopcock, reflux condenser, and magnetic stirrer, mPDA (0.108 g, 1.00 mmol), lithium chloride (5 wt%), and dry NMP (5.0 mL) were added at room temperature. To this solution, mPDA2CyC2 (0.439 g, 1.00 mmol) was added, and the mixture was stirred for 10 minutes, followed by stirring at 150 °C for 6 hours. The polymerization solution was then cooled to room temperature and poured into an excess amount of methanol (300 mL) containing NH3 (aq.) (3 mL) to remove HCl and inorganic salts produced during the reaction. The precipitate was washed with hot methanol and dried at 180 °C for 12 hours under reduced pressure. This procedure yielded 0.403 g (85%) of the title polymer. FT-IR [KBr (cm−1)]: 3616, 3381 (N–H), 3330–2896 (N–H, H-bonded), 1576 (CN), 1508 (CC), 1441 (CN), 1400 (CC), 1185, 1161 (C–N), 868, 801, 768, 677 (C–H). 1H-NMR (400 MHz, DMSO-d6, ppm) δ 6.76 (CH, 4H), 7.17 (CH, 3H), 7.58 (CH, 2H), 7.80 (aromatic CH, 2H), 8.23 (aromatic CH, 1H), 8.83 (guanamine NH, 2H), 8.90 (ring guanamine NH, 4H).
Poly(mPDA2CyC2-pPDA) (c-PG(pPDA))
A similar procedure described above was used except for using pPDA (0.108 g, 1.00 mmol) instead of mPDA. Yield 0.260 g (55%). FT-IR[KBr (cm−1)]: 3616, 3377 (N–H), 3330–3110 (N–H, H-bonded), 1579 (CN), 1485 (CC), 1441 (CN), 1398 (CC), 1200 (C–N), 864, 806, 768, 677 (C–H).
Poly(mPDA2CyC2-ODA) (c-PG(ODA))
A similar procedure described above was used except for using ODA (0.200 g, 1.00 mmol) instead of mPDA. Yield 0.515 g (91%). FT-IR[KBr (cm−1)]: 3612, 3389 (N–H), 3330–3110 (N–H, H-bonded), 1574 (CN), 1494 (CC), 1439 (CN), 1405 (CC), 1205 (C–O), 1159 (C–N), 869, 802, 764, 675 (C–H). 1H-NMR (400 MHz, DMSO-d6, ppm) δ 6.75 (aromatic CH, 4H), 6.92 (aromatic CH, 4H), 7.15 (aromatic CH, 2H), 7.77 (aromatic CH, 2H), 7.85 (aromatic CH, 4H), 8.89 (ring guanamine NH, 4H), 9.03 (guanamine NH, 2H).
Poly(mPDA2CyC2-BAFL) (c-PG(BAFL))
A similar procedure described above was used except for using BAFL (0.348 g, 1.00 mmol) instead of mPDA. Yield 0.657 g (92%). FT-IR[KBr (cm−1)]: 3623, 3384 (N–H), 3330–3110 (N–H, H-bonded), 1573 (CN), 1509 (CC), 1445 (CN), 1412 (CC), 1228, 1188, 1165 (C–N), 873, 808, 745, 680 (C–H). 1H-NMR (400 MHz, DMSO-d6, ppm) δ 6.71 (aromatic CH, 4H), 7.03 (aromatic CH, 4H), 7.12 (aromatic CH, 2H), 7.32–7.45 (aromatic CH, 6H), 7.73 (aromatic CH, 6H), 7.91 (aromatic CH, 2H), 8.85 (ring guanamine NH, 4H), 9.05 (guanamine NH, 2H).
Poly(mPDA2CyC2-TFMB) (c-PG(TFMB))
A similar procedure described above was used except for using TFMB (0.320 g, 1.00 mmol) instead of mPDA. Yield 0.570 g (83%). FT-IR[KBr (cm−1)]: 3645, 3402 (N–H), 3330–3110 (N–H, H-bonded), 1571 (CN), 1513 (CC), 1445 (CN), 1404 (CC), 1315, 1164 (C–N), 1113 (C–F), 870, 804, 760, 684 (C–H). 1H-NMR (400 MHz, DMSO-d6, ppm) δ 6.80 (aromatic CH, 4H), 7.23 (aromatic CH, 4H), 7.76 (aromatic CH, 2H), 8.04 (aromatic CH, 2H), 8.53 (aromatic CH, 2H), 9.06 (ring guanamine NH, 4H), 9.49 (guanamine NH, 2H).
Poly(mPDA2CyC2-BisAAF) (c-PG(BisAAF))
A similar procedure described above was used except for using BisAAF (0.334 g, 1.00 mmol) instead of mPDA. Yield 0.588 g (84%). FT-IR[KBr (cm−1)]: 3604, 3399 (N–H), 3330–3110 (N–H, H-bonded), 1576 (CN), 1510 (CC), 1447 (CN), 1406 (CC), 1238 (C–F), 1196, 1164 (C–N), 963, 924 (C–CF3), 802 (C–H). 1H-NMR (400 MHz, DMSO-d6, ppm) δ 6.76 (aromatic CH, 4H), 7.16 (aromatic CH, 2H), 7.23 (aromatic CH, 4H), 7.78 (aromatic CH, 2H), 8.03 (aromatic CH, 4H), 8.99 (ring guanamine NH, 4H), 9.36 (guanamine NH, 2H).
Film preparation
The polymer sample was dissolved in DMAc (5 wt%), and the homogeneous solution was drop-cast onto a glass plate. After drying in an oven at room temperature for 12 hours, the film was sequentially dried at temperatures of 50 °C, 80 °C, and 120 °C for 3 hours each. Finally, the film was dried at 150 °C for 12 hours.
Measurements
FT-IR spectra were recorded on a JASCO FT/IR-4200 using the KBr pellet method. NMR spectroscopy was performed on a Bruker AC-400P spectrometer at 400 MHz for 1H and 100 MHz for 13C measurements. Deuterated dimethyl sulfoxide (DMSO-d6) was used as the solvent, with tetramethylsilane as the internal reference. The Mn, Mw, and Mw/Mn values were determined using a Tosoh HLC-8220 gel permeation chromatograph equipped with refractive index and UV detectors and a consecutive polystyrene gel column (TSK-GEL α-M ×2) at 40 °C and eluted with NMP containing 0.01 mol L−1 LiBr at a flow rate of 1.0 mL min−1. Thermogravimetric analysis was performed on a Hitachi TG/DTA7220 system at a heating rate of 10 °C min−1 under exposure to air or N2. Differential scanning calorimetry (DSC) was performed on a Hitachi X-DSC7000 system at a heating rate of 20 °C min−1 under nitrogen. Thermal mechanical analysis (TMA) was performed on a Hitachi TMA/SS7100 at a heating rate of 10 °C min−1 under nitrogen. Dynamic mechanical analysis (DMA) was performed on a Hitachi DMS7100 at a heating rate of 2 °C min−1 under nitrogen. Polymer films were cut into pieces of 20 mm × 9 mm × 0.12 mm dimensions, and a load was applied to the films with a strain amplitude of 5 μm and a frequency of 1 Hz. The tensile properties of the films were measured on an AGS-X (Shimadzu) at an extension rate of 10 mm min−1. The optical transparency of the PG films was measured using a Shimadzu UV-vis spectrophotometer (UV-1800). Refractive index and birefringence were measured by Metricon Prism Coupler Model 2010/M. Fast-atom-bombardment mass spectroscopy was performed using a JEOL JMS-700 mass spectrometer. FAB was generated using xenon as the primary beam at 6 keV energy. The ion-accelerating voltage was 10 kV and 3-nitrobenzyl alcohol was used for the FAB ionization matrix.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Ms Shiduko Nakajo (Iwate University) for FAB MS measurements.
Notes and references
- S. Kopolow, T. E. Hogen Esch and J. Smid, Macromolecules, 1971, 4, 359–360 CrossRef.
- M. J. Marsella and T. M. Swager, J. Am. Chem. Soc., 1993, 115, 12214–12215 CrossRef CAS.
- H. Cheng, L. Ma, Q. S. Hu, X. F. Zheng, J. Anderson and L. Pu, Tetrahedron: Asymmetry, 1996, 7, 3083–3086 CrossRef CAS.
- N. Yamaguchi and H. W. Gibson, Angew. Chem., Int. Ed., 1999, 38, 143–147 CrossRef CAS.
- J. Morgado, F. Cacialli, R. H. Friend, B. S. Chuah, S. C. Moratti and A. B. Holmes, Synth. Met., 2000, 111–112, 449–452 CrossRef CAS.
- S. H. Liao, Y. L. Li, T. H. Jen, Y. S. Cheng and S. A. Chen, J. Am. Chem. Soc., 2012, 134, 14271–14274 CrossRef CAS PubMed.
- M. Furue, A. Harada and S. Nozakura, J. Polym. Sci., Part C: Polym. Lett., 1975, 13, 357–360 CAS.
- S. Shinkai, H. Kawaguchi and O. Manabe, J. Polym. Sci., Part C: Polym. Lett., 1988, 26, 391–396 CrossRef CAS.
- Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic, Dordrecht, 2001 Search PubMed.
- C. D. Gutsche, Calixarenes: An Introduction, in Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008 Search PubMed.
- J. Ueda, M. Kamigaito and M. Sawamoto, Macromolecules, 1998, 31, 6762–6768 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 1998, 31, 7218–7225 CrossRef CAS.
- A. I. Costa, L. F. V. Ferreira and J. V. Prata, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6477–6488 CrossRef CAS.
- P. D. Barata, A. I. Costa, L. F. V. Ferreira and J. V. Prata, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5040–5052 CrossRef CAS.
- P. D. Barata, A. I. Costa and J. V. Prata, React. Funct. Polym., 2012, 72, 627–634 CrossRef CAS.
- J. H. Wosnick and T. M. Swager, Chem. Commun., 2004, 2744–2745 RSC.
- A. I. Costa, H. D. Pinto, L. F. V. Ferreira and J. V. Prata, Sens. Actuators, B, 2012, 161, 702–713 CrossRef CAS.
- P. D. Barata and J. V. Prata, ChemPlusChem, 2014, 79, 83–89 CrossRef CAS.
- J. V. Prata, A. I. Costa, G. Pescitelli and H. D. Pinto, Polym. Chem., 2014, 5, 5793–5803 RSC.
- A. Dondoni, C. Ghiglione, A. Marraa and M. Scoponi, Chem. Commun., 1997, 673–674 RSC.
- S. I. Kim, T. J. Shin, M. Ree, G. T. Hwang, B. H. Kim, H. Han and J. Seo, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2013–2026 CrossRef CAS.
- H. H. Yu, B. Xu and T. M. Swager, J. Am. Chem. Soc., 2003, 125, 1142–1143 CrossRef CAS PubMed.
- Y. Yang and T. M. Swager, Macromolecules, 2006, 39, 2013–2015 CrossRef CAS.
- Y. Yang and T. M. Swager, Macromolecules, 2007, 40, 7437–7440 CrossRef CAS.
- M. X. Wang and H. B. Yang, J. Am. Chem. Soc., 2004, 126, 15412–15422 CrossRef CAS PubMed.
- B. Y. Hou, D. X. Wang, H. B. Yang, Q. Y. Zeng and M. X. Wang, J. Org. Chem., 2007, 72, 5218–5226 CrossRef CAS PubMed.
- D. W. Wang and M. M. Fisher, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 671–677 CrossRef CAS.
- N. Irles, J. Puiggalí and J. A. Subirana, Macromol. Chem. Phys., 2001, 202, 3316–3322 CrossRef CAS.
- Y. Shibasaki, T. Koizumi, N. Nishimura and Y. Oishi, Chem. Lett., 2011, 40, 1132–1134 CrossRef CAS.
- K. Saito, N. Nishimura, S. Sasaki, Y. Oishi and Y. Shibasaki, React. Funct. Polym., 2013, 73, 756–763 CrossRef CAS.
- A. Fujimori, S. Miura, T. Kikkawa and Y. Shibasaki, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 999–1009 CrossRef CAS.
- T. Kotaki, N. Nishimura, M. Ozawa, A. Fujimori, H. Muraoka, S. Ogawa, T. Korenaga, E. Suzuki, Y. Oishi and Y. Shibasaki, Polym. Chem., 2016, 7, 1297–1308 RSC.
- S. Miura, Y. Shidara, T. Yunoki, M. A. A. Mamun, Y. Shibasaki and A. Fujimori, Macromol. Chem. Phys., 2017, 218, 1600520 CrossRef.
- Y. Shibasaki, T. Kotaki, T. Bito, R. Sasahara, N. Idutsu, A. Fujimori, S. Miura, Y. Shidara, N. Nishimura and Y. Oishi, Polymer, 2018, 146, 12–20 CrossRef CAS.
- R. Wang, C. Pellerin and O. Lebel, J. Mater. Chem., 2009, 19, 2747–2753 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09136j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
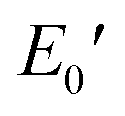 ) of 1.72 GPa. This value started to sharply decrease from 137 °C (Ts), reaching a rubbery plateau from 228 °C (Tr) at an E′ value (
) of 1.72 GPa. This value started to sharply decrease from 137 °C (Ts), reaching a rubbery plateau from 228 °C (Tr) at an E′ value (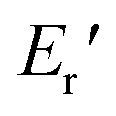 ) of 0.0135 GPa. For the l-PG(BAFL) film, the DMA curve showed better thermal stability owing to its rigid and bulky fluorene pendant effect, i.e., the values of Ts, Tr,
) of 0.0135 GPa. For the l-PG(BAFL) film, the DMA curve showed better thermal stability owing to its rigid and bulky fluorene pendant effect, i.e., the values of Ts, Tr, 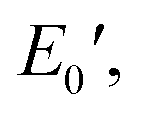 and
and 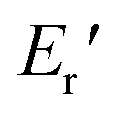 were 199.7 °C, 288.8 °C, 4.79 GPa, and 0.0257 GPa, respectively. For the c-PG films, the values of Ts, Tr,
were 199.7 °C, 288.8 °C, 4.79 GPa, and 0.0257 GPa, respectively. For the c-PG films, the values of Ts, Tr, 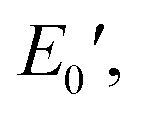 and
and 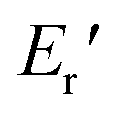 were significantly improved to 372.4 °C, 423.6 °C, 5.18 GPa, and 0.506 GPa for c-PG(ODA) and 387.1 °C, > 425.5 °C, 5.98 GPa, and > 0.615 GPa for c-PG(BAFL), respectively. The plot of tan
were significantly improved to 372.4 °C, 423.6 °C, 5.18 GPa, and 0.506 GPa for c-PG(ODA) and 387.1 °C, > 425.5 °C, 5.98 GPa, and > 0.615 GPa for c-PG(BAFL), respectively. The plot of tan