DOI:
10.1039/C9RA09228E
(Paper)
RSC Adv., 2020,
10, 533-540
Gold dipyrrin-bisphenolates: a combined experimental and DFT study of metal–ligand interactions†
Received
7th November 2019
, Accepted 18th December 2019
First published on 2nd January 2020
Abstract
Given that noninnocent and metalloradical-type electronic structures are ubiquitous among dipyrrin-bisphenolate (DPP) complexes, we synthesized the gold(III) derivatives as potentially innocent paradigms against which the properties of other metallo-DPP derivatives can be evaluated. Electronic absorption spectra, electrochemical studies, a single-crystal X-ray structure, and DFT calculations all suggest that the ground states of the new complexes indeed correspond to an innocent AuIII–DPP3−, paralleling a similar description noted for Au corroles. Interestingly, while DFT calculations indicate purely ligand-centered oxidations, reduction of AuDPP is predicted to occur across both the metal and the ligand.
1. Introduction
The dipyrrin-bisphenols (H3DPP) are an emerging class of ligands that share a number of similarities with corroles (Fig. 1).1 Thus, they are triprotic, afford a square-planar environment for coordinated metals, and even exhibit redox-active behavior remarkably similar to that of corroles.2,3 Thus, like their corrole analogues,4–6 a number of M[DPP] derivatives (M = Co,7,8 Ni,7 Cu9) are best regarded as MII–L˙2−, as opposed to MIII–L3−. These similarities promise applications of M[DPP] derivatives in catalytic transformations, in which metallocorroles have already proved useful.10,11 To better understand the role of metalloradical or noninnocent states in DPP chemistry, we have for some time sought innocent M[DPP] complexes, whose properties can serve as standards against which other DPP derivatives can be evaluated. Given that gold(III) corroles have emerged as archetypes of innocent metallocorroles,12–15 we synthesized a series of gold(III) dipyrrin-bisphenolate derivatives, which, as described below, also appear to exhibit innocent AuIII–DPP3− ground states.
 |
| | Fig. 1 Free-base dipyrrin-bisphenol and meso-triarylcorrole ligands. | |
2. Results and discussion
2.1 Physical measurements
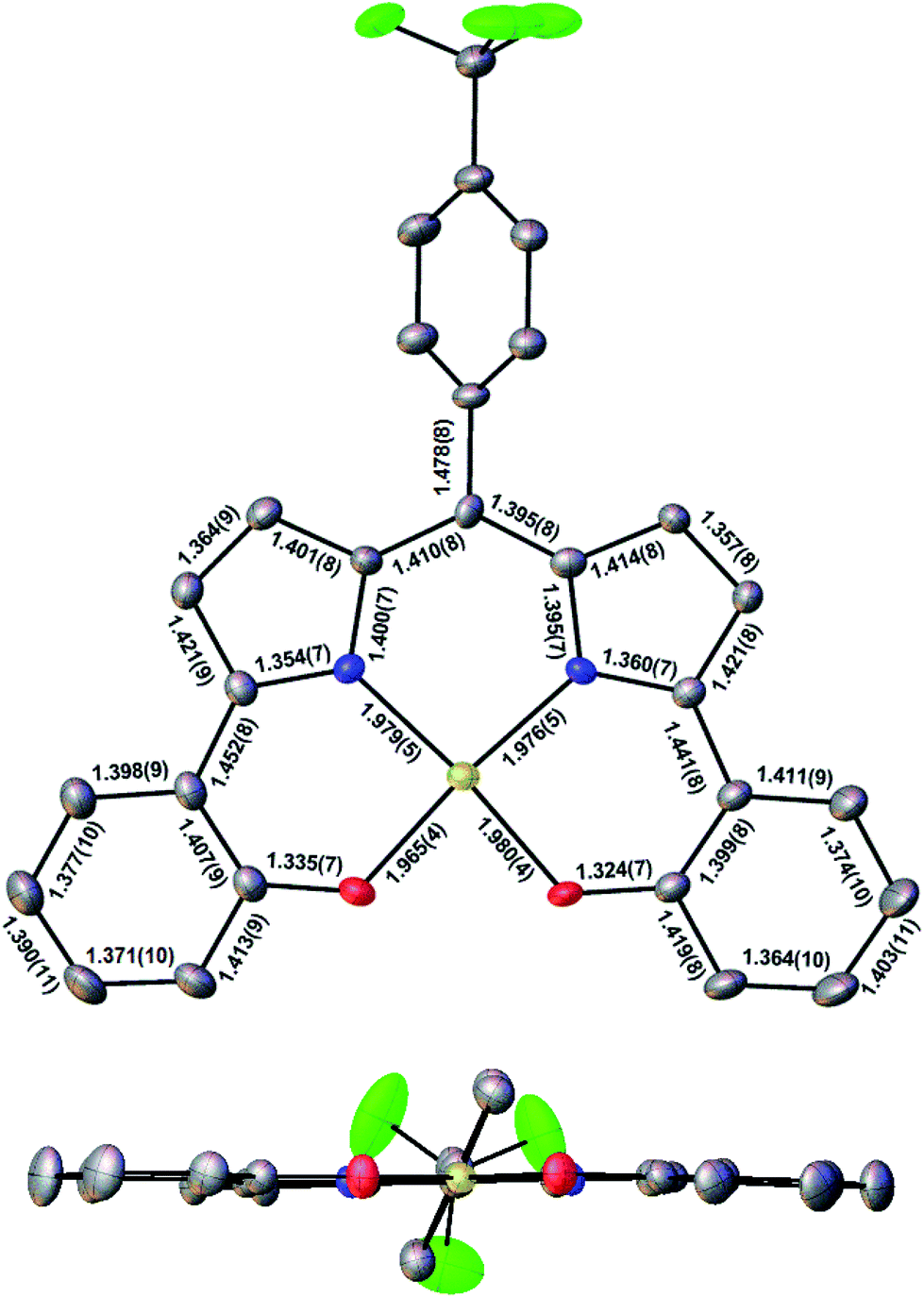
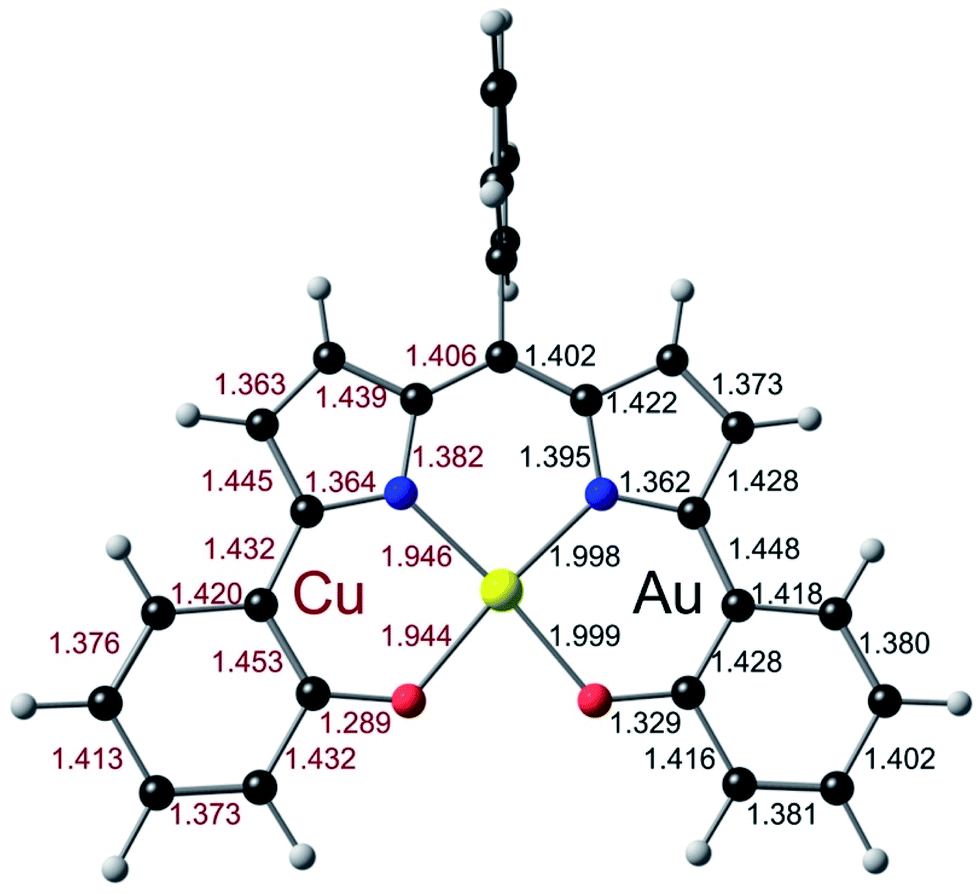
Three Au(III) meso-para-X-phenyl dipyrrin-bisphenolate complexes Au[XDPP] with X = CF3, H, and Me were obtained as blue solids in 50–77% yields via the interaction of the corresponding free ligands and a threefold molar excess of Au(III) acetate in pyridine.12 A single-crystal X-ray structure (Table 1 and Fig. 2) could be obtained for one of the complexes (X = CF3). The X-ray structure reveals Au–N/O distances of around 1.97 Å, which are approximately 0.02–0.03 Å longer than Au–N distances typically observed for Au(III) corroles.12–15 An examination of the skeletal bond distances of Au[CF3DPP] led to the interesting observation that the C–C bonds in the phenolate moieties span a narrower range (∼0.04 Å) relative to M(DPP) derivatives (∼0.08 Å) that are unambiguously describable as metalloradicals, as for M = Cu (CCSD: FICCEC, FICCUS)16 and Pt (LACCUQ, LACDAX).17 Similarly narrow C–C bond distance ranges are also observed for nonradicaloid Ge (VIVNAR, PONGOQ, SIRFOQ),18 Mn (UTOVEF19 and EXOBAV20), Al (NABFII),21–23 Ga (WOMPAS),24 and In (WOMPIA)24 DPP complexes.
Table 1 Crystal data and structure refinement for Au[CF3DPP]
| Sample |
Name |
| Chemical formula |
C28H16AuF3N2O2 |
| Formula mass |
666.39 |
| Crystal system |
Monoclinic |
| Crystal size (mm3) |
0.090 × 0.040 × 0.020 |
| Space group |
P21/c |
| λ (Å) |
0.7288 |
| a (Å) |
6.8844(4) |
| b (Å) |
16.4488(8) |
| c (Å) |
19.8240(10) |
| α (°) |
90 |
| β (°) |
98.802(2) |
| γ (°) |
90 |
| Z |
4 |
| V (Å3) |
2218.4(2) |
| Temperature (K) |
100(2) |
| Density (calculated) |
1.995 Mg m−3 |
| Measured reflections |
89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 167 167 |
| Unique reflections |
8107 |
| Parameters |
0 |
| Restraints |
325 |
| Rint |
0.0502 |
| θ range (°) |
2.482 to 33.562 |
| R1, wR2 all data |
0.0462, 0.1131 |
| S (GooF) all data |
1.332 |
| Max/min res. dens. (e Å−3) |
3.137/−2.242 |
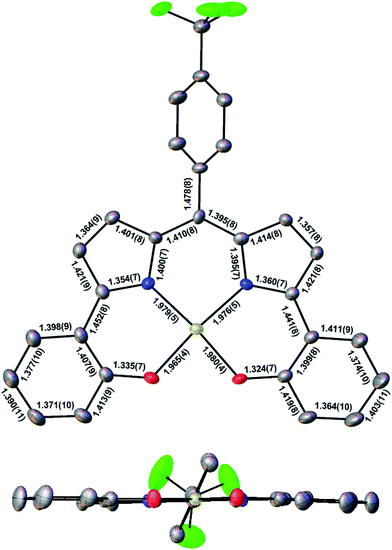 |
| | Fig. 2 Molecular structure of Au[CF3DPP]: top view (above) with selected distances (Å) and side view (below). | |
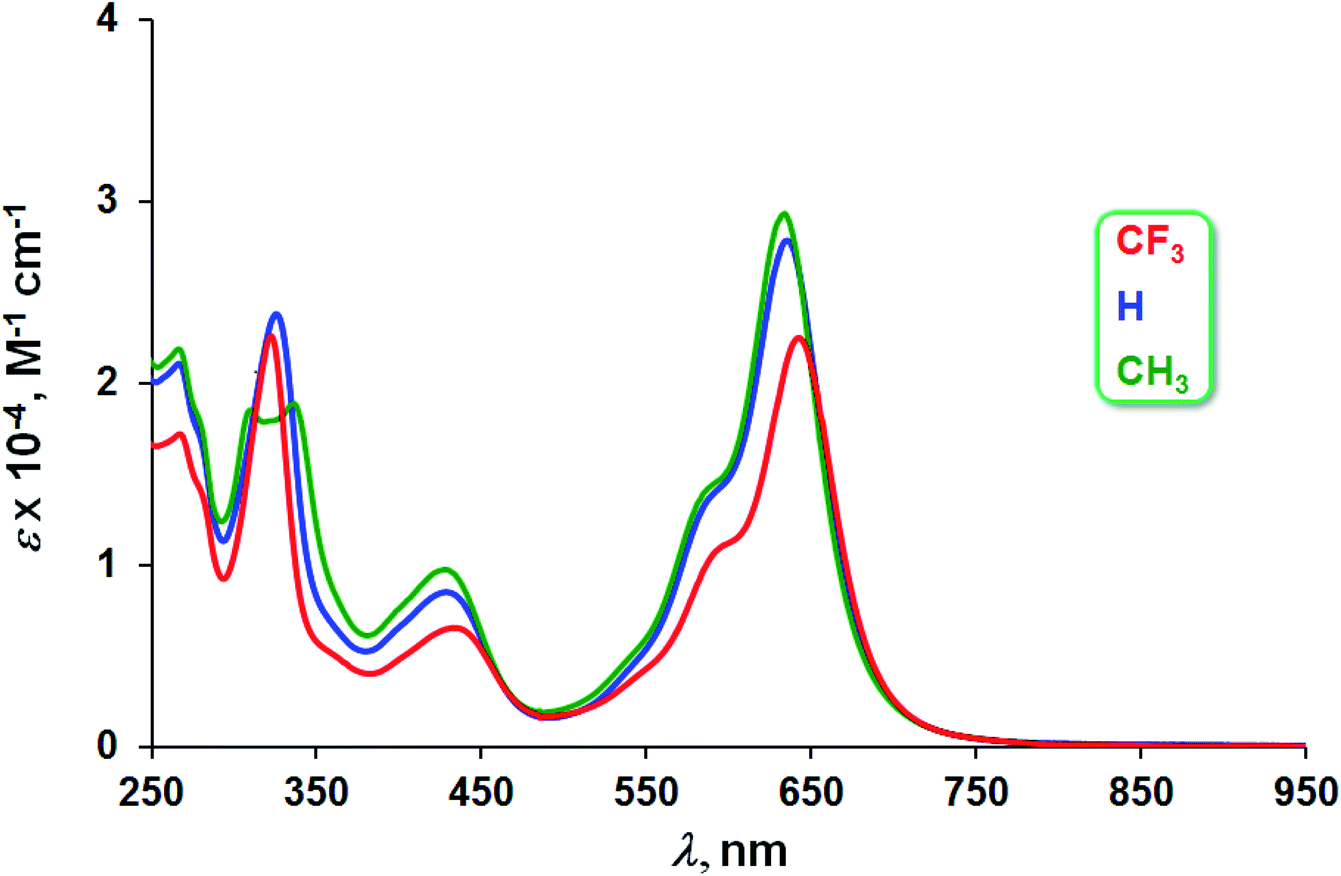
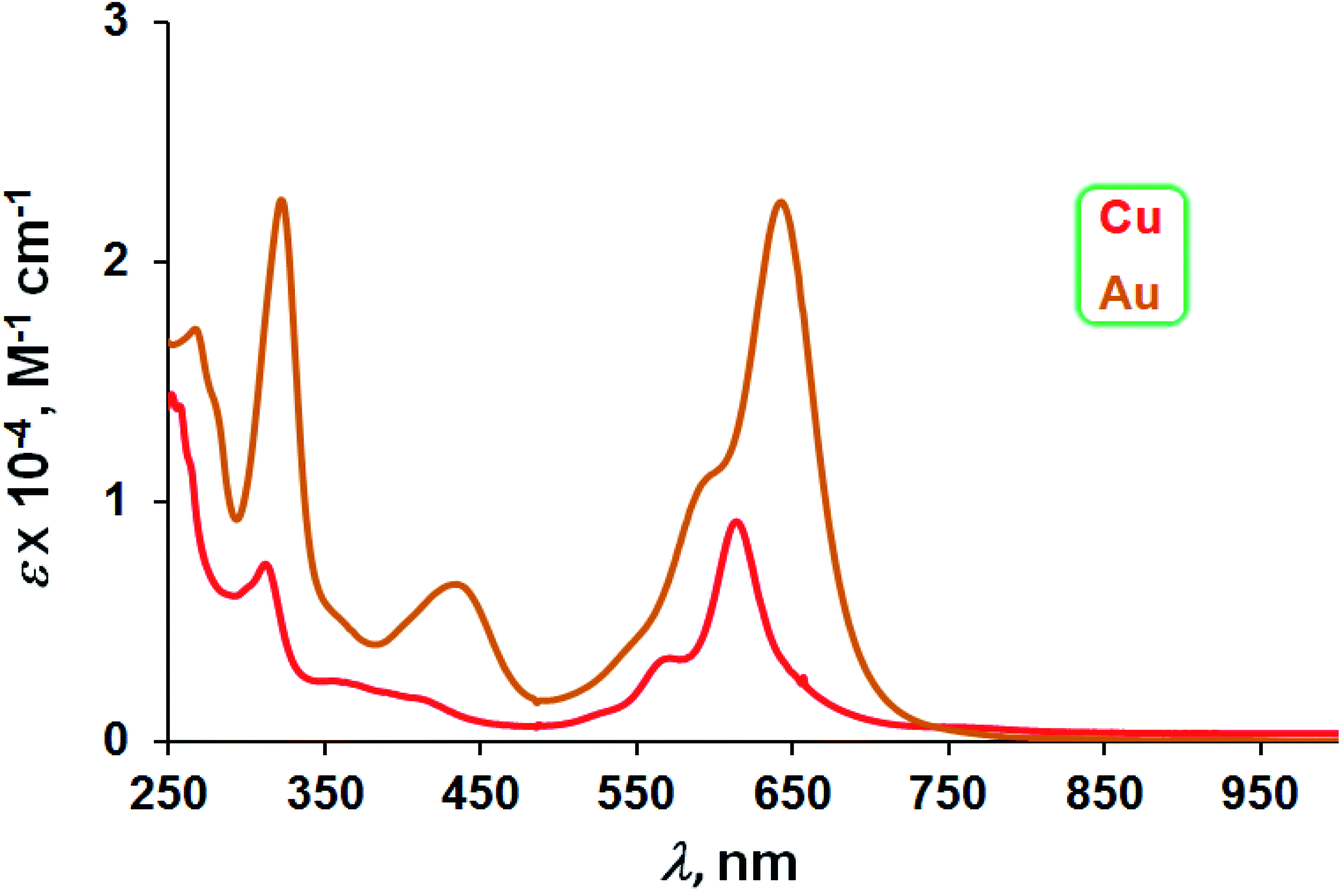
The three Au complexes exhibit similar optical spectra, with the strongest absorption occurring in the red at 639 ± 5 nm (Fig. 3). The molar absorptivities turned out to be around 3.0 × 104 M−1 cm−1, over three times the value observed for Cu[CF3DPP], consistent with an innocent electronic-structural description for the Au complexes and a radical description for the Cu complex (Fig. 4).25,26
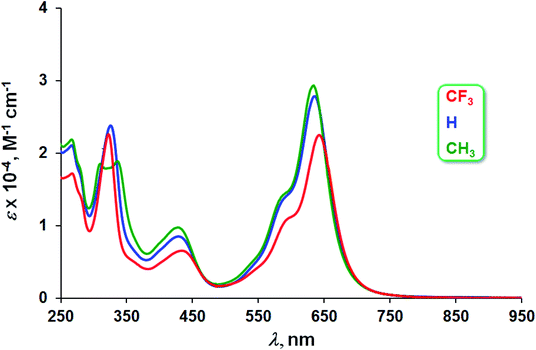 |
| | Fig. 3 UV-vis spectra of Au[XDPP] (X = CF3, H, and CH3) in dichloromethane. | |
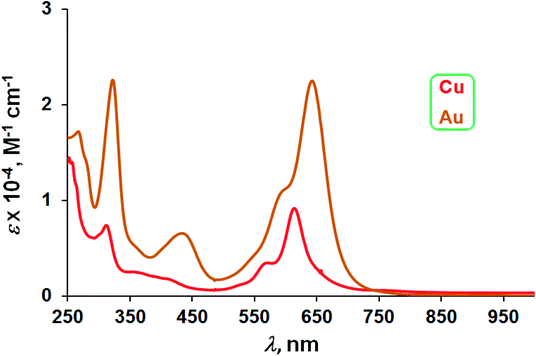 |
| | Fig. 4 UV-vis spectra of M[CF3DPP] (M = Cu and Au) in dichloromethane. | |
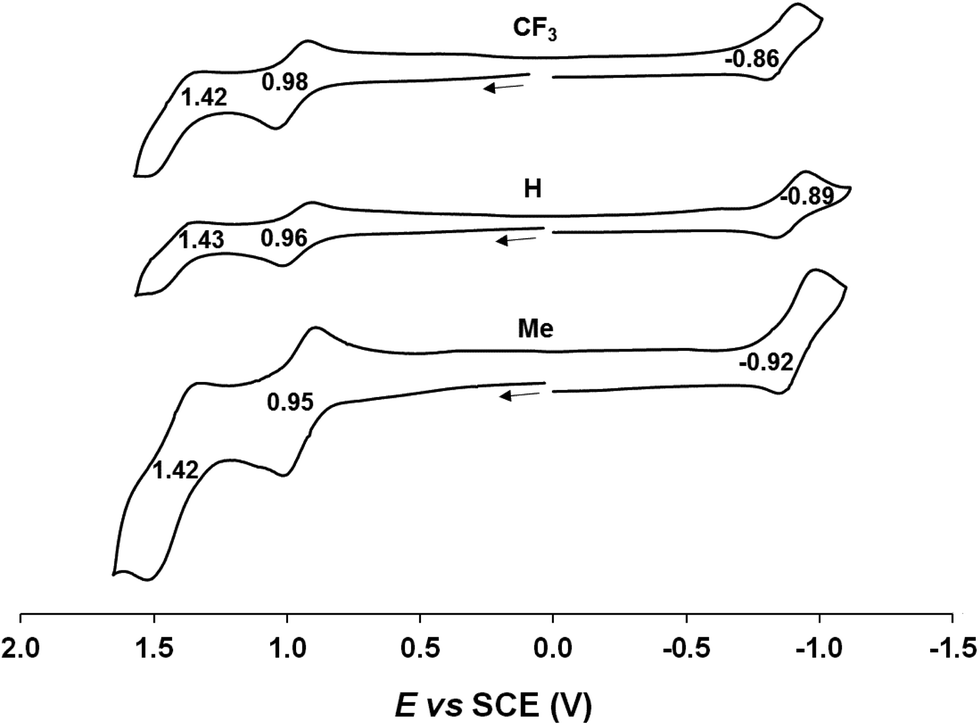
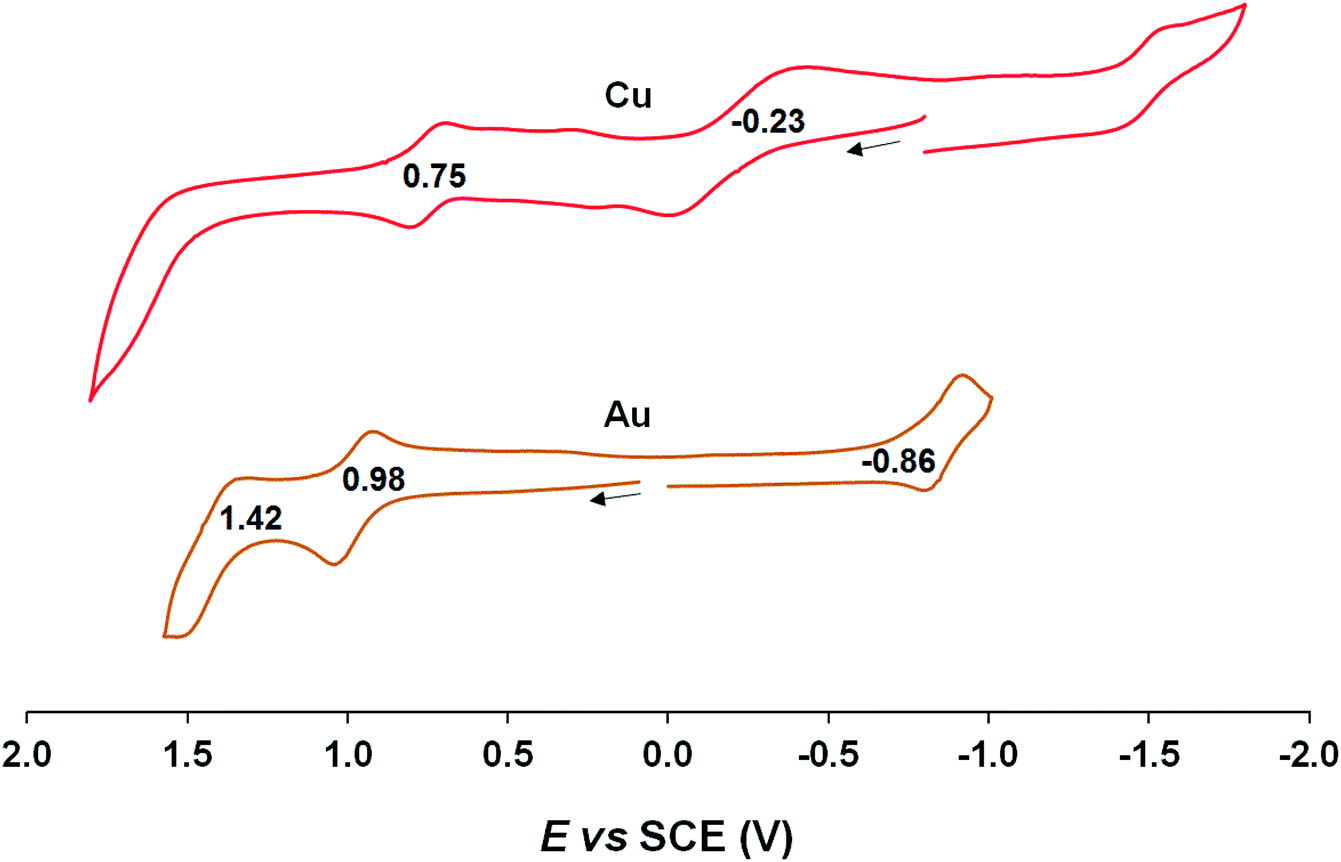
Cyclic voltammetric measurements, indicating relatively high oxidation potentials of around +0.95 V against the saturated calomel electrode (SCE),27 relatively low reduction potentials of around −0.90 V, and substantial electrochemical HOMO–LUMO gaps of around 1.85 V, are also suggestive of an innocent description for the Au[XDPP] complexes (Fig. 5). In contrast, Cu[CF3DPP] (Fig. 6) was found to exhibit an electrochemical HOMO–LUMO gap of just under 1.0 V (qualitatively consistent with similar observations elsewhere9). These electrochemical trends closely mirror those observed for analogous coinage metal corroles.6a,d,g,h,12,15b
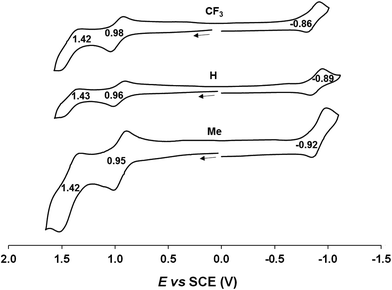 |
| | Fig. 5 Cyclic voltammograms of Au[XDPP] (X = CF3, H and CH3) in benzonitrile. Scan rate: 0.1 V s−1. | |
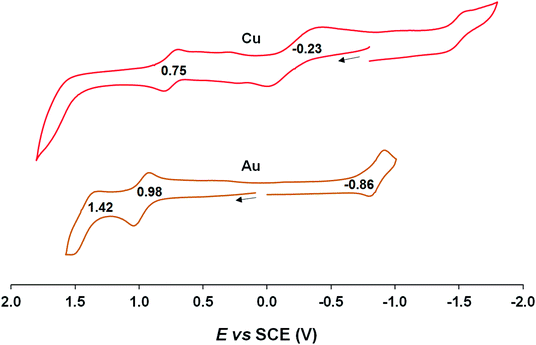 |
| | Fig. 6 Cyclic voltammograms of M[CF3DPP] (M = Cu and Au) in benzonitrile. Scan rate: for Cu is 0.05 V s−1 and 0.1 V s−1 for Au. | |
2.2 DFT calculations
To obtain a unified interpretation of the above findings, we carried out scalar-relativistic DFT (OLYP28,29/STO-TZ2P) calculations on the para-unsubstituted complexes M(DPP) for M = Cu and Au. For Cu, the ground state turned out to be a triplet, consistent with a CuII–L˙2− description.9 For Au, the ground state was unambiguously a singlet, with the triplet approximately 1.1 eV higher in energy. Fig. 7, which juxtaposes the optimized skeletal bond distances in the Cu- and Au-DPP complexes, confirms that the phenolate C–C bonds in the Au complex span a significantly smaller range than those in Cu[DPP].
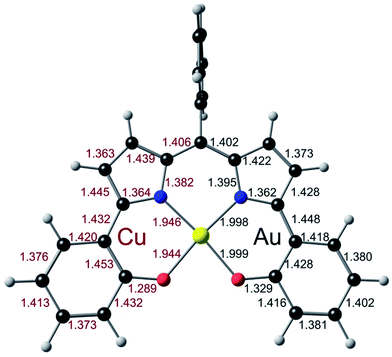 |
| | Fig. 7 Juxtaposition of the OLYP/STO-TZ2P optimized geometries (Å) Cu (left) and Au (right) DPP complexes. | |
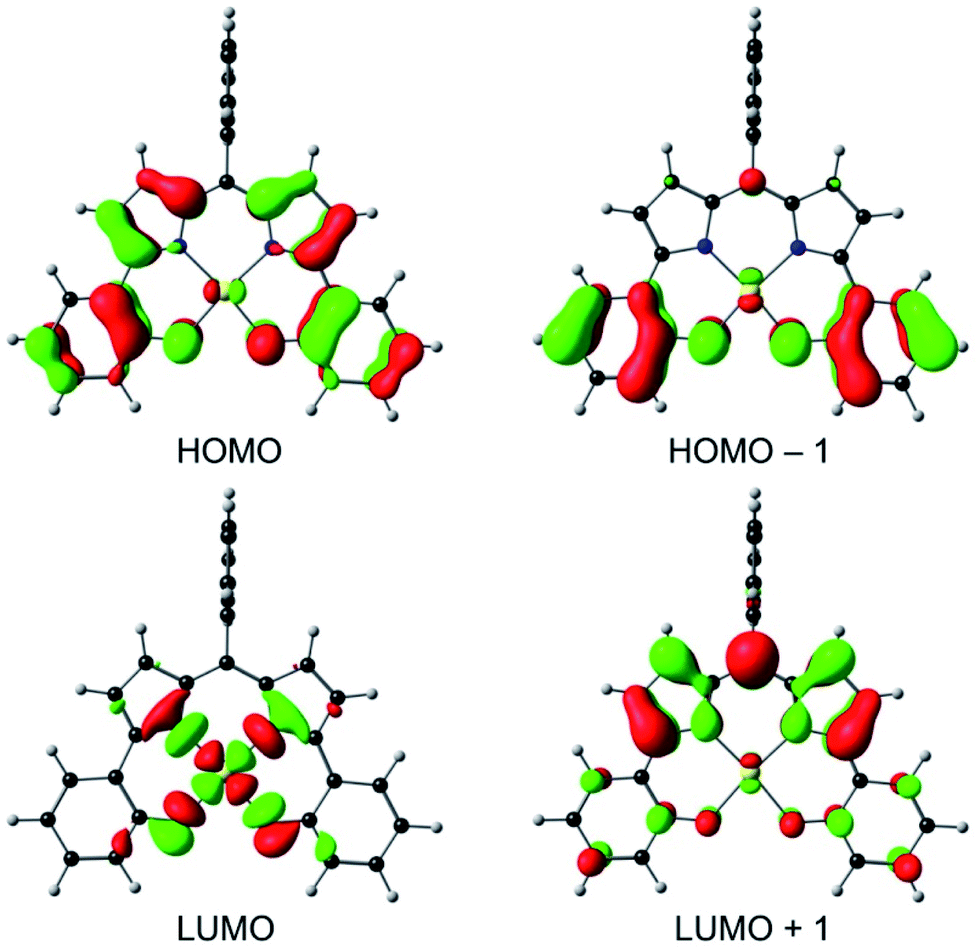
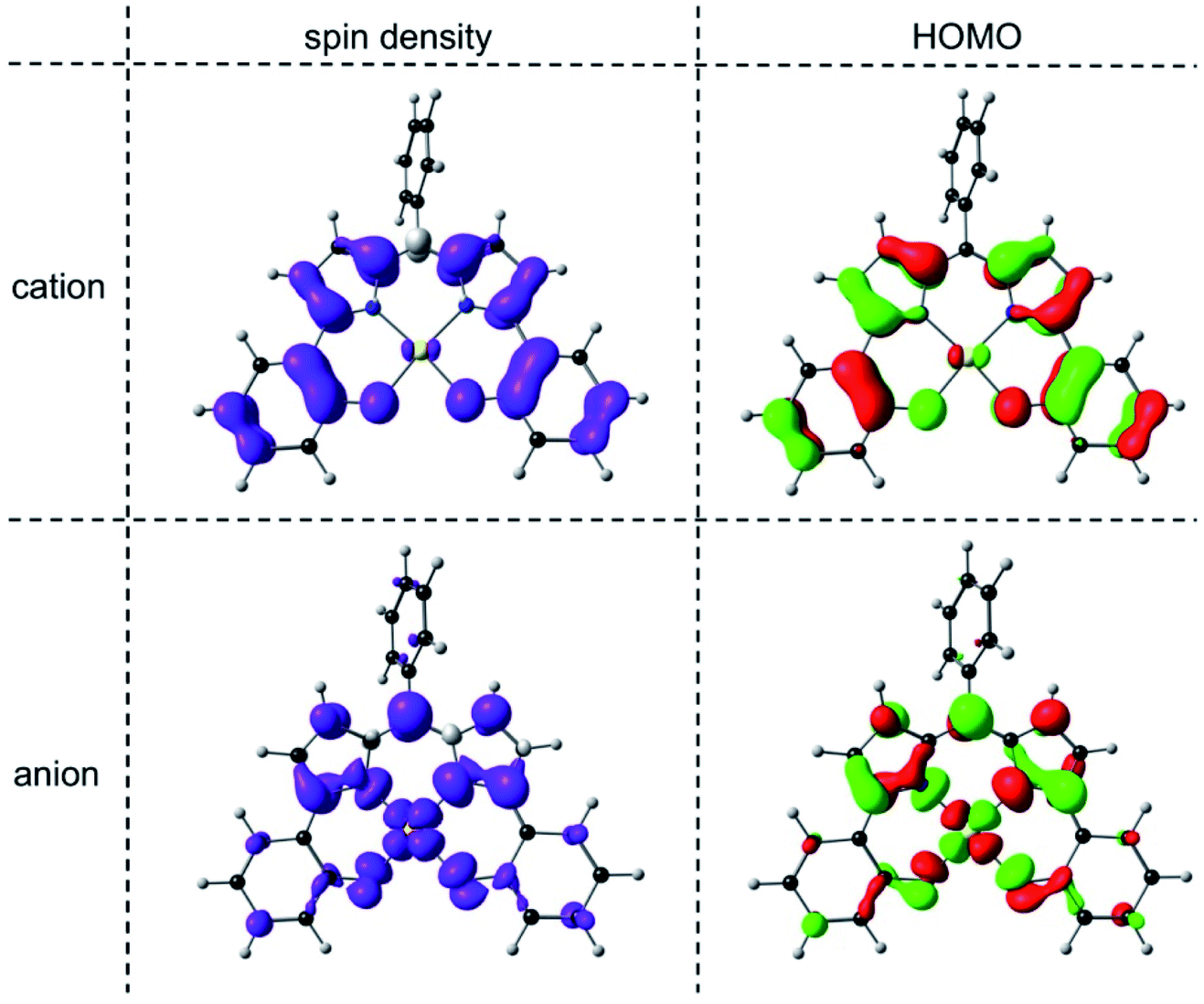
An examination of the Kohn–Sham frontier orbitals of Au[DPP] and their eigenvalues showed that while the ligand-based HOMO is energetically well-separated from the other MOs, the two LUMOs – a DPP-based π-symmetry MO and an Au 5dx2−y2-based σ-symmetry MO – are near-degenerate under C2v symmetry (Fig. 8). Relaxing the point group symmetry to C2 allows the two LUMOs to mix, as is clear from both the HOMO and spin density profiles of the Au[DPP]− anion. Thus, while confirming pure ligand-centered oxidation,30 the calculations provide a more nuanced picture of the reduction process, which occurs over both the Au and the DPP ligand (Fig. 9). In this respect, the Au–DPP complexes differ from simple Au corroles, which exhibit purely ligand-centered reduction.31 Presumably, the open-chain DPP ligands afford a more flexible coordination cavity for a larger, reduced Au center relative to the much more sterically constrained corroles.
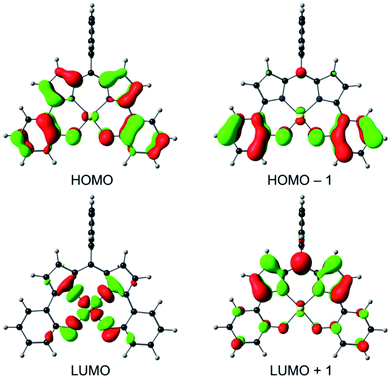 |
| | Fig. 8 OLYP/STO-TZ2P frontier MOs of Au[DPP] under a C2v symmetry constraint. | |
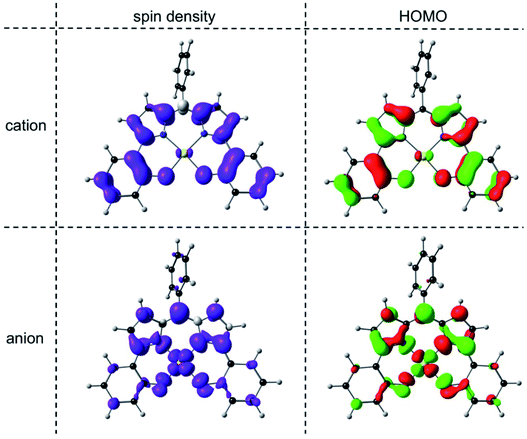 |
| | Fig. 9 OLYP/STO-TZ2P spin density and HOMO profiles of the cationic and anionic states of Au[DPP] optimized with a C2 symmetry constraint. | |
3. Conclusion
The first Au(III) dipyrrin-bisphenolate complexes, Au[XDPP] with X = CF3, H, and Me, have been synthesized in fair to good yields (50–77%) yields via the interaction of the corresponding free ligands and a threefold molar excess of Au(III) acetate in pyridine. X-ray structure analysis, optical spectroscopy, electrochemistry and DFT calculations all suggest an innocent AuIII–DPP3− description for the complexes. The calculations, however, also suggest that while the compounds undergo ligand-centered oxidation, reduction occurs across both the Au(5dx2−y2) orbital and the DPP π-LUMO. In the latter respect, the Au–DPP complexes differ from simple Au corroles, which undergo exclusively corrole-centered reductions.
4. Experimental section
4.1. Materials and instruments
All reagents and solvents were used as purchased unless noted otherwise. Benzonitrile was distilled from P4O10 and stored over activated 4 Å molecular sieves. Ultraviolet-visible (UV-vis) spectra were recorded in CH2Cl2 on an HP 8454 or a Varian Cary 50 spectrophotometer. Unless otherwise mentioned, 1H (400 MHz) and 19F (376 MHz) NMR spectra were recorded in CDCl3 on a 400 MHz Bruker Avance III HD spectrometer equipped with a 5 mm BB/1H (BB = 19F, 31P, and 15N) SmartProbe and referenced to residual CHCl3 (δ = 7.26 ppm) and 2,2,2-trifluoroethanol-d3 (δ = −77.8 ppm), respectively. In the case of the free ligands, 1H NMR spectra were recorded on a Bruker Avance III 500 spectrometer operating at 500 MHz and 19F NMR spectra were recorded on a Bruker Avance III 600 spectrometer operating at 564 MHz and available at the PACSMUB-WPCM technological platform, which relies on the “Institut de Chimie Moléculaire de l’Université de Bourgogne” and Satt Sayens “TM”, a Burgundy University private subsidiary. All NMR shift values are expressed as ppm. 1H and 19F spectra were calibrated using the residual peak of chloroform at 7.26 ppm or acetone-d6 at 2.05 ppm. High-resolution electrospray ionization mass spectra were recorded on an LTQ Orbitrap XL spectrometer. MALDI-TOF mass spectra were recorded on a Bruker Ultraflex Extreme MALDI Tandem TOF Mass Spectrometer using dithranol as the matrix. Cyclic voltammetry was performed with an EG&G Model 263A potentiostat having a three-electrode system, including a glassy carbon working electrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE). Tetra(n-butyl)ammonium perchlorate (TBAP) was recrystallized three times from absolute ethanol and dried in vacuo for at least one week prior to use as supporting electrolyte. The SCE was separated from the bulk solution by a fritted-glass bridge filled with the solvent/supporting-electrolyte mixture. Sample solutions in dry benzonitrile were purged with argon for at least 5 min prior to electrochemical measurements, which were also carried out under an argon blanket. All potentials are referenced to the SCE. The dipyrrin-bisanisole H3[HDPPOMe] and the corresponding dipyrrin-bisphenol H3[HDPP] were synthesized as described in the literature.32
4.2. General synthetic procedure for dipyrrin-bisanisoles
To a stirred solution of the appropriate benzaldehyde (0.86 mmol, 1.0 eq.) and 2-(2-methoxyphenyl)pyrrole (synthesized according to a literature procedure,33 299 mg, 1.73 mmol, 2.0 eq.) in CH2Cl2 (15 mL) under argon, trifluoroacetic acid (22 μL) was added and the mixture was stirred for 2 h at room temperature. 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 200 mg, 0.88 mmol) was then added and the resulting solution stirred overnight at room temperature. The reaction mixture was washed with saturated NaHCO3 aqueous solution and extracted with CH2Cl2. The organic phase was dried over MgSO4, evaporated to dryness, and subjected to column chromatography as described below.
4.3. H3[CF3DPPOMe]
This compound was purified by silica gel column using CH2Cl2 and a 9:1 mixture of CH2Cl2/MeOH as eluent and by alumina using a 2:3 mixture of CH2Cl2/heptane. Yield 281 mg (0.56 mmol, 65%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 526 (2.62), 610 (0.27). 1H NMR δ (CDCl3, δ = 7.26 ppm): 13.66 (s, 1H, NH); 8.04 (d, 2H, J = 7.5 Hz, phenylmethoxy); 7.72 (d, 2H, J = 8.0 Hz, m or o 10-pCF3C6H4); 7.67 (d, 2H, J = 8.0 Hz, m or o 10-pCF3C6H4); 7.35 (t, 2H, J = 7.5 Hz, phenylmethoxy); 7.05 (t, 2H, J = 7.5 Hz, phenylmethoxy); 7.00 (d, 2H, J = 7.5 Hz, phenylmethoxy); 6.94 (d, 2H, J = 4.5 Hz, β-H); 6.54 (d, 2H, J = 4.5 Hz, β-H); 3.87 (s, 6H, OMe). 19F NMR δ: −62.96 (s, 3F, 10-pCF3C6H4). LRMS (MALDI/TOF) [M]+˙: 500.65 (expt), 500.17 (calcd). HRMS (ESI) [M + H]+: 501.1781 (expt), 501.1784 (calcd).
4.4. H3[MeDPPOMe]
This compound was purified by silica gel column using a 4:1 mixture of heptane/AcOEt as eluent. Yield 212 mg (0.47 mmol, 55%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 316 (2.08), 521 (2.96), 601 (0.60). 1H NMR δ (CDCl3, δ = 7.26 ppm): 13.75 (s, 1H, NH); 8.06 (d, 2H, J = 7.5 Hz, phenylmethoxy); 7.45 (d, 2H, J = 8.0 Hz, m or o 10-pMeC6H4); 7.35 (t, 2H, J = 7.5 Hz, phenylmethoxy); 7.27 (d, 2H, J = 8.0 Hz, m or o 10-pMeC6H4); 7.05 (t, 2H, J = 7.5 Hz, phenylmethoxy); 7.01 (d, 2H, J = 7.5 Hz, phenylmethoxy); 6.95 (d, 2H, J = 4.5 Hz, β-H); 6.67 (d, 2H, J = 4.5 Hz, β-H); 3.87 (s, 6H, OMe); 2.47 (s, 3H, CH3). LRMS (MALDI/TOF) [M]+˙: 446.58 (expt), 446.20 (calcd). HRMS (ESI) [M + H]+: 447.2059 (expt), 447.2067 (calcd).
4.5. General synthetic procedure for dipyrrin-bisphenols
The experimental procedure was adapted from methodology described in the literature for the preparation of the unsubstituted phenyl ligand H3[HDPP].32 To a stirred solution of the corresponding dipyrrin-bisanisole (0.282 mmol) in CH2Cl2 (14 mL) under an argon atmosphere, BBr3 (1.0 M in heptane, 5.63 mL, 5.63 mmol) was added at 0 °C. The reaction mixture was stirred and allowed to warm up to room temperature and left for 3 days before quenching with methanol (14 mL). Concentrated HCl (37%, 1.35 mL) was then added and the resulting mixture was refluxed for 3 h. After cooling, the mixture was neutralized with saturated aqueous NaHCO3 and extracted with ethyl acetate. The organic layer was dried over MgSO4 and evaporated to dryness; the residue was then subjected to column chromatography to yield the desired free dipyrrin-bisphenol ligands.
4.6. H3[CF3DPP]
The compound was purified by silica gel column using 3:1 heptane/ethyl acetate as eluent. Yield 58.6 mg (0.12 mmol, 44%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 301 (1.17), 545 (1.53), 612 (0.47). 1H NMR δ (acetone-d6, δ = 2.05 ppm): 7.90 (d, 2H, J = 8.0 Hz, m or o 10-pCF3C6H4); 7.87 (d, 2H, J = 7.5 Hz, phenoxy); 7.80 (d, 2H, J = 8.0 Hz, m or o 10-pCF3C6H4); 7.30 (t, 2H, J = 7.5 Hz, phenoxy); 7.16 (m, 2H, β-H); 7.06 (d, 2H, J = 7.5 Hz, phenoxy); 6.98 (t, 2H, J = 7.5 Hz, phenoxy); 6.67 (m, 2H, β-H). 19F NMR δ: −63.01 (s, 3F, 10-pCF3C6H4). LRMS (MALDI/TOF) [M]+˙: 472.64 (expt), 472.14 (calcd). HRMS (ESI) [M + H]+: 473.1459 (expt), 473.1471 (calcd).
4.7. H3[MeDPP]
The compound was purified by silica gel column using a 4:1 heptane/ethyl acetate as eluent. Yield 89.2 mg (0.213 mmol, 75%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 323 (1.27), 373 (0.53), 545 (2.04), 601 (0.52). 1H NMR δ (acetone-d6, δ = 2.05 ppm): 7.86 (d, 2H, J = 7.5 Hz, phenoxy); 7.46 (d, 2H, J = 8.0 Hz, m or o 10-pMeC6H4); 7.37 (t, 2H, J = 8.0 Hz, m or o 10-pMeC6H4); 7.27 (t, 2H, J = 7.5 Hz, phenoxy); 7.13 (d, 2H, J = 4.5 Hz, β-H); 7.03 (d, 2H, J = 7.5 Hz, phenoxy); 6.95 (t, 2H, J = 7.5 Hz, phenoxy); 6.75 (m, 2H, β-H); 2.48 (s, 3H, CH3). LRMS (MALDI/TOF) [M]+˙: 418.55 (expt), 418.17 (calcd). HRMS (ESI) [M + H]+: 419.1748 (expt), 419.1754 (calcd).
4.8. General synthetic procedure for gold dipyrrin-bisphenolates
Gold acetate (3 equiv.) was added to a pink solution of the free dipyrrin-bisphenol ligand (30 mg) in pyridine (6 mL). The resulting suspension was stirred for 24 h and monitored with TLC and mass spectrometry. The blue suspension that was finally obtained was passed through Celite, the resulting solution was filtered, and the filtrate was rotary-evaporated to dryness. The brown residue obtained was dissolved in THF or CHCl3 and filtered twice through a double-layer of filter paper. The resulting blue filtrate was rotary-evaporated to yield a blue solid, which was thoroughly washed with n-hexane and dried under vacuum. Unfortunately, the compounds proved quite light-sensitive, especially in the presence of air, preventing us from obtaining satisfactory elemental analyses. Fortunately, X-ray quality crystals could be obtained for Au[CF3DPP] via slow diffusion of methanol into a concentrated chloroform solution in about 2 weeks.
4.9. Au[CF3DPP]
Yield 21 mg (0.031 mmol, 50%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 322 (2.26), 435 (0.65), 643 (2.25). 1H NMR δ (1,1,2,2-tetrachloroethane-d2, δ = 6.00 ppm): 7.82 (d, 2H, J = 8.0 Hz, m or o-10-pCF3C6H4); 7.78 (d, 2H, J = 7.8 Hz, phenoxy); 7.71 (d, 2H, J = 8.0 Hz, o or m-10-pCF3Ph); 7.39 (t, 2H, J = 7.8 Hz, phenoxy); 7.32 (d, 2H, J = 8.2 Hz, phenoxy); 7.25 (d, 2H, J = 4.8 Hz, β-H); 6.96 (t, 2H, J = 7.4 Hz, phenoxy); 6.83 (d, 2H, J = 4.8 Hz, β-H). 19F NMR δ: −63.16 (s, 2F, 10-pCF3C6H4); −63.18 (s, 1F, 10-pCF3C6H4). HRMS (ESI, major isotopomer) [M]+: 666.0789 (expt), 666.0824 (calcd).
4.10. Au[HDPP]
Yield 23 mg (0.038 mmol, 52%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 325 (2.38), 429 (0.85), 636 (2.79). 1H NMR δ: 7.71 (d, 2H, J = 8.0 Hz, phenoxy); 7.56 to 7.50 (d, 5H, Ph), 7.33 to 7.28 (m, 4H, phenoxy); 7.16 (d, 2H, J = 4.8 Hz, β-H), 6.91 to 6.85 (m, 2H, phenoxy), 6.83 (d, 2H, J = 4.8 Hz, β-H). HRMS (ESI, major isotopomer) [M]+: 598.0926 (expt), 598.0950 (calcd).
4.11. Au[MeDPP]
Yield 34 mg (0.055 mmol, 77%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 310 (1.85), 336 (1.89), 427 (0.97), 634 (2.93). 1H NMR δ (1,1,2,2-tetrachloroethane-d2, δ = 6.00 ppm): δ 7.77 (d, 2H, J = 7.8 Hz, phenoxy); 7.44 (d, 2H, J = 7.9 Hz, m or o-10-pCH3C6H4), 7.39 to 7.30 (m, 4H, phenoxy; 2H, o or m-10-pCH3C6H4); 7.24 (d, 2H, J = 4.7 Hz, β-H), 6.97 to 6.92 (overlapping d, 2H, J = 4.7 Hz, β-H and t, 2H, J = 7.8 Hz, phenoxy), 2.49 (3H, CH3, 10-pCH3C6H4). HRMS (ESI, major isotopomer) [M]+: 612.1096 (expt), 612.1107 (calcd).
4.12. Synthesis of Cu[CF3DPP]
Copper acetate (11 mg, 0.055 mmol, 5 equiv.) was added to a pink solution of the H3[CF3DPP] ligand (5 mg, 0.011 mmol) in pyridine (2 mL). The suspension was stirred for 1 h, at the end of which the reaction was complete, as indicated by TLC (CHCl3-2% CH3OH) and mass spectrometry. The blue suspension obtained was filtered through Celite and the resulting solution was filtered twice before evaporation under vacuum. Yield 5.5 mg (0.010 mmol, 91%). UV-vis λmax [nm, ε × 104 (M−1 cm−1)]: 312 (0.74), 572 (0.35), 614 (0.92). HRMS (ESI, major isotopomer) [M + H]+ = 533.0527 (expt), 533.0533 (calcd).
4.13. X-ray structure determination
X-ray diffraction data were collected on beamline 12.2.1 at the Advanced Light Source of Lawrence Berkeley National Laboratory, Berkeley, California. The samples were mounted on MiTeGen® kapton loops and placed in a 100(2) K nitrogen cold stream provided by an Oxford Cryostream 700 Plus low temperature apparatus on the goniometer head of a Bruker D8 diffractometer equipped with PHOTONII CPAD detector. Diffraction data were collected using synchrotron radiation monochromated with silicon(111) to a wavelength of 0.7288(1) Å. In each case, an approximate full-sphere of data was collected using 1° ω scans. Absorption corrections were applied using SADABS.34 The structure was solved by intrinsic phasing (SHELXT)35 and refined by full-matrix least squares on F2 (SHELXL-2014)36 using the ShelXle GUI.37 Appropriate scattering factors were applied using the XDISP38 program within the WinGX suite.39 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were geometrically calculated and refined as riding atoms.
4.14. Computational methods
DFT calculations were carried out at the scalar-relativistic level with the ZORA (Zeroth Order Regular Approximation to the Dirac equation)40–42 Hamiltonian, the OLYP28,29 exchange-correlation functional, and all-electron ZORA STO-TZ2P relativistic basis sets, all as implemented in the ADF program system.43,44
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the Research Council of Norway (grant no. 263332 to AG), the CNRS (UMR UB-CNRS 6302), the “Université Bourgogne Franche-Comté”, the FEDER-FSE Bourgogne 2014/2020 (European Regional Development Fund), the “Conseil Régional de Bourgogne” (through the PARI II CDEA project), the National Research Fund of the Republic of South Africa (grant numbers 113327 and 96111 to JC), and the Advanced Light Source, Berkeley, California. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors warmly thank Mrs Sandrine Pacquelet for technical assistance.
References
- W. Shan, N. Desbois, S. Pacquelet, S. Brandès, Y. Rousselin, J. Conradie, A. Ghosh, C. P. Gros and K. M. Kadish, Inorg. Chem., 2019, 58, 7677–7689 CrossRef PubMed.
-
(a) K. E. Thomas, A. B. Alemayehu, J. Conradie, C. M. Beavers and A. Ghosh, Acc. Chem. Res., 2012, 45, 1203–1214 CrossRef CAS PubMed;
(b) A. Ghosh, Chem. Rev., 2017, 117, 3798–3881 CrossRef CAS PubMed;
(c) S. Ganguly and A. Ghosh, Acc. Chem. Res., 2019, 52, 2003–2014 CrossRef CAS PubMed.
- S. Nardis, F. Mandoj, M. Stefanelli and R. Paolesse, Coord. Chem. Rev., 2019, 388, 360–405 CrossRef CAS.
-
(a) S. Ganguly, D. Renz, L. J. Giles, K. J. Gagnon, L. J. McCormick, J. Conradie, R. Sarangi and A. Ghosh, Inorg. Chem., 2017, 56, 14788–14800 CrossRef CAS PubMed;
(b) S. Ganguly, J. Conradie, J. Bendix, K. J. Gagnon, L. J. McCormick and A. Ghosh, J. Phys. Chem. A, 2017, 121, 9589–9598 CrossRef CAS PubMed.
- X. Jiang, W. Shan, N. Desbois, V. Quesneau, S. Brandès, E. Van Caemelbecke, W. R. Osterloh, V. Blondeau-Patissier, C. P. Gros and K. M. Kadish, New J. Chem., 2018, 42, 8220–8229 RSC.
- For selected references to ligand noninnocence in Cu corroles, see:
(a) I. H. Wasbotten, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8104–8116 CrossRef CAS PubMed;
(b) C. Brückner, R. P. Briñas and J. A. K. Bauer, Inorg. Chem., 2003, 42, 4495–4497 CrossRef PubMed;
(c) M. Bröring, F. Bregier, E. C. Tejero, C. Hell and M. C. Holthausen, Angew. Chem., Int. Ed., 2007, 46, 445–448 CrossRef PubMed;
(d) K. E. Thomas, I. H. Wasbotten and A. Ghosh, Inorg. Chem., 2008, 47, 10469–10478 CrossRef CAS PubMed;
(e) A. B. Alemayehu, E. Gonzalez, L.-K. Hansen and A. Ghosh, Inorg. Chem., 2009, 48, 7794–7799 CrossRef CAS PubMed;
(f) A. B. Alemayehu, L.-K. Hansen and A. Ghosh, Inorg. Chem., 2010, 49, 7608–7610 CrossRef CAS PubMed;
(g) K. E. Thomas, J. Conradie, L.-K. Hansen and A. Ghosh, Eur. J. Inorg. Chem., 2011, 1865–1870 CrossRef CAS;
(h) S. Berg, K. E. Thomas, C. M. Beavers and A. Ghosh, Inorg. Chem., 2012, 51, 9911–9916 CrossRef CAS PubMed;
(i) K. E. Thomas, L. J. McCormick, D. Carrié, H. Vazquez-Lima, G. Simmoneaux and A. Ghosh, Inorg. Chem., 2018, 57, 4270–4276 CrossRef CAS PubMed;
(j) I. K. Thomassen, L. J. McCormick and A. Ghosh, ACS Omega, 2018, 3, 5106–5110 CrossRef CAS PubMed;
(k) H. Lim, K. E. Thomas, B. Hedman, K. O. Hodgson, A. Ghosh and E. I. Solomon, Inorg. Chem., 2019, 58, 6722–6730 CrossRef CAS PubMed.
- A. Kochem, L. Chiang, B. Baptiste, C. Philouze, N. Leconte, O. Jarjayes, T. Storr and F. Thomas, Chem.–Eur. J., 2012, 18, 14590–14593 CrossRef CAS PubMed.
- Y. Feng, L. A. Burns, L.-C. Lee, C. D Sherrill and C. W. Jones, Inorg. Chim. Acta, 2015, 430, 30–35 CrossRef CAS.
- L. Lecarme, A. Kochem, L. Chiang, J. Moutet, F. Berthiol, C. Philouze, N. Leconte, T. Storr and F. Thomas, Inorg. Chem., 2018, 57, 9708–9719 CrossRef CAS PubMed.
- Z. Gross and H. B. Gray, Adv. Synth. Catal., 2004, 346, 165–170 CrossRef CAS.
- N. Levy, A. Mahammed, M. Kosa, D. T. Major, Z. Gross and L. Elbaz, Angew. Chem., Int. Ed., 2015, 54, 14080–14084 CrossRef CAS PubMed.
- K. E. Thomas, A. B. Alemayehu, J. Conradie, C. M. Beavers and A. Ghosh, Inorg. Chem., 2011, 50, 12844–12851 CrossRef CAS PubMed.
- First reports of Au corroles:
(a) A. B. Alemayehu and A. Ghosh, J. Porphyrins Phthalocyanines, 2011, 15, 106–110 CrossRef CAS;
(b) E. Rabinovich, I. Goldberg and Z. Gross, Chem.–Eur. J., 2011, 17, 12294–12301 CrossRef CAS PubMed.
-
(a) K. E. Thomas, C. M. Beavers and A. Ghosh, Mol. Phys., 2012, 110, 2439–2444 CrossRef CAS;
(b) J. Capar, J. Zonneveld, S. Berg, J. Isaksson, K. J. Gagnon, K. E. Thomas and A. Ghosh, J. Inorg. Biochem., 2016, 162, 146–153 CrossRef CAS PubMed;
(c) W. Sinha, M. G. Sommer, M. van der Meer, S. Plebst, B. Sarkar and S. Kar, Dalton Trans., 2016, 45, 2914–2923 RSC;
(d) K. Sudhakar, A. Mizrahi, M. Kosa, N. Fridman, B. Tumanskii, M. Saphier and Z. Gross, Angew. Chem., Int. Ed., 2017, 56, 9837–9841 CrossRef CAS PubMed;
(e) K. E. Thomas, K. J. Gagnon, L. J. McCormick and A. Ghosh, J. Porphyrins Phthalocyanines, 2018, 22, 596–601 CrossRef CAS.
- Note that silver corroles may be innocent or noninnocent, depending on the substitution pattern:
(a) R. Sarangi, L. J. Giles, K. E. Thomas and A. Ghosh, Eur. J. Inorg. Chem., 2016, 3225–3227 CrossRef CAS;
(b) K. E. Thomas, H. Vazquez-Lima, Y. Fang, Y. Song, K. J. Gagnon, C. M. Beavers, K. M. Kadish and A. Ghosh, Chem.–Eur. J., 2015, 21, 16839–16847 CrossRef CAS PubMed.
- L. Lecarme, A. Kochem, L. Chiang, J. Moutet, F. Berthiol, C. Philouze, N. Leconte, T. Storr and F. Thomas, Inorg. Chem., 2018, 57, 9708–9719 CrossRef CAS PubMed.
- M. Yamamura, H. Takizawa, Y. Gobo and T. Nabeshima, Dalton Trans., 2016, 45, 6834–6838 RSC.
- M. Yamamura, M. Albrecht, M. Albrecht, Y. Nishimura, T. Arai and T. Nabeshima, Inorg. Chem., 2014, 53, 1355–1360 CrossRef CAS PubMed.
- S. Rausaria, A. Kamadulski, N. P. Rath, L. Bryant, Z. Chen, D. Salvemini and W. L. Neumann, J. Am. Chem. Soc., 2011, 133, 4200–4203 CrossRef CAS PubMed.
- S. El Ghachtouli, K. Wójcik, L. Copey, F. Szydlo, E. Framery, C. Goux-Henry, L. Billon, M.-F. Charlot, R. Guillot, B. Andrioletti and A. Aukauloo, Dalton Trans., 2011, 40, 9090–9093 RSC.
- M. Saikawa, M. Daicho, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 4014–4017 RSC.
- T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728–10735 CrossRef CAS PubMed.
- M. Yamamura, H. Takizawa, N. Sakamoto and T. Nabeshima, Tetrahedron Lett., 2013, 54, 7049–7052 CrossRef CAS.
- A. Sumiyoshi, Y. Chiba, R. Matsuoka, T. Noda and T. Nabeshima, Dalton Trans., 2019, 48, 13169–13175 RSC.
- Unlike Au corroles,26 the compounds do not exhibit NIR phosphorescence in common organic solvents, including toluene and dichloromethane.
- A. B. Alemayehu, N. U. Day, T. Mani, A. B. Rudine, K. E. Thomas, O. A. Gederaas, S. A. Vinogradov, C. C. Wamser and A. Ghosh, ACS Appl. Mater. Interfaces, 2016, 8, 18935–18942 CrossRef CAS PubMed.
- The relative constancy of the oxidation potential with respect to the substituent X suggests that the HOMO does not have much of an amplitude at the meso position. This is indeed the case, as shown later in the paper.
- The OPTX exchange functional: N. C. Handy and A. J. Cohen, Left-right correlation energy, Mol. Phys., 2001, 99, 403–412 CrossRef CAS.
- The LYP correlation functional: C. T. Lee, W. T. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- Early studies from one of our laboratories established that DFT calculations generally do an excellent job of describing the ionization potentials and cationic states of porphyrin-type molecules:
(a) A. Ghosh and J. Almlöf, Chem. Phys. Lett., 1993, 213, 519–521 CrossRef CAS;
(b) A. Ghosh, J. Am. Chem. Soc., 1995, 117, 4691–4699 CrossRef CAS;
(c) A. Ghosh and T. Vangberg, Theor. Chem. Acc., 1997, 97, 143–149 Search PubMed;
(d) A. Ghosh, Acc. Chem. Res., 1998, 31(4), 189–198 CrossRef CAS;
(e) A. Ghosh, T. Wondimagegn and A. B. J. Parusel, J. Am. Chem. Soc., 2000, 122, 5100–5104 CrossRef CAS;
(f) A. Ghosh and E. Steene, J. Biol. Inorg Chem., 2001, 6, 739–752 CrossRef CAS PubMed.
- The electron affinities and anionic states of porphyrin-type molecules have been less explored by means of DFT calculations. Nevertheless, the available data suggest that DFT does a good job of tackling the question of metal versus ligand-centered reduction. See, e.g.: H. Ryeng, E. Gonzalez and A. Ghosh, J. Phys. Chem. B, 2008, 112, 15158–15173 CrossRef CAS PubMed.
- C. Ikeda, S. Ueda and T. Nabeshima, Chem. Commun., 2009, 2544–2546 RSC.
- R. D. Rieth, N. P. Mankad, E. Calimano and J. P. Sadighi, Org. Lett., 2004, 6, 3981–3983 CrossRef CAS PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- L. Kissel and R. H. Pratt, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 170–175 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, S. J. A. van Gisbergen, C. F. Guerra, E. J. Baerends, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1964008. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09228e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ac,
Simon J. Teat
ac,
Simon J. Teat