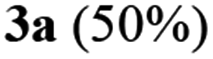DOI:
10.1039/C9RA09236F
(Paper)
RSC Adv., 2020,
10, 289-297
Novel isatin–indole derivatives as potential inhibitors of chorismate mutase (CM): their synthesis along with unexpected formation of 2-indolylmethylamino benzoate ester under Pd–Cu catalysis†
Received
7th November 2019
, Accepted 18th December 2019
First published on 2nd January 2020
Abstract
A series of novel isatin–indole derivatives has been designed as potential inhibitors of chorismate mutase (CM) that is known to be present in bacteria, fungi and higher plants but not in human. The design was supported by in silico docking studies that predicted strong interactions of these molecules with CM. The target compounds were synthesized via the one-pot coupling/cyclization method involving the reaction of an isatin based terminal alkyne with 2-iodosulfanilides under Pd–Cu catalysis. A number of isatin–indole derivatives were prepared using this method. A side product e.g. 2-indolylmethylamino benzoate ester derivative was obtained as a result of isatin ring opening (ethanolysis) of products in certain cases. Additionally, regioselective reduction of selected compounds afforded the corresponding C-3 hydroxy derivatives. All isatin–indole derivatives showed good to high inhibition of CM in vitro among which two compounds (3e and 3f) showed inhibition at nanomolar concentration.
Introduction
The chorismate mutase or CM (EC 5.4.99.5) catalyzes the Claisen rearrangement of chorismate to prephenate in the shikimate biosynthetic pathway to form the essential amino acids, phenylalanine and tyrosine. The CM is considered as an attractive target for the identification of effective antibacterial agents due to a number of reasons, e.g. (i) its inhibition may hinder the supply of nutrients to the organism, (ii) low sequence homology among the known CMs, and more importantly (iii) it exists in bacteria, fungi and higher plants but not in humans.1–3 Thus efforts have been devoted to the identification of inhibitors of CM.2
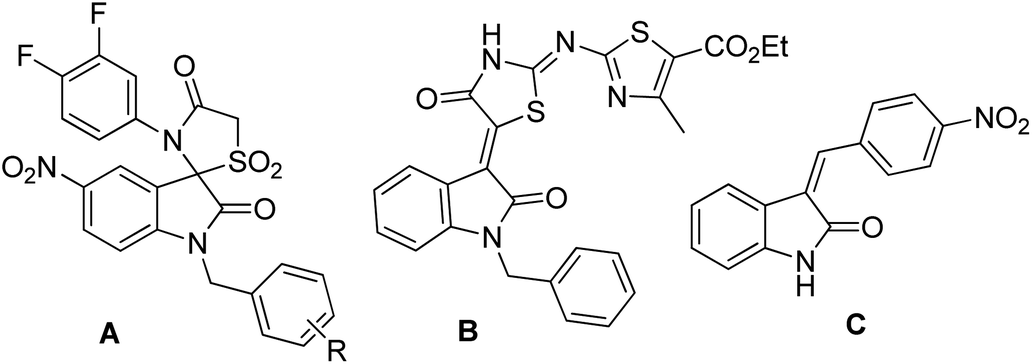
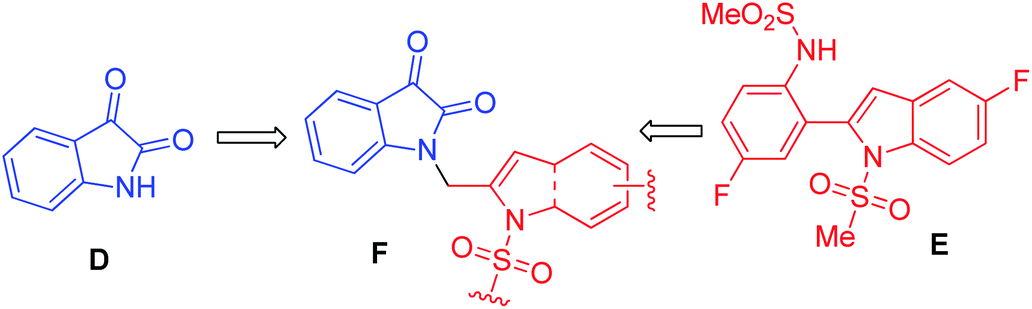
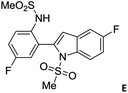
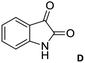
Over the years, the isatin (or indoline-2,3-dione) framework have been explored for the identification of potential anti-tubercular agents (Fig. 1).4–13 Indeed, a simple isatin derivative C (Fig. 1) has shown an IC50 of 1.01 ± 0.22 μM when tested against purified CM.14 All these reports and our longstanding interest in the discovery and development of novel inhibitors of CM prompted us to focus on isatin framework14 (D, reported MIC = 340.13 μM, Fig. 2). Earlier we have reported a series of 2-substituted N-sulfonyl indole derivatives one of which i.e. E (Fig. 2) showed weak inhibition of CM in vitro with an IC50 value 17.02 μM.15 In order to identify more potent inhibitors of CM we decided to explore the isatin–indole conjugate F (Fig. 2). Our design mainly involved the introduction of a linker ‘‘CH2” at C-2 of the indole ring of known inhibitor E and replacing the aryl moiety by an isatin ring. It was anticipated that besides introducing the structural flexibility through the linker “CH2” integrating the structural features of isatin D and E in a single molecular entity i.e. F could improve the overall potency. Notably, while the linker could be chosen from a variety of groups to afford a range of corresponding molecules we focused on simpler moiety like “CH2” group at the initial stage.
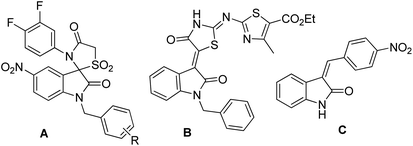 |
| | Fig. 1 Representative isatin derivatives reported as anti-tubercular agents. | |
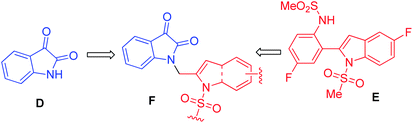 |
| | Fig. 2 Design of new isatin–indole framework F based on isatin D and the known inhibitor E. | |
Results and discussion
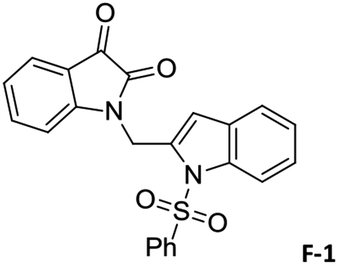
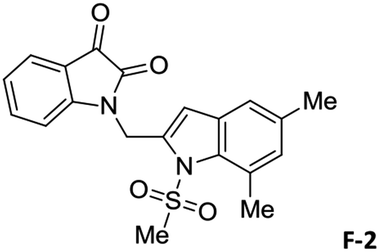
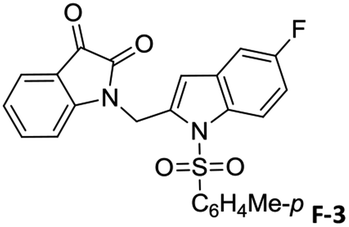
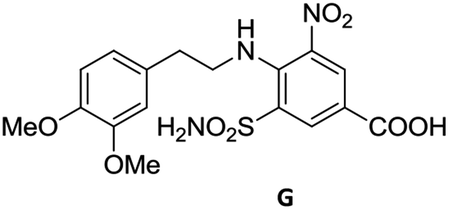
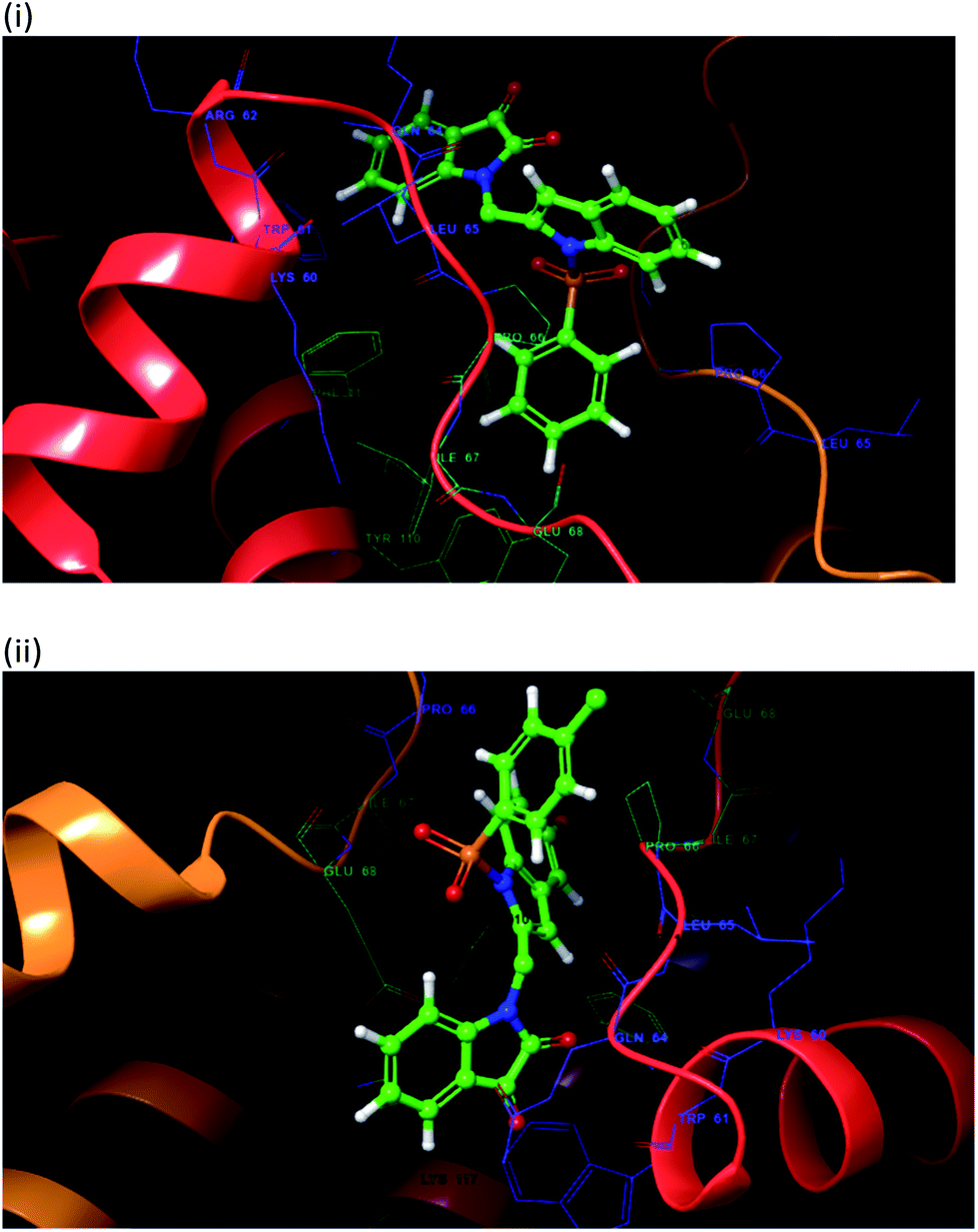
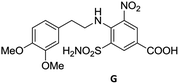
To justify our focus on isatin–indole framework for obtaining potential inhibitor of CM the docking study of three representative molecules e.g. F-1, F-2 and F-3 was performed using AutoDock Vina and the binding energies are presented in Table 1. Since it was noted earlier16 that the binding energy of studied ligands at the interface site was lower than the other possible sites of MtbCM (indicating preferred stabilization of ligands at the interface site of MtbCM) we considered the interface site of MtbCM for docking of our molecules. All molecules showed better binding energies than the reference compound E 15 and G![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 [i.e. 4-(3,4-dimethoxyphenethylamino)-3-nitro-5-sulfamoylbenzoic acid] as well as isatin D suggesting their possible superior inhibitory properties of CM. The molecule F-1 was stabilized (Fig. 3(i), see also Fig. S-1 in ESI†) by several hydrophobic interactions with the hydrophobic residues (LEU65, PRO66, ILE67, TYR110, PHE113 of A chain and PRO66, ILE67, TYR110 of B chain) and hydrophobic regions of polar and charged residues (GLN64, LYS60, GLU68 of A chain and GLU68 of B chain). Further, the molecule participated in a pi–pi interaction with TRP61 of A chain. Indeed, the high docking score of F-1 was mainly contributed by the maximum number of hydrophobic contacts shown by this molecule. The molecule F-2 though participated in similar hydrophobic interactions (mainly with ILE67, PRO66, etc. of both A and B chain) but comparatively fewer in number perhaps accounted its relatively lower docking score. It also participated in pi–pi interactions with TRP61A and TYR110B (see Fig. S-2 in ESI†). Like F-1 the molecule F-3 too was stabilized by several hydrophobic contacts with hydrophobic residues (ILE67, PRO66, TYR110 etc, of both A and B chain) and hydrophobic region of polar/charged residues (GLU68B, GLN64A, and LYS60A) (Fig. 3(ii), see also Fig. S-3 in ESI†).
17 [i.e. 4-(3,4-dimethoxyphenethylamino)-3-nitro-5-sulfamoylbenzoic acid] as well as isatin D suggesting their possible superior inhibitory properties of CM. The molecule F-1 was stabilized (Fig. 3(i), see also Fig. S-1 in ESI†) by several hydrophobic interactions with the hydrophobic residues (LEU65, PRO66, ILE67, TYR110, PHE113 of A chain and PRO66, ILE67, TYR110 of B chain) and hydrophobic regions of polar and charged residues (GLN64, LYS60, GLU68 of A chain and GLU68 of B chain). Further, the molecule participated in a pi–pi interaction with TRP61 of A chain. Indeed, the high docking score of F-1 was mainly contributed by the maximum number of hydrophobic contacts shown by this molecule. The molecule F-2 though participated in similar hydrophobic interactions (mainly with ILE67, PRO66, etc. of both A and B chain) but comparatively fewer in number perhaps accounted its relatively lower docking score. It also participated in pi–pi interactions with TRP61A and TYR110B (see Fig. S-2 in ESI†). Like F-1 the molecule F-3 too was stabilized by several hydrophobic contacts with hydrophobic residues (ILE67, PRO66, TYR110 etc, of both A and B chain) and hydrophobic region of polar/charged residues (GLU68B, GLN64A, and LYS60A) (Fig. 3(ii), see also Fig. S-3 in ESI†).
Table 1 Docking of molecules into MtbCM a
| Molecules |
AutoDock Vina score (kcal mol−1) |
| E, G and D are known/reference compounds. |
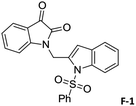 |
−9.7 |
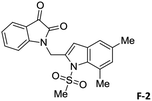 |
−8.8 |
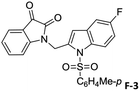 |
−9.3 |
 |
−6.9 |
 |
−7.3 |
 |
−6.2 |
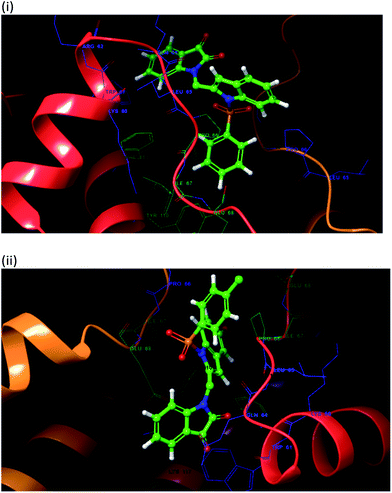 |
| | Fig. 3 The 3D interaction diagram of (i) molecule F-1 and (ii) molecule F-3 with interface residues of chorismate mutase (PDB code: 2FP2) (the A chain is in red color, and B chain is orange color) that were prepared in Maestro visualizer (Schrödinger, LLC). | |
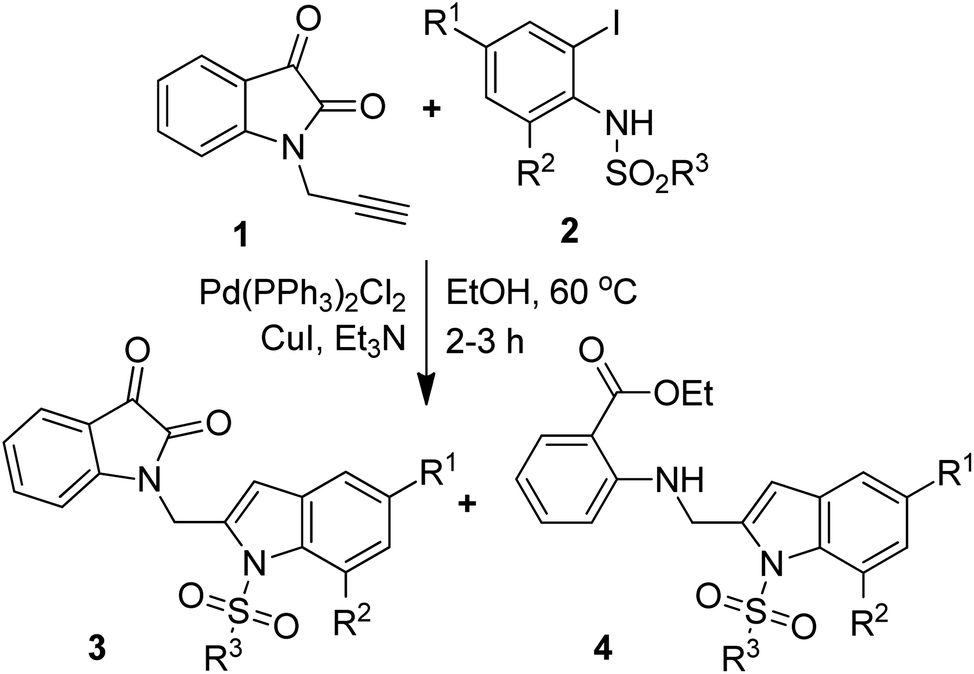
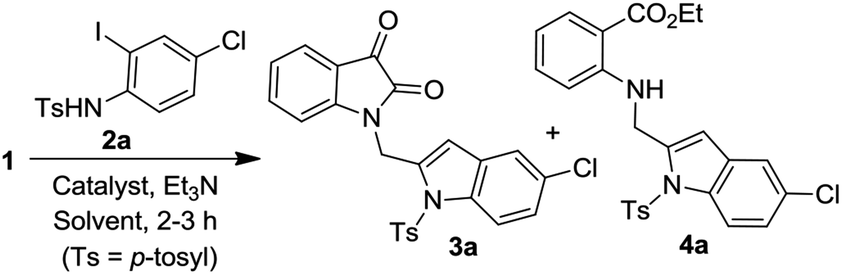
The one-pot coupling/cyclization strategy under the catalysis of transition metals has become a common and versatile approach for the synthesis of 2-substituted indole derivatives.15,18–20 We also adopted a similar strategy for the synthesis of designed isatin–indole derivatives. Thus the required alkyne 1 prepared from isatin D was coupled with 2-iodosulfanilides 2 under Pd/Cu-catalysis in ethanol to afford the desired compound 3 (Scheme 1). Interestingly, a side product e.g. 2-indolylmethylamino benzoate ester derivative 4 [formed as a result of ring opening (ethanolysis) of the isatin moiety of 3] was also isolated in certain cases. The details of this study along with the pharmacological evaluation of synthesized compounds are presented. To the best of our knowledge the synthesis and CM inhibitory potential of the present class of compounds21 has not been explored earlier.
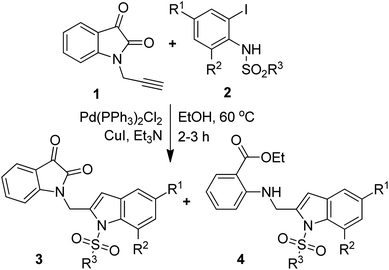 |
| | Scheme 1 Coupling of isatin based alkyne 1 with 2-iodosulfanilides (2) under Pd/Cu-catalysis. | |
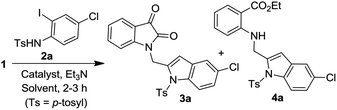
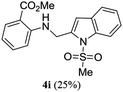
To establish the optimized reaction conditions the coupling of alkyne 1 with 2-iodosulfanilide 2a was examined under various reaction conditions and the results are summarized in Table 2. The reaction proceeded well when carried out using Pd(PPh3)2Cl2/CuI as a catalyst system and Et3N as a base in EtOH (entry 1, Table 2). However, a mixture of compounds e.g. the desired product 3a along with a side product 4a was obtained in this case. Based on the spectral data the compound 4a was identified as a 2-indolylmethylamino benzoate ester derivative. In order to improve the yield of 3a further the reaction was carried out by varying a number of parameters. The change of base from Et3N to K2CO3 (entry 2, Table 2) or Cs2CO3 (entry 3, Table 2) suppressed the formation of 4a but afforded 3a in low yield. Except in case of MeOH a similar observation was noted when EtOH was replaced by other solvents e.g. tBuOH or PEG-400 (entries 4–6, Table 2). Omission of CuI or Pd-catalyst was found to be counterproductive (entry 7 and 8, Table 2). While reaction proceeded in the presence of Pd(PPh3)2Cl2/Cu(OAc)2 (entry 9) however the use of Cu(OAc)2 alone or other Pd-catalyst e.g. Pd(OAc)2 or combination of Pd(OAc)2/CuI or 10%Pd–C/CuI/PPh3 was found to be ineffective (entries 10–13, Table 2). Overall, though 4a was obtained as side product however the best yield of 3a was obtained under the conditions of entry 1 and hence the combination of Pd(PPh3)2Cl2/CuI, Et3N and EtOH was used to prepare the other analogues of 3a.
Table 2 The coupling of alkyne 1 with 2-iodosulfanilide 2a under various reaction conditionsa
|
| Entry |
Catalyst |
Solvent |
Yieldb (%) |
| 3a |
4a |
| Reaction conditions: all the reactions were carried out using 1 (1 mmol), 2 (1.2 mmol), a Pd-catalyst (5 mol%) and Cu-catalyst (5 mol%) in the presence of Et3N (3 mmol) in a solvent (10 mL) at 60 °C for 2–3 h. Isolated yield. K2CO3 was used as a base. Cs2CO3 was used as a base. Corresponding methyl ester was obtained. The reaction was performed for 6–8 h. |
| 1 |
Pd(PPh3)2Cl2/CuI |
EtOH |
60 |
30 |
| 2c |
Pd(PPh3)2Cl2/CuI |
EtOH |
25 |
0 |
| 3d |
Pd(PPh3)2Cl2/CuI |
EtOH |
30 |
0 |
| 4 |
Pd(PPh3)2Cl2/CuI |
MeOH |
50 |
35e |
| 5 |
Pd(PPh3)2Cl2/CuI |
tBuOH |
20 |
0 |
| 6 |
Pd(PPh3)2Cl2/CuI |
PEG-400 |
55 |
0 |
| 7f |
Pd(PPh3)2Cl2 |
EtOH |
0 |
0 |
| 8f |
CuI |
EtOH |
0 |
0 |
| 9 |
Pd(PPh3)2Cl2/Cu(OAc)2 |
EtOH |
50 |
25 |
| 10 |
Cu(OAc)2 |
EtOH |
0 |
0 |
| 11 |
Pd(OAc)2 |
EtOH |
0 |
0 |
| 12 |
Pd(OAc)2/CuI |
EtOH |
0 |
0 |
| 13 |
10%Pd–C/PPh3/CuI |
EtOH |
0 |
0 |
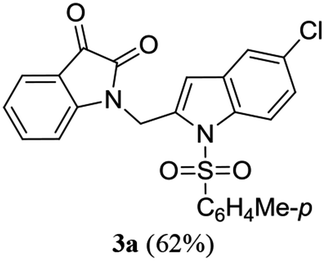
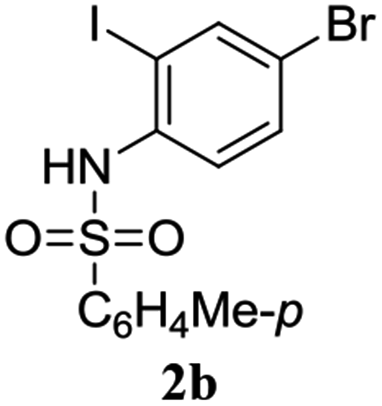
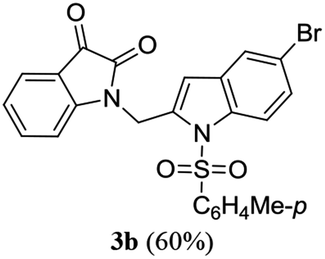
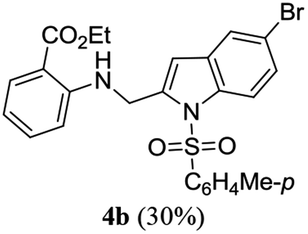
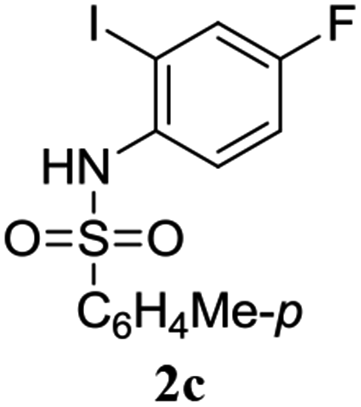
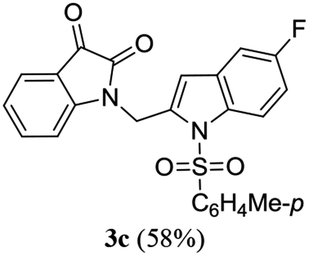
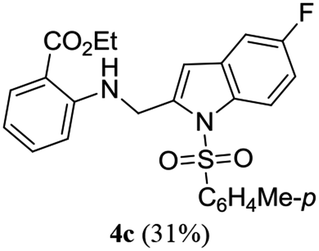
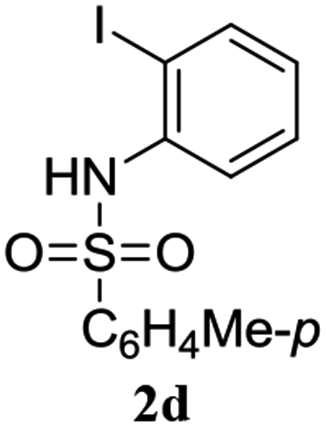
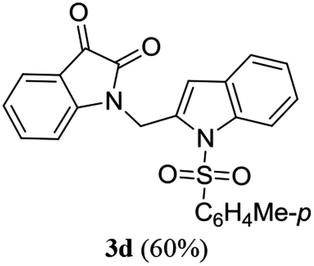
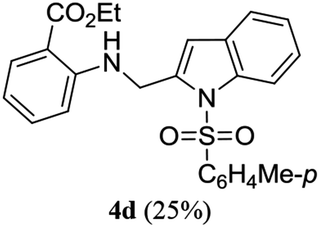
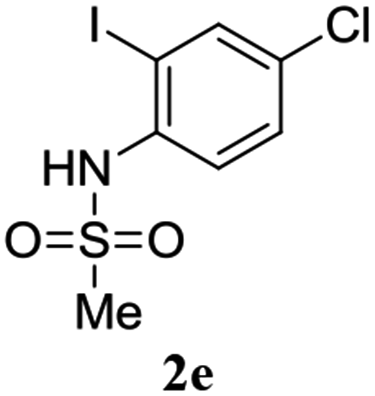
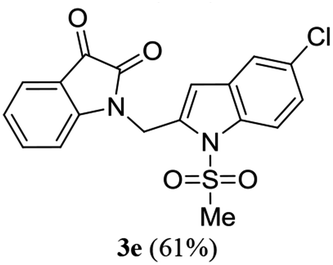
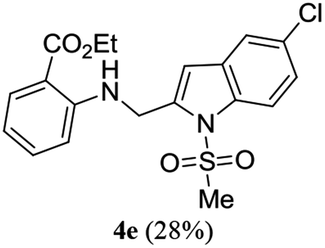
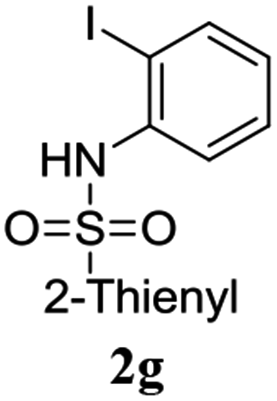
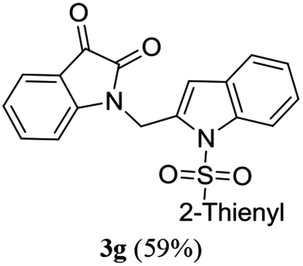
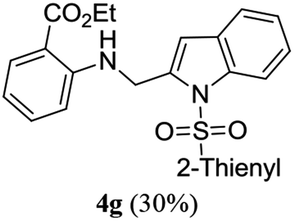
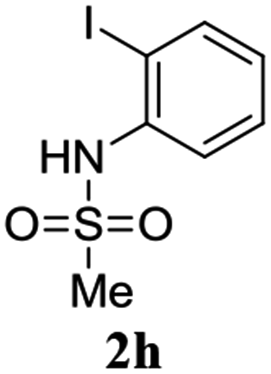
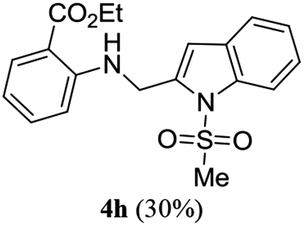
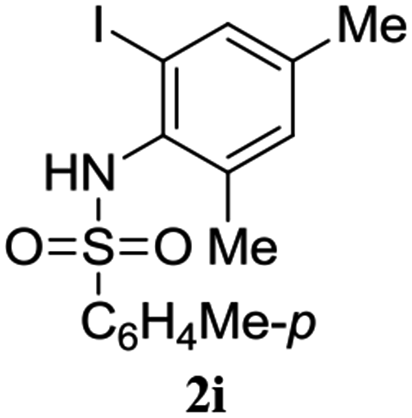
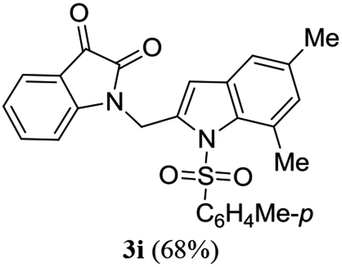
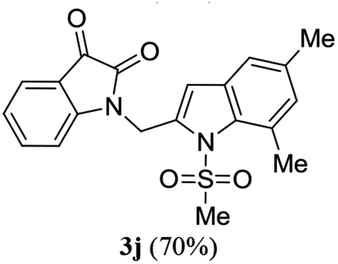
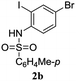
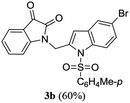
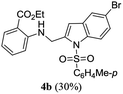
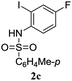
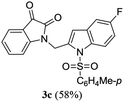
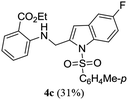
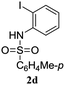
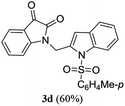
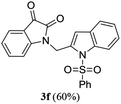
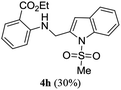
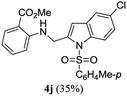
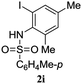
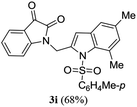
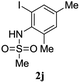
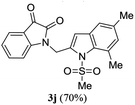
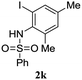
A range of compounds (3) based on isatin–indole framework F (Fig. 2) were prepared via the coupling-cyclization of 1 with a variety of 2-iodosulfanilides (2) (Table 3). The coupling-cyclization reaction proceeded well in all these cases affording the desired product in acceptable yields. Substituents like F, Cl, Br and Me on the sulfanilide ring and groups such as mesyl, p-tosyl, benzenesufonyl and 2-thienosulfonyl at the sulfanilide nitrogen were tolerated under the conditions employed. As mentioned earlier, the 2-indolylmethylamino benzoate ester derivative (4) was obtained in several cases especially when 2-iodosulfanilides having a substituent at C-4 position was employed. Notably, formation of this type of side product was almost suppressed (or formed in traces quantity) when 2-iodosulfanilides having a substituent at C-2 position in addition to C-4 were employed. Nevertheless, separation of compound 3 and 4 via column chromatography was found to be a straightforward process and both compounds were isolated in pure form without encountering with any difficulties. It is worthy to mention that the use of an appropriate internal alkyne e.g. 1-(3-phenylprop-2-ynyl)indoline-2,3-dione in place of terminal alkyne 1 under the current reaction conditions was not successful. The use of 2-iodoaniline or 2,2,2-trifluoro-N-(2-iodophenyl)acetamide in place of anilide 2 was also not successful (see ESI† for further details) indicating certain drawbacks of this methodology.
Table 3 Synthesis of isatin–indole derivatives (3) along with 2-indolylmethylamino benzoate ester derivative 4 (Scheme 1)a
| Entry |
Anilides (2) |
Products (3) |
Products (4) |
| Figure in the bracket indicates isolated % yield. MeOH was used as a solvent in place of EtOH. |
| 1 |
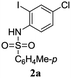 |
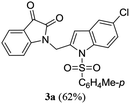 |
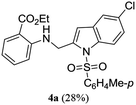 |
| 2 |
 |
 |
 |
| 3 |
 |
 |
 |
| 4 |
 |
 |
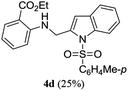 |
| 5 |
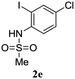 |
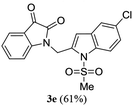 |
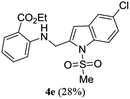 |
| 6 |
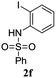 |
 |
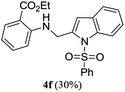 |
| 7 |
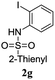 |
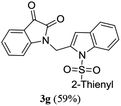 |
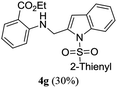 |
| 8 |
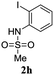 |
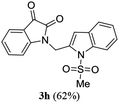 |
 |
| 9b |
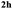 |
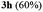 |
 |
| 10b |
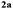 |
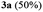 |
 |
| 11 |
 |
 |
Trace |
| 12 |
 |
 |
Trace |
| 13 |
 |
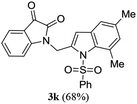 |
Trace |
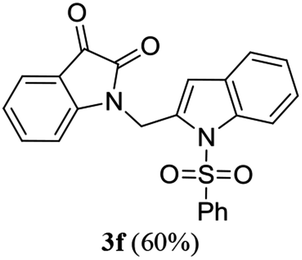
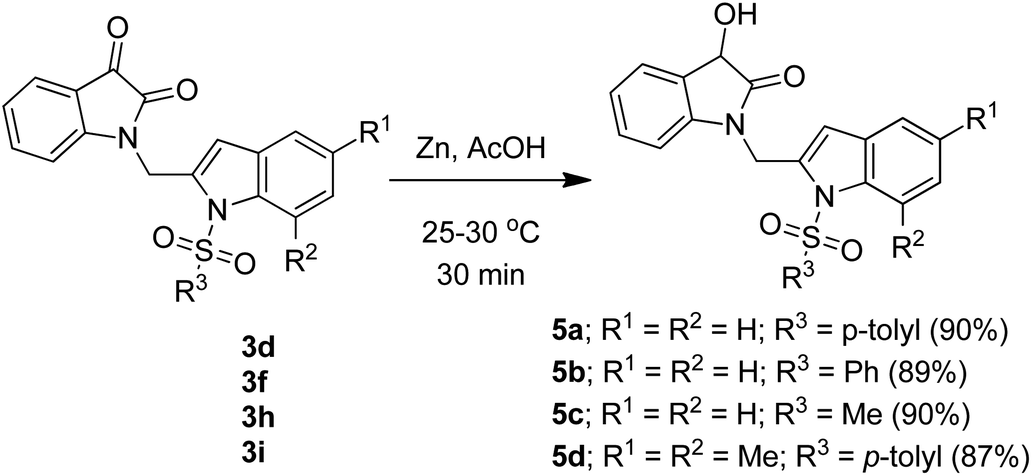
To expand the scope and applicability of this methodology some of the synthesized isatin–indole derivatives were treated with Zn in acetic acid when selective reduction of C-3 keto group (isatin ring) was observed. Indeed the corresponding C-3 hydroxy derivatives were obtained in excellent yield (Scheme 2). Moreover, the synthesis of compound 3d was undertaken in gram scale when this compound was obtained in 67% yield.
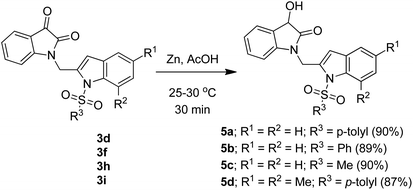 |
| | Scheme 2 Regioselective reduction of compound 3d, 3f, 3h and 3i. | |
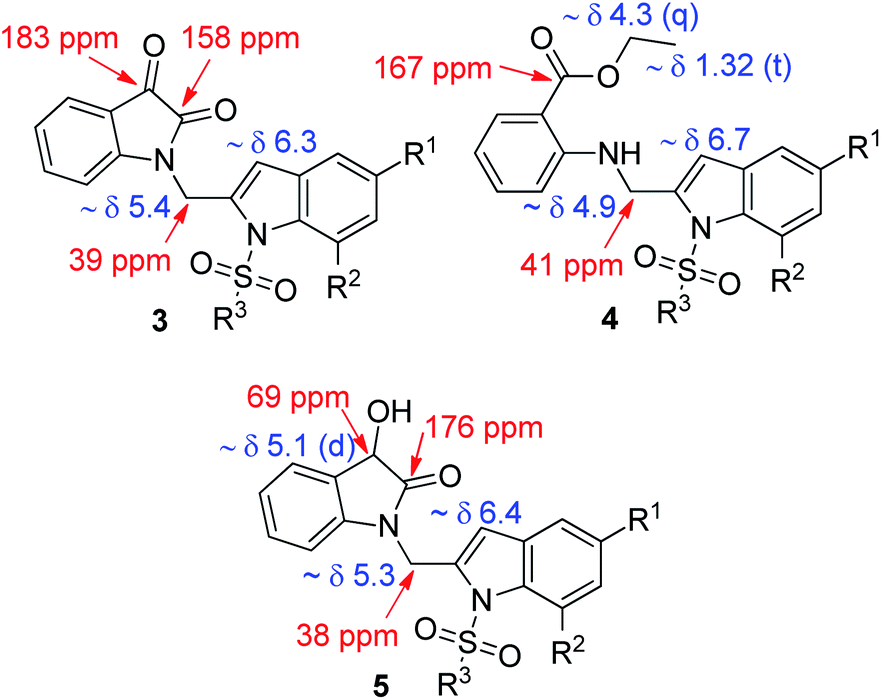
All the synthesized compounds (3, 4 and 5) were characterized by (1H and 13C NMR, and MS) spectral data (Fig. 4). Briefly, a singlet in the region δ 6.3–6.7 in the 1H NMR spectra of these compounds accounted for C-3 indole proton whereas a signal in the region δ 5.0–5.4 was due to the “CH2” moiety of 3 or 4 or 5, respectively. In 13C NMR, the “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” group appeared near 183 ppm (ketone) and 158 ppm (amide) in case of 3, 167 ppm (ester) in case of 4 and 176 ppm (amide) in case of 5. Additionally, the presence of OEt group in case of 4 and benzylic “CHOH” group in case of 5 was indicated by the corresponding 1H NMR spectra.
O” group appeared near 183 ppm (ketone) and 158 ppm (amide) in case of 3, 167 ppm (ester) in case of 4 and 176 ppm (amide) in case of 5. Additionally, the presence of OEt group in case of 4 and benzylic “CHOH” group in case of 5 was indicated by the corresponding 1H NMR spectra.
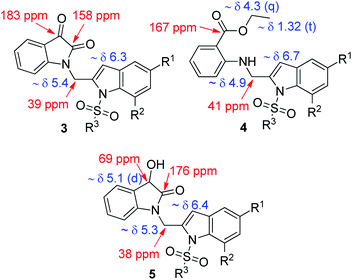 |
| | Fig. 4 Partial 1H and 13C NMR data of compound 3, 4 and 5. | |
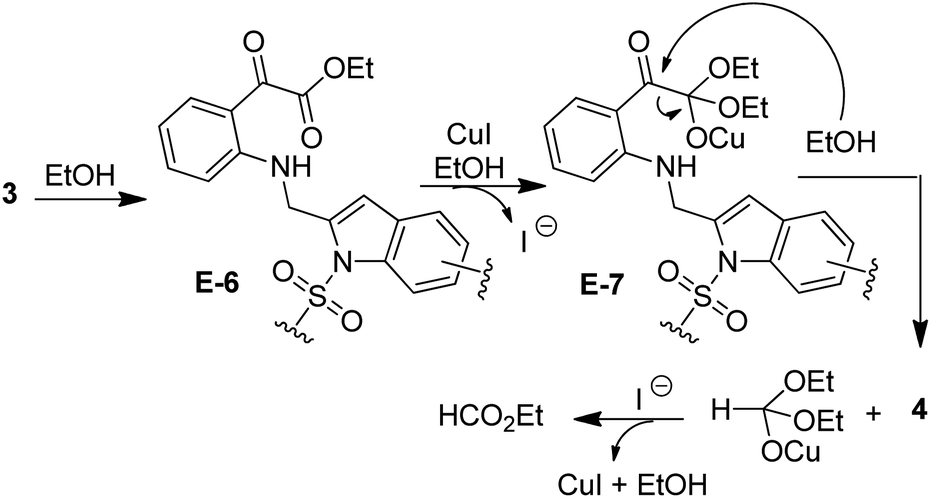
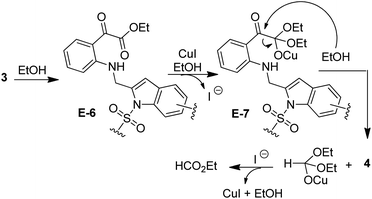
A plausible mechanism18,19,21 for the formation of isatin–indole derivatives 3 via coupling/cyclization is depicted in Scheme 3. Thus the coupling proceeded via the generation of organo-Pd(II) species E-1 [as a result of oxidative addition of Pd(0) to 2] that on transmetalation with Cu(I)-acetylide (generated in situ from 1) afforded E-2. On reductive elimination E-2 produced E-3 with the regeneration of Pd(0)-catalyst thereby completing the first catalytic cycle (the Sonogashira coupling). The second catalytic cycle, facilitated by the Cu(I)-catalyst involved intramolecular cyclization of E-3. Indeed, the polarization of the alkyne moiety of E-3 by the Cu(I) species seemed to be favoured by the coordination with the proximate “CO” group. Thus the intermediate E-4 followed by E-5 was formed that finally afforded the desired isatin–indole derivative 3. At the same time Cu(I)-catalyst was regenerated to complete the second catalytic cycle. It is evident from Table 3 that the side product 4 was isolated in several cases (though traces in some cases) as a result of opening of isatin ring. Indeed, the ring opening of isatins in MeOH or EtOH leading to the formation of the corresponding 2-aminobenzoate esters has been reported earlier but the reaction was performed in the presence of an oxidizing agent e.g. TBHP.22a,b The simple alcoholysis of isatin on the other hand is known to produce the α-keto ester derivative.22c Based on these report we propose (Scheme 4) that the isatin–indole 3 afforded E-6 in the presence of EtOH that on further reaction with EtOH aided by CuI provided E-7. Finally, the compound 4 was obtained via a C–C bond cleavage of E-7 with the regeneration of CuI. To gain further evidence the compound 3a was treated with CuI and Et3N in EtOH for 3 h under the conditions of entry 1 of Table 1 (without any Pd-catalyst). The ester 4a was obtained albeit in low yield (11%) in this case.
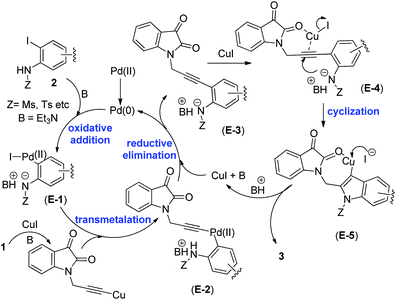 |
| | Scheme 3 The plausible reaction mechanism for the formation compound 3 under Pd/Cu-catalysis. | |
 |
| | Scheme 4 The proposed reaction mechanism for the formation of compound 4. | |
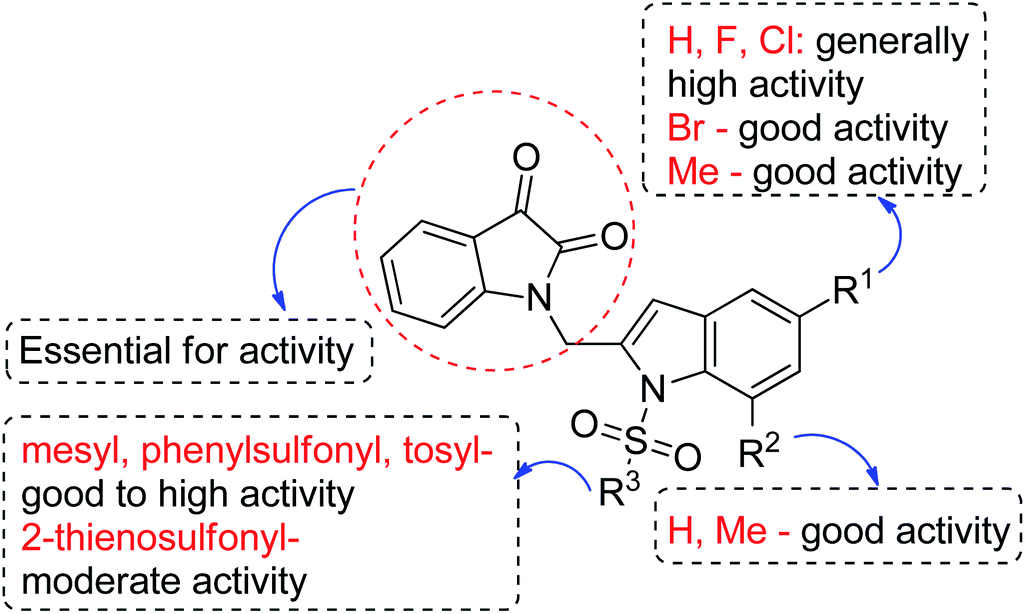
We then tested all the synthesized compounds i.e. 3, 4 and 5 for their CM inhibitory properties in vitro23 using an assay that involved measurement of catalytic activity of enzyme (CM) in the conversion of chorismate (substrate) to prephenate. The known inhibitor17 G (Table 1) was used as a reference compound. All compounds were tested at an initial concentration of 30 μM. In case of isatin–indole series (3) most of the compounds showed good to high inhibition (>65%, Table 4) that was better than the reference compound G. Notably, none of the compound 4 and 5 showed any significant inhibition of CM (Table 4) indicating chemical modification of isatin ring was not favourable for CM inhibition in case of current series of compounds. An overview of SAR (Structure–Activity-Relationship) is presented in Fig. 5. Briefly, the H, F and Cl group at C-5 of indole ring was beneficial for CM inhibition whereas other substituents like “Br” or “Me” at this position was also helpful. The “H” or “Me” group at C-7 position was generally favourable. Among the sulfonyl substituents at indole N-1 position the mesyl, phenylsulfonyl and p-tosyl group appeared to be favourable for high activity whereas the 2-thienosulfonyl group was found to be inferior. Nevertheless, the two best active compounds e.g. 3e and 3f were selected for concentration dependent study. It is evident from Table 5 that both these compounds maintained >50% inhibition till the concentration of 100 nM whereas inhibition was still observed at low concentrations. Thus the IC50 of these compounds appeared to be ≤ 100 nM that was superior to the IC50 of known compound G i.e. ∼5.3 μM. This was further supported by the docking of 3e and 3f (or F-1, Table 1) into the active site of CM. In addition to the hydrophobic contacts as observed in case of other isatin–indole derivatives (Table 1), a halogen bond incorporated by Cl with TRP61 of A chain was observed in case of 3e (AutoDock Vina score −8.9 kcal mol−1) (see ESI†). Notably, docking of ester 4f and 5b showed decreased number of hydrophobic contacts with CM (see Fig S-7 and S-8, ESI†) compared to 3e and 3f thereby providing the possible reasons for their low inhibition of CM observed. Overall, both 3e and 3f were identified as highly potent compounds and their further profiling is ongoing.
Table 4 In vitro evaluation of isatin–indole derivatives (3) against CM
| Compounds |
% Inhibition @ 30 μMa |
Compounds |
% Inhibition @ 30 μMa |
| Data represent the mean values of three independent determinations. |
| 3a |
66.83 |
4a |
4.54 |
| 3b |
67.53 |
4b |
9.59 |
| 3c |
71.36 |
4c |
1.38 |
| 3d |
67.67 |
4e |
14.18 |
| 3e |
73.43 |
4f |
15.34 |
| 3f |
74.20 |
4h |
26.29 |
| 3g |
47.93 |
4j |
14.12 |
| 3h |
70.28 |
5a |
18.65 |
| 3i |
71.61 |
5b |
10.26 |
| 3j |
72.92 |
5c |
5.93 |
| 3k |
67.30 |
|
|
| Compound G |
56.97 |
|
|
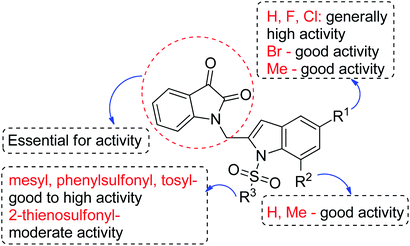 |
| | Fig. 5 Summary of SAR for CM inhibitory activities of compound 3. | |
Table 5 Concentration dependent study of compound 3e and 3f against CM
| Compound concentration |
% Inhibitiona |
| 3e |
3f |
| Data represent the mean values of three independent determinations. |
| 30 μM |
73.43 |
74.20 |
| 10 μM |
61.32 |
68.91 |
| 3 μM |
55.18 |
54.23 |
| 1 μM |
51.87 |
57.96 |
| 300 nM |
52.07 |
55.40 |
| 100 nM |
50.46 |
54.27 |
| 30 nM |
42.04 |
37.98 |
| 10 nM |
31.70 |
24.93 |
Having identified the hit molecules it was become essential to perform the early detection of ADME (absorption, distribution, metabolism, and excretion) or pharmacokinetic properties of NCEs (new chemical entities). Indeed, this is known to be a helpful strategy in the initial stage of the drug discovery process. Hence the computational ADME prediction of compounds 3e and 3f along with the known inhibitor G was performed using SwissADME web-tool24 and results are presented in Table 6 (among the various descriptors only notable one are listed in the table). To our satisfaction desirable ADME was predicted for both the compound 3e and 3f. Indeed, high GI absorption, no BBB (Blood Brain Barrier) penetration and no P-gp substrate potential have been predicted for both molecules with superior bioavailability score over the reference molecule G. Overall, the compound 3e and 3f could be better inhibitors than G and appeared to have medicinal value especially from the viewpoint of developing potential anti-tubercular agents.
Table 6 Computational ADME prediction of 3e, 3f and G
| Properties |
Molecules |
| 3e |
3f |
G |
| logP: lipophilicity. logS (ESOL): water solubility, calculated by ESOL method which quantitative structure–property relationship (QSPR) based model. GI: gastrointestinal. BBB: blood brain barrier. P-gp: permeability glycoprotein. |
| (i) Physicochemical |
| Molecular weight (g mol−1) |
388.82 |
416.45 |
425.41 |
| Consensus logPa |
2.68 |
3.28 |
0.90 |
| logS (ESOL)b |
−4.25 (moderately soluble) |
−5.03 (moderately soluble) |
−3.63 (soluble) |
|
| (ii) Pharmacokinetics |
| GIc absorption |
High |
High |
Low |
| BBBd permeation |
No |
No |
No |
| P-gpe substrate |
No |
No |
Yes |
|
| (iii) Druglikenss |
| Lipinski rule |
No violation |
No violation |
No violation |
| Veber rule |
No violation |
No violation |
1 Violation (TPSA >140; i.e. 182.15 Å2) |
| Bioavailability score |
0.55 |
0.55 |
0.11 |
Conclusions
In conclusion, a series of novel isatin–indole derivatives has been designed as potential inhibitors of chorismate mutase (CM). The proposed design was supported by in silico docking studies that predicted strong interactions of these molecules with CM. The target compounds were then synthesized via the one-pot coupling/cyclization strategy involving the reaction of an isatin based terminal alkyne with 2-iodosulfanilides under Pd–Cu catalysis. A number of isatin–indole derivatives (3) were prepared using this method in acceptable yields (58–70%). A side product e.g. 2-indolylmethylamino benzoate ester derivative (4) was obtained as a result of isatin ring opening (ethanolysis) of main products in certain cases. Additionally, regioselective reduction of selected compounds afforded the corresponding C-3 hydroxy derivatives (5). Notably, all isatin–indole derivatives but not the esters (4) or C-3 hydroxy derivatives (5) showed good to high inhibition of CM in vitro. Two isatin–indole derivatives (3e and 3f) showed inhibition at nanomolar concentration. Thus the potent CM inhibitory properties along with the predicted desirable ADME properties in silico highlighted potential of these compounds for further Med Chem effort. Overall, the current research related to isatin/indole chemistry could attract further interest.
Conflicts of interest
The authors confirm that there are no conflicts to declare.
Acknowledgements
GSR thanks DST, India for a INSPIRE fellowship (IF160590). Authors thank the Management of DRILS, Hyderabad, India and Manipal University, Manipal, India for encouragement and support and DBT, New Delhi, India for financial assistance (grant no. BT/PR12817/COE/34/23/2015).
Notes and references
- E. Haslam, Shikimic Acid: Metabolism and Metabolites, Wiley, New York, 1993 Search PubMed.
- For a review, see: M. Khanapur, M. Alvala, M. Prabhakar, K. S. Kumar, R. K. Edwin, P. S. V. K. S. Saranya, R. K. Patel, G. Bulusu, P. Misra and M. Pal, Bioorg. Med. Chem., 2017, 25, 1725–1736 CrossRef CAS PubMed.
- End to killer TB? Scientists identify an enzyme that may hold clue, https://researchmatters.in/article/end-killer-tb-scientists-identify-enzymemay-hold-clue Search PubMed.
- N. Karali, A. Gürsoy, F. Kandemirli, N. Shvets, F. B. Kaynak, S. Özbey, V. Kovalishyn and A. Dimoglo, Bioorg. Med. Chem., 2007, 15, 5888–5904 CrossRef CAS PubMed.
- T. Aboul-Fadl, H. A. Abdel-Aziz, M. K. Abdel-Hamid, T. Elsaman, J. Thanassi and M. J. Pucci, Molecules, 2011, 16, 7864–7879 CrossRef CAS PubMed.
- M. F. Abo-Ashour, W. M. Eldehna, R. F. George, M. M. Abdel-Aziz, M. M. Elaasser, N. M. Abdel Gawad, A. Gupta, S. Bhakta and S. M. Abou-Seri, Eur. J. Med. Chem., 2018, 160, 49–60 CrossRef CAS PubMed.
- A. Dandia, R. Singh and K. Arya, Phosphorus, Sulfur, Silicon Relat. Elem., 2004, 179, 551–564 CrossRef CAS.
- V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hübet, D. Rauh and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 5902–5905 CrossRef CAS PubMed.
- V. V. Vintonyak, K. Warburg, B. Over, K. Hübel, D. Rauh and H. Waldmann, Tetrahedron, 2011, 67, 6713–6729 CrossRef CAS.
- A. Dandia, A. K. Jain and A. K. Laxkar, RSC Adv., 2013, 3, 8422–8430 RSC.
- A. Dandia, R. Singh and D. Saini, J. Chem. Sci., 2013, 125, 1045–1053 CrossRef CAS.
- S. J. Tantry, G. Degiacomi, S. Sharma, L. K. Jena, A. Narayan, S. Guptha, G. Shanbhag, S. Menasinakai, M. Mallya, D. Awasthy, G. Balakrishnan, P. Kaur, D. Bhattacharjee, C. Narayan, J. Reddy, C. N. Naveen Kumar, R. Shandil, F. Boldrin, M. Ventura, R. Manganelli, R. C. Hartkoorn, S. T. Cole, M. Panda, S. D. Markad, V. Ramachandran, S. R. Ghorpade and N. Dinesh, Bioorg. Med. Chem. Lett., 2015, 25, 3234–3245 CrossRef CAS PubMed.
- M. A. Borad, M. N. Bhoi, S. K. Rathwa, M. S. Vasava, H. D. Patel, C. N. Patel, H. A. Pandya, E. A. Pithawala and J. J. Georrge, Interdiscip. Sci.: Comput. Life Sci., 2018, 10, 411–418 CrossRef CAS PubMed.
- V. U. Jeankumar, R. Alokam, J. P. Sridevi, P. Suryadevara, S. S. Matikonda, S. Peddi, S. Sahithi, M. Alvala, P. Yogeeswari and D. Sriram, Chem. Biol. Drug Des., 2014, 83, 498–506 CrossRef CAS PubMed.
- A. Nakhi, B. Prasad, U. Reddy, R. M. Rao, S. Sandra, R. Kapavarapu, D. Rambabu, G. Rama Krishna, C. M. Reddy, K. Ravada, P. Misra, J. Iqbal and M. Pal, RSC Med. Chem., 2011, 2, 1006–1010 RSC.
- P. Prakash, B. Aruna, A. A. Sardesai and S. E. Hasnain, J. Biol. Chem., 2005, 280, 19641–19648 CrossRef CAS PubMed.
- H. Agrawal, A. Kumar, N. C. Bal, M. I. Siddiqi and A. Arora, Bioorg. Med. Chem. Lett., 2007, 17, 3053–3058 CrossRef CAS PubMed.
-
(a) S. Cacchi, G. Fabrizi and L. M. Parisi, Org. Lett., 2003, 5, 3843–3846 CrossRef CAS PubMed;
(b) S. Cacchi, G. Fabrizi, L. M. Parisi and R. Bernini, Synlett, 2004, 287–290 CrossRef CAS;
(c) G. A. Slough, V. Krchňák, P. Helquist and S. M. Canham, Org. Lett., 2004, 6, 2909–2912 CrossRef CAS PubMed;
(d) F. Liu and D. Ma, J. Org. Chem., 2007, 72, 4844–4850 CrossRef CAS PubMed;
(e) H. A. Oskooie, M. M. Heravi and F. K. Behbahani, Molecules, 2007, 12, 1438–1446 CrossRef CAS PubMed.
-
(a) M. Pal, V. Subramanian, V. R. Batchu and I. Dager, Synlett, 2004, 1965–1969 CrossRef CAS;
(b) M. Layek, U. Lakshmi, D. Kalita, D. K. Barange, A. Islam, K. Mukkanti and M. Pal, Beilstein J. Org. Chem., 2009, 5, 46, DOI:10.3762/bjoc.5.46;
(c) R. M. Rao, U. Reddy Ch, A. Nakhi, N. Mulakayala, M. Alvala, M. K. Arunasree, R. R. Poondra, J. Iqbal and M. Pal, Org. Biomol. Chem., 2011, 9, 3808–3816 RSC;
(d) B. Prasad, R. Adepu, S. Sandra, D. Rambabu, G. R. Krishna, C. M. Reddy, G. S. Deora, P. Misra and M. Pal, Chem. Commun., 2012, 48, 10434–10436 RSC.
- For selected reviews, see:
(a) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed;
(b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2309 CrossRef CAS PubMed;
(c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed;
(d) H. J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4427 CrossRef PubMed.
- For other class of isatin-indole derivatives, see:
(a) B. V. S. Reddy, N. Rajeswari, M. Sarangapani, Y. Prashanthi, R. J. Ganji and A. Addlagatta, Bioorg. Med. Chem. Lett., 2012, 22, 2460–2463 CrossRef PubMed;
(b) R. I. Al-Wabli, A. S. Zakaria and M. I. Attia, Molecules, 2017, 22, E1958, DOI:10.3390/molecules22111958.
-
(a) Y. W. Wang, L. Zheng, F. C. Jia, Y. F. Chen and A. X. Wu, Tetrahedron, 2019, 75, 1497–1503 CrossRef CAS;
(b) A. H. Kalbandhe, A. C. Kavale, P. B. Thorat and N. N. Karade, Synlett, 2016, 27, 763–768 CAS;
(c) For a review, see: J. F. M. Da Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273–324 CrossRef CAS.
-
(a) S. K. Kim, S. K. Reddy, B. C. Nelson, G. B. Vasquez, A. Davis, A. J. Howard, S. Patterson, G. L. Gilliland, J. E. Ladner and P. T. Reddy, J. Bacteriol., 2006, 188, 8638–8648 CrossRef CAS PubMed;
(b) S. Sasso, C. Ramakrishnan, M. Gamper, D. Hilvert and P. Kast, FEBS J., 2005, 272, 375–389 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures, Tables and Schemes, Experimental procedures, spectral data of all new compounds and copies of spectra. See DOI: 10.1039/c9ra09236f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Parimal Misraa and
Manojit Pal
b,
Parimal Misraa and
Manojit Pal