
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Investigation of yellow horn (Xanthoceras sorbifolia Bunge) transcriptome in response to different abiotic stresses: a comparative RNA-Seq study†
Yanhe Lang *,
Zhi Liu and
Zhimin Zheng
*,
Zhi Liu and
Zhimin Zheng
State Key Laboratory of Tree Genetics and Breeding Laboratory, Ministry of Education, Alkali Soil Natural Environmental Science Center (ASNESC), College of Life Science, Northeast Forestry University, Harbin, Heilongjiang Province, China. E-mail: langyanhe@163.com; Tel: +86-151-0453-8096
First published on 12th February 2020
Abstract
Yellow horn (Xanthoceras sorbifolia Bunge) is a well-known oil-rich seed shrub which can grow well in barren and arid environments in the northern part of China. Yellow horn has received worldwide attention because of its excellent economic and environmental value. However, because of its limited genetic data, little information can be found regarding the molecular defense mechanisms of yellow horn exposed to various abiotic stresses. In view of this, the current study aims to investigate the impact of different abiotic stresses (i.e. NaCl, ABA and low temperature) on the transcriptome of yellow horn using RNA-Seq. Based on the transcriptome sequencing data, approximately 27% to 45% of stress-responsive genes were found highly expressed after stress treatment for 24 h. In addition, these genes were found to be still expressed after stress treatment for 48 h. However, many additional genes were stress-regulated after 48 h treatment compared with the 24 h treatment. GO enrichment analysis revealed that the expression patterns of the stress-responsive, type-specific terms were generally down-regulated. Most shared GO terms were primarily involved in protein folding, unfolding protein binding, protein transport and protein modification. Further, transcription factors (TFs), such as ERFs, bHLH, GRAS and NAC, were found to be enriched only in the low temperature treatment group, particularly the ERF TFs families. These combined results suggested that yellow horn may have developed specific molecular defense systems against diverse abiotic stresses.
Introduction
Yellow horn (Xanthoceras sorbifolium) is an endemic oil-rich, woody deciduous shrub, which is widely distributed in the northern part of China.1 Yellow horn is the only species of Xanthoceras belonging to the family of Sapindaceae, which has attracted worldwide attention in recent years owing to its economic and environmental value.2 As one kind of oil-rich seed plant, yellow horn has been widely used to produce renewable and environmentally friendly bioactive oils.3,4 Previous publications have addressed the unsaturated fatty acids in the seeds of yellow horn, including gondoic acid, oleic acid, nervonic acid and linoleic acid, ranging from 85% to 93%.5,6 Furthermore, the Chinese government has launched yellow horn breeding programs in order to achieve food security and ease the energy crisis in a sustainable manner.7 Unlike most energy-resource trees that can only survive in farmland, yellow horn can not only survive in barren, drought and cold environment, but also grow well in the saline-alkali environment.8 As a result, yellow horn can be cultivated in deserts, semi-arid and arid zones in the northwest of China, thereby having the potential for future vegetation restoration of barren, deserts, semi-arid and arid regions.9The growth of plants is susceptible to various abiotic stresses.10,11 When individual abiotic stress happens, plants often exhibit reduced growth and productivity.12 Therefore, the elucidation of the possible molecular responses of plants under different abiotic stresses would be helpful to understand the general defense mechanisms of plants when exposed to unfavorable environmental conditions. However, as a non-model plant, there is no information available regarding the molecular defense responses of yellow horn subjected to various abiotic stresses, since the whole-genome sequences of yellow horn has not been released until June, 2019.13,14
The rapid development of the next generation sequencing (NGS) can facilitate comprehensive analyses of complex genome based on the large-scale sequencing data.15,16 The applications of NGS technologies are not only contributable to a deep investigation into the genomic resources for yellow horn, but also conducive to a quick identification of differentially expressed genes (DEGs), as well as the key metabolic pathways of yellow horn in response to abiotic stresses.17,18 As one type of NGS, RNA-sequencing (RNA-Seq) can provide unprecedented opportunities for the generation of genomic information in uncharacterized earlier systems.19,20 Recent literatures have also reported the transcriptome of many plants performed using RNA-Seq, such as tomato,21 Rosa chinensis,22 watermelon23 as well as tobacco.24 However, only a few publications focused on the investigations into yellow horn transcriptome.8,25 In addition, little research has been dedicated to the molecular defensive responses of yellow horn in response to different abiotic stresses.
In this study, we investigated the transcriptome of yellow horn exposed to different abiotic stresses (i.e. NaCl, ABA and low temperature) using RNA-Seq based on the Illumina Nova Seq6000 2 × 150 bp platform, including the functional annotations of transcripts and characterization of DEGs. These transcripts would represent the first transcriptome sequence dataset of yellow horn responsive to different abiotic stresses. These transcriptome data would provide new perspectives for the exploration of possible molecular defensive responses of yellow horn against different abiotic stresses.
Materials and methods
Plant material and treatment
In this study, the yellow horn seeds were obtained from the Research Center of Yellow Horn Engineering Technology, State Forestry and Grassland Administration, Chifeng city, China. The germinated seeds were randomly selected and cultivated at 25 °C in order to obtain the yellow horn seedlings (about 15 cm long). These seedings of yellow horn were soil cultured in the greenhouse with 16 h-light–8 h-dark light conditions under 50–60% relative humidity (RH), and irrigated with moderate water. After cultivation for 30 days, twelve 15 cm-long seedlings under same growing conditions were randomly selected for leave sampling. These seedlings were randomly divided into four groups, including the control group and three treatment groups. Three yellow horn seedlings in each treatment group were subjected to different abiotic stresses, i.e. 2% NaCl (w/v) treatment, 100 μm ABA treatments and low temperature (4 °C) treatment, respectively. Leave sampling were conducted after 24 h and 48 h stress applications. The two time points were selected according to the phenotypic changes of yellow horn seedings after 24 h and 48 h treatments as compared to the control groups. Leaves samples were collected in triplicate for RNA-Seq throughout this investigation. These freshly leave samples were snapped frozen using liquid nitrogen and then preserved at −80 °C for further analysis.RNA extraction, cDNA library construction and RNA-sequencing
The extraction of total RNA from the collected leaves of yellow horn was performed using the TRIzol® Reagent (Invitrogen) following the manufacturer's instructions. Afterwards, the genomic DNA was digested using RNase-free DNase I (TaKara, Dalian, China) following the manufacturer's protocols. The integrity of total RNA were detected using 1% agarose gels and then quantified using Nanodrop ND-2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). Total RNA extracts with the OD260/280 value higher than 2.0 were selected for the construction of the cDNA library using the TruSeq™ RNA sample preparation kit (San Diego, CA) on the basis of the Illumina manufacturer's instructions (Illumina, San Diego, CA, USA).26The synthesis of cDNA, end repair, A-base addition as well as the ligation of the Illumina-indexed adaptors were carried out according to Illumina's protocols.26 Briefly, the cleaved RNA fragments were used for the synthesis of the first-strand of cDNA using the random hexamers and reverse transcriptase. Then the synthesis of the second-strand of cDNA was performed using dNTPs, RNase H and DNA polymerase I. The resultant dscDNA fragments were purified using AMPure XP beads (Beckman Coulter Genomics, Danvers, MA, USA), and then subjected to end repair through the addition of a single “A” base and the ligation of Illumina multiplex barcode adapters. The adaptor-modified cDNA fragments were separated by gel purification using AMPure XP beads and amplified via the PCR reactions using Phusion DNA polymerase (NEB) to enrich the adapter-ligated of fragments. The resultant PCR products were purified with AMPure XP beads again, and then the final cDNA libraries were constructed.
The quality of different cDNA libraries was examined via the TBS380 system before the proceeding of the subsequent analysis with the aim to ensure the assembly of sequences. Afterwards, the clustering of these index-coded samples was performed on a cBot Cluster Generation System using the HiSeq PE Cluster Kit v4 (Illumina, San Diego, CA, USA) according to the manufacturer's protocols. After cluster generation, the transcriptome sequencing of the paired-end cDNA libraries derived from both control and treated groups was performed on the llumina Nova Seq6000 2 × 150 bp platform by Shanghai Biozeron Biotechnology Co. Ltd (Shanghai, China). All raw data have been submitted to the NCBI database.
Quality control and mapping
The quality of the paired-end raw reads generated by Illumina platform was firstly examined using the Fastp (version 0.20.0) (https://www.jianshu.com/p/6f492058da5b). Then these raw reads were trimmed before assembly by eliminating the adaptors and primers with the Cutadapt software (version 1.11)27 in order to get the high-quality reads. Then the LUCY2 software28 was applied in order to remove the low-quality regions and bases. Clean reads were finally obtained by discarding the reads with adapters, poly-N, together with the low-quality reads. All subsequent downstream analysis were conducted in terms of the high-quality clean reads. The clean reads were separately mapped to yellow horn reference genome in NCBI (accession number: PRJNA483857) with orientation mode using the HISAT2 software (version 2.0.5).29,30 Then the read count for each gene was obtained according to the mapping results.Identification of differentially expressed genes
In order to characterize the DEGs between the control and treatment groups, gene expression level was estimated using feature Counts (version 1.5.0-P1).31 Briefly, the measurement of gene expression level for each transcript was implemented via the fragments with per kilo bases or per million bases of transcripts using the mapped (TPM) approach.32 The differential expression analysis of yellow horn samples subjecting to different abiotic stresses were performed using DESeq2 in the R software (version 3.3.2).33 The resultant P values adjusted to the Benjamini and Hochberg's approach in order to control the false discovery rate (FDR) to correct for multiple testing.34 The genes with FDR value ≤ 0.05 and |log2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) FC| ≥ 1 were considered the threshold of differential expression as reported earlier.35
FC| ≥ 1 were considered the threshold of differential expression as reported earlier.35
Go and KEGG annotation
To better understand the functions of differentially expressed genes (DEGs) and obtain better insights into the stress-responsive genes and pathways, DEGs were annotated in the GO databases (E-value ≤1 × 10−6) and the KEGG databases with an E-value ≤1 × 10−10 using the eggNOG-mapper software.36 In addition, GO functional classifications were performed using the hypergeometric distribution algorithm in the context of Molecular Function (MF), Biological Processes (BP) and Cellular Component (CC) ontologies performed with WEGO software,37 while the KEGG pathway assignments were carried out using the online KEGG Automatic Annotation Server (KAAS), as described in a previous study.38GO and KEGG enrichment analysis
To investigate the BF aspect of the screened DEGs, GO enrichment and KEGG enrichment were conducted using Goatools39 and KOBAS,40 respectively. GO enrichment analysis was conducted using the GOseq package in the R software on the basis of Wallenius non-central hyper-geometric distribution.41 The KOBAS software was used to conduct the KEGG pathway enrichment analysis of the DEGs and then examine the statistical significance of DEGs enriched in the KEGG pathways.Multidimensional scaling analysis
In order to provide a visual representation of transcriptomic relationships between different RNA-Seq samples by spatial arrangement and further evaluate the quality of the transcriptomic sequencing data, the clustering of RNA-Seq samples were implemented using the multidimensional scaling (MDS) analysis performed using SPSS19.0 software (IBM, Armonk, NY, USA) by inputting the abundance of genes as variables.Results
Transcriptomic relationships of RNA-Seq samples based on the MDS approach
The reliability of transcriptomic sequencing was verified as presented in Fig. S1.† The mapping result of RNA-sequencing data to the yellow horn reference genome was shown in ESI 1 Table S1.† The transcriptomic relationships between the type and duration of diverse abiotic stresses (i.e. NaCl stress, ABA stress and low temperature stress) were determined using the MDS method as presented in Fig. 1. The MDS patterns of the replicated yellow horn samples exposed to different abiotic stresses were explored in the context of 24 h and 48 h treatment, as illustrated in Fig. 1A and B. It is obvious that RNA-Seq samples exposed to different abiotic stresses for 24 h and 48 h were prone to be separable in the MDS plot. This demonstrated that the observed divergence in transcriptome was strongly related to the type and duration of abiotic stresses. However, within each treatment group at 24 h and 48 h, RNA-Seq samples tended to be grouped closely together. In addition, control samples and low temperature treated samples positioned most closely after treatment for 24 h (Fig. 1A), whereas control samples and ABA stressed samples positioned closely after treatment for 48 h (Fig. 1B). However, the control samples and low temperature treated samples positioned most distantly apart, indicating the differential MDS patterns may owe much to the stress durations.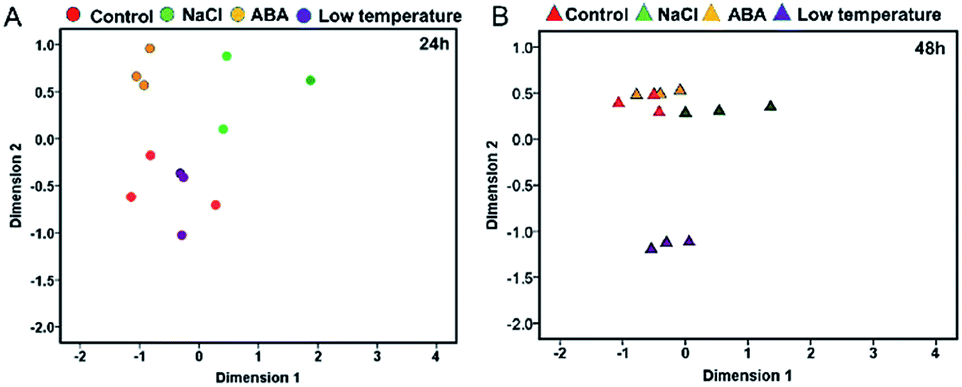 | ||
| Fig. 1 The MDS clustering patterns of the replicated RNA-Seq samples. Features represent libraries from control group, NaCl, ABA and low temperature groups after 24 h treatment (A) and 48 h treatment (B). RNA-Seq samples were arranged in terms of their calculated Euclidean distances. | ||
Identification of differentially expressed genes
In this study, the DEGs related to salt stress, ABA stress and low temperature stress were identified in three pairwise contrasts between the control and stress-treated samples after treatment for 24 h and 48 h. The number of DEGs which have FDR value ≤ 5% and |log2FC| ≥ 1 was illustrated for three treatment groups in the context of 24 h and 48 h and presented as volcano plots (Fig. 2A). A full list of the DEGs was displayed as in ESI Table S2.† After stress exposure for 24 h and 48 h, salt stress treatment led to the smallest number of DEGs (1119 at 24 h and 770 at 48 h). However, the most significant impact on gene expression can be found in the low temperature treatment group, with 2628 DEGs at 24 h and 3480 DEGs after 48 h treatment. The total number of DEGs involved in the low temperature treatment was substantially higher than those regulated by salt stress or ABA stress alone. This result may ascribe to the type and duration of abiotic stresses.
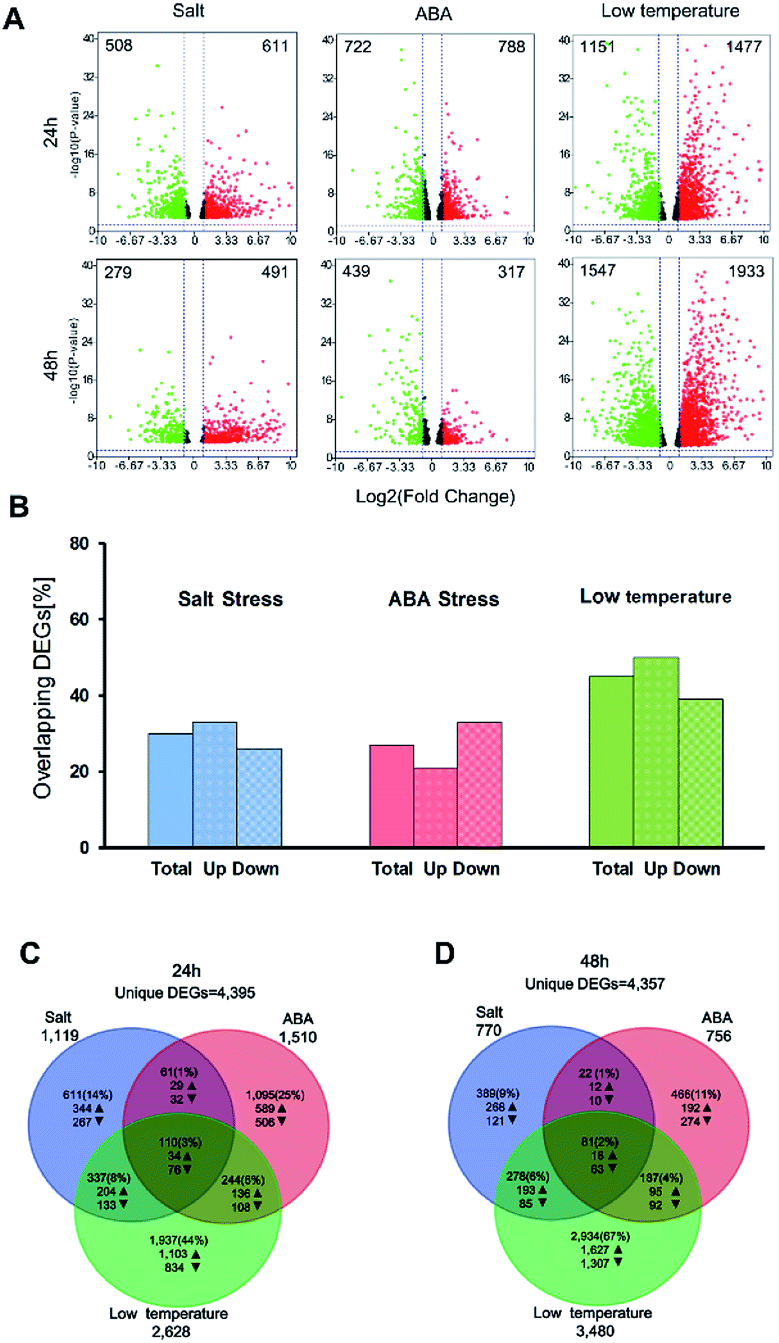 | ||
| Fig. 2 Summary of the DEGs between control group and stress-treated groups. (A) Volcano plots show the DEGs for each treatment over time. The up-regulated DEGs are symbolized by red dots, while the down-regulated DEGs are symbolized by green dots. The number of total DEGs are displayed in the upper left and right corner for the significant up- or down-regulated DEGs. The DEGs that do not exceed the threshold (|log2FC| ≥ 5 and FDR ≤ 5%) are illustrated in black color; (B) overlapped DEGs at 24 h that also expressed at 48 h for each treatment in percent. Bars indicate the all overlaps of DEGs, the up-regulated and the down-regulated DEGs, respectively; (C and D) Venn diagrams show the overlaps between DEGs in response to salt stress, ABA stress and low temperature stress after 24 h and 48 h of stress applications. Arrows indicate the number of up- or down-regulated DEGs. | ||
The DEGs found between 27% (ABA treatment) and 45% (low-temperature treatment) of after 24 h were also expressed after 48 h of treatment (Fig. 2B). 21–33% of the up-regulated over-lapped DEGs and 26–39% of the down-regulated over-lapped DEGs were found conserved as a function of time among each treatment. Cross-comparison was made between the DEGs sets after 24 h, which showed that the highest proportion of DEGs (44%) was unique to the low temperature treatment, while salt stress (14%) and ABA stress (25%) treatment led to less DEGs (Fig. 2C). This indicated the low temperature treatment cannot lead to the additive regulation of DEGs than the other two individual stress treatment. Instead, a substantial number of DEGs were merely regulated by the low temperature treatment but not by the two individual stress factors. 110 sets of the overlap DEGs (3%) responded to all three treatments after 24 h, indicating that the regulatory mechanism that were not affected by the stress type. In contrast, 48 h stress response showed lower overlap genes (2%) in response to all treatments. 67% of DEGs were unique to low temperature stress at 48 h (Fig. 2D), while only 9–11% of DEGs were unique to the salt treatment and ABA treatment, respectively.
Evaluation of stress-responsive pathways
In order to study the biological functions of DEGs, GO enrichment analysis were performed. GO terms were designated to DEGs to identify the stress-responsive biological processes and functions. The obtained results were cross-compared using the agriGO software (version 2.0).42 A whole list of the enriched GO terms in all treatment-by-time groups was given as presented in ESI Table S3† and S4.† There remained 57 GO terms in total in response to different abiotic stresses treatment at 24 h after filtering using REVIGO43 as shown in Fig. 3. GO terms were shared between two or more treatments. None of these GO terms were treatment-specific. Most GO terms were commonly enriched and shared between different abiotic stresses treated groups. The shared terms of BP and MF in response to salt stress and low temperature stress were mostly down-regulated, including metabolic processes such as ‘protein folding’ (GO:0006457) and ‘intracellular protein transmembrane transport’ (GO:0065002). Only a few shared GO terms were found to be up-regulated, including not only catalytic activities such as ‘glucosyltransferase activities’ (GO:0046527) but also metabolic processes involved in a series of regulatory processes (e.g. GO:0060255 and GO:0051252). Mutual DEGs between ABA stress and low temperature stress were all down-regulated, which were identified to be involved in general stimulus responses and protein folding process. These shared DEGs were similar to those between salt stress and low temperature, featured by the unique GO terms ‘response to hydrogen peroxide’ (GO:0042542), ‘negative regulation of biological process’ (GO:0048519) and ‘protein transmembrane import into intracellular organelle’ (GO:004743). The last set of treatment-independent terms (i.e. salt stress and ABA stress) also showed down-regulation in most biological processes. Twenty-two mutual shared terms were found while eleven of them were unique to the first two sets. Only two shared terms were found up-regulated, which mainly involved in general stimulus responses, i.e. ‘response to water deprivation’ (GO:0009414) and ‘response to water’ (GO:0009415). Ten shared terms were found down-regulated in all treatment groups at 24 h.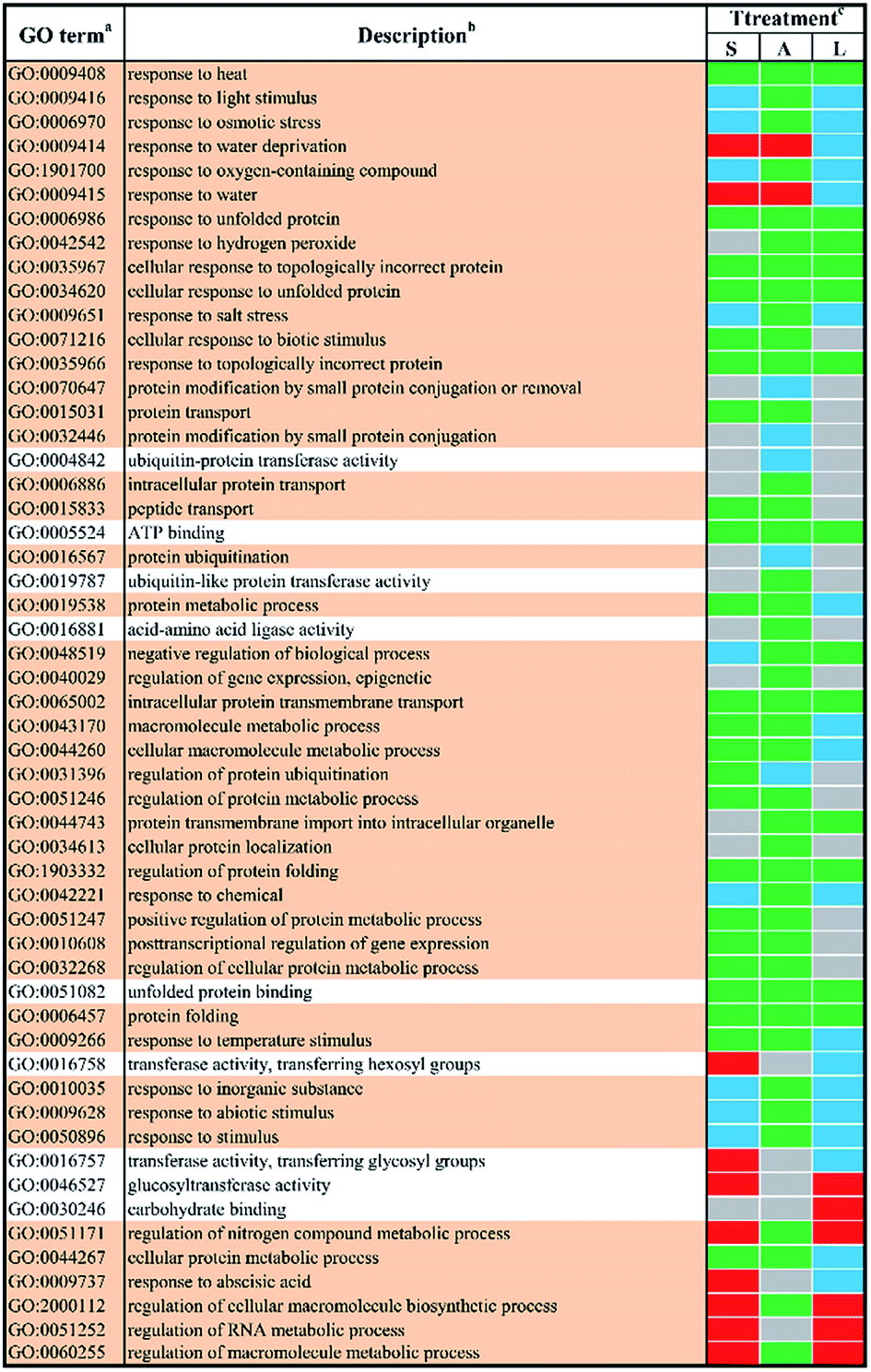 | ||
| Fig. 3 The enriched l GO terms among different DEGs after 24 h stress applications. a Non-redundant GO terms (FDR ≤ 5% and similarity ≤ 0.5) are shown for the identified biological processes (BP) (light khaki background) and molecular functions (MF) (white background). b The identified GO terms belonging to the same cluster are listed higher-ranking term. c Treatments are salt stress (S), ABA stress (A) and low temperature stress (L). The regulation direction is represented by green (down-regulation), red (up-regulation), blue (both down-regulation and up-regulation included) and grey (not significantly regulated). | ||
In total, 44 GO terms in response to different abiotic stresses treatment after 48 h exposure remained after filtering (Fig. 4). GO terms assigned to DEGs in response to salt stress and ABA stress were mostly down-regulated with only four terms up-regulated, i.e. ‘regulation of cellular process’ (GO:0050794) and ‘regulation of cellular metabolic process’ (GO:0031323) in response to salt stress, ‘cellular response to extracellular stimulus’ (GO:0031668) as well as ‘negative regulation of organelle organization’ (GO:0010639) in response to ABA stress. In contrast, the GO terms in response to low temperature stress remained mainly up-regulated. The shared GO terms were found to be largely down-regulated between salt stress and low temperature stress, with only two shared terms (i.e. regulation of cellular process, GO:0050794; regulation of cellular metabolic process, GO:0031323) involved in metabolic regulation processes remaining up-regulated. These shared GO terms were mainly involved in the biological process, such as protein folding (GO:0006457), protein complex assembly (GO:0006461) and protein complex biogenesis (GO:0070271); the molecular functions like unfolded protein binding (GO:0051082) as well as several general stimulus responses. Mutual DEGs between ABA stress and low temperature were found to be all down-regulated after treatment for 48 h, which were identified to be involved in general stimulus responses and protein folding process, which correlated well with the mutual DEGs in response to ABA stress and low temperature treatment for 24 h. The last set of treatment-independent terms (i.e. salt stress and ABA stress) showed largest number of down-regulated terms and down-regulation in most biological processes. This also agreed well with the mutual DEGs in response to salt stress and ABA treatment for 24 h. Eight shared terms were found down-regulated in all abiotic stress treatments at 48 h. Six shared terms involved in general stimulus responses and only two shared terms involved in protein folding (GO:0006457) and the regulation of reactive oxygen species metabolic process (GO:2000379).
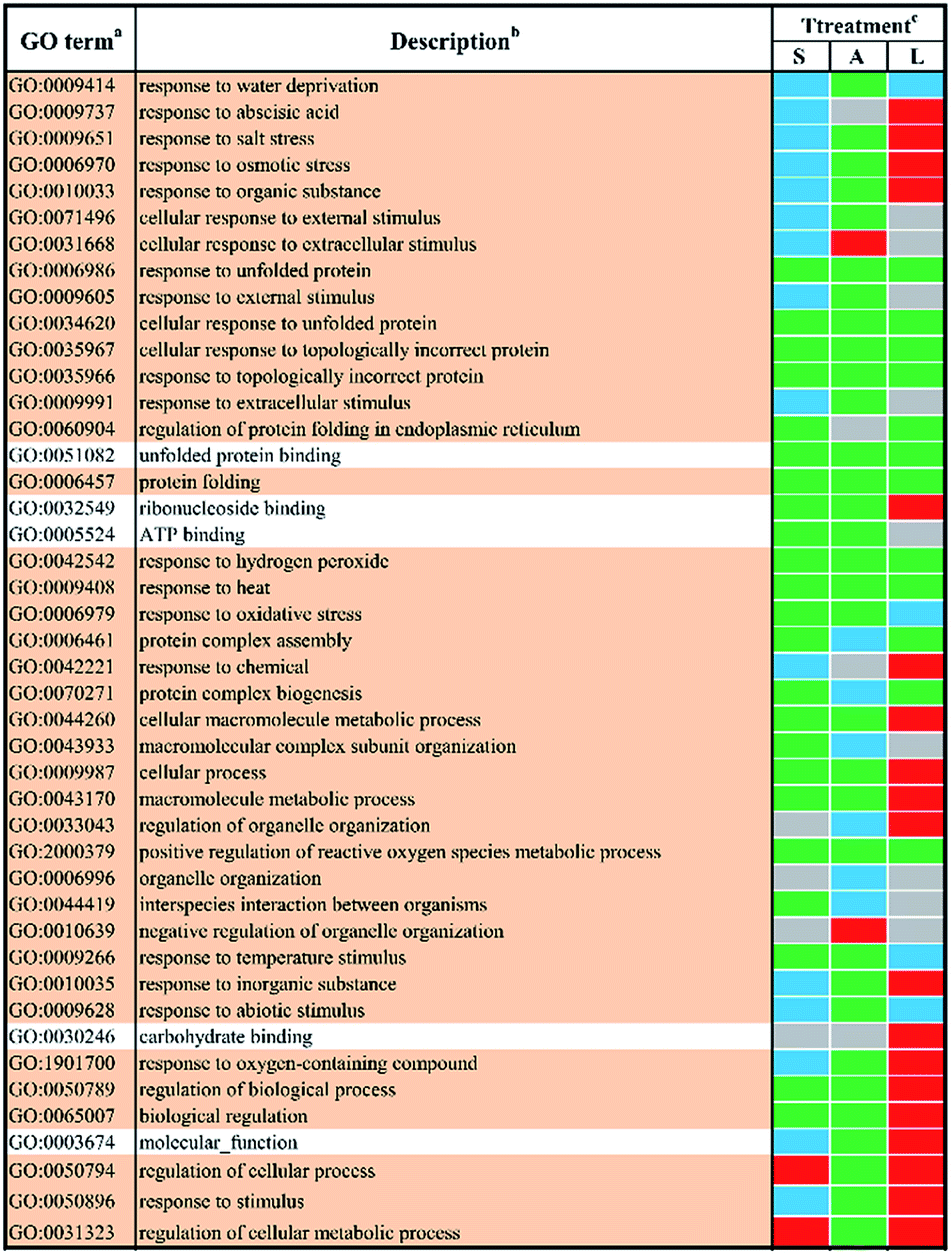 | ||
| Fig. 4 The enriched l GO terms among different DEGs after 48 h stress applications. a Non-redundant GO terms (FDR ≤ 5% and similarity ≤ 0.5) are shown for the identified biological processes (BP) (light khaki background) and molecular functions (MF) (white background). b The identified GO terms belonging to the same cluster are listed higher-ranking term. c Treatments are salt stress (S), ABA stress (A) and low temperature stress (L). The regulation direction is represented by green (down-regulation), red (up-regulation), blue (both down-regulation and up-regulation included) and grey (not significantly regulated). | ||
Distribution of TFs in the DEG sets
In total, 1599 yellow horn transcription factors (TFs) were expressed in the present RNA-Seq dataset which belong to 58 TF families. They were used as a reference distribution to identify the deviations in the family distributions of the DEGs for each treatment at different points in time (Fig. 5). All selected TFs were found to be reduced after the salt treatment and ABA treatment at 24 h and 48 h. No salt treatment-specific and ABA treatment-specific enrichment of TF families was observed at both time points. In contrast to the other two treatments, four TF families, including ERF, NAC, GRAS and B3, were found to be generally enriched after low temperature treatment at 24 h and 48 h. The ERF and NAC families were enriched after 24 h after salt treatment particularly the ERF TF family, which was over-represented after low temperature treatment. At 24 h, only bHLH and C3H TF families were found enriched specifically upon low temperature treatment. At 48 h, the bZIP, G2-like, WRKY and MIKC_MADS TF families were enriched upon low temperature treatment.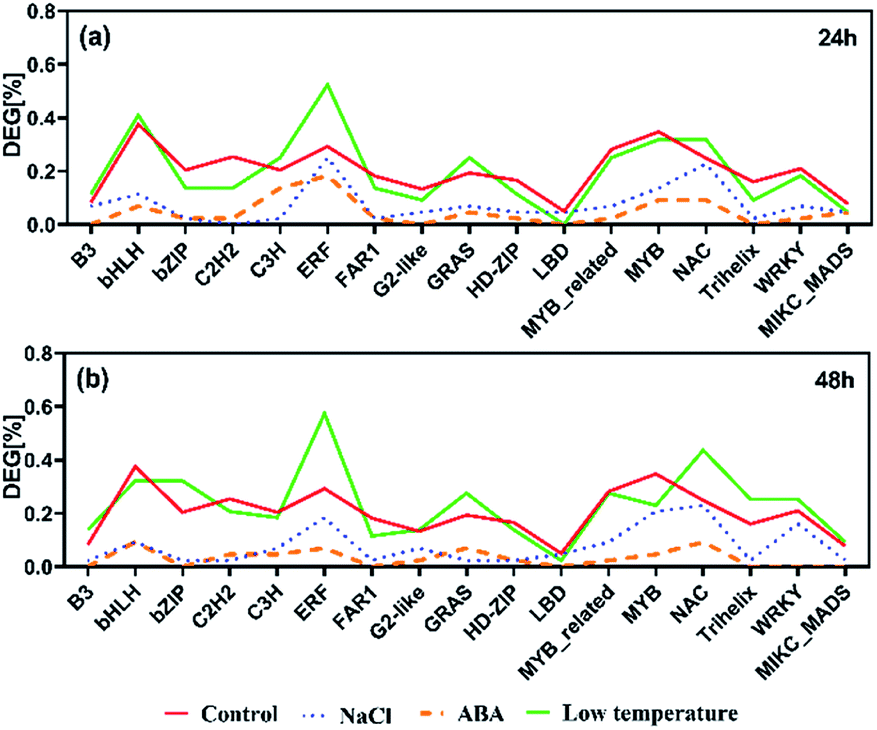 | ||
| Fig. 5 The prevalence of transcription factor (TF) families after salt, ABA and low temperature stress for 24 h (a) and 48 h (b). Families with ≥30 expressed members are shown. | ||
Discussion
Abiotic stresses are considered key factors which can negatively affect the growth and productivity of plants.44–46 Yellow horn (Xanthoceras sorbifolia Bunge) is a well-known oilseed shrub of important economic value in China and also considered major contributor for the Chinese oilseed production.4 In addition, owing to its strong adaption to various abiotic stress conditions such as saline-alkali environment, deserts as well as arid regions, yellow horn has been deemed as potential pioneer tree species for future vegetation and restoration.9 Thus, there is an urgent need to be felt to reveal the molecular responses of yellow horn exposed to various abiotic stresses at present as well as in the years to come, since the genomic annotation of yellow horn has become publicly available recently. Furthermore, recent advances in biotechnology have also accelerated the molecular understanding of plants in response to different abiotic stresses.38 As a result, a huge number of stress-responsive genes have been screened and functionally annotated in model plants such as Arabidopsis,47 Oryza sativa48 and Zea mays.49 Nevertheless, little information can be available on the molecular responses of yellow horn with regard to salt stress, ABA stress and low temperature stress. Considering this, we investigate the impact of different individual abiotic stresses, including salt stress (2% NaCl), ABA stress (100 mM ABA) and low temperature stress (4 °C) on the global transcriptome profiles of yellow horn.In order to screen the transcriptomic patterns of yellow horn and its strong adaptions to diverse abiotic stresses, young seedings were exposed to different stress conditions for 24 h and 48 h were analyzed using RNA-Seq. There were visible phenotypic changes developmental differences in yellow horn after exposure for 24 h and 48 h (data not shown). This result agreed well with previous literature, which highlighted that although the transcriptomic adaptions can be detectable only after treatment for a few hours, the observed phenotypic effects appeared only after treatment for a couple of days.50 A full number of identified DEGs between control group and treated groups varied slightly between the duration of treatment with 4395 DEGs after 24 h (Fig. 2C) and 4357 DEGs after 48 h (Fig. 2D). However, clear differences in the number of DEGs can be found between different stress types.
Salt treatment and ABA treatment led to a lower number of DEGs than low temperature treatment at 24 h and 48 h as presented in Fig. 2A (1119 vs. 2628 at 24 h and 770 vs. 3480 at 48 h for salt treatment; 1510 vs. 2628 at 24 h and 756 vs. 3480 at 48 h for ABA treatment). It can be seen that the individual stress type and duration have important impacts on the number of DEGs, which correlated well with a previous study.51,52 In addition, more DEGs can be identified for yellow horn in response to low temperature stress than to salt treatment and ABA treatment, implying yellow horn may have strong defensive systems in response to low temperature environment. Meanwhile, DEGs regulated by different abiotic stresses at different time points exhibited a conservation of 27% to 45% to large extent (Fig. 2B). This suggested that the established short-term molecular stress responses for plants still needs to be verified via longer-term stress treatment.53,54 Further study is currently in progress to verify the molecular stress responses for yellow horn using long-term stress treatment using RNA-Seq. A comparative study of the DEG sets for each time point across treatments revealed that 44% of all stress-responsive genes treated after 24 h were unique to the low temperature treatment, while 67% of DEGs were unique to the low temperature stress after 48 h. A substantial overlapped genes with low temperature regulated genes can be observed (17% after 24 h and 12% after 48 h). This implied that the stress responses and adaptive processes of yellow horn in response to different stresses may be regulated through a complex network of cross-talk among different signaling pathways.55
In order obtain further insights into the biological processes and molecular functions shown as stress-responsive, GO terms were assigned to the DEGs and analyzed for enrichment. The enriched GO terms for the two time points shown in Fig. 1 and 2 confirmed the complexities of stress responses involved in a host number of signaling pathways. Salt stress and ABA stress shared the enriched GO terms probably caused by the lack of water, thereby leading to similar responses at both time points.56 The regulation direction was highly conserved within eight mutually enriched terms involving in protein metabolic process such as protein folding (GO:0006457), unfold protein binding (GO:0051082) and general stimulus responses to unfolded protein (GO:0006986) etc. Under salt stress after 24 h, seven shared GO terms were found to be up-regulated, which mainly involve in general stimulus responses to water (GO:0009415) and water deprivation (GO:0009414); catalytic activity, including transferase activities (GO:0016758) and glucosyltransferase activity (GO:0046527); some regulatory metabolic process (GO:0009737, GO:2000112, GO:0051252 and GO:0060255). However, after 48 h of salt stress, only two up-regulated shared terms were identified, which involved in cellular response to extracellular stimulus (GO:0065002) and negative regulation of organelle organization (GO:0010639). This indicated a better adaptation of yellow horn in response to salt exposure. In contrast, all shared terms between ABA stress and low temperature stress were down-regulated as observed at both time points. Exposure to salt and low temperature would bring about a significant down-regulation of stress-responsive shared terms. Only a few shared terms were found to be up-regulated. Previous studies have examined the expressional changes of genes involved in different abiotic stresses.57–60 These combined results suggested that genes involved in response to various abiotic stress were regulated by developmental stages and therefore displayed different regulation patterns over time. According to the GO enrichment, a host of genes mainly involved in protein metabolic process such as protein folding, unfold protein binding, protein transport and protein modification. This suggested that the molecular mechanism of yellow horn in response to abiotic stresses may be strongly related to protein-related biological processes. Further studies are still needed to identify the key proteins in yellow horn responsive to various abiotic stresses.
TFs can be helpful to control the activity of the downstream target genes. In yellow horn, 1599 TFs in total are classified into 58 families, among which 1165 TFs were found to be active in response to NaCl, ABA and low temperature stresses in this study. In this study, the ERF families were found over-represented among all treatment, with a major proportion of these TFs belongs to the ERF families. The ERFs have been reported to actively participate in a host of plant biological processes (e.g. developmental and physiological processes), but also act in response to wounding61,62 and various abiotic stresses.63,64 Previous publication has found that the over-expressed ERFs are more resistant to abiotic stresses such as salinity, cold and water stress.65
In the present study, genes identified as ERFs were both up-and down-regulated among different treatment groups. Exposure of yellow horn to high salinity and ABA stress can lead to the reduction of ERFs, while the low treatment can result in the over-expression of ERFs. This indicated a swift induction of abiotic stresses which can increase ethylene production. A moderate change can lead to the inhibitory effect on the biosynthesis of ethylene, thereby resulting in the differential regulatory directions in gene expression patterns. In addition, previous literature has found that the bHLH66 and b-ZIP67 families were ABA-dependent which can be induced in the presence of ABA. Both of them have important regulatory functions in developmental and physiological processes in plants. However, the slight enrichment of bHLH and b-ZIP can be observed only in low temperature after treatment for 24 h and 48 h, respectively. No clear enrichment of bHLH and b-ZIP among DEGs can be found in yellow horn in response to ABA treatment. Thus, the functions of bHLH and b-ZIP genes in yellow horn were still required to be elucidated in our further studies.
Conclusions
We demonstrated for the first time the complex molecular responses of yellow horn exposed to multiple abiotic stresses using RNA-Seq. These combined results would give a deep understanding of the molecular defensive mechanisms of yellow horn in response to diverse abiotic stresses. Our findings would make a substantial contribution to the elucidation of the defense mechanisms of yellow horn at the molecular level. In addition, these identified candidate genes would also be the major resources for further detailed genetic studies. Further studies are currently in progress in order to reveal the molecular defense mechanisms of yellow horn in response to combinational abiotic stresses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Fundamental Research Funds for the Chinese Central Universities (grant number 2572018AA28). We thank to Biozeron Biotechnology Co. Ltd (Shanghai, China) for their assistance in RNA sequencing. The authors thanked Zhi Liu of Northeast Forestry University for his assistance in the bioinformatic analysis.References
- Z. Y. Yao, J. H. Qi and L. M. Yin, Renewable Sustainable Energy Rev., 2013, 24, 57–65 CrossRef CAS.
- S. Buerki, I. Lowry, P. Porter, N. Alvarez, S. G. Razafimandimbison, P. Küpfer and M. W. Callmander, Plant Ecology and Evolution, 2010, 143, 148–159 CrossRef.
- Z. Shen, K. Zhang, Y. Ao, L. Ma and J. Duan, J. For. Res., 2019, 30, 869–877 CrossRef CAS.
- J. Li, Y. J. Fu, X. J. Qu, W. Wang, M. Luo, C. J. Zhao and Y. G. Zu, Bioresour. Technol., 2012, 108, 112–118 CrossRef CAS.
- S. Zhang, Y. G. Zu, Y. J. Fu, M. Luo, W. Liu, J. Li and T. Efferth, Bioresour. Technol., 2010, 101, 2537–2544 CrossRef CAS PubMed.
- H. Yu, S. Fan, Q. Bi, S. Wang, X. Hu, M. Chen and L. Wang, Ind. Crops Prod., 2017, 97, 425–430 CrossRef CAS.
- A. Ebert, Sustainability, 2014, 6, 319–335 CrossRef.
- Y. Liu, Z. Huang, Y. Ao, W. Li and Z. Zhang, PLoS One, 2013, 8, e74441 CrossRef CAS PubMed.
- C. J. Ruan, R. Yan, B. X. Wang, S. Mopper, W. K. Guan and J. Zhang, Ecol. Eng., 2017, 99, 504–512 CrossRef.
- M. Tester and A. Bacic, Plant Physiol., 2005, 137, 791–793 CrossRef CAS PubMed.
- I. Rejeb, V. Pastor and B. Mauch-Mani, Plants, 2014, 3, 458–475 CrossRef PubMed.
- N. Suzuki, R. M. Rivero, V. Shulaev, E. Blumwald and R. Mittler, New Phytol., 2014, 203, 32–43 CrossRef PubMed.
- Q. Bi, Y. Zhao, W. Du, Y. Lu, L. Gui, Z. Zheng, H. Yu, Y. Cui, Z. Liu and T. Cui, GigaScience, 2019, 8, giz070 CrossRef.
- Q. Liang, H. Li, S. Li, F. Yuan, J. Sun, Q. Duan, Q. Li, R. Zhang, Y. L. Sang and N. Wang, GigaScience, 2019, 8, giz071 CrossRef PubMed.
- S. Goodwin, J. D. McPherson and W. R. McCombie, Nat. Rev. Genet., 2016, 17, 333 CrossRef CAS PubMed.
- W. R. McCombie, J. D. McPherson and E. R. Mardis, Cold Spring Harbor Perspect. Med., 2018, a036798 Search PubMed.
- K. Moustafa and J. M. Cross, 2018.
- L. Skøt, R. Kelly and M. W. Humphreys, in Genomics Assisted Breeding of Crops for Abiotic Stress Tolerance, vol. II, Springer, 2019, pp. 91–103 Search PubMed.
- T. Dresselhaus and R. Hückelhoven, Agronomy, 2018, 8, 267 CrossRef CAS.
- A. Rai, M. Yamazaki and K. Saito, Curr. Opin. Cell. Biol., 2019, 15, 58–67 Search PubMed.
- J. Pirrello, C. Deluche, N. Frangne, F. Gevaudant, E. Maza, A. Djari, M. Bourge, J. P. Renaudin, S. Brown and C. Bowler, Plant J., 2018, 93, 387–398 CrossRef CAS.
- X. Tian, Z. Wang, Q. Zhang, H. Ci, P. Wang, L. Yu and G. Jia, Russ. J. Plant Physiol., 2019, 1–9 Search PubMed.
- H. Jiang, H. Tian, C. Yan, L. Jia, Y. Wang, M. Wang, C. jun Jiang, Y. Li, J. Jiang and L. Fang, Sci. Hortic., 2019, 248, 248–255 CrossRef CAS.
- W. Li and Y. Guo, in Plant Senescence, Springer, 2018, pp. 331–337 Search PubMed.
- L. Wang, C. Ruan, L. Liu, W. Du and A. Bao, Int. J. Mol. Sci., 2018, 19, 3071 CrossRef.
- R. Kumar, Y. Ichihashi, S. Kimura, D. H. Chitwood, L. R. Headland, J. Peng, J. N. Maloof and N. R. Sinha, Front. Plant Sci, 2012, 3, 202 CAS.
- M. Martin, EMBnet J., 2011, 17, 10–12 CrossRef.
- K. i. Tamura and J. i. Yonemaru, Grassl. Sci., 2010, 56, 230–239 CrossRef CAS.
- G. Wen, Proceedings of the 2017 International Conference on Biomedical Engineering and Bioinformatics, 2017, pp. 11–15 Search PubMed.
- M. Taschuk and G. Wilson, PLoS Comput. Biol., 2017, 13, e1005412 CrossRef.
- Y. Liao, G. K. Smyth and W. Shi, Bioinformatics, 2013, 30, 923–930 CrossRef PubMed.
- E. Góngora-Castillo, G. Fedewa, Y. Yeo, J. Chappell, D. DellaPenna and C. R. Buell, in Methods in enzymology, Elsevier, 2012, vol. 517, pp. 139–159 Search PubMed.
- M. I. Love, W. Huber and S. Anders, Genome Biol., 2014, 15, 550 CrossRef PubMed.
- Y. Benjamini and Y. Hochberg, J. Roy. Stat. Soc. B Stat. Methodol., 1995, 57, 289–300 Search PubMed.
- Y. Zhao, H. Wang, C. Wu, M. Yan, H. Wu, J. Wang, X. Yang and Q. Shao, Oncol. Rep., 2018, 39, 1197–1206 CAS.
- J. Huerta-Cepas, K. Forslund, L. P. Coelho, D. Szklarczyk, L. J. Jensen, C. von Mering and P. Bork, Mol. Biol. Evol., 2017, 34, 2115–2122 CrossRef CAS.
- J. Ye, L. Fang, H. Zheng, Y. Zhang, J. Chen, Z. Zhang, J. Wang, S. Li, R. Li and L. Bolund, Nucleic Acids Res., 2006, 34, W293–W297 CrossRef CAS PubMed.
- V. Kumar, S. Datir, T. Khare and V. Shriram, in Advances in Rice Research for Abiotic Stress Tolerance, Elsevier, 2019, pp. 615–632 Search PubMed.
- D. Klopfenstein, L. Zhang, B. S. Pedersen, F. Ramírez, A. W. Vesztrocy, A. Naldi, C. J. Mungall, J. M. Yunes, O. Botvinnik and M. Weigel, Sci. Rep., 2018, 8, 10872 CrossRef CAS.
- C. Xie, X. Mao, J. Huang, Y. Ding, J. Wu, S. Dong, L. Kong, G. Gao, C.-Y. Li and L. Wei, Nucleic Acids Res., 2011, 39, W316–W322 CrossRef CAS PubMed.
- Y. Fang, C. Zhang, T. Wu, Q. Wang, J. Liu and P. Dai, PLoS One, 2017, 12, e0170609 CrossRef PubMed.
- Z. Du, X. Zhou, Y. Ling, Z. Zhang and Z. Su, Nucleic Acids Res., 2010, 38, W64–W70 CrossRef CAS PubMed.
- F. Supek, M. Bošnjak, N. Škunca and T. Šmuc, PLoS One, 2011, 6, e21800 CrossRef CAS PubMed.
- C. Chikkaputtaiah, J. Debbarma, I. Baruah, L. Havlickova, H. P. D. Boruah and V. Curn, Plant Biotechnol. Rep., 2017, 1–20 Search PubMed.
- M. Fujita, Y. Fujita, Y. Noutoshi, F. Takahashi, Y. Narusaka, K. Yamaguchi-Shinozaki and K. Shinozaki, Curr. Opin. Plant Biol., 2006, 9, 436–442 CrossRef.
- R. Mittler, Trends Plant Sci., 2006, 11, 15–19 CrossRef CAS.
- M. Safaeizadeh and T. Boller, Plant Signaling Behav., 2019, 14, e1590094 CrossRef PubMed.
- W. Kong, H. Zhong, X. Deng, M. Gautam, Z. Gong, Y. Zhang, G. Zhao, C. Liu and Y. Li, Plants, 2019, 8, 30 CrossRef CAS PubMed.
- L. Zhai, F. Teng, K. Zheng, J. Xiao, W. Deng and W. Sun, Hereditas, 2019, 156, 27 CrossRef PubMed.
- N. Opitz, C. Marcon, A. Paschold, W. A. Malik, A. Lithio, R. Brandt, H. P. Piepho, D. Nettleton and F. Hochholdinger, J. Exp. Bot., 2015, 67, 1095 CrossRef PubMed.
- A. Osthoff, P. D. dalle Rose, J. A. Baldauf, H.-P. Piepho and F. Hochholdinger, BMC Genomics, 2019, 20, 325 CrossRef.
- R. Mitra, S. Jadhav, B. S. McEwen, A. Vyas and S. Chattarji, Proc. Natl. Acad. Sci., 2005, 102, 9371–9376 CrossRef CAS.
- K. Urano, Y. Kurihara, M. Seki and K. Shinozaki, Curr. Opin. Plant Biol., 2010, 13, 132–138 CrossRef CAS PubMed.
- H.-W. Koyro, P. Ahmad and N. Geissler, in Environmental adaptations and stress tolerance of plants in the era of climate change, Springer, 2012, pp. 1–28 Search PubMed.
- L. Chalker-Scott, Photochem. Photobiol., 1999, 70, 1–9 CrossRef CAS.
- J. Zhang, W. Jia, J. Yang and A. M. Ismail, Field Crop. Res., 2006, 97, 111–119 CrossRef.
- X. Duan, X. Chen, S. Wang and X. Zhang, Russ. J. Plant Physiol., 2019, 66, 110–118 CrossRef CAS.
- M. Mansouri, M. R. Naghavi, H. Alizadeh, G. Mohammadi-Nejad, S. A. Mousavi, G. H. Salekdeh and Y. Tada, Funct. Integr. Genomics, 2019, 19, 13–28 CrossRef CAS PubMed.
- A. Teshome, S. Byrne, T. Didion, J. De Vega, C. Jensen, M. Klaas and S. Barth, BMC Res. Notes, 2019, 12, 311 CrossRef CAS PubMed.
- Z. Zhang, B. Cao, N. Li, Z. Chen and K. Xu, Environ. Exp. Bot., 2019, 103814 CrossRef CAS.
- U. J. Phukan, G. S. Jeena, V. Tripathi and R. K. Shukla, Front. Plant Sci, 2017, 8, 150 Search PubMed.
- U. Druege, P. Franken and M. R. Hajirezaei, Front. Plant Sci, 2016, 7, 381 Search PubMed.
- Z. Xie, T. M. Nolan, H. Jiang and Y. Yin, Front. Plant Sci, 2019, 10, 228 CrossRef PubMed.
- J. Debbarma, Y. N. Sarki, B. Saikia, H. P. D. Boruah, D. L. Singha and C. Chikkaputtaiah, Mol. Biotechnol., 2019, 61, 153–172 CrossRef CAS PubMed.
- K. Thirugnanasambantham, S. Durairaj, S. Saravanan, K. Karikalan, S. Muralidaran and V. I. H. Islam, Plant Mol. Biol. Rep., 2015, 33, 347–357 CrossRef CAS.
- S. Jones, Genome Biol., 2004, 5, 226 CrossRef PubMed.
- L. G. G. Corrêa, D. M. Riaño-Pachón, C. G. Schrago, R. V. dos Santos, B. Mueller-Roeber and M. Vincentz, PLoS One, 2008, 3, e2944 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09535g |
| This journal is © The Royal Society of Chemistry 2020 |