
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of a multiple and global analytical indicator of batch consistency: traditional Chinese medicine injection as a case study†
Libing Chena,
Fang Zhaoa,
Wenzhu Lia,
Zeqi Chena,
Jianyang Pana,
Difeifei Xiongb,
Bailing Lib,
Qingjie Zhangb and
Haibin Qu *a
*a
aPharmaceutical Informatics Institute, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China. E-mail: quhb@zju.edu.cn
bGuizhou Baite Pharmaceutical Co., LTD, Guizhou 550008, China
First published on 11th March 2020
Abstract
This paper evaluates a multiple and global analytical indicator of batch consistency in traditional Chinese medicine injections (TCMIs) via a chemometrics tool, which is more comprehensive to appraise quality consistency of different batches of injections than the traditional method of fingerprint similarity. A commonly used TCMI, Salviae miltiorrhizae and ligustrazine hydrochloride injection (SLI), was employed as a model. With the aid of a chemometrics tool (principal component analysis, PCA), evaluation of multiple and global analytical indicators of batch consistency, which included saccharides, phenolic acids and inorganic salts (18 indicators in total), was carried out to appraise the quality consistency of 13 batches of injection provided by the Guizhou Baite Pharmaceutical Co., Ltd. (Guizhou, China). Compared with the traditional HPLC-UV fingerprint similarity evaluation, the method proposed in the paper can more comprehensively and correctly reflect the quality consistency of different batches of injections. In this paper, the multi-index evaluation result showed poor batch consistency, which was more consistent with the determination results, while the fingerprint similarity evaluation results still showed good batch consistency. The HPLC-UV fingerprint reflects only substances with UV absorption, but it is not able to reflect substances without UV absorption or weak UV absorption, which leads to inappropriate conclusions. Therefore, quality consistency of injections can be effectively appraised by evaluation of multiple and global analytical indicators, instead of HPLC-UV fingerprint only. For visualizing the batch consistency of the multiple and global analytical indicators, a heat map was used to represent the fluctuation. Furthermore, critical indicator identification was also applied to select several indicators that should be paid more attention during the process of quality control of injection. And the analysis result showed that Na+, fructose (Fru), glucose (Glc), manninotriose (Man), danshensu (DSS) and salvianolic acid B (SAB) are the indicators that should be given more attention when controlling the quality of injections, also called critical quality control indicators. The proposed method provides a reference for the quality control of TCMIs and has broad application potential.
1. Introduction
Traditional Chinese Medicine (TCM) is a substance used in the prevention, treatment, and diagnosis of diseases as well as in rehabilitation and health care under the guidance of TCM theory. TCMs are mainly derived from natural medicines and processed products, including botanicals, animal medicines, mineral medicines, and some chemical and biological products. A traditional Chinese medicine injection (TCMI) is a formulation prepared from effective substances extracted from the TCM product. TCMIs are found in many different forms, such as sterile solution, aseptic powder or concentrated liquid that is formulated into a solution before use for injection. In addition, they play an important role in rescuing critically ill patients because of their quick and reliable effectiveness.However, safety issues related to TCMIs are gradually worsening due to the frequent occurrence of adverse reactions. This is mainly because the composition of TCMs is very complicated and there are still many ingredients that have not been confirmed with current technical analysis methods. These ingredients are composed of active substances, sugars, and inorganic salts, as well as some unknown ingredients, which can include some macromolecule impurities such as proteins, tannins, and resins if the purification technology is not effective. For medication safety, the sugars as well as macromolecule impurities of the injection should be also analyzed to prevent adverse reactions caused by intravenous injection.1
Regarding the quality control of TCMIs, ensuring product consistency has always been a challenge due to the complexity of TCM ingredients and the many chemical reactions that occur during the process of high temperature boiling purification, and pH adjustment. Chemical evaluation methods are widely used in ensuring quality consistency,2–4 although biological evaluation methods have also been employed.5–9 HPLC-UV chemical fingerprint analysis has broad applications in quality control due to its high specificity, selectivity and accessibility.10–13 However, the HPLC-UV method can detect only organics with UV absorption; it is unable to do in organics with no UV absorption or weak UV absorption. Although HPLC-ELSD analysis can compensate for this limitation and is used to detect substances with weak UV absorption, such as saccharides, as well as to develop saccharide fingerprints for quality control.14,15 HPLC-UV fingerprint analysis is still a commonly used method to evaluate the quality consistency of TCMIs. Due to the shortcomings mentioned above, it may be limited with regards to drawing an accurate conclusion on the product consistency with few indicators. Although there is a multi-fingerprint method proposed to identify qualified Chinese medicinal materials,16–18 it is seldom used in evaluating the consistency of injection. Due to the attention TCMIs have recently received due to their safety issues, a comprehensive method to evaluate the quality consistency of TCMIs and effectively control their quality is urgently needed.
Salvia miltiorrhiza ligustrazine injection (SLI) is prepared from ligustrazine hydrochloride, Salvia miltiorrhiza extract, glycerin and water for injection in a certain ratio, and it is a commonly used TCMI treatment for occlusive cerebrovascular disease. Salvia miltiorrhiza is the dried root and rhizome of Salvia miltiorrhiza Bge.19 It is a long-standing traditional Chinese medicine and was used from 220 AD. It was recorded in the Shennong's Materia Medica and the Compendium of Materia Medica (both are well-known medicinal books in China) because of its significant medicinal effects such as promoting blood circulation and removing blood stasis. Salvia miltiorrhiza can be used for the treatment of irregular menstruation, dysmenorrhea, amenorrhea, cardiovascular diseases and so on. The compositions of Salvia miltiorrhiza are classified into hydrophobic components and water-soluble ones and are very complicated. The hydrophobic substances of Salvia miltiorrhiza are mainly composed of tanshinone I (Tan I), tanshinone IIA (Tan IIA), tanshinone IIB (Tan IIB), cryptotanshinone (CPT), dihydrotanshinone (DHT), etc.20 The water-soluble components of Salvia miltiorrhiza mainly include saccharides, such as glucose (Glc), maltose, and triose,21 and phenolic acids, including danshensu (DSS), protocatechuic aldehyde (PA), protocatechuic acid (PCA), rosmarinic acid (RA), lithospermic acid (LA), salvianolic acid A, B, (SAA, SAB), etc.22,23 These components exert different effects, on the antioxidant, microcirculation, tissue repair and antithrombosis activities in cells.24–30 And there are many Salvia miltiorrhiza related preparations, including Salvia miltiorrhiza polyphenols acid salt for injection, Salvia miltiorrhiza injection, compound Salvia miltiorrhiza capsules, SLI, Danhong injection, etc. They are widely used in cardiovascular diseases treatment. In the quality standard of SLI, only the characteristic chromatogram of the injection and the contents of two kinds of active substances, DSS and ligustrazine hydrochloride, are specified,31 which is not complete enough. On the one hand, the quantification analysis for seldom active substances would not adequately reflect the quality of the injection, on the other hand, the characteristic map includes only several characteristic peaks detected with HPLC-UV, which is very disadvantageous for drug safety. Therefore, methods for detection of different kinds of indicators such as the active substances, saccharides, impurities, inorganic salts, etc. are proposed in this paper to evaluate the batch consistency of multiple and global analytical indicators with a multivariate statistical tool, providing a reference for the appraisement of quality consistency of TCMIs.
HPLC-ELSD plays an important role in the analysis of esters, surfactants, sugars, amino acids, quaternary ammonium salts, polymers, steroids, which are hard to be detected by HPLC-UV method. In this article, HPLC-ELSD analysis was applied for detecting the concentrates of saccharides, which were used in the evaluation. And ion chromatography as well as atomic absorption spectroscopy analysis was also carried out for detection of inorganic salts.
Multivariate statistical projection methods such as principal component analysis (PCA)32 and partial least squares (PLS),33 which compress multiple related variables into a few statistics for analysis, are the technical core of multivariate data analysis. With the development of multivariate statistical analysis, its application in the quality control of TCM is becoming more extensive.34–36 Huang et al. reviewed the current application of chemometrics in TCM for the determination, identification and discrimination of the bioactive or marker components.37 Zeng et al. used DART-MS combined with PCA to provide a reliable method for the identification of analytical markers and batch consistency evaluation in the quality control of TCM preparations.38,39 Yu et al. used PCA to evaluate the consistency of 20 batches of Folium isatidis with the content of the active components as input, which provides a reference method for quality control.4
Owing to many components in the injection that have not been identified, there are many adverse reactions in the clinical application of SLI, such as rashes, itching, chest tightness and so on. Therefore, impurity detection and batch consistency evaluation of indicators are still important parts of the quality control of SLI. For this, we conducted an evaluation of multiple and global analytical indicators of batch consistency for SLI, followed by critical indicator identification, providing a methodology for the quality control of TCMIs. For the evaluation of multiple and global analytical indicators of batch consistency, it was carried out as follows: first, different analysis methods to detect different kinds of substances in TCMI such as phenolic acids (with UV absorption), saccharides (without UV absorption), inorganic salts were developed. After the contents of indicators were obtained, a multivariate statistical tool was used to analyze the content data to evaluate the batch consistency.
The article is constructed as follows: first, a HPLC-UV fingerprint analysis method, a macromolecule impurity detection method and quantitative analysis methods for the substances in the injection, including saccharides and phenolic acids were established. Second, we analyzed the HPLC-UV fingerprints of 13 batches of SLI using the “Similarity Evaluation System for Chromatographic Fingerprint of TCM (2012 Version, Committee of Chinese Pharmacopeia)”. The similarity evaluation results of HPLC-UV fingerprints were all above 0.985, suggesting good batch consistency. For the reason mentioned above and to appraise the quality consistency of TCMIs more comprehensively and accurately, evaluation of multiple and global analytical indicators of batch consistency, which included saccharides, phenolic acids and inorganic salts (18 indicators in total) was subsequently carried out. The contents of phenolic acids (with UV absorption) were detected by the established HPLC-UV method. The HPLC-ELSD method (for saccharides analysis) and phenol sulfuric acid assay (for total sugars analysis) were also developed with good method validation performance to detect the contents of related saccharides. And inorganic salts were detected by ion chromatography and atomic absorption spectroscopy. Thus, information of important substances in injections was obtained for further multivariate statistical analysis. In addition, a heat map representing the fluctuations in the indicators in 13 batches of injection was drawn to visualize the batch consistency. Finally, critical indicator identification was applied to select the indicators of interest. The indicators Na+, fructose (Fru), Glc, manninotriose (Man), DSS and SAB were identified as those that should be given more serious attention in the quality control of SLI.
2. Experimental
2.1. Materials and reagents
The SLI was provided by the Guizhou Baite Pharmaceutical Co., Ltd. (Guizhou, China). The information regarding the 13 batches of injection is shown in Table 1. For convenience, the injections of the corresponding batch were numbered 1–13, as shown in the Table 1, and used consistently thereafter. Somatostatin (Som), thymosin (Thy), insulin (Ins), and ribonuclease A (Rib A) were purchased from the National Institutes for Food and Drug Control (Beijing, China); Fru was purchased from ACROS Organic (Belgium). Phenol was obtained from Aladdin (Shanghai, China). Glc and sulfuric acid were obtained from the Sinopharm Group Chemical Reagent Co., Ltd. (Shanghai, China). Melibiose (Mel), Man, sodium danshensu (DSS-Na), PA, RA, SAB, and SAA were all purchased from the Shanghai Ronghe Pharmaceutical Technology Co., Ltd., and HPLC-grade acetonitrile and methanol were purchased from Merck (Darmstadt, Germany). Formic acid (FA) and acetic acid (AA) were obtained from Roe Scientific Inc. (Newark, USA). Trifluoroacetic acid (TFA) was purchased from J&K (Beijing, China). All of the acids used for analysis were of HPLC-grade. Deionized water was prepared by a Millipore Milli-Q Plus system (Millipore, Bedford, MA, USA). The other reagents were of analytical grade.| Number | Batch number | Number | Batch number |
|---|---|---|---|
| 1 | 20180333 | 8 | 20180404 |
| 2 | 20180334 | 9 | 20180405 |
| 3 | 20180335 | 10 | 20180406 |
| 4 | 20180336 | 11 | 20180407 |
| 5 | 20180401 | 12 | 20180410 |
| 6 | 20180402 | 13 | 20180411 |
| 7 | 20180403 |
2.2. Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol:deionized water (1:550:450) mixture that was selected based on our previous study at a final concentration of 200 μg mL−1. The stock solution was stored at 4 °C.
:methanol:deionized water (1:550:450) mixture that was selected based on our previous study at a final concentration of 200 μg mL−1. The stock solution was stored at 4 °C.Injection samples: first, 10 mL of SLI was added to the ultrafiltration centrifuge tube for concentration by centrifugation (2800×g for 10 min) to 0.25 mL. Then, the concentrated solution in the ultrafiltration centrifuge tube was transferred with the TFA:methanol:deionized water (1:550:450) mixture to a 2.0 mL volumetric flask and then diluted to scale. After centrifugation (10000 rpm for 10 min), the supernatant was collected for analysis.
Injection samples: the SLI was diluted to 10 mL in a volumetric flask with deionized water at a final concentration of 20.0 mg mL−1.
Injection samples: the SLI was diluted with acetonitrile (60%, v/v) to a final concentration of 0.4 g mL−1. After centrifugation (10000 rpm for 10 min), the supernatant was collected for analysis.
:methanol:deionized water (20:200:780) mixture at final concentrations of 0.526 mg mL−1, 0.0739 mg mL−1, 0.0751 mg mL−1, 0.148 mg mL−1, and 0.129 mg mL−1 respectively.Injection samples: the SLI was diluted with an AA:methanol:deionized water (20:200:780) mixture to a final concentration of 0.55 g mL−1. After centrifugation (10000 rpm for 10 min), the supernatant was collected for analysis.
All of the other working solutions were obtained by diluting the stock solutions with the corresponding solvent and were stored at 4 °C. The determination of the inorganic salt ion concentration of the samples was carried out by testing center, and thus, the preparation of standard working solutions and injection samples is not shown.
2.3. Analytical conditions for the multi-indicator analysis
HPLC-UV analysis for the establishment of chemical fingerprint chromatography and similarity evaluation. We also conducted a fingerprint study for the injection with the same HPLC-UV analysis method to evaluate batch consistency. Fingerprints were obtained by the above HPLC-UV analysis method, and the similarity between two fingerprints was calculated. The correlation coefficient and congruence coefficient are the main algorithms in the calculation of similarity between two fingerprints.40,41 X(x1,x2,…xn) and Y(y1,y2,…yn) are assumed to be two sets of fingerprints. n is the number of each set of variables, i.e., the number of finger peaks. cov(X,Y) is the covariance of the vectors, Sx or Sy is the standard deviation, and ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) or ȳ is the mean. The correlation coefficient r (−1 ≤ r ≤ 1) is calculated by formula (1).
or ȳ is the mean. The correlation coefficient r (−1 ≤ r ≤ 1) is calculated by formula (1).
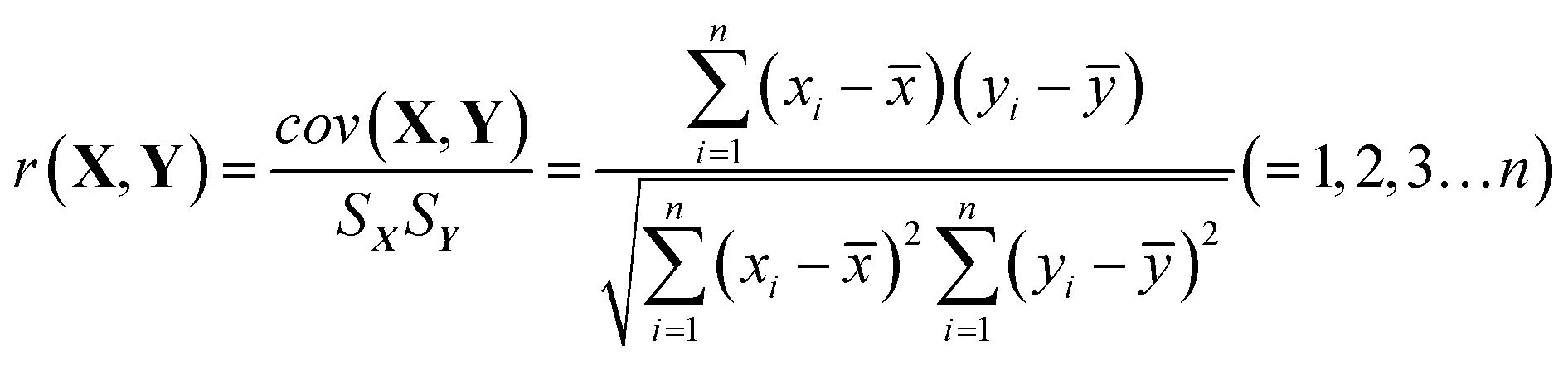 | (1) |
The congruence coefficient c (0 < c < +1) is the cosine of the angle between two fingerprints or the scalar product of the normed fingerprints, while r is the scalar product of the normed mean centered fingerprints. X(x1,x2,…xn) and Y(y1,y2,…yn) are assumed to be two sets of measured fingerprints. The congruence coefficient c is calculated by formula (2).
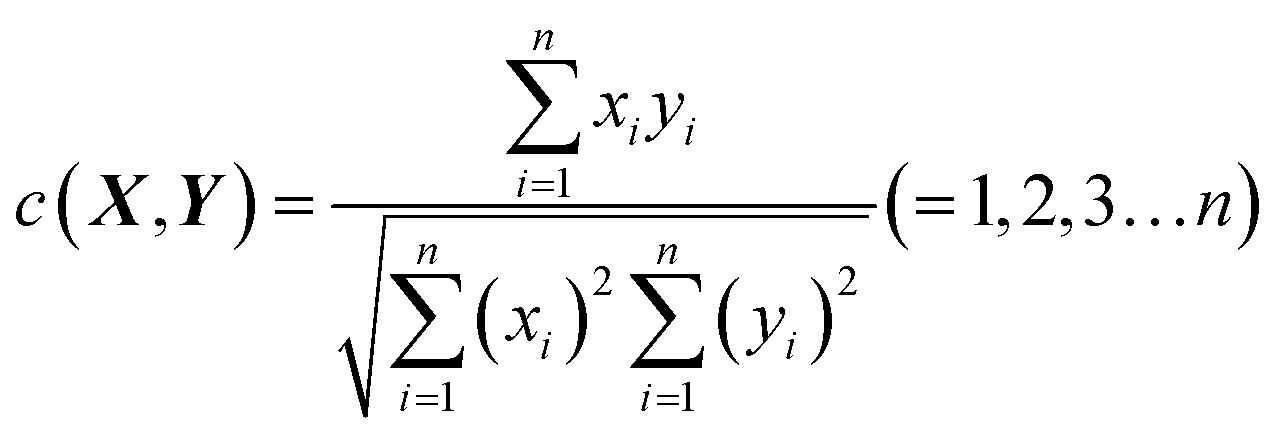 | (2) |
The two fingerprints are highly correlated if r or c is close to 1. However, the algorithms applied in the evaluation of fingerprint similarity, such as the correlation coefficient and congruence coefficient, are easily affected by peaks with a large area.41 The SLI was prepared from ligustrazine hydrochloride, Salvia miltiorrhiza extract, glycerin and water in a certain ratio, and the content of ligustrazine hydrochloride was much higher than the substances in the Salvia miltiorrhiza extract. Peaks with large peak areas account for a large weight in the similarity calculation. Similarity statistics are more sensitive when there is a lack of a peak with a large area. And the inclusion of ligustrazine hydrochloride with a large peak area as a fingerprint peak to evaluate batch-to-batch consistency may be less convincing. Therefore, two kinds of evaluation modes were applied: one was the inclusion of ligustrazine hydrochloride as a fingerprint peak to evaluate the consistency between injection batches, and the other was the exclusion of ligustrazine hydrochloride as a fingerprint peak to evaluate the consistency between injection batches.
Ion chromatography analysis: the analysis was performed on a thermoelectric AQUION ion chromatograph. The AS11-HC (50 × 4 mm, 4 μm) and AS11-HC (250 × 4 mm, 4 μm) were used as a protection column and analysis column, respectively. The analysis was performed in the inhibition conductance mode. The sample injection volume was set at 25 μL. The KOH solution was used as an eluent with a volume flow rate of 1 mL min−1, and the eluent gradient was set as follows: 0–5 min, 5 mM; 5–20 min, 5–30 mM; 20–23 min, and 30 mM; 23–31 min, 5 mM.
Atomic absorption spectroscopy analysis: the analysis was performed on Agilent 200 series AA (Agilent Technologies Inc., USA). The detection conditions were set as shown in Table 2.
| Elementa | Air (L min−1) | Acetylene (L min−1) | Wavelength (nm) | Slit width (nm) | Measurement time (s) | Lamp current (mA) |
|---|---|---|---|---|---|---|
| a The element exists in the form of ions in the injection, and thus, the ion forms instead of element forms in the following is used and is used consistently thereafter. | ||||||
| K | 13.5 | 2.0 | 766.5 | 1.0 | 5.0 | 5.0 |
| Na | 13.5 | 2.0 | 589.0 | 0.5 | 5.0 | 5.0 |
| Ca | 13.5 | 2.0 | 422.7 | 0.5 | 5.0 | 10.0 |
| Mg | 13.5 | 2.0 | 285.2 | 0.5 | 5.0 | 4.0 |
| Fe | 13.5 | 2.0 | 248.3 | 0.2 | 5.0 | 5.0 |
Validation of the method accuracy, precision, repeatability and stability: the recovery (accuracy) of the method was calculated from the nominal concentration (cn) and the observed concentration (c0) as follows:
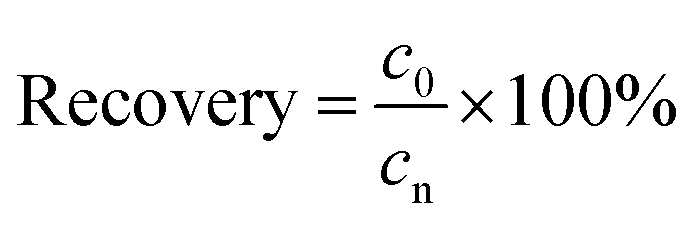 | (3) |
For the macromolecular analysis, saccharide analysis and total sugar analysis, the ratio of the amount of the reference substance to the amount of the component to be determined in the sample is 1:1, and 6 parts of the solution were prepared in parallel. The recovery rate and RSD were therefore calculated. Meanwhile, for the phenolic acid analysis, three concentration ratios were set. The high, medium and low ratios of the amount of the reference substance to the amount of the component to be tested in the sample were controlled at 1.5:1, 1:1, and 0.5:1, respectively. In the validation of the methods, the instrument precision, intraday and interday precision as well as stability were investigated according to the drug quality standard analysis method verification guiding principle “9101” of the Chinese Pharmacopoeia.43
In addition, the precision, repeatability and stability of the HPLC-UV fingerprint method were also tested. The repeatability as well as the stability of the HPLC fingerprint method were determined and expressed by the relative standard deviation (RSD) of the average relative retention times and relative peak areas of the 18 characteristic common peaks with respect to the reference peak (peak 3) at a retention time (tR) of 6.271 min.
The determination of the inorganic salt ion concentration of the samples was carried out by testing center, and thus, the validation of this method is not shown.
2.4. Data analysis
PCA is the technique for finding a transformation that transforms an original set of correlated variables to a new set of uncorrelated variables, called principal components. The first principal component explains the largest variation of origin data, and the variance of subsequent principal components explain successively decreased. Commonly the first K principle components are selected. Then the high-dimensional data can be replaced by a set of low-dimensional data (K dimensions), and most of the data information can still be retained. After PCA analysis, the original data can be represented by a formula followed:
| X = TP′ + E |
The PCA algorithm can be implemented by decomposing the covariance matrix based on SVD and eigenvalue decomposition.
Third, heat map analysis was also applied to visually describe the variation of indicators between batches. When using the heat map to describe the variation in indicators between batches, a unit variance preprocess was applied as follows:
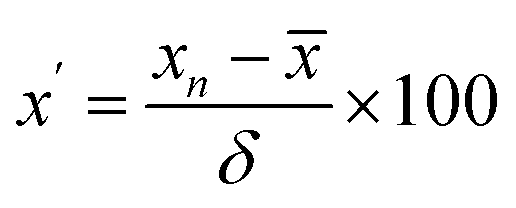 | (4) |
is the average value of the indicator in the 13 batches of SLI, and δ is the standard deviation of the indicator in the 13 batches of SLI.
Finally, critical indicator identification was carried out by using a loading plot, which can help select the key quality control indicators that deserve serious attention in the quality control (see Part 3.7.3).
3. Results and discussion
3.1. Method validation
The LODs and LOQs, recovery of the phenol sulfuric acid assay, HPLC-ELSD method and HPLC-UV method for the determination of the substances in the injection are summarized in Table 3, 4 and 5. The results show that the methods had good sensitivity and excellent accuracy.| Codes | LOD (μg mL−1) | LOQ (μg mL−1) | Recovery | RSD of recovery |
|---|---|---|---|---|
| DSS | 0.084 | 0.280 | 98.01 | 1.51 |
| PA | 0.034 | 0.068 | 95.10 | 4.18 |
| RA | 0.065 | 0.324 | 99.50 | 0.91 |
| SAB | 0.106 | 0.254 | 98.74 | 1.21 |
| SAA | 0.260 | 0.521 | 104.49 | 0.92 |
The RSDs of the instrument precision, intraday precision, interday precision, stability and equation of linear regression of all of the indicators are provided in ESI,† which are with good performance. The linear regression models of the methods fitted good and all had high correlation coefficients.
The HPLC-UV fingerprint of the injection including 18 common peaks is shown in Fig. 1. The results of the method validation are also provided in ESI.† The HPLC-UV fingerprint method performed well in methodological verification.
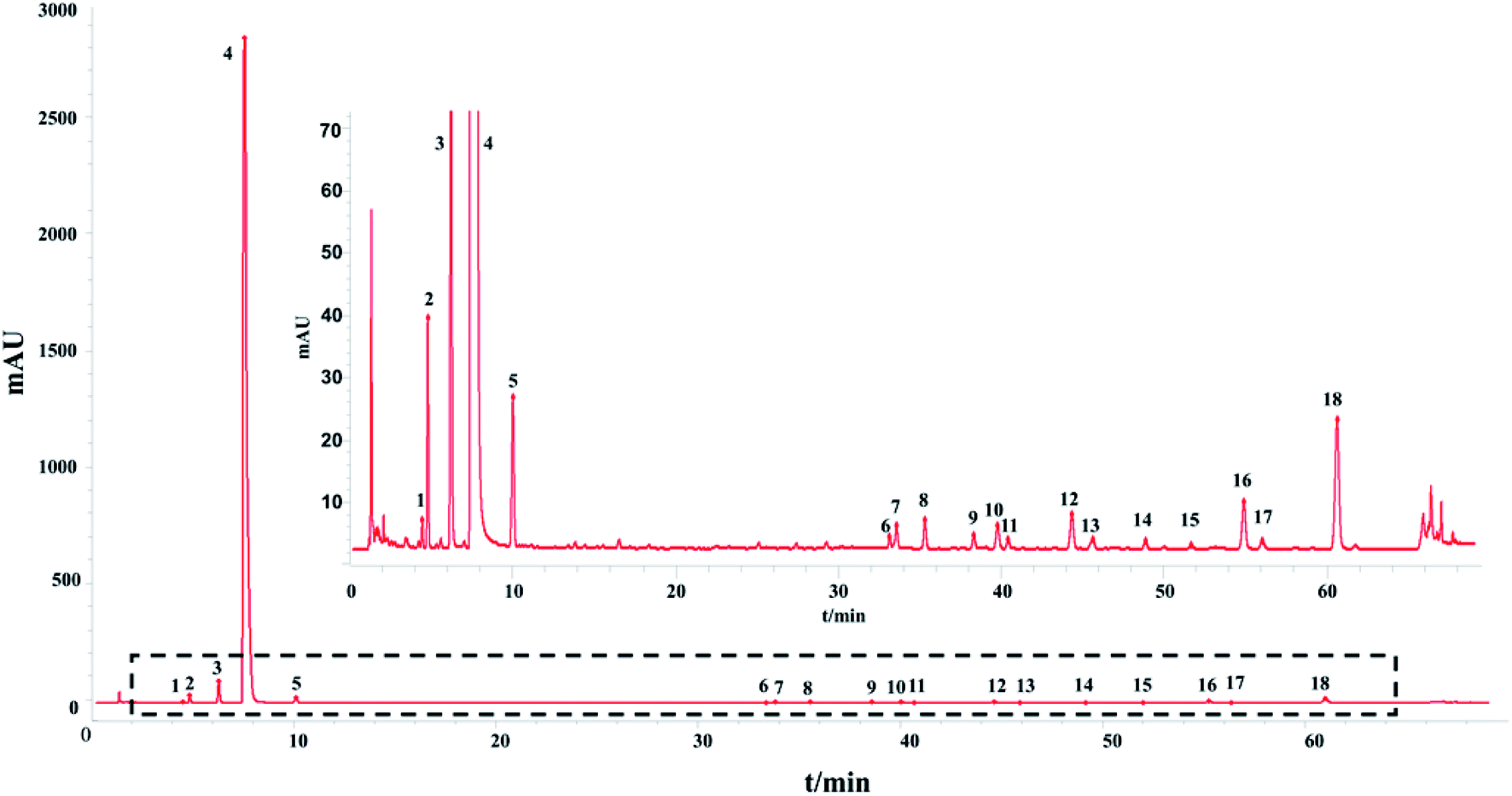 | ||
| Fig. 1 The control fingerprint of the 13 batches of SLI: 2, 5-HMF; 3, DSS; 4, ligustrazine hydrochloride; 5, PA; 12, RA; 16, SAB; 18, SAA; and the rest are unknown. (The figure in the upper right corner is an enlarged view of the contents of the dashed box.) | ||
The accuracy validation was carried out at the same time. The test of the sample recovery for the accuracy validation of the method was carried out according to the methods given in Section 2.9. The recoveries of the analytes are summarized in the tables above. The sample recoveries being close to 100% and low RSDs showed that the accuracy of the methods was good. The recovery test of Somatostatin was not carried out because of its small molecular weight and it could not be intercepted completely by the film used during sample preparation.
3.2. HPLC-UV analysis for determination of macromolecule impurities
In this section, the determination of macromolecule impurities in the SLI was conducted. Compounds with large molecular weights were the first ones eluted, and the compounds with small molecular weights were eluted later. A good linear relationship was presented in the range of molecular weights from 1.683 kDa to 13.700 kDa (see the ESI†). The HPLC-UV chromatograms of the standard solution and sample solutions are shown in Fig. 2(A) and (B). (The analysis of macromolecule impurities for 13 batches of the SLI has been conducted, the remaining 12 HPLC-UV chromatograms of the injections are attached in ESI†).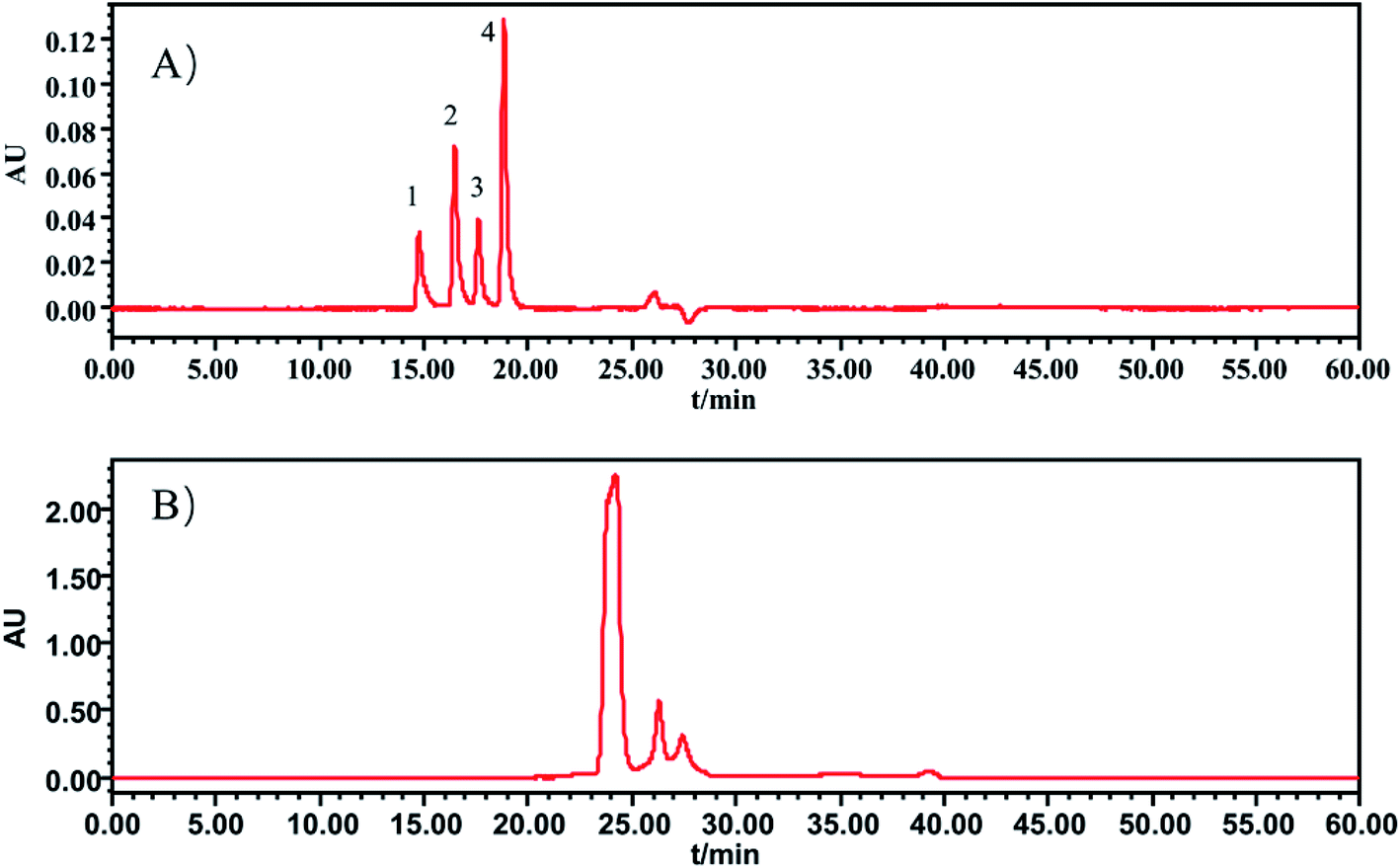 | ||
| Fig. 2 HPLC-UV chromatograms of (A) standard working solutions, 1: Rib A, 2: Ins, 3: Thy; 4: Som. (B) sample solution of SLI. | ||
Macromolecules whose molecular weights were greater than 1.638 kDa all peaked within the first 20 min, while there were not any peaks presented during that period in the test of the injection. This finding indicated that there were no macromolecule impurities (within a molecular weight ranging from 1.683 kDa to 13.700 kDa) in the SLI. This is good news with regards to the absence of macromolecule impurities in the injection, considering their harmfulness to the human body.
3.3. Carbohydrate analysis
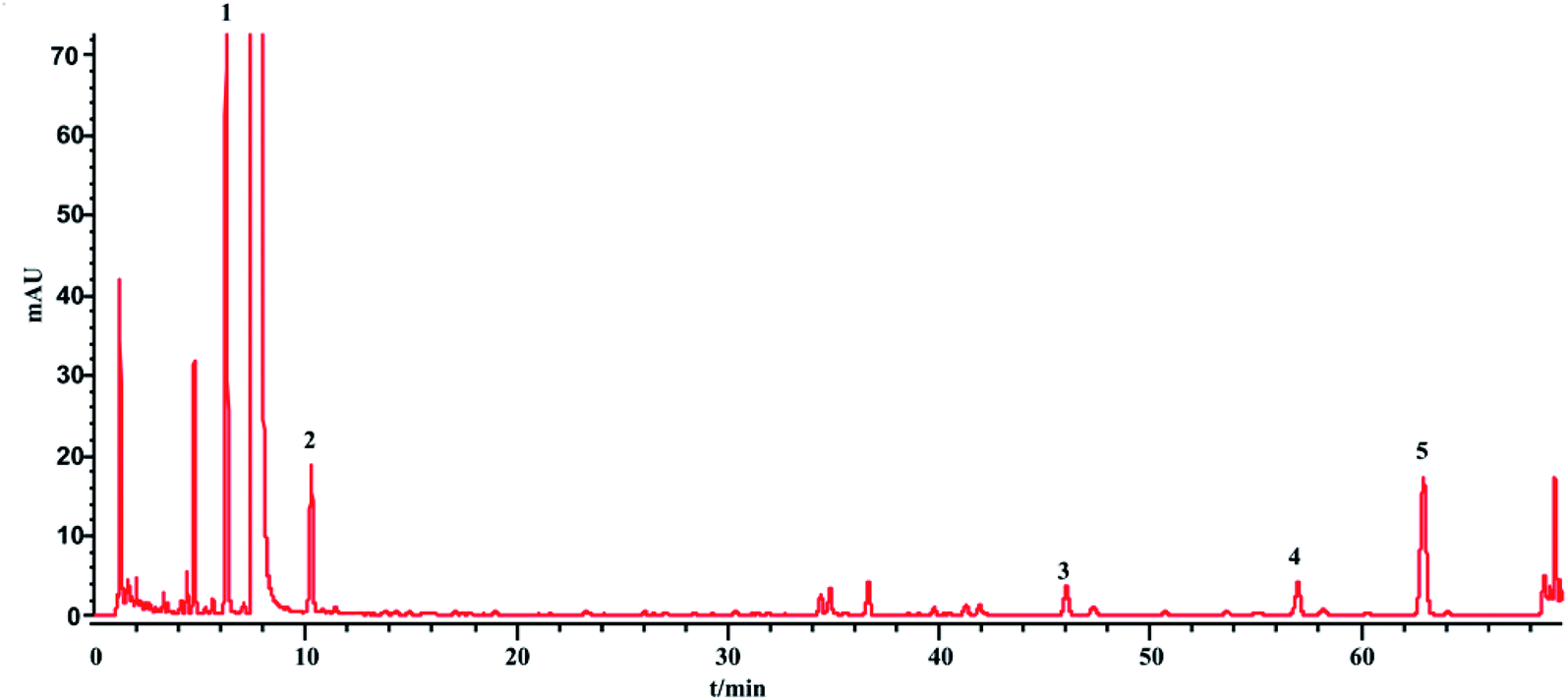
The phenol sulfuric acid assay was used to determine the total sugar and HPLC-ELSD for the saccharides (monosaccharides and oligosaccharides). The contents of the saccharides in SLI were analyzed according to Sections 2.4 and 2.5. The HPLC-ELSD chromatograms of the injection are shown in Fig. 3. Four kinds of saccharides were identified with the references, which were Fru, Glc, Mel, and Man, respectively.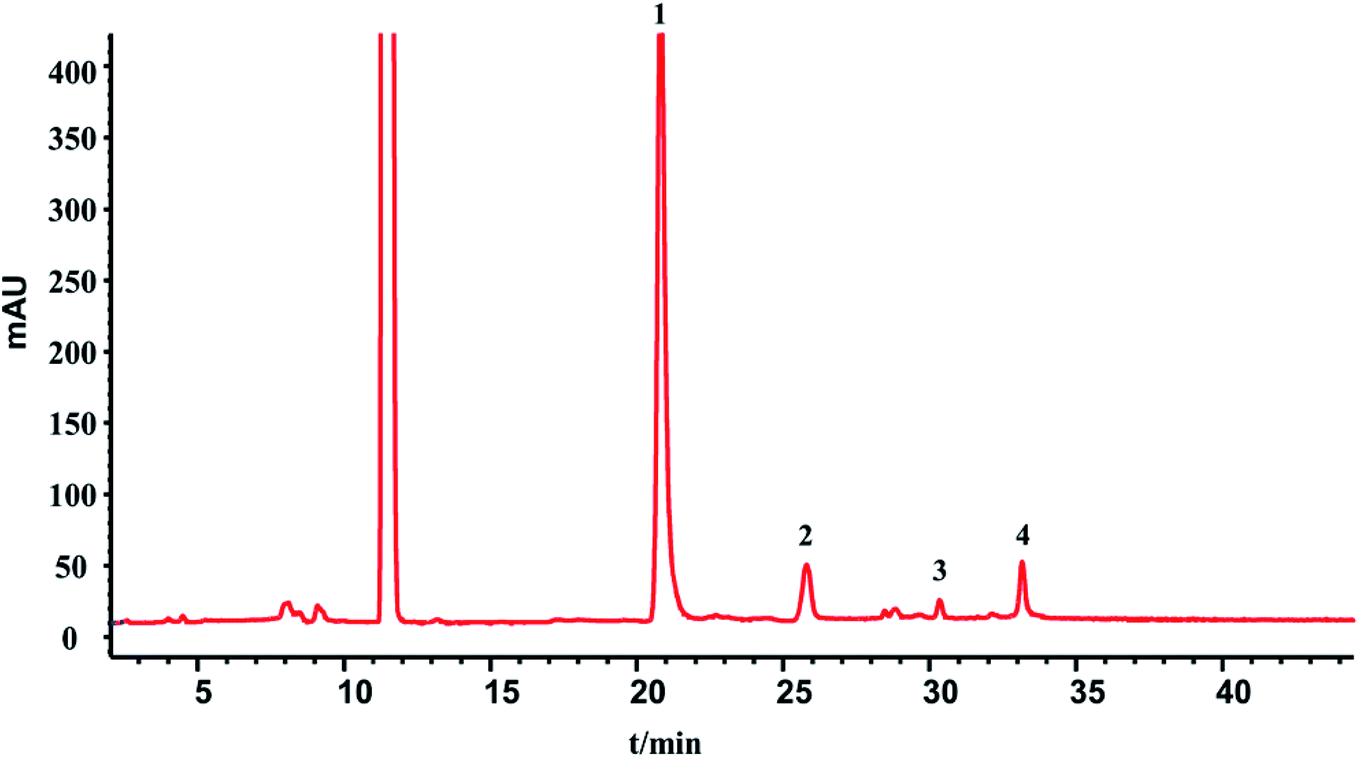 | ||
| Fig. 3 HPLC-ELSD chromatograms of 1, Fru; 2, Glc; 3, Mel; 4, Man. | ||
In addition, the determination of the sugar contents of 13 batches of injection samples by the two methods was conducted, and the differences in contents detected by the two methods were compared. The comparison results are shown in Table 6.
| Batch number | Fru (%) | Glc (%) | Mel (%) | Man (%) | The sum of the four kinds of saccharides (%) | Total sugar (%) |
|---|---|---|---|---|---|---|
| 1 | 0.15 | 0.06 | 0.02 | 0.07 | 0.30 | 0.31 |
| 2 | 0.15 | 0.06 | 0.02 | 0.07 | 0.30 | 0.30 |
| 3 | 0.15 | 0.06 | 0.02 | 0.06 | 0.28 | 0.31 |
| 4 | 0.16 | 0.06 | 0.02 | 0.07 | 0.30 | 0.31 |
| 5 | 0.16 | 0.06 | 0.02 | 0.07 | 0.30 | 0.30 |
| 6 | 0.15 | 0.06 | 0.02 | 0.06 | 0.29 | 0.31 |
| 7 | 0.16 | 0.06 | 0.02 | 0.07 | 0.31 | 0.31 |
| 8 | 0.15 | 0.06 | 0.02 | 0.07 | 0.30 | 0.30 |
| 9 | 0.15 | 0.06 | 0.02 | 0.06 | 0.29 | 0.30 |
| 10 | 0.15 | 0.05 | 0.02 | 0.08 | 0.31 | 0.31 |
| 11 | 0.14 | 0.06 | 0.02 | 0.08 | 0.30 | 0.30 |
| 12 | 0.14 | 0.06 | 0.02 | 0.08 | 0.30 | 0.31 |
| 13 | 0.14 | 0.06 | 0.02 | 0.08 | 0.29 | 0.30 |
The total sugar content (%) determined by the phenol sulfuric acid assay was nearly equal to the sum of the saccharide contents determined by the HPLC-ELSD method. This result suggests that the sugars in the injection were almost all detected by the HPLC-ELSD method. In other words, the HPLC-ELSD method is effective and accurate in determining saccharide content. As shown in Table 6, the sugar in the injection is mainly fructose, which has the highest percentage, followed by manninotriose and Glc, while melibiose has the smallest content. Furthermore, there were some fluctuations among the 13 batches of SLI.
3.4. HPLC-UV analysis for the quantitative determination of phenolic acid and fingerprint chromatography consistency evaluation
 | ||
| Fig. 4 HPLC chromatogram of the injection: 1 – DSS, 2 – PA, 3 – RA, 4 – SAB, 5 – SAA. | ||
| Batch number | Content (%) | ||||
|---|---|---|---|---|---|
| DSS | PA | RA | SAB | SAA | |
| 1 | 0.0435 | 0.0024 | 0.0022 | 0.0051 | 0.0066 |
| 2 | 0.0428 | 0.0023 | 0.0022 | 0.0049 | 0.0064 |
| 3 | 0.0430 | 0.0024 | 0.0022 | 0.0049 | 0.0062 |
| 4 | 0.0432 | 0.0024 | 0.0022 | 0.0049 | 0.0062 |
| 5 | 0.0433 | 0.0024 | 0.0022 | 0.0052 | 0.0066 |
| 6 | 0.0432 | 0.0024 | 0.0022 | 0.0050 | 0.0063 |
| 7 | 0.0448 | 0.0025 | 0.0023 | 0.0053 | 0.0068 |
| 8 | 0.0440 | 0.0025 | 0.0023 | 0.0051 | 0.0066 |
| 9 | 0.0436 | 0.0024 | 0.0022 | 0.0051 | 0.0066 |
| 10 | 0.0436 | 0.0023 | 0.0022 | 0.0052 | 0.0067 |
| 11 | 0.0438 | 0.0019 | 0.0016 | 0.0056 | 0.0040 |
| 12 | 0.0441 | 0.0019 | 0.0016 | 0.0057 | 0.0041 |
| 13 | 0.0436 | 0.0018 | 0.0016 | 0.0055 | 0.0038 |
| Average | 0.0436 | 0.0023 | 0.0021 | 0.0052 | 0.0059 |
| RSD% | 1.20 | 10.63 | 13.21 | 5.12 | 19.06 |
From Table 7, the contents of DSS has a good batches consistency with a RSD values 1.20%. While for PA, RA, SAA and SAB, there are big fluctuations, especially in the last three batches. It may be due to that phenolic acids are easily degraded or reacted under strong acid and alkali conditions. And, production fluctuations and storage time factors would also cause the differences.
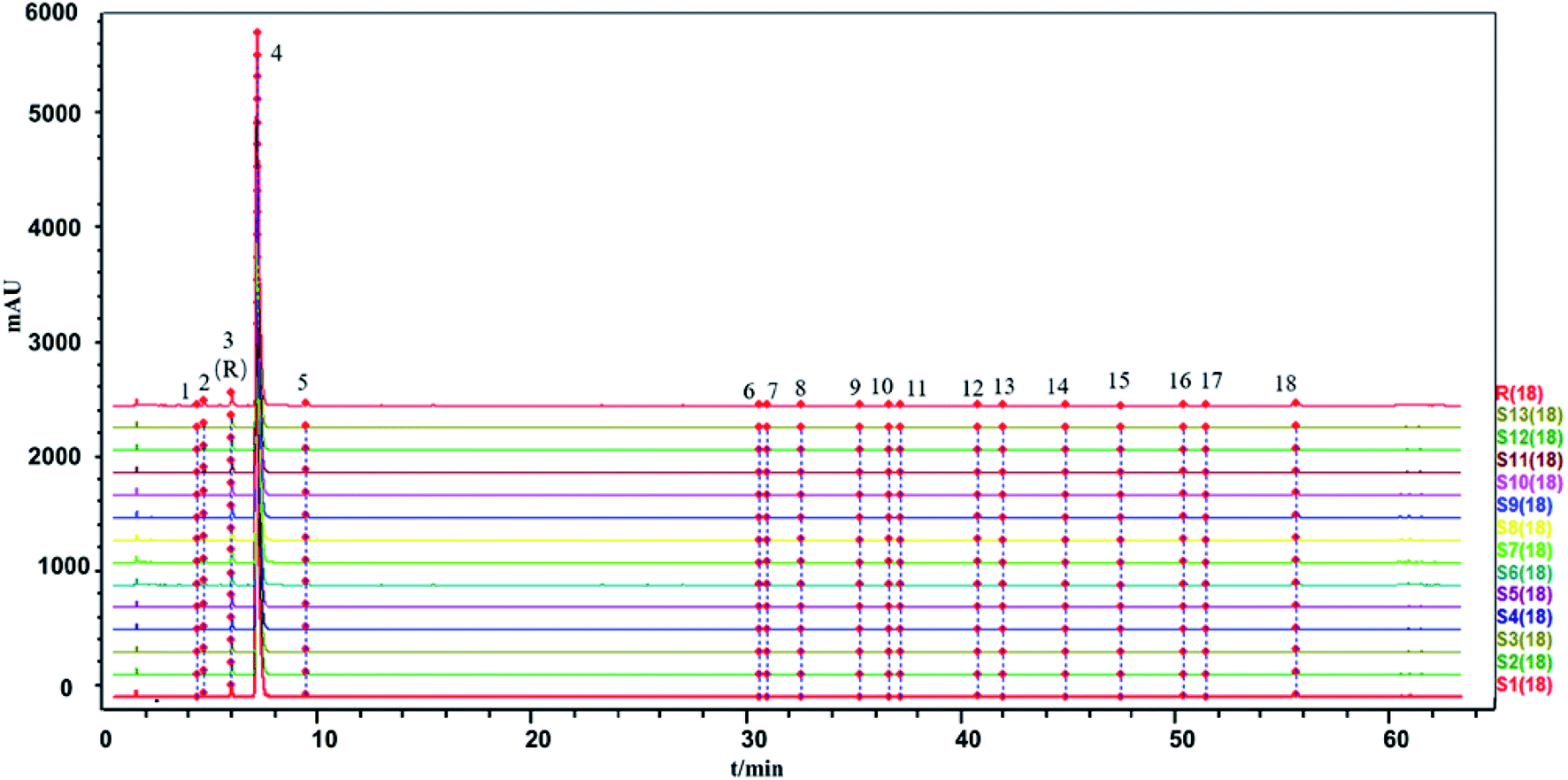 | ||
| Fig. 5 The HPLC-UV fingerprints of the 13 batches of SLI and generated control fingerprint (R): 2, 5-HMF; 3, DSS; 4, ligustrazine hydrochloride; 5, PA; 12, RA; 16, SAB; 18, SAA; and the rest are unknown. | ||
The similarity between the fingerprints of different batches of injection and the control fingerprint were all equal to 1 when ligustrazine hydrochloride was involved as a common peak, and the similarity statistics were all above 0.985 when ligustrazine hydrochloride was excluded, indicating good consistency between batches, as seen in Fig. 6. However, it may not be comprehensive to evaluate the batch consistency by the HPLC-UV fingerprint only, which is mentioned above. Thus, an evaluation of a multiple and global analytical indicator of batch consistency was employed, (see Part 3.6).
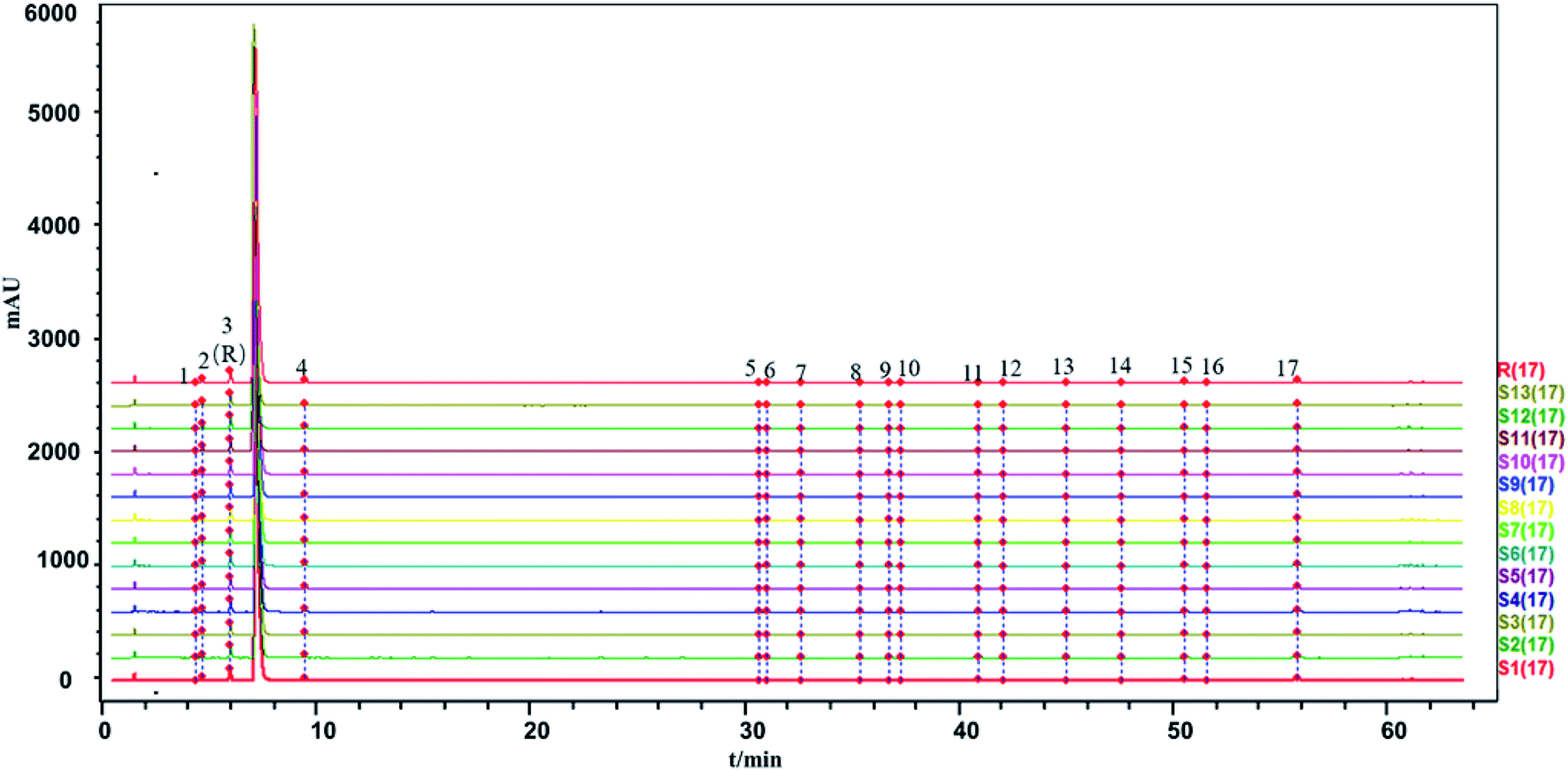 | ||
| Fig. 6 The HPLC-UV fingerprints of the 13 batches of SLI and generated control fingerprint (R) (not including the ligustrazine hydrochloride): 2,5-HMF; 3, DSS; 4, PA; 11, RA; 15, SAB; 17, SAA; and the rest are unknown. | ||
3.5. Inorganic salt analysis
The ion concentrations of the 13 batch injections are shown in Table 8, including the anions SO42−, Cl−, and NO3− detected by ion chromatography and the cations K+, Ca2+, Na+, Mg2+, and Fe+ (since the iron element has a valence of +2 and +3, the iron element in the sample is represented by Fe+) detected by atomic absorption spectrometry.| Batch number | Contents of inorganic salts (mg L−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| Cl− | NO3− | SO42− | Na+ | K+ | Ca2+ | Mg2+ | Fe+ | |
| 1 | 3.2779 | 0.0721 | 0.1171 | 318.7 | 162.6 | 7.38 | 41.79 | 0.109 |
| 2 | 3.1748 | 0.0694 | 0.1225 | 275.3 | 119.0 | 7.35 | 41.75 | 0.103 |
| 3 | 3.2534 | 0.072 | 0.1492 | 277.3 | 129.9 | 6.92 | 41.76 | 0.152 |
| 4 | 3.2827 | 0.0707 | 0.1187 | 387.9 | 157.2 | 8.46 | 58.80 | 0.106 |
| 5 | 3.2052 | 0.0706 | 0.1265 | 273.7 | 116.5 | 7.05 | 41.78 | 0.118 |
| 6 | 3.2927 | 0.0717 | 0.1341 | 267.1 | 98.6 | 7.04 | 42.5 | 0.075 |
| 7 | 3.1201 | 0.0747 | 0.1167 | 261.5 | 133.7 | 7.38 | 42.59 | 0.105 |
| 8 | 3.2347 | 0.0768 | 0.1052 | 266.6 | 127.8 | 6.98 | 42.21 | 0.099 |
| 9 | 3.2533 | 0.0728 | 0.1242 | 266.7 | 127.0 | 7.02 | 41.84 | 0.106 |
| 10 | 3.2353 | 0.0716 | 0.123 | 292.9 | 143.7 | 6.87 | 42.21 | 0.079 |
| 11 | 3.2878 | 0.0679 | 0.1025 | 277.9 | 124.8 | 7.49 | 43.00 | 0.097 |
| 12 | 3.2311 | 0.0687 | 0.0945 | 267.1 | 118.9 | 7.27 | 40.96 | 0.066 |
| 13 | 3.3242 | 0.0683 | 0.1013 | 299.4 | 140.9 | 7.85 | 45.24 | 0.067 |
| RSD% | 1.68 | 3.56 | 12.53 | 11.96 | 13.18 | 6.04 | 10.76 | 23.62 |
The Cl−, NO3− and SO42− contents ranged from 3.1201 to 3.3242 mg L−1, 0.0679 to 0.0768 mg L−1 and 0.0945 to 0.1492 mg L−1, respectively. The Na+, K+, Ca2+, Mg2+ and Fe+ contents ranged from 261.5 to 387.9 mg L−1, 98.60 to 162.6 mg L−1, 6.87 to 8.46 mg L−1, 40.96 to 58.80 mg L−1, and 0.066 to 0.152 mg L−1, respectively. The RSD value of the ion detection results, suggests that the differences in inorganic salts between batches of injection were large, such as SO42−, Na+, K+, Mg2+, and Fe+. The fluctuation in the inorganic salt ions showed that the production process of the injections indicates a lack of control and that more attention should be paid to this indicator.
3.6. Multivariate statistical analysis
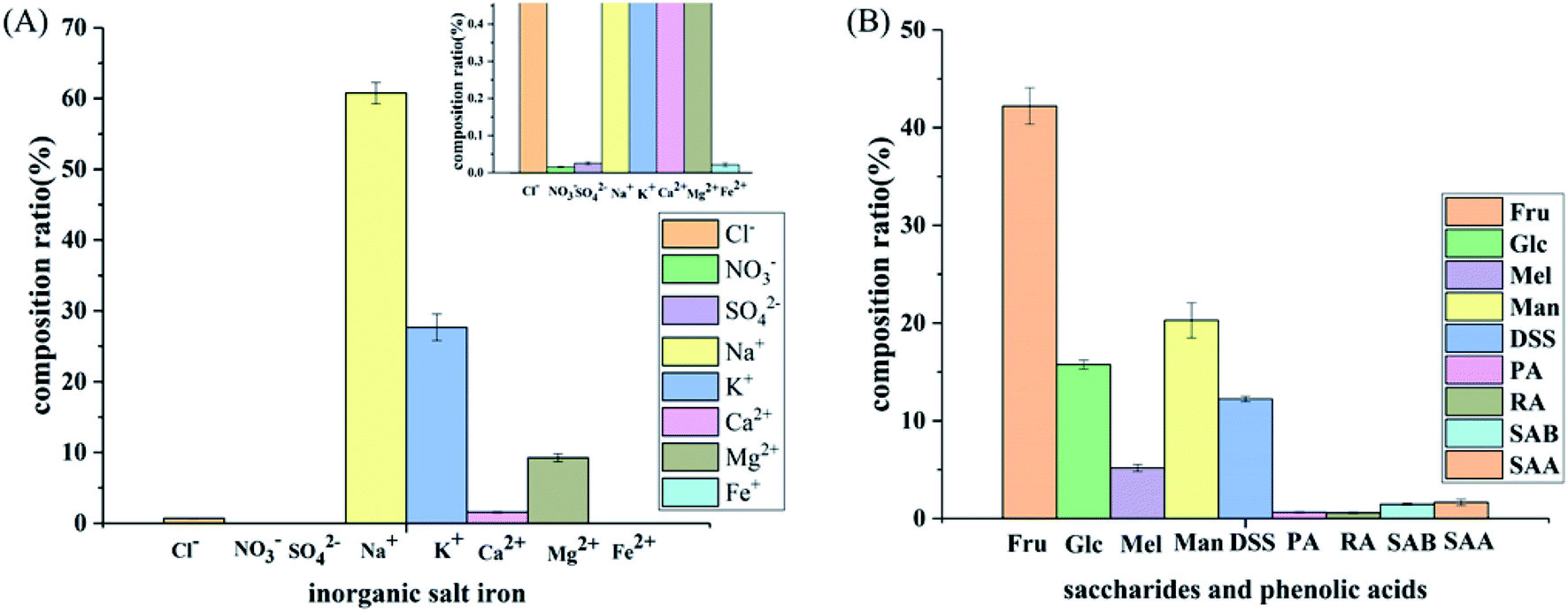 | ||
| Fig. 7 Composition of the SLI: (A) histogram of the inorganic salt ion composition in the SLI and (B) histogram of the composition of carbohydrates and organic phenolic acids. | ||
As shown in Fig. 7(A), the cation concentration was generally higher than that of the anions, and the contents of Na+ and K+ in the cations were relatively high. Since these two ions have an important influence on the cell osmotic pressure and myocardial activity, strict control is required to ensure the safety of the drug. In addition, as seen in Fig. 7(B), sugars accounted for the largest proportion, followed by DSS, and the phenolic acids accounted for the smallest proportion.
Principal component analysis: the content data of the indicators of 13 batches of injection were imported into SIMCA13.0 to establish a PCA model. According to the cross-validation result and the total variance explained by the principal component, the first four principal components were selected, accounting for 83.6% of the total variance. And the variance explained by the first two principal components is 63.8% (Principal component 1 (PC1): 43.9%, Principal component 2 (PC2): 19.9%). PC1 mainly accounts for the variation of sugars, phenolic acids and SO42−, Fe+ and NO3−. And PC2 does for the Na+, K+, Ca2+, Mg2+ as well as Cl−(see in Fig. 8). A biplot co-charting scores and loadings together for their simultaneous display and interpretation is presented in Fig. 8. The first two principle components are shown, and PC1 mainly accounted for the variance of the inorganic salts and PC2 accounted for the saccharides and phenolic acids. Green dots represent variables, corresponding to the points of loading plot. Brown dots represent the sample points of different batches, corresponding to the scores plot. For the batch consistency evaluation, the smaller the distance between the samples is, the more similar their chemical composition.
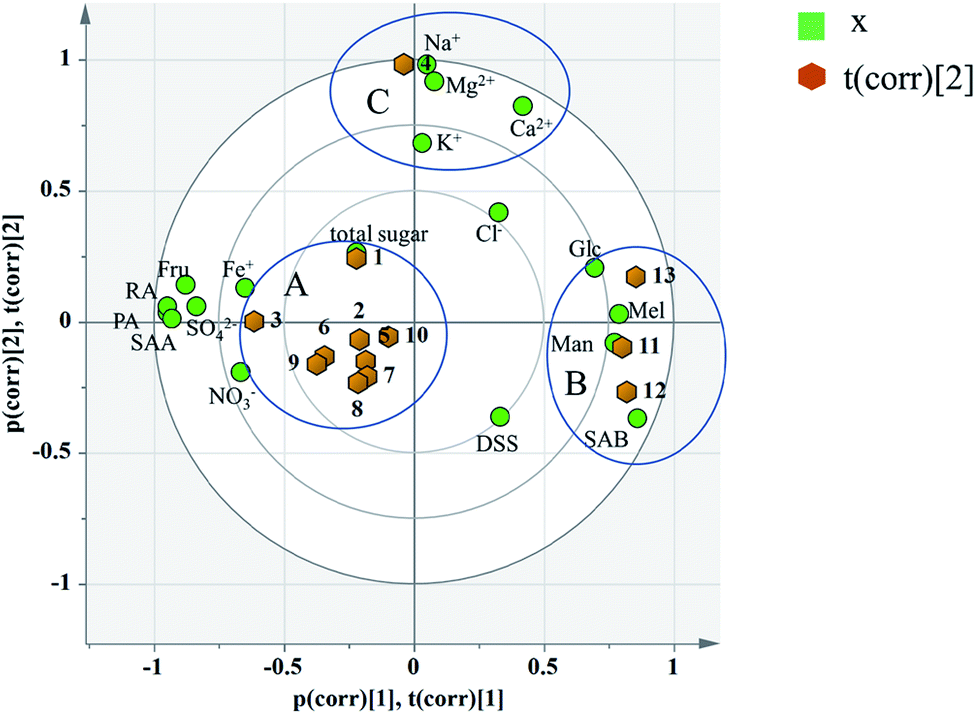 | ||
| Fig. 8 Biplot of 13 batches of injection: co-charting scores and loadings together for their simultaneous display and interpretation. Green dots represent variables, corresponding to the points of loading plot. Brown dots represent the sample points of different batches, corresponding to the scores plot. | ||
According to the distribution characteristics of the sample, it is divided into 3 groups (A, B and C). And the distribution characteristics indicated that the quality consistency was not good among batches. Variables situate near the observation implies that the observation is high for these variables. Observations close to the plot origin have average properties. Variables close to the plot origin do not contribute to the formation of the scores in question. As shown in Fig. 8, the indices of NO3−, SO42−, Fe+, Fru, Glc, Mel, Man, PA, RA, SAB, and SAA contributed more to the differences between group A and group B. It was consistent with the content data. The content of SAB in group B was significantly higher than that in the other groups, and the contents of PA and SAA were also significantly lower than the others (see the Table 7). This may be due to the process fluctuations and differences in the sample storage time. SAB will degrade to produce PA and SAA at a specific temperature, pH and storage time.44 The longer the storage time is, the more the SAB degrades. In fact, the production date of group B was later than that of the other groups, which can fully explain the difference. Group C was different from group A and group B due to the variation in its inorganic salts, which is attributed to PC2, and may be due to the fluctuations in the raw materials and processes.
Heatmap analysis: a heat map analysis was used to describe the batch consistency more visually than the raw data form. The heat map of 13 batches of injection is shown in Fig. 9. A commonly used method, unit variance, was applied to preprocess different dimensional data to eliminate unit differences. After preprocessing, the mean and standard deviation of the preprocessed data were 0 and 1, respectively. As shown in Fig. 9, the darker the color is, the larger the deviation from the mean. If the data distribution is concentrated or the dispersion is small, then the color of the indicator should be relatively fewer and lighter, suggesting that the batch consistency of the indicator is good. If the dispersion of the data is large, then the color is relatively more and darker, which implies that the batch consistency of the indicator is poor. As shown in Fig. 9, the batch consistencies of Cl−, NO3−, total sugar, Glc, and DSS were good despite the deviation in one batch being relatively large, which did not affect the global consistency, while indicators including SO42−, Na+, K+, Ca2+, Mg2+, Fe+, Man, Mel, PA, RA, SAB and SAA varied considerably among the batches. Comparing the PCA model and heat map analysis, the PCA model can quantify sample similarity while the heat map has a high degree of visualization of batch consistency. And a more comprehensive understanding of batch-to-batch consistency can be obtained by combining the PCA and heatmap analysis.
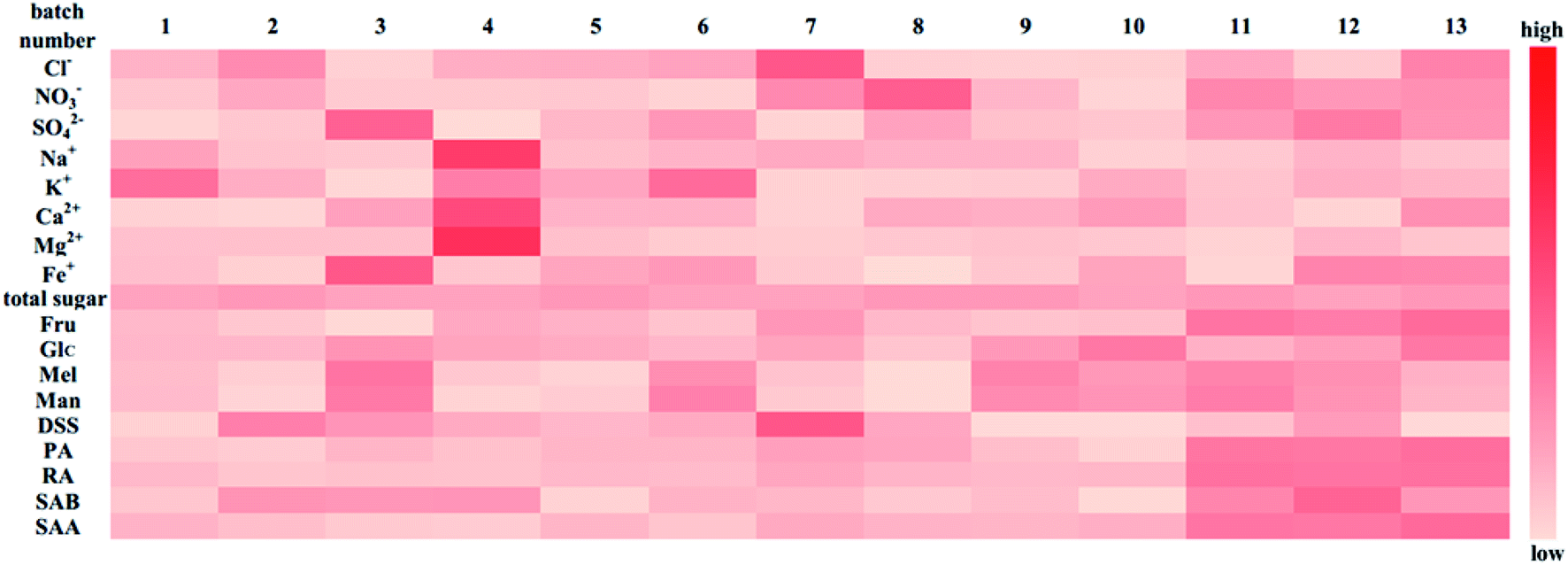 | ||
| Fig. 9 Indicators heat map of 13 batches of injection for evaluating the batch consistency. | ||
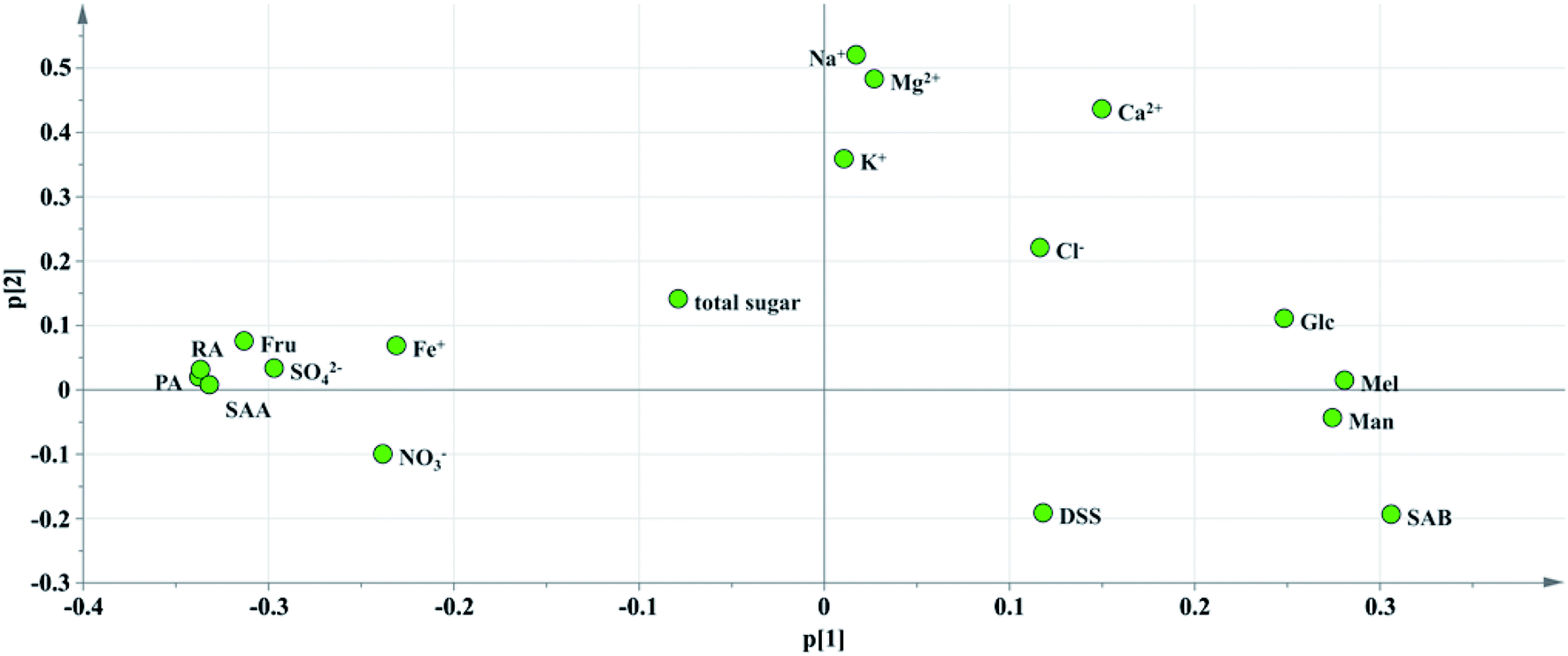 | ||
| Fig. 10 Loading diagram of PC1 and PC2. | ||
Critical inorganic salt indicator identification: as shown in Fig. 10, the position of Na+ was close to those of Mg2+, Ca2+ and K+, and they were considered positively correlated. According to variables relation as well as the content distribution of chemicals, Na+ with a high content could be selected as a key quality control indicator. If the content of Na+ is at a reasonable level, the content of the other cations would be within the specification as well. The anions were not selected as key quality control indicators because the batch-to-batch consistency of Cl− was good and the contents of the other anions, such as SO42− and NO3− were low. And it would be futile to control indicators with a low content that have no influence on health.
Critical saccharide indicator identification: in the contents of saccharides, the Fru content was the highest, followed by Man and Glc, and the Mel content was the lowest. The loading diagram showed that the load scores of PC1 and PC2 of Mel and Man were very similar indicating that the two indicators were highly correlated, which might be related to the degradation of stachyose in Salvia miltiorrhiza. As stachyose is a common sugar in Salvia miltiorrhiza,45 it is surprising that stachyose was not found in the SLI. A reasonable explanation would be that the stachyose has degraded. As reported, stachyose can hydrolyze to form Man under acidic conditions.46 In addition, studies have shown that stachyose can decompose in the body to form Mel and Man. Mel and Man correlated strongly to each other showed that the same chemical changes may have occurred during in the process. Finally, Fru, Glc and Man were identified as the critical quality control indicators according to loading plot and the content distributions of the chemicals.
Critical phenolic acid indicator identification: the load scores of PC1 and PC2 of PA, RA and SAA were very similar, indicating that they were highly correlated with each other. In addition, the degradation law of SAB mentioned above indicated that SAB was strongly correlated to PA and SAA. To control the quality of SLI more effectively, SAB was selected as a key quality control indicator to monitor the content variations in PA, RA,and SAA. Finally, SAB and the active substance DSS were identified as key indicators for the quality control of phenolic acids according to the same rule mentioned above.
In summary, Na+, Fru, Glc, Man, DSS and SAB were identified as the key quality control indicators that deserve more attention according to the content levels, batch consistency evaluation results, and variables correlated to each other as well as the quality requirements of SLI to improve product consistency.
4. Conclusion
Quality control, especially the batch consistency of TCMIs, is an important task for pharmaceutical companies. The HPLC-UV fingerprint method is not an effective way to evaluate the batch consistency because it cannot detect substances without UV absorption. Therefore, a more comprehensive method to apply batch consistency analysis to TCMI, namely, a multi-indicator global analysis, is urgently needed. This paper took SLI as an example to systematically study the key quality attributes of the injections, including macromolecule impurities, carbohydrate components (total sugars and saccharides), active substances (phenolic acids), and inorganic salt ions as well as the HPLC-UV fingerprint, and all of the analytical methods were validated with good performance. Then, the batch consistency of SLI was evaluated with a global analysis by applying PCA. The result was compared with the traditional similarity evaluation. The comprehensiveness and accuracy of the result was fully discussed. To make the batch consistency evaluation more visually, a heat map was applied at the same time. Last, Na+, Fru, Glc, Man, DSS and SAB were selected as key quality control indicators according to critical indicator identification, providing a reference for the quality control of the injection. The evaluation of a multiple and global analytical indicator of batch consistency was carried out from the perspective of the chemical quality of the injection, providing a methodology for the quality control of injections. Although this method cannot directly evaluate the safety of the preparation, it can also guarantee it indirectly (by evaluating the chemical quality of injection). And the established analytical methods also make it operable when evaluating quality consistency for injections. The evaluation of a multiple and global analytical indicator of batch consistency employed in this article would provide a reference for the quality consistency appraisement of a TCMI because of its comprehensiveness and may have broad application potential.Conflicts of interest
There are no conflicts to declare.References
- H. X. Zhang, Master thesis, University of Chinese Medicine, 2014.
- Y. Li, T. Wu, J. Zhu, L. Wan, Q. Yu, X. Li, Z. Cheng and C. Guo, J. Pharm. Biomed. Anal., 2010, 52, 597–602 CrossRef CAS PubMed.
- H. S. Xiong, X. C. Gong and H. B. Qu, J. Pharm. Biomed. Anal., 2012, 70, 178–187 CrossRef CAS PubMed.
- Y. P. Yu, D. D. Gong, Y. Zhu, W. Wei and G. X. Sun, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1095, 149–156 CrossRef CAS PubMed.
- Y. S. Ren, P. Zhang, D. Yan, J. B. Wang, X. X. Du and X. H. Xiao, J. Pharm. Biomed. Anal., 2011, 56, 436–442 CrossRef CAS PubMed.
- Y. M. Zhang, D. Yan, P. Zhang, Y. S. Ren, S. F. Zhang, X. Feng and X. H. Xiao, Acta Pharm. Sin., 2010, 45, 93–97 CAS.
- L. L. Zhang, L. N. Ma, W. W. Feng, C. E. Zhang, F. Y. Sheng, Y. Zhang, C. Xu, G. Dong, X. P. Dong, X. H. Xiao and D. Yan, Anal. Bioanal. Chem., 2014, 406, 5009–5018 CrossRef CAS PubMed.
- X. S. Wang, Master thesis, Beijing University Of Chinese Medicine, 2010.
- X. J. Feng, Master thesis, Hebei Medical University, 2016.
- R. Dong, J. Su, H. Nian, H. Shen, X. Zhai, H. Xin, L. Qin and T. Han, J. Funct. Foods, 2017, 37, 513–522 CrossRef CAS.
- X. He, J. Li, W. Zhao, R. Liu, L. Zhang and X. Kong, J. Funct. Foods, 2015, 171, 405–411 CAS.
- J. X. Ke, Q. Yuan, S. S. Li, G. H. Shen, A. J. Chen, L. Q. Ying, L. X. Yan, W. H. Jun, L. M. Liang, P. Biao, Y. Meng and Z. Z. Qing, Ind. Crops Prod., 2018, 119, 267–276 CrossRef CAS.
- G. Y. Xie, Q. H. Xu, R. Li, L. Shi, Y. Han, Y. Zhu, G. Wu and M. J. Qin, J. Pharm. Biomed. Anal., 2018, 164, 283–295 CrossRef PubMed.
- D. D. Luo, Y. Liu, Y. P. Wang, X. C. Zhang, L. F. Huang and B. Z. Duan, Biochem. Syst. Ecol., 2018, 76, 46–51 CrossRef CAS.
- H. L. Wang, W. F. Yao, D. N. Zhu and Y. Z. Hu, Chin. J. Nat. Med., 2010, 8, 343–348 CrossRef CAS.
- C. Y. Li, H. Y. Chen, W. P. Liu and W. Rui, J. Biol. Macromol., 2019, 123, 766–774 CrossRef CAS PubMed.
- Y. F. Wang, J. H. Xian, X. G. Xi and X. L. Wei, J. Biol. Macromol., 2013, 92, 583–590 CAS.
- H. Wang, H. X. Xin, J. F. Cai, F. B. Li, Y. Jin and F. Qing, Chin. J. Chromatogr., 2016, 34, 726–736 CrossRef CAS.
- C. Dong, S. L. Hui, J. Jian and Y. M. Zhi, Chin. Tradit. Pat. Med., 2019, 41, 34–38 Search PubMed.
- Q. Zhang, H. T. Fan, Y. Zhang and W. Hou, Chin. Arch. Tradit. Chin. Med., 2019 Search PubMed , http://kns.cnki.net/kcms/detail/21.1546.r.20190815.1015.042.html.
- T. Ge, Y. F. Zheng, J. Cui, Y. M. Xia, L. Huang and G. P. Peng, Med. Mod. Chin., 2014, 16, 989–991 CAS.
- L. J. Ma, Z. X. Zhu, H. Guo and Y. Gan, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 833, 260–263 CrossRef CAS PubMed.
- L. N. Li, J. Chin. Pharm. Sci., 1997, 6, 3–10 Search PubMed.
- R. F. Zhou, H. S. Long, B. Zhang, Z. Z. Lao, Q. Y. Zheng, T. C. Wang, Y. X. Zhang, Q. G. Wu, X. P. Lai, G. Li and L. Z. Lin, Mol. Med. Rep., 2019, 19, 1309–1317 CAS.
- Y. J. Cui, Y. Q. Li, W. Shi, F. M. F. Yang, X. M. Zhao and Z. L. Xia, Chin. J. Chromatogr., 2007, 25, 686–689 CAS.
- L. Chao, L. Z. Guo, Q. Xing, Y. Yuan, X. Bin and B. H. Ming, Chin. Pharmacol. Bull., 2015, 31, 449–452 Search PubMed.
- H. L. Liu, J. Lv, Z. M. Zhao, L. Shen and C. H. Liu, Chin. Tradit. Herb. Drugs, 2016, 47, 938–943 CAS.
- S. H. Li, Y. L. Liang, Y. J. Yang, W. X. Lai and L. Cui, Chin. Pharmacol. Bull., 2016, 32, 902–905 Search PubMed.
- H. G. Wang, R. Wang, X. Y. Zhao, L. Yu and G. H. Du, Chin. J. Pharmacol. Toxicol., 2018, 32, 302 Search PubMed.
- X. L. Zhou, S. W. Chan, H. L. Tseng, Y. Deng, P. M. Hoi, P. S. Choi, P. M. Y. Or, J.-m. Yang, F. F. Y. Lam, S. M. Y. Lee, G. P. H. Leung, S. K. Kong, H. P. Ho, Y. W. Kwan and J. H. K. Yeung, Phytomedicine, 2012, 19, 1263–1269 CrossRef CAS PubMed.
- National Pharmacopoeia Committee, Salvia miltiorrhiza Ligustrazine Injection Standard Public Draft, 2017 Search PubMed.
- S. Wold, Chemom. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17 CrossRef CAS.
- S. N. Duan, W. Qi, S. W. Zhang, K. K. Huang and D. Yuan, J. Food Drug Anal., 2019, 27, 275–283 CrossRef CAS PubMed.
- L. Q. Hu, S. Ma, C. L. Yin and Z. M. Liu, J. Sci. Food Agric., 2019, 99, 1413–1424 CrossRef CAS PubMed.
- B. Ankit, C. Vikas, R. K. Rawal and S. Sharma, J. Pharm. Anal., 2014, 4, 223–233 CrossRef PubMed.
- Y. P. Huang, Z. W. Wu, R. H. Su, G. H. Ruan, F. Y. Du and G. K. Li, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1026, 27–35 CrossRef CAS PubMed.
- S. S. Zeng, L. Wang, T. Chen, Y. F. Wang, H. B. Mo and H. B. Qu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 733, 38–47 CrossRef CAS PubMed.
- S. S. Zeng, T. Chen, L. Wang and H. B. Qu, J. Pharm. Biomed. Anal., 2013, 76, 87–95 CrossRef CAS PubMed.
- M. Goodarzi, P. J. Russell and Y. Vander Heyden, Anal. Chim. Acta, 2013, 804, 16–28 CrossRef CAS PubMed.
- D. Liang, Y. H. Yin, L. Y. Miao, X. Zheng, W. Gao, X. D. Chen, M. Wei, S. J. Chen, S. Li, G. Z. Xin, P. Li and H. J. Li, J. Pharm. Biomed. Anal., 2019, 169, 1–10 CrossRef CAS PubMed.
- Environmental Protection Department, Environmental Monitoring-Technical Guideline on Drawing and Revising Analytical Method Standards, 2010 Search PubMed.
- National Pharmacopoeia Committee, Chinese Pharmacopoeia, 2015 Search PubMed.
- S. C. Huang and H. B. Qu, Chin. J. Mod. Appl. Pharm., 2015, 32, 644–648 CAS.
- Y. F. Zheng, T. Ge and Y. M. Xia, J. Nanjing Univ. Tradit. Chin. Med., 2014, 30, 573–575 CAS.
- W. H. Wu, X. S. Wen and Y. Zhao, J. Chin. Med. Mater., 2007, 30, 848 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10065b |
| This journal is © The Royal Society of Chemistry 2020 |