
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effects of the crosslinking position and degree of conjugation in perylene tetraanhydride bisimide microporous polymers on fluorescence sensing performance†
Chen Hu,
Ying-Chun Gao,
Can Zhang,
Min Liu and
Tong-Mou Geng *
*
AnHui Province Key Laboratory of Optoelectronic and Magnetism Functional Materials, School of Chemistry and Chemical Engineering, Anqing Normal University, Anqing 246011, China. E-mail: gengtongmou@aqnu.edu.cn
First published on 31st January 2020
Abstract
In this study, two fluorescence conjugated microporous polymers based on perylene tetraanhydride bisimide (DP4A0 and DP4A2) were prepared via Sonogashira–Hagihara cross-coupling polymerization for the efficient detection of o-nitrophenol (o-NP). They were well characterized via FT-IR, solid state 13C NMR, elemental analysis, and other material characterization techniques. The experiments proved that both CMPs possess high thermal and chemical stability and a porous nature with Brunauer–Emmett–Teller (BET) specific surface areas of 41.3 and 402.1 m2 g−1. Importantly, owing to signal amplification by the conjugated skeleton, DP4A0 and DP4A2 exhibit extremely high sensitivity to o-NP with Ksv values of 1.83 × 104 and 1.69 × 104 L mol−1 and limits of detection of 5.73 × 10−9 and 7.36 × 10−9 mol L−1, respectively. The sensing performance of DP4A0 and DP4A2 was dependent on the position of crosslinking points and crosslinking density. Finally, super amplified quenching was considered the electron transfer mechanism and hydrogen bond interactions were also present.
Introduction
The determination of nitroaromatic compounds (NACs) has increased rapidly over the years because of its significance in environment, national defense, and public safety.1–6 There are some techniques and devices used for detecting NACs, such as metal detectors, trained canines, gas chromatography coupled with mass spectrometry (GC-MS),7 electrochemical methods,8,9 ion mobility spectroscopy (IMS),10 high performance liquid chromatography,11 quartz crystal microbalance (QCM), colorimetry, and surface enhanced Raman spectroscopy (SERS).6,12 Fluorescence detection methods have emerged as one of the most promising approaches because of the quick response time, moderate price, high sensitivity and selectivity, simple operation, and portability for on-site testing.4,6 Although traditional fluorescent substances (such as organic fluorescent dyes) have made some progress, they have low limits of detection (LODs) and may result in more environmental pollution due to their toxicity, poor biodegradability, presence of redundant heavy metals as well as inferior chemical stability and photobleaching.6 Because of the “aggregation-induced quenching (ACQ)” effects, most organic dyes deplete their excitation energy in their aggregated state. By covalently incorporating dye-based monomers into conjugated microporous polymers (CMPs), fluorophores can be separated spatially through porosity, thereby remarkably increasing the fluorescent intensity of the monomers, which are initially affected by ACQ.13,14CMPs are worth the consideration for a variety of reasons: first, compared with similar building blocks, CMPs become prominent electron donors due to the expansion of π-conjugation. Furthermore, in virtue of the π* delocalization, the enhanced donating ability of CMPs in the excited state provides a platform for the facile migration of excitation. Therefore, the interaction between CMPs and NACs will augment and the fluorescence will be quenched, which is found by Swager et al., known as ‘molecular wire effect’.15 The extended π-conjugation of conjugated polymers was shown to be an up-and-coming feature of amplified signal transduction.3,16 Second, because they are constructed by carbon–carbon or carbon–nitrogen bonds, CMPs are thermally and chemically stable.3,17 Third, the structure and porosity of the CMPs may be adjusted by judiciously selecting building blocks, since high porosity can enhance the rapid diffusion of NACs in porous frameworks and impactful host–guest interactions, leading to improved fluorescence quenching efficiency and sensitivity.3
Since the discovery of the first CMP in 2007,18 there has been much interest in the synthesis and possible applications of these materials.17,19,20 Numerous CMPs have been successfully synthesized and applied as chemosensors, particularly fluorescent CMPs, which have been employed for the detection of NACs.21,22 NACs have strong electron acceptance ability. Therefore, adding NACs will affect the excited states of CMPs, which leads to fluorescence quenching.23 Recently, we developed a fluorescent CMP based on perylene tetraanhydride bisimide (DP2A2) via a Sonogashira–Hagihara cross-coupling polymerization for sensing o-nitrophenol (o-NP).14 In a continuation of our previous study, this paper reports the synthesis of two CMPs (DP4A0 and DP4A2) with high surface areas and microporosity (Scheme 1). The effects of different cross-linking points and densities on the fluorescence sensing properties of the CMPs were studied.
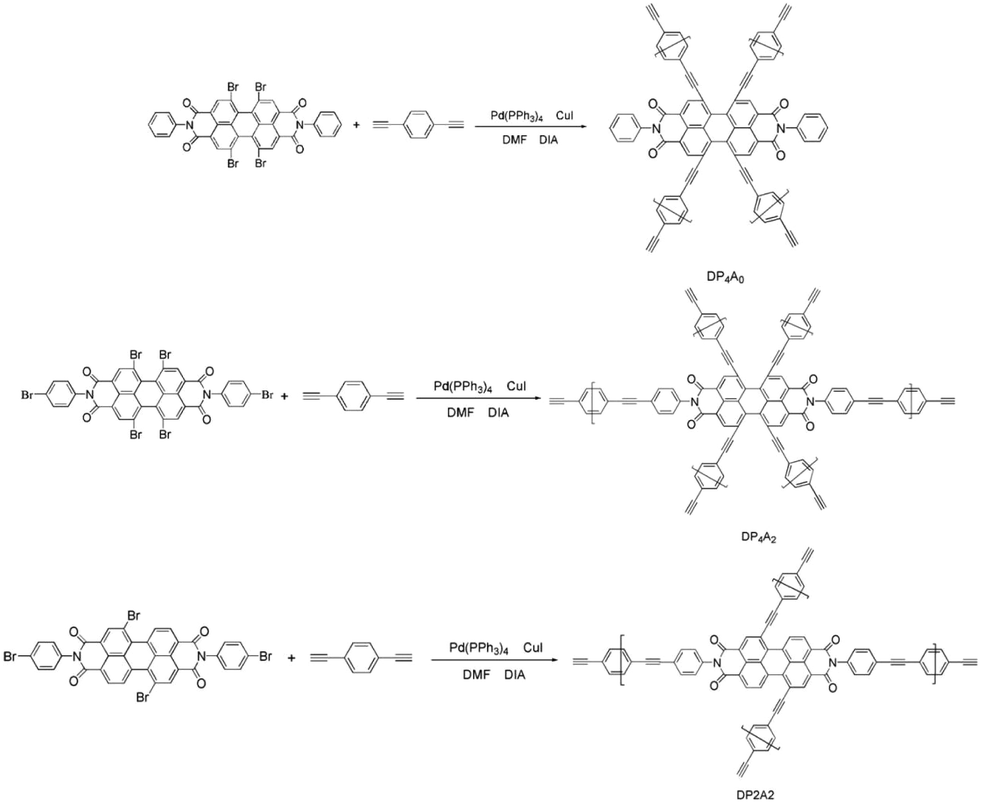 | ||
| Scheme 1 Synthesis of DP4A0, DP4A2, and DP2A2 via Sonogashira–Hagihara cross-coupling reactions. | ||
According to the serial numbers in Scheme 2, the cross-linking points of DP4A0 should be located at 1, 6, 7, and 12. The cross-linking points of DP4A2 should be located at 1, 6, 7, 12, 13, and 14 and the cross-linking points of DP2A2 should be located at 1, 7, 13, and 14.
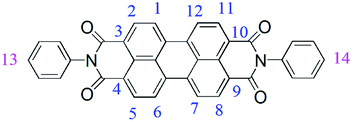 | ||
| Scheme 2 The serial numbers of N,N′-diphenyl-perylene-3,4:9,10-tetracarboxydiimide. | ||
Experimental section
Materials
Perylene-3,4:9,10-tetracarboxylic dianhydride (PTCDA, 98%), copper(I) iodide (CuI) (99.5%), 1,4-diethynylbenzene (DEB), and tetrakis(-triphenylphosphine) palladium (Pd(PPh3)4) were bought from Aladdin Chemistry Co. Ltd. p-Bromoaniline (99%) was purchased from J & K scientific Ltd. Bromine, 1,4-dioxane (DOX), acetonitrile (ACN), methanol, N,N-dimethylformamide (DMF, 99.0%), chloroform, ethanol (EtOH), tetrahydrofuran (THF) sodium hydrate, acetone, toluene, hydrochloric acid, diisopropylamine (DIA), o-NP, 4-nitrotoluene (p-NT), picric acid (PA), phenol (PhOH), dinitrotoluene (DNT), 1,3-dinitrobenzene (m-DNB), and 1,4-dinitrobenzene (p-DNB) were purchased commercially and used without further purification. N,N-Diphenyl-1,4,7,12-tetrabromoperylene-3,4:9,10-tetracarboxydiimide (PBr4ABr0) and N,N′-di(4-bromophenyl)-1,6,7,12-tetrabromoperylene-3,4:9,10-tetracarboxydiimide (PBr4ABr2) were synthesized in accordance with previous reports.23,24Characterization, morphology analysis and methods
Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Nicolet Nexus 8700 Fourier transform infrared spectrometer in KBr pellets. Solid-state 13C cross polarization magic angle spinning NMR (ss 13C NMR) spectra were recorded on a Bruker Avance III 400 NMR spectrometer, using a contact time of 2.0 ms and a relaxation delay of 10.0 s. Elemental analysis (EA) was performed on an analyzer (model VarioELIII). Solid UV-Vis absorption spectra were recorded using a PerkinElmer Lambda 950 UV-Vis spectrophotometer.The thermal gravimetric analysis (TGA) measurements were performed using a Mettler ATA409PC thermo-gravimetric analysis instrument between room temperature to 800 °C at a heating rate of 10 °C min−1 under a N2 atmosphere. Powder X-ray diffraction (PXRD) was examined on a Rigaku model XRD600 diffractometer equipped with Ni-filtered Cu Kα radiation (40 kV, 100 mA) by depositing powder on glass substrates at room temperature. The scanning speed was 5° min−1 and scanning ranges are from 2θ = 5° to 60° with 0.02° increments. Scanning electron microscopy (SEM) was performed on a JEOL-3400LV and S-3400N microscope (Japan) with an accelerating voltage of 5.0 kV.
The specific surface area and pore size distributions were measured using a Bel Japan Inc. model BELSORP-mini II sorption analyzer with N2 adsorption and desorption at 77 K, and the pore parameters (BET specific surface area, pore size, and pore volume) could be estimated from the adsorption–desorption isotherms. Samples were degassed at 150 °C in a vacuum for 10 h before each measurement. Fluorescence spectra were recorded at room temperature using Hitachi F-4500 spectrophotometers. A 0.1 mol L−1 THF–NACs stock solution was prepared. A 1.0 mg mL−1 solution of the dispersion colloid was obtained by adding DP4A0 or DP4A2 (25 mg) to THF (25 mL), followed by ultrasonic dispersion. As soon as the NAC solution was added, the fluorescence intensity of the dispersion was determined. The LODs were obtained using the equation: LOD = 3s/ρ, where s is the standard deviation of the blank measurement and ρ is the slope of the relative fluorescence intensity (I0/I) over the sample concentration.
Results and discussion
DP4A0 and DP4A2 as well as their corresponding building blocks were prepared by following approaches found in previous reports.14,24,25 The structures of DP4A0 and DP4A2 were confirmed via FT-IR spectroscopy, ss 13C NMR spectroscopy, elemental analysis (EA), and UV-Vis absorption spectroscopy. The FT-IR spectra of both DP4A0 and DP4A2 (Fig. 1) showed peaks at 2189 and 2200 cm−1, which are the characteristic of bis-substituted acetylenes.14,26 The polyimide rings were confirmed by the characteristic peaks of the carbonyl groups in six-membered polyimide rings (∼1670 cm−1 and ∼1710 cm−1).14,27–30 The bands at 1331 and 1335 cm−1 are attributed to C–N stretching vibration peaks.14,27,30 The bands at 1591, 1485/1501, and 1405 cm−1 are the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching peaks of phenyl rings. The bands at 744 and 755 cm−1 are the absorption peaks of C–H plane deformations of singly substituted phenyl rings.14
C stretching peaks of phenyl rings. The bands at 744 and 755 cm−1 are the absorption peaks of C–H plane deformations of singly substituted phenyl rings.14
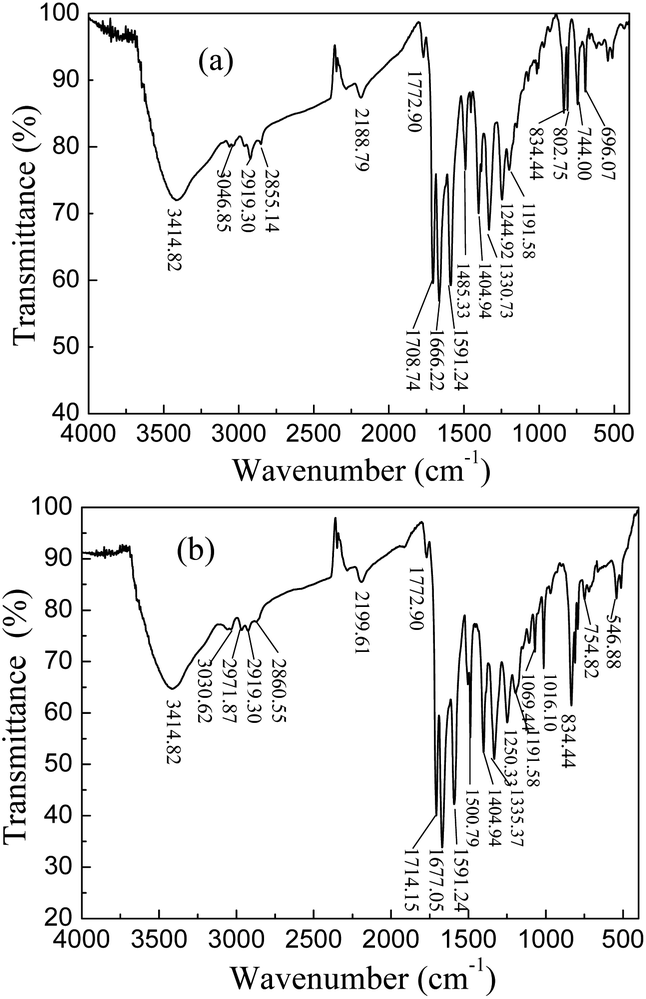 | ||
| Fig. 1 FT-IR spectra of DP4A0 and DP4A2. | ||
The chemical structure of DP4A0 and DP4A2 was further analyzed via ss 13C NMR spectrometry (Fig. 2). The peaks at about 162 ppm pertained to the CO bond in the six-membered polyimide rings.14,27–29 The signals near 132.37/130.81 and 128.18, 122.23/122.03 ppm pertained to other aromatic carbon atoms (benzene, perylene) from the two building blocks.14,27 The signals at 93.40/90.97 (Ar–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ar), 82.83/83.05, and 77.87/77.64 ppm (Ar–CC–H) correspond to the alkynyl units in the CMPs.14,25,31 The elemental analysis of the CMPs indicated that there is a deviation from the theoretical values, which is common for CMPs probably because of the incomplete combustion or adsorption of water and gases.32 Fig. S1a† shows the solid UV-Vis spectra of DP4A0 and the corresponding building block PBr4ABr0. The DP4A0 powder shows an extra peak at 288 nm, which is not found in PBr4ABr0. The relative intensity of this peak decreases in the polymer. Compared to PBr4ABr0 with a peak at 272 nm, there is an obvious red-shift.19 There were two strong peaks for both DP4A2 and PBr4ABr2, in which a red-shift was observed for the main peak (Fig. S1b†).29 The results clearly indicated that products with new optical performance were formed.28
C–Ar), 82.83/83.05, and 77.87/77.64 ppm (Ar–CC–H) correspond to the alkynyl units in the CMPs.14,25,31 The elemental analysis of the CMPs indicated that there is a deviation from the theoretical values, which is common for CMPs probably because of the incomplete combustion or adsorption of water and gases.32 Fig. S1a† shows the solid UV-Vis spectra of DP4A0 and the corresponding building block PBr4ABr0. The DP4A0 powder shows an extra peak at 288 nm, which is not found in PBr4ABr0. The relative intensity of this peak decreases in the polymer. Compared to PBr4ABr0 with a peak at 272 nm, there is an obvious red-shift.19 There were two strong peaks for both DP4A2 and PBr4ABr2, in which a red-shift was observed for the main peak (Fig. S1b†).29 The results clearly indicated that products with new optical performance were formed.28
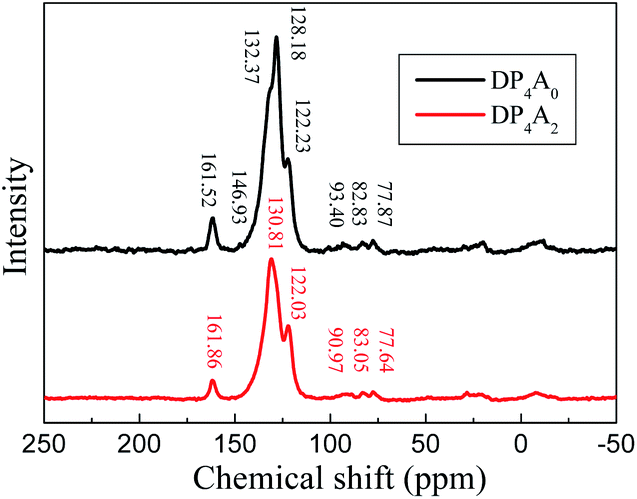 | ||
| Fig. 2 The ss 13C NMR spectra of DP4A0 and DP4A2. | ||
To ascertain the porosity of DP4A0 and DP4A2, nitrogen adsorption–desorption experiments were performed at 77 K (Fig. 3). DP4A0 assumes a Type I isotherm in the range of p/p0 = 0.05–0.15 according to IUPAC classifications and then takes on a very flat adsorption plateau.30 The gas absorption decreases sharply at lower relative pressures.27 In sharp contrast, DP4A2, although similar to DP2A2, displays a combination of type I and type II isotherms. With the increase in the nitrogen sorption at high relative pressure, there is a palpable hysteresis phenomenon, which might be due to the existence of a mesoporous or interparticulate void in the sample.14 The isotherm of DP4A2 exhibits sharp N2 adsorption under low relative pressure (p/p0 < 0.01), which means that there are substantial micropores and ultramicropores.14,24,29,30 Using the BET model, these isotherms give apparent surface areas of 41.3 m2 g−1 and 402.1 m2 g−1 for DP4A0 and DP4A2, respectively (Table 1). The BET surface area of DP4A2, similar to that of DP2A2 (378 m2 g−1),14 is higher than that of DP4A0 although the chemical composition of DP2A2 and DP4A0 is the same. A reasonable explanation for this is the difference in the point position and degree of crosslinking.27,29,30 The pore volumes were obtained using DFT methods from the nitrogen sorption isotherms. DP4A2 shows a larger total pore volume when compared to DP4A0 and is similar to DP2A2 (Table 1). The ratio of Vmic/Vtot can be used as a scale for the degree of microporosity in the CMPs. DP4A2 shows a high degree of microporosity, of which 63.27% of the total pore volume consists of micropores. The low volume ratio of DP4A0 indicates clearly that there is a certain amount of large pores or interparticulate porosity.29,30 The degree of crosslinking in DP4A2 is 6, which is larger than that of DP4A0 and DP2A2 (4); therefore, DP4A2 has a higher specific surface area and microporosity compared to DP4A0 and DP2A2. Although the crosslinking degree of both DP4A0 and DP2A2 is 4, their crosslinking point position is different. The positions of the crosslinking points in DP4A0 are located at the 1, 6, 7, and 12 positions, while that of DP2A2 are located at the 1,7,13, and 14 positions. Because the positions of the crosslinking points in DP4A0 are more crowded than that of DP2A2, thus the specific surface area and microporosity of DP4A0 are much lower than that of DP2A2. The pore volume distributions as a function of the pore width for DP4A0 and DP4A2 were estimated via the nonlocal DFT (NLDFT) method (Fig. 3b). It can be seen that DP4A0 possesses a narrow pore size distribution in the minor region with a pore size centered at 2.15 nm. DP4A0 has a very low total pore volume and there are both micropores and pores in the mesopore region.27,29 The N2 sorption isotherm of DP4A2 indicated that DP4A2 includes a certain amount of pores with a relatively broad diameter distribution from 1 to 4 nm and a large proportion of the pores are centered at 1.22 nm. Although micropores dominate in DP4A2, there is a certain proportion of mesopores in the DP4A2 network.14,27
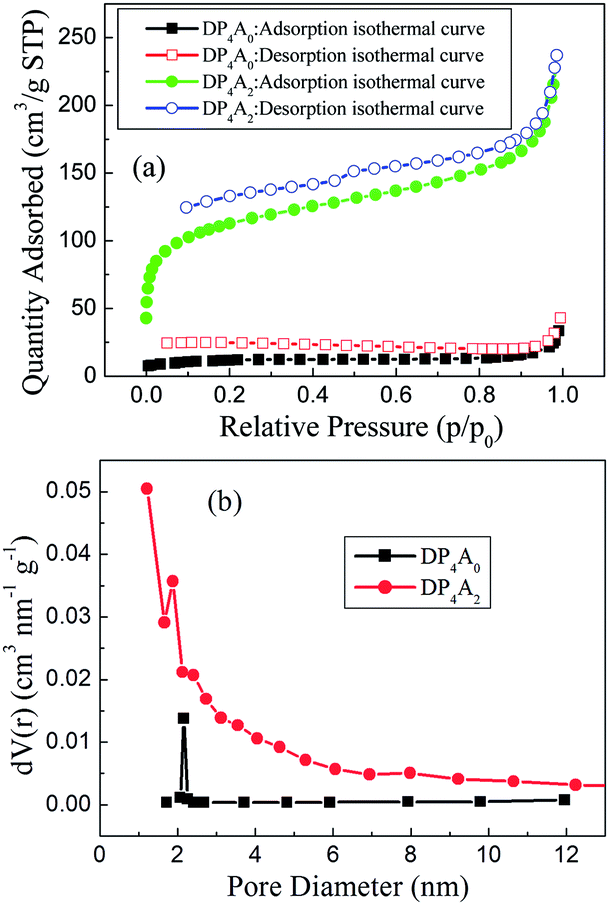 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms measured at 77 K for DP4A0 and DP4A2; (b) the BJH pore size distribution profiles of DP4A0 and DP4A2 calculated using the NLDFT method. | ||
| CMPs | SBETa (m2 g−1) | Vtot (tpv)b (cm3 g−1) | Vmicroc (cm3 g−1) | Vmicro/Vtot | Smicroc (m2 g−1) | Sexternalc (m2 g−1) |
|---|---|---|---|---|---|---|
| a Specific surface area calculated from the adsorption branch of the nitrogen isotherm using the BET method with a relative pressure (p/p0) range from 0.01 to 0.10.b The total pore volume was obtained from the BET data up to p/p0 = 0.99 and is defined as the sum of the micropore volume and the volumes of larger pores.c The micropore volume was calculated from the nitrogen adsorption isotherm using the t-plot method. | ||||||
| DP4A0 | 41.3 | 0.03772 | 0.00743 | 0.1971 | 16.9 | 24.4 |
| DP4A2 | 402.1 | 0.3665 | 0.2319 | 0.6327 | 115.9 | 170.4 |
| DP2A2 | 378.0 | 0.2804 | 0.1008 | 0.3595 | 217.8 | 160.2 |
DP4A0 and DP4A2 are all black powders. They are not soluble in water and common organic solvents and are also chemically stable in dilute aqueous solutions of acids or bases due to their highly crosslinked structures. There are a few weight losses at 275 and 362 °C due to escaping solvent that was caught inside the polymer networks. They have excellent thermal stability with decomposition temperatures (Tdec) of 453 and 547 °C determined at 5% mass loss and char yields of 60.01% and 90.26% at 800 °C under a N2 atmosphere (Fig. 4). The high thermal stability is due to the covalent bonds, the high crosslinking density of the networks, and the good stability of the perylene imide backbones.14,27–30 PXRD measurements confirmed the amorphous feature of the CMPs (Fig. S2†),27,30 which are expected for a typical non-dynamic covalently cross-linked CMP materials.33 Morphologies monitored by SEM demonstrate that DP4A0 displays a Tremella fuciformis-like morphology.30 DP4A2 exhibits a floppy surface and an interconnected spherical particle-like morphology (Fig. 5).27–29
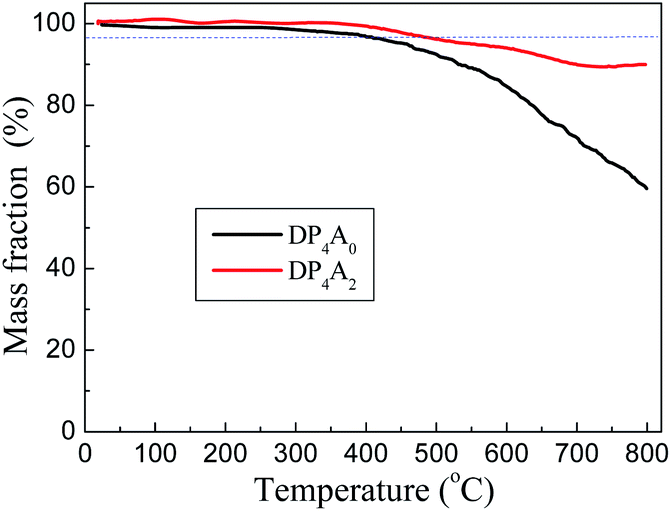 | ||
| Fig. 4 TGA thermograms of DP4A0 (black) and DP4A2 (red), measured in a nitrogen atmosphere at a heating rate of 10 °C min−1. | ||
 | ||
| Fig. 5 SEM images of (a) DP4A0 and (b) DP4A2. | ||
We investigated the emission spectra of DP4A0 and DP4A2 in different polar solvents (Fig. 6), including ACN, DMF, acetone, THF, chloroform, DOX, and EtOH.3,4,34 It was found that DP4A0 and DP4A2 disperse liquid in DMF, DOX, chloroform, and THF and exhibited purple or yellow emission when excited with 365 nm UV light.6,35,36 Thanks to the completely π-conjugated skeleton from the connection of the phenyl rings, DP4A0 and DP4A2 exhibited the strongest fluorescence in THF (λex = 370 nm) and DOX (λex = 365 nm) suspensions, respectively.22,35,36 Their ability to use fluorescence sensing to detect NACs (such as o-NP) was discussed. As shown in Fig. S3,† a rapid and clearly visible fluorescence turn-off response was observed upon increasing the concentration of o-NP in the solution. As the concentration of o-NP in the solution increased, a fast fluorescence turn-off response occurred, which signified that DP4A0 and DP4A2 enable effective “real-time” detection of o-NP.4,37
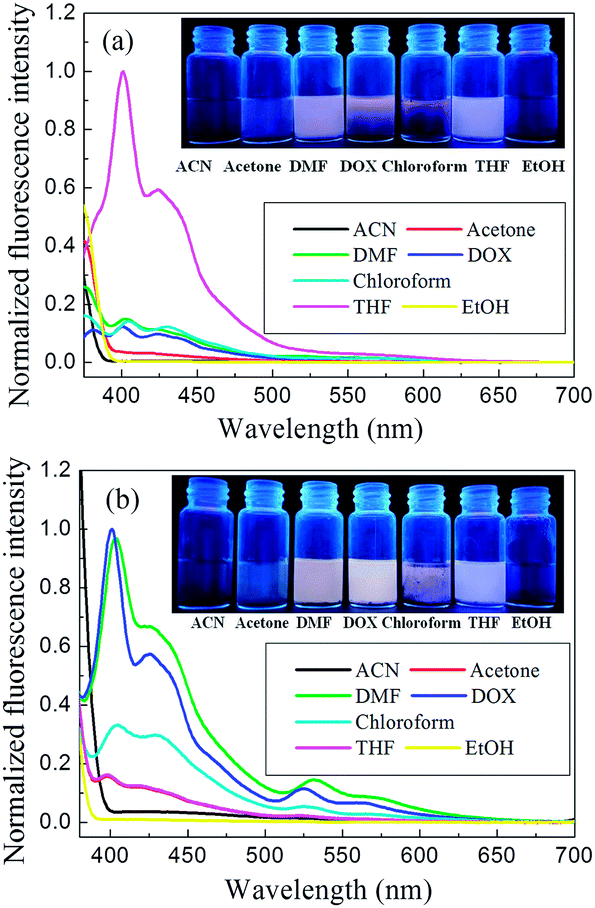 | ||
| Fig. 6 Fluorescent emission spectra of (a) DP4A0 (λex = 370 nm) and (b) DP4A2 (λex = 365 nm) dispersed in various solvents. Insets: photographs of solvent dispersed DP4A0 and DP4A2 excited with UV light at 365 nm. | ||
We investigated DP4A0 and DP4A2 as fluorescent probes for sensing NACs by successively adding NACs, such as NB, PA, DNT, p-DNB, o-NP, p-NT, m-DNB, and non-nitroaromatic analytes including PhOH. Fig. 7 and S4–S6† show the fluorescence spectra and I0/I plots of DP4A0 and DP4A2 before and after adding NACs and different degrees of quenching were observed after the addition of the NACs. The fluorescence of DP4A0 and DP4A2 was almost completely quenched after the addition of o-NP, while the fluorescence was not obviously quenched after the addition of other NACs (PA, NB, DNT, p-NT, p-DNB, m-DNB, and PhOH), indicating that DP4A0 and DP4A2 exhibit extremely high selectivity for o-NP.37,38 Furthermore, the fluorescence intensity of DP4A0 and DP4A2 were overwhelmingly affected by the molar concentration of o-NP. To qualitatively comprehend the quenching influence of o-NP on DP4A0 and DP4A2, the quenching coefficients (Ksv) were calculated using the Stern–Volmer (S–V) equation: I0/I = 1 + Ksv[Q] (I0 and I are the fluorescent intensities of DP4A0 or DP4A2 before and after the addition of the analyte and [Q] is the concentration of the analyte). As shown in Fig. 7c, good linear S–V relationships were observed for DP4A0 and DP4A2, and the Ksv were up to 4 orders of magnitude greater than before the addition of o-NP, which indicated that DP4A0 and DP4A2 have high sensitivity to o-NP.5,23,34,38,39 In addition, the LODs of DP4A0 and DP4A2 to o-NP are 5.73 × 10−9 and 7.36 × 10−9 mol L−1, respectively, also indicating a high sensitivity to o-NP.4–6 Fig. 8 shows that as more o-NP was added, the tighter the aggregates of both CMPs were, meaning that there are intense interactions between both CMPs and o-NP.14,40,41
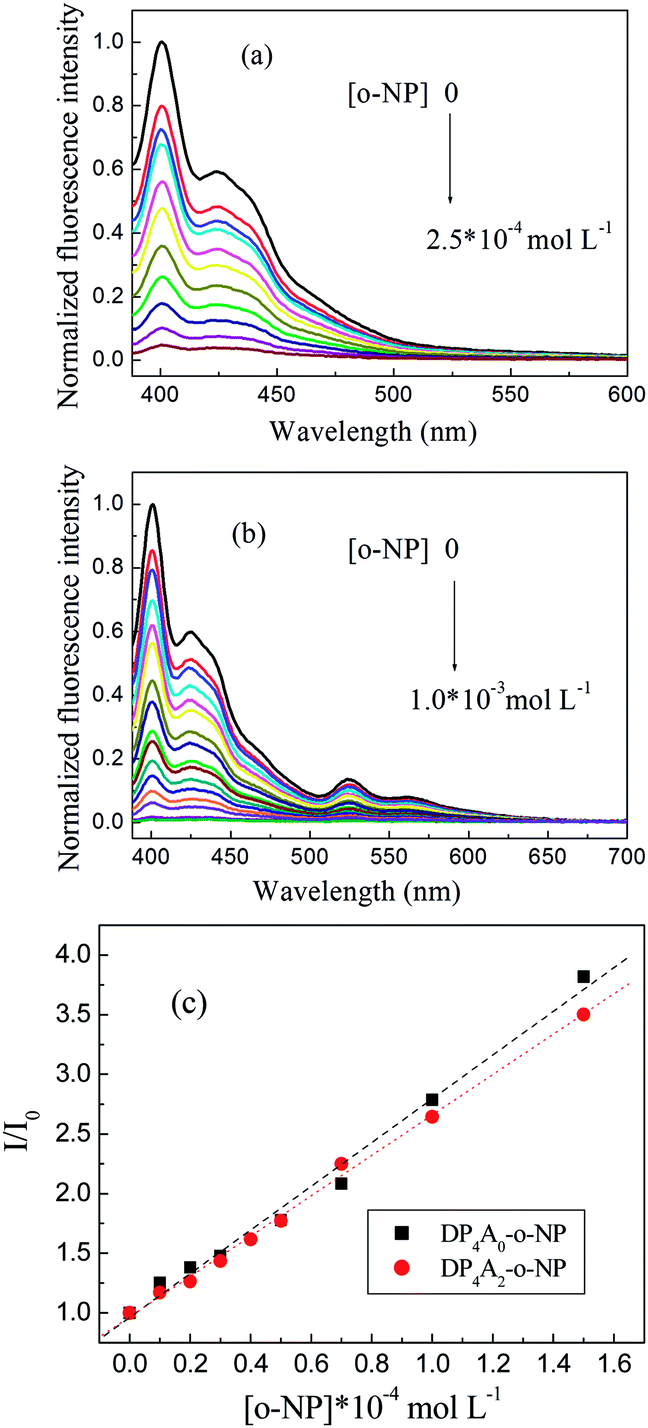 | ||
| Fig. 7 The changes in fluorescence spectra of (a) DP4A0 and (b) DP4A2 dispersions in THF and DOX upon the addition of o-NP; (c) S–V plots of DP4A0 and DP4A2 with various concentrations of o-NP (1.0 mg mL−1, excited at 370 and 365 nm). | ||
 | ||
| Fig. 8 The SEM images of (a) DP4A0 (top) and (b) DP4A2 (bottom) in the absence and presence of o-NP at various concentrations. | ||
Again it can be seen from Tables 2 and S1† that the sensitivity to o-NP is ordered DP2A2 > DP4A0 > DP4A2. There are similar trends for the sensitivity to PA and DNT.14 We speculated that the former is caused by the different positions of the crosslinking points, and the latter is caused by the excessive crosslinking density, which limits the conjugation effects of DP4A2. Since the crosslinking density of DP4A2 (6) is greater than that of DP2A2 and DP4A0 (4), the degree of conjugation in the DP4A2 network is lower than that of DP2A2 and DP4A0, which is not conducive to the transfer of electrons. Therefore, although the specific surface area, pore volume, and micropore volume of DP4A2 are larger than that of DP2A2 and DP4A0, the sensitivity of DP4A2 to o-NP is lower than that of DP2A2 and DP4A0. The crosslinking density of DP4A0 and DP2A2 is the same but the positions of the crosslinking points are different. Because the position of crosslinking points in DP4A0 is closer than that of DP2A2, the conjugate property of DP4A0 is lower than that of DP2A2. Moreover, the specific surface area and micropore properties of DP4A0 are much lower than that of DP2A2 and the chance of contact between the active sites of DP4A0 and o-NP is less than that of DP2A2, thus the sensitivity of DP4A0 to o-NP is lower than that of DP2A2.
| CMPs | The equation | Regression coefficient (R) | Concentration range of NACs (mol L−1) | Limits of detection (mol L−1) |
|---|---|---|---|---|
| DP4A0 | I0/I = 0.962 + 1.83 × 104 [o-NP] | 0.9948 | 0 to 1.5 × 10−5 | 5.73 × 10−9 |
| DP4A2 | I0/I = 0.966 + 1.69 × 104 [o-NP] | 0.9983 | 0 to 1.5 × 10−4 | 7.36 × 10−9 |
| DP2A2 | I0/I = 1.001 + 1.998 × 104 [o-NP] | 0.9924 | 0 to 3.5 × 10−5 | 1.50 × 10−9 |
To investigate the cross-effects of other NACs, we also conducted competitive experiments to further explore the selectivity of DP4A0 and DP4A2 to o-NP. The I0/I values of DP4A0 and DP4A2 in different NACs mixtures with and without o-NP are recorded in Fig. 9. After adding other NACs (e.g. NB, m-DNB, p-DNB, p-NT, DNT, and phenol), the I0/I values for DP4A0 or DP4A2 did not change significantly, with the exception of the PA experiment. When PA was added to a solution of DP4A0 or DP4A2 containing o-NP, the I0/I value increased significantly. There are signs that DP4A0 and DP4A2 have unusually high selectivity to o-NP.4,5
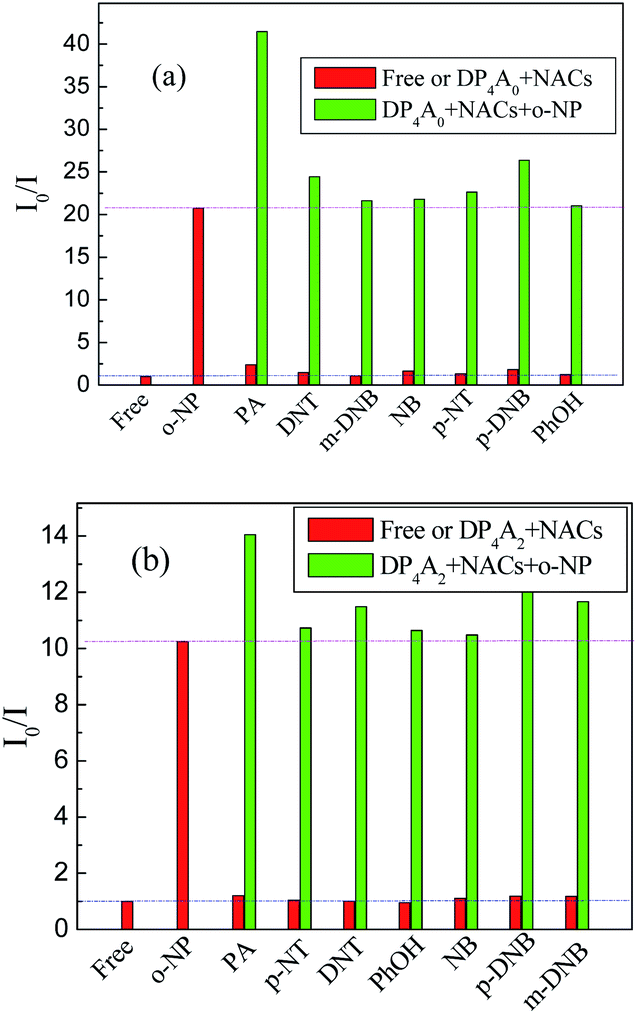 | ||
| Fig. 9 The selectivity and competitiveness of DP4A0 and DP4A2 (1.0 mg mL−1) for sensing NACs at the same concentration of 2.5 × 10−4 mol L−1 of o-NP in THF and DOX (λex = 370 and 365 nm). | ||
At higher concentrations of NACs the shapes of the curves are due to a superamplified quenching effect.42 As shown in Fig. S5,† the S–V plots exhibited hyperbolic curves, which suggests that they are both static and dynamic quenching phenomena occurring at the same time in the detecting process.4,39,43
Fluorescence resonance energy transfer (FRET) and photoinduced electron transfer (PET) mechanisms were explored to understand the sensing performance of DP4A0 and DP4A2. The absorption spectra of the NACs have almost no overlap with the emission spectra of DP4A0 (besides p-NT, m-DNB) and DP4A2 (except for o-NP, PA), indicating that the fluorescence quenching mainly resulted from PET process between electron-rich DP4A0 or DP4A2 and the electron deficient NACs (Fig. S7†).34,44–47 In order to assess the feasibility of PET from DP4A2 and DP4A2 to NACs, the lowest-unoccupied molecular orbital (LUMOs) energy levels and the highest occupied molecular orbital (HOMO) energy levels of DP4A0 and DP4A2 were calculated.34 As shown in Fig. S8 and Table S2,† the LUMO levels of DP4A0 and DP4A2 are higher than that of the NACs, which allows for electron transfer from DP4A0 and DP4A2 to the NACs.4 For example, the LUMO energy levels of PA and p-DNB are lower than that of DP4A0 and DP4A2.34,48 However, the LUMO levels are not the only factor affecting the PET process, it is also necessary to take into account the HOMO energy level of the NACs if it is lower than that of the CMPs. Hence, although the LUMO energies of DP4A0 and DP4A2 are lower than that of NB, DNT, p-NT, m-DNB, o-NP, and PhOH, a PET process can occur as well.34,49 Generally speaking, nitro groups play an important part in fluorescence quenching because their contribution to the LUMO energies is achieved by utilizing their electron withdrawing ability. Therefore, with increasing numbers of nitro groups, lower LUMO energy levels are obtained, thus improving electron transfer efficiency.6,39 The interaction of the analytes with DP4A0 and DP4A2, as well as efficient PET are considered the key parameters in the quenching mechanism.
However, the fluorescence quenching phenomena cannot exclude the possibility of a FRET mechanism. O–H⋯π and OH⋯N interactions may also impact the sensitivity of DP4A0 and DP4A2 for detecting o-NP.39 The NAC-containing hydroxyl groups (PA, NP, and PhOH) can effectively cause fluorescence quenching. This may be due to weak interactions such as hydrogen bonding of hydroxyl groups with nitrogen, oxygen, and π-electrons in the framework.39,50,51 The results indicated that there are three important factors for the detection of NACs, i.e., nitro groups, hydrogen bonding, and N, O⋯π interactions (Fig. S9†).6 High quenching constants and quenching rates, together with extremely low LOD values, make DP4A0 and DP4A2 highly sensitive chemical sensors for detecting NACs, particularly o-NP.39
In order to study the fluorescence stability of DP4A0 and DP4A2, annealing experiments were carried out. Solid powders of DP4A0 and DP4A2 were heated to 50 °C for half an hour in the atmosphere. Then, the temperature was increased to 100 °C where it was maintained for another half an hour. The temperature was then increased to 150 °C and 200 °C. After cooling, the samples were scattered in DOX or THF at a polymer concentration of 1.0 mg mL−1. Fig. S10† shows the fluorescence spectra of DP4A0 and DP4A2 after annealing in the atmosphere. As can be seen that they were still quite stable after heating to 100 °C for half an hour, and the fluorescence spectra were nearly the same as before heating. As the temperature increased, the fluorescence peak intensities decreased but there was no red-shift observed. Below 100 °C, fluorescence emission is stable. This result is due to the chemical structures of DP4A0 and DP4A2 and their stable perylene units.14,52,53
Conclusions
In summary, we have succeeded in developing solvent dispersible, fluorescent conjugated microporous polymers by taking advantage of Sonogashira–Hagihara cross-coupling reactions. We employed these systems as chemosensors for sensing nitroaromatic compounds. The two CMPs exhibit high BET specific surface areas and total pore volumes. They emitted strong purple and yellow fluorescence under UV light. It was found that DP4A0 dispersion in THF and DP4A2 dispersion in DOX can be effectively utilized to detect o-NP by fluorescence quenching in real-time. Specifically, DP4A0 and DP4A2 exhibited high selectivity to o-NP over other NACs. The relationship between the fluorescence intensity and o-NP concentration can be quantified. The detection limits of o-NP using DP4A0 and DP4A2 as probes were estimated to be 5.73 × 10−9 and 7.36 × 10−9 mol L−1. The possible fluorescence sensing mechanism of both CMPs was deemed to be an electron transfer as well as hydrogen bonding and N, O⋯π interactions. The sensing performance of DP4A0 and DP4A2 was dependent on the position of the crosslinking points and crosslinking density.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of the Anhui Education Department (under Grant No. KJ2018A0319).Notes and references
- Y. Salinas, R. Martınez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- V. S. Mothika, A. Räupke, K. O. Brinkmann, T. Riedl, G. Brunklaus and U. Scherf, ACS Appl. Nano Mater., 2018, 11, 6483–6492 CrossRef.
- N. Jiang, G. Li, W. Che, D. Zhu, Z. Su and M. R. Bryce, J. Mater. Chem. C, 2018, 41, 11162–11169 Search PubMed.
- M. Wang, H. Zhang, L. Guo and D. Cao, Sens. Actuators, B, 2018, 274, 102–109 CrossRef CAS.
- R. Sun, X. Huo, H. Lu, S. Feng, D. Wang and H. Liu, Sens. Actuators, B, 2018, 265, 476–487 CrossRef CAS.
- K. M. Roscioli, E. Davis, W. F. Siems, A. Mariano, W. Su, S. K. Guharay and H. H. Hill, Anal. Chem., 2011, 83, 5965–5971 CrossRef CAS PubMed.
- H. Ma, F. Li, P. Li, H. Wang, M. Zhang, G. Zhang, M. Baumgarten and K. Müllen, Adv. Funct. Mater., 2016, 26, 2025–2031 CrossRef CAS.
- C. Gu, N. Huang, Y. Wu, H. Xu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 11540–115404 CrossRef CAS PubMed.
- M. Martin, M. Crain, K. Walsh, R. A. Mcgill, E. Houser, J. Stepnowski, S. Stepnowski, H. D. Wu and S. Ross, Sens. Actuators, B, 2007, 126, 447–454 CrossRef CAS.
- M. Rahimi-Nasrabadi, M. M. Zahedi, S. M. Pourmortazavi, R. Heydari, H. Rai, J. Jazayeri and A. Javidan, Microchim. Acta, 2012, 177, 145–152 CrossRef CAS.
- J. M. Sylvia, J. A. Janni, J. D. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834–5840 CrossRef CAS PubMed.
- Y. K. Li, S. M. Bi, F. Liu, S. Y. Wu, J. Hu, L. M. Wang, H. L. Liu and Y. Hu, J. Mater. Chem. C, 2015, 3, 6876–6881 RSC.
- T. M. Geng, D. K. Li, Z. M. Zhu, W. Y. Zhang, S. N. Ye, H. Zhu and Z. Q. Wang, Anal. Chim. Acta, 2018, 1011, 77–85 CrossRef CAS PubMed.
- J. S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- W. Huang, M. Bender, K. Seehafer, I. Wacker, R. R. Schröder and U. H. F. A. Bunz, Macromolecules, 2018, 51, 1345–1350 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J. X. Jiang, F. B. Su, A. F. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- B. Bonillo, R. S. Sprick and A. I. Cooper, Chem. Mater., 2016, 10, 3469–3480 CrossRef.
- X. Li, Z. Li and Y. W. Yang, Adv. Mater., 2018, 30, 1800177 CrossRef PubMed.
- L. Sun, Y. Zou, Z. Liang, J. Yu and R. Xu, Polym. Chem., 2014, 5, 471–478 RSC.
- S. Wang, Y. Liu, Y. Yu, J. Du, Y. Cui, X. Song and Z. Liang, New J. Chem., 2018, 42, 9482–9487 RSC.
- W. Donga, Z. Ma, Q. Duan and T. Fei, Dyes Pigm., 2018, 159, 128–134 CrossRef.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267–14273 CrossRef CAS PubMed.
- P. Wang, H. L. Zhang and L. Zhang, Chem. Ind. Eng. Prog., 2000, 3, 460–463 Search PubMed.
- S. J. Ren, R. Dawson, D. J. Adams and A. I. Cooper, Polym. Chem., 2013, 4, 5585–5590 RSC.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970–980 CrossRef CAS.
- Y. Liao, J. Weber and C. F. J. Fau, Macromolecules, 2015, 48, 2064–2073 CrossRef CAS.
- K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. Georg, Chem. Mater., 2012, 24, 969–971 CrossRef CAS.
- S. Wu, S. Gu, A. Zhang, G. Yu, Z. Wang, J. Jian and C. Pan, J. Mater. Chem. A, 2015, 3, 878–885 RSC.
- L. Chen, Y. Honsho, S. Seki and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- X. Y. Wang, C. Zhang, Y. Zhao, S. J. Ren and J. X. Jiang, Macromol. Rapid Commun., 2016, 37, 323–329 CrossRef CAS PubMed.
- S. Bhunia, N. Dey, A. Pradhana and S. Bhattacharya, Chem. Commun., 2018, 54, 7495–7498 RSC.
- F. Wei, X. Cai, J. Nie, F. Wang, C. Lu, G. Yang, Z. Chen, C. Ma and Y. Zhang, Polym. Chem., 2018, 27, 3832–3839 RSC.
- A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., 2018, 57, 8579–8583 CrossRef.
- Y. Yang, L. Feng, J. Ren, Y. Liu, S. Jin, L. Su, C. Wood and B. Tan, Macromol. Rapid Commun., 2018, 1, 1800441 CrossRef PubMed.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed.
- M. W. Zhu, S. Q. Xu, X. Z. Wang, Y. Chen, L. Dai and X. Zhao, Chem. Commun., 2018, 54, 2308–2311 RSC.
- K. Saumya and S. C. Veettil, J. Photochem. Photobiol., A, 2019, 371, 414–422 CrossRef.
- N. Niamsa, C. Kaewtong, W. Srinonmuang, B. Wanno, B. Pulpokab and T. Tuntulani, Polym. Chem., 2013, 4, 3039–3046 RSC.
- Y. D. Hang, J. Wang, T. Jiang, N. N. Lu and J. L. Hua, Anal. Chem., 2016, 88, 1696–1703 CrossRef CAS PubMed.
- J. Liu, Y. Zhong, P. Lu, Y. Hong, J. W. Y. Lam, M. Faisal, Y. Yu, K. S. Won and B. Z. Tang, Polym. Chem., 2010, 1, 426–429 RSC.
- X. G. Hou, Y. Wu, H. T. Cao, H. Z. Sun, H. B. Li, G. G. Shan and Z. M. Su, Chem. Commun., 2014, 50, 6031–6034 RSC.
- N. Sang, C. X. Zhan and D. P. Cao, J. Mater. Chem. A, 2015, 3, 92–96 RSC.
- L. Guo, M. Wang, X. Zeng and D. Cao, Mater. Chem. Front., 2017, 1, 2643–2650 RSC.
- T. M. Geng, S. N. Ye, Y. Wang, H. Zhu, X. Wang and X. Liu, Talanta, 2017, 165, 282–288 CrossRef CAS PubMed.
- S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
- X. J. Qi, Y. H. Jin, N. Li, Z. Wang, K. C. Wang and Q. H. Zhang, Chem. Commun., 2017, 53, 10318–10321 RSC.
- C. L. Zhang, S. M. Zhang, Y. H. Yan, F. Xia, A. N. Huang and Y. H. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- G. He, H. Peng, T. Liu, M. Yang, Y. Zhang and Y. Fang, J. Mater. Chem., 2009, 19, 7347–7453 RSC.
- W. B. Wu, S. H. Ye, G. Yu, Y. Q. Liu, J. G. Qin and Z. Li, Macromol. Rapid Commun., 2012, 33, 164–171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of DP4A0, DP4A2, solid UV-vis spectra, XRD, SEM, fluorescence spectra, Stern–Volmer plots, HOMO and LUMO values. See DOI: 10.1039/c9ra10384h |
| This journal is © The Royal Society of Chemistry 2020 |