
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic transitions and ESIPT kinetics of the thienyl-3-hydroxychromone nucleobase surrogate in DNA duplexes: a DFT/MD-TDDFT study†
Alain Sougnabéa,
Daniel Lissouckab,
Fabien Fontaine-Vivec,
Mama Nsangoud,
Yves Mély e,
Alain Burgerc and
Cyril A. Kenfack*a
e,
Alain Burgerc and
Cyril A. Kenfack*a
aLaboratoire d'Optique et Applications, Centre de Physique Atomique Moléculaire et Optique Quantique, Faculté des Sciences Université de Douala, B. P. 8580 Douala, Cameroon. E-mail: ckenf@yahoo.com
bDepartment of Renewable Energy, Higher Technical teachers' Training College, University of Buea, P. O. Box 249, Buea Road, Kumba, Cameroon
cInstitut de Chimie de Nice, UMR 7272, Université Côte d'Azur, CNRS, Parc Valrose, 06108 Nice Cedex 2, France
dDépartement de Physique, Faculté des Sciences Physiques, Ecole Normale Supérieure de Maroua, Université de Maroua, Cameroon
eLaboratoire de Bioimagerie et Pathologies, UMR 7021 du CNRS, Faculté de Pharmacie Faculté de Pharmacie, Université de Strasbourg, 74, Route du Rhin, 67401, Illkirch Cedex, France
First published on 20th February 2020
Abstract
The fluorescent nucleobase surrogate M (2-thienyl-3-hydroxychromone fluorophore) when imbedded in DNA opposite an abasic site exhibits a two colour response highly sensitive to environment changes and base composition. Its two colour emission originates from an excited state intramolecular proton transfer (ESIPT), which converts the excited normal N* form into its T* tautomer. To get deeper insight on the spectroscopic properties of M in DNA duplexes, quantum chemical calculations were performed on M stacked with different base pairs in model trimers extracted from MD simulations. The photophysics of M in duplexes appeared to be governed by stacking interactions as well as charge and hole transfer. Indeed, stacking of M in DNA screens M from H-bonding with water molecules, which favours ESIPT and thus, the emission of the T* form. With A and T flanking bases, the electronic densities in the frontier MOs were localized on M, in line with its effective absorption and emission. In addition, reduction of the free rotation between the thienyl and chromone groups together with the shielding of the dye from water molecules largely explain its enhanced quantum yield in comparison to the free M in solution. By contrast, the localisation of the electron density on the flanking G residues in the ground state and the energetically favorable hole transfer from M to G in the excited state explains the reduced quantum yield of M sandwiched between CG pairs. Finally, the much higher brightness of M as compared to 2-aminopurine when flanked by A and T residues could be related to the much stronger oscillator strength of its S0 → S1 transition and the ineffective charge transfer from M to A or T residues.
Introduction
Site-specific fluorescent DNA labels are of high demand in order to sense DNA hybridization, conformation changes and activities of DNA-binding proteins and enzymes.1–8 Valuable information can be obtained from these labels by analysing their fluorescence intensity, anisotropy, excited state lifetime and emission band position. Among the different types of fluorescent labels, the environment-sensitive ones that minimally perturb the structure and function of DNA are of upmost interest. As a typical example, 2-aminopurine (2AP) a popular adenine fluorescent analogue has been successfully used in a large range of applications. However, 2AP is limited by its extremely low quantum yield when incorporated in oligonucleotides (ODNs).9 This low quantum yield results from both static quenching due to electron delocalization in the ground state with weak oscillator strength and dynamic quenching through efficient non-radiative relaxation to a charge transfer state.10,11More recently, the 2-thienyl-3-hydroxychromone (3HC) dye (Fig. 1), referred to as M in this manuscript, has been introduced as a new nucleobase surrogate for DNA labelling, which could replace 2AP and other fluorescent nucleoside analogues.1,2,6–8,13,14 M is highly attractive because it is a rather flat molecule and its size corresponds well to the size of an AT or GC base pair. The excellent stacking properties of M with its neighboring base pairs as well as its preferential syn–anti conformation in DNAs were recently evidenced by NMR and MD simulations.15
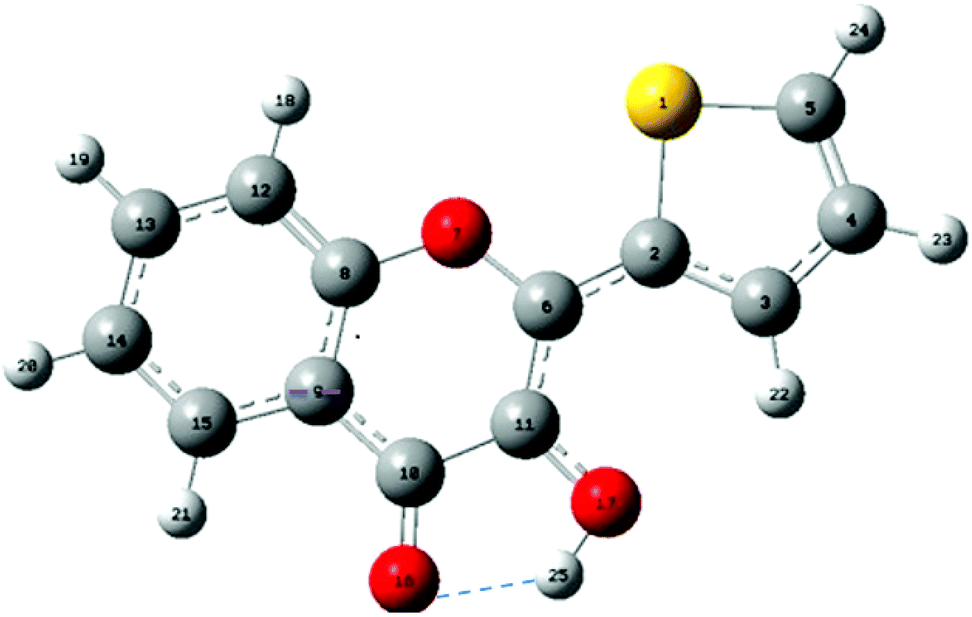 | ||
| Fig. 1 Structure and atom numbering of 2-thienyl-3HC (M). The intramolecular H-bond between O16 and H25 is drawn as a dashed line. | ||
The spectroscopic properties of M in solvents and oligonucleotides (ODN) have also been extensively studied.7,16–18 This dye undergoes an excited state intramolecular proton transfer (ESIPT) between the 3-hydroxyl and 4-carbonyl groups. As a result, M exhibits two fluorescent bands in the visible region that are sensitive to hydration and polarity; the short-wavelength band being attributed to the normal form (N*) and the long-wavelength band to the tautomer form (T*), the product of the ESIPT reaction.7,11
The quantum yield (QY) of M in labelled ODNs7 is remarkably enhanced as compared to that of the free dye M when sandwiched by AT pairs. This increase in QY is accompanied by a strong decrease in the N*/T* intensity ratio (IN*/IT*) and a red shift of the T* band. The behaviour of M was noticeably different when sandwiched between CG pairs, showing a lower QY and a higher N*/T* intensity ratio. Interestingly, the QY of this probe is about 2–25-fold larger than that of 2AP in corresponding ODN sequences. Since M absorbs two times more, it thus appears up to 50-fold brighter than 2AP in ODNs.11,18
In this work, the structural and spectroscopic properties of the ESIPT M dye as a free probe and included in M-containing trimers were investigated by combining quantum mechanics (QM) calculations and molecular dynamics (MD) simulations to understand how the electronic properties of M are influenced in DNA duplexes. A deeper insight in the photophysics of M in DNA is mandatory for interpreting protein/DNA interactions and designing new 3HC derivatives with optimized photophysical properties. MD and QM are powerful tools for unravelling the complex interplay between fluorescent nucleoside analogues or intercalating agents and DNA19–21 and clarifying the contribution of the surrounding nucleotides to the photophysics of the fluorescent reporter. The MD/QM approach was applied with success to single band emitters such as 2AP and 8-vinyl-deoxyadenosine;5,9,10 but to the best of our knowledge, application to a dual emissive fluorescent dye, such as the ESIPT dye M, is unprecedented. The most stable conformations of M in protic and aprotic media as well as in the ODN context were investigated. The absorption and emission wavelengths of M and the energy diagram of the different states that intervene in its photophysics were calculated by using the dispersion-corrected PCM-DFT and PCM-TDDFT. Calculations on M-containing trimers extracted from MD simulations frames were found to correlate well with previously reported experimental data of a series of 15-mer ODNs differing by the nature of the base pairs flanking the central M opposite an abasic site.
Computational details
In solution, the electronic properties of M were investigated by using a dispersion-corrected density functional theory22 at PBE0-D/TZVP level. The dispersion corrections are mandatory to correctly handle the electronic transitions with CT. The solvent effect was taken into account by the PCM solvation theory. The PBE0 functional was chosen because it gives a good performance in the calculations with H-bonding systems23,24 and the prediction of vertical transition energies.25,26 The TZVP basis set from the Ahlrichs group is well adapted for the investigation of nonbonding interactions and electronic properties of synthetic DNA nucleoside.27 The transition state of M was optimised by a procedure developed by Ayala and Schlegel.28 A similar procedure with different functionals and basis sets was also used by Yamazaki and Taketsugu.29 To account for the impact of H-bonding in water, a complex of M in interaction with explicit water molecules was considered. The most stable conformation of M was then retrieved and used for the TDDFT calculation of the electronic transitions with the same hybrid functional and basis set. For our calculations, different 3-HC forms were considered in both ground and excited states. For each state, we considered the normal (N) and tautomer (T) forms, as well as their corresponding hydrated species (NH and TH). Taking into account that the ESIPT reaction (>10 ps) is much slower than the solvent relaxation processes (<1 ps),30,31 we assumed that the ESIPT reaction occurs between solvent-relaxed excited states. All calculations were performed on g16.32In the DNA context, MD simulation was used to sample the possible conformations of the M labelled duplexes. Their photophysics was calculated by collecting an equilibrium MD simulation and then by post processing the resulting trajectory similarly to the work of Furse and Corcelli.33 To obtain the labelled duplexes, we started from regular B-form duplexes with the sequence d(CGT TTT XMX TTT TGC) where X = A, T or C are the flanking bases of M. The complementary sequence contains the central motif YAbY, where Y = T, A or G corresponds to the base complementary to X and Ab is the abasic site opposite M. The duplexes were built starting with an AT central base pair by using the NAB module of AMBER14 package.34 The central base A was manually replaced by the most stable ground state of M in water optimised at the D-PBE0/TZVP level. Finally, T opposite M was cut out, and only the sugar was left in order to obtain an abasic site and maintain the DNA double helix. The restrained electrostatic potential (RESP)35–37 was used to derive the partial charges on M in the ground state in order to reproduce its DFT electrostatic potential. This approach was proven to satisfactorily describe dynamic processes in DNA.33 The Na+ counter ions were placed around the ODNs to neutralise the negative charges of the phosphate backbone, and the whole system was solvated with a box of 10 Å TIP3 water molecules. The ff99SB force field was used for DNA natural bases, while the GAFF force field was used for M. Water molecules were energy minimized for 500 steps using the Steepest Descent algorithm (SD) and further 1500 steps using the Conjugate Gradient algorithm (CG), while keeping the solute as frozen. Then, the solvated solute was energy minimized for 1000 steps using the SD and 1500 steps using the CG before being heated from 0 to 300 K during 10 ps in the NVT (constant number of particles N, volume V and temperature T) ensemble, T is regulated via a Langevin thermostat. A density equilibration was carried out for 100 ps in the NPT (constant number of particles N, pressure P and temperature T) ensemble, before running the production of 100 ns MD trajectories in the ground state. Their structural parameters were obtained from web 3DNA program38 and the relative enthalpy change ΔH values were calculated by solving the Poisson–Boltzmann equation (PBE) and the General Born Model (GBM) by using an approach combining MM energies with continuum solvent approaches commonly referred to as (MM-PBSA) and (MM-GBSA) for the two methods, respectively.39,40 The wavelength positions of the N* and T* emissions were estimated by using the above mentioned functional and basis sets. The relative energy between these states was obtained from single point calculation on these structures.
Electronic transitions in duplexes generally occur between molecular orbitals (MOs) that are either localised on the fluorescent probe or delocalised on the flanking nucleobases.10,11 As a consequence, the trimer supermolecule approach41 was adopted to predict the photophysics of M in the duplexes. For each considered duplex, a trimer composed of the central motif XMX + YAbY was excised in order to calculate the electronic transitions of M in a DNA. The dispersion corrected functional PBE0-D was used at this purpose to properly describe the stacking interactions that occur when M is embedded in a DNA duplex. To characterize the excited-state transitions, a localized orbital picture was used in which transitions were described as linear combinations of localized (excitonic) and intermolecular (CT) transitions. In the present study, we have focused the calculations only on singlet transitions that occur at low energy (E < 3.3 eV; λ > 350 nm).
Results and discussion
Calculation of the geometry and photophysics of free M in acetonitrile and water
To rationalise the photophysics of M in DNA duplexes, we first investigated the electronic transitions and relative energies of its different states in the free (non-incorporated) dye in acetonitrile and water, taken as models of polar aprotic and protic media, respectively.Representative geometric parameters obtained in acetonitrile from the DFT calculations at PBE0-D/TZVP level for the most stable conformation of M in its S0 ground and relaxed SN1(R) and ST1(R) excited states, are given in Table 1. Though M adopts a planar conformation in both states, significant differences in the interatomic distances can be observed between the two states. The most prominent are the shortening of the C(2)–C(6) bond joining the thienyl ring to the chromone and the C(11)–O(17) bond of the hydroxyl group by 0.04 and 0.03 Å, respectively, as well as the lengthening of the C(6)–C(11) bond by 0.04 Å. Noticeably, the affected bonds are consecutive. This behaviour suggests an electron delocalisation from the thienyl ring and proton donor oxygen O17 to the proton acceptor O16, as already mentioned in other 3HC derivatives.42 This electron delocalisation is thought to render O17H more acidic (as supported by the Mulliken charge variation, Δq = +0.06 a.u) and O16 more basic (Δq = −0.06 a.u), thus favouring the ESIPT. Interestingly, the distance between the transferring proton and the carbonyl oxygen is 1.976 Å, thus supporting the formation of an intramolecular H-bond (Fig. 1) which is known to facilitate the proton transfer from the donor O(17) to the acceptor O(16). The excited T* form shows a remarkable shortening of the C(11)–O(17) bond by 0.075 Å, and a lengthening of the C(10)–O(16) bond by 0.092 Å, which are involved in the proton transfer process. The geometry modifications between N* and T* forms indicate that M undergoes structural rearrangement during the ESIPT process. PCM-TDDFT calculations with the PBE0-D functional on the TZVP optimised geometry further predict the S0 → S1 absorption peak (and oscillator strength) at 356 nm (0.6), the N* emission band at 417 nm, and the T* emission at 536 nm, respectively (Fig. 2). The predicted transitions are close to the previously reported experimental values.12
| Bonds | Bonds lengths | Angles | Bonds angles | ||||
|---|---|---|---|---|---|---|---|
| S0 | SN1(R) | ST1(R) | S0 | SN1(R) | ST1(R) | ||
| S(1)–C(2) | 1.740 | 1.768 | 1.764 | C(2)–S(1)–C(5) | 91 | 91 | 91 |
| S(1)–C(5) | 1.717 | 1.720 | 1.718 | S(1)–C(2)–C(3) | 111 | 113 | 110 |
| C(2)–C(3) | 1.377 | 1.410 | 1.402 | S(1)–C(2)–C(6) | 119 | 120 | 119 |
| C(2)–C(6) | 1.437 | 1.390 | 1.395 | C(3)–C(2)–C(6) | 127 | 130 | 129 |
| C(3)–C(4) | 1.411 | 1.390 | 1.396 | C(2)–C(3)–C(4) | 113 | 113 | 112 |
| C(4)–C(5) | 1.364 | 1.383 | 1.377 | C(3)–C(4)–C(5) | 112 | 113 | 113 |
| C(6)–O(7) | 1.350 | 1.360 | 1.367 | S(1)–C(5)–C(4) | 112 | 114 | 112 |
| C(6)–C(11) | 1.374 | 1.405 | 1.433 | C(2)–C(6)–O(7) | 112 | 115 | 114 |
| O(7)–C(8) | 1.350 | 1.376 | 1.365 | C(2)–C(6)–C(11) | 126 | 127 | 126 |
| C(8)–C(9) | 1.394 | 1.410 | 1.413 | O(7)–C(6)–C(11) | 121 | 118 | 119 |
| C(8)–C(12) | 1.392 | 1.376 | 1.377 | C(6)–O(7)–C(8) | 121 | 122 | 122 |
| C(9)–C(10) | 1.450 | 1.428 | 1.400 | O(7)–C(8)–C(9) | 121 | 122 | 121 |
| C(9)–C(15) | 1.404 | 1.407 | 1.412 | O(7)–C(8)–C(12) | 117 | 116 | 116 |
| C(10)–C(11) | 1.440 | 1.452 | 1.433 | C(9)–C(8)–C(12) | 122 | 122 | 121 |
| C(10)–O(16) | 1.248 | 1.258 | 1.330 | C(8)–C(9)–C(10) | 119 | 119 | 117 |
| C(11)–O(17) | 1.344 | 1.314 | 1.262 | C(8)–C(9)–C(15) | 119 | 118 | 118 |
| C(12)–C(13) | 1.380 | 1.405 | 1.396 | C(10)–C(9)–C(15) | 122 | 123 | 124 |
| C(13)–C(14) | 1.405 | 1.390 | 1.393 | C(9)–C(10)–C(11) | 125 | 116 | 121 |
| C(14)–C(15) | 1.375 | 1.384 | 1.381 | C(9)–C(10)–O(16) | 116 | 128 | 122 |
| C(11)–C(10)–O(16) | 121 | 116 | 116 | ||||
| C(6)–C(11)–C(10) | 121 | 123 | 118 | ||||
| C(6)–C(11)–O(17) | 119 | 123 | 124 | ||||
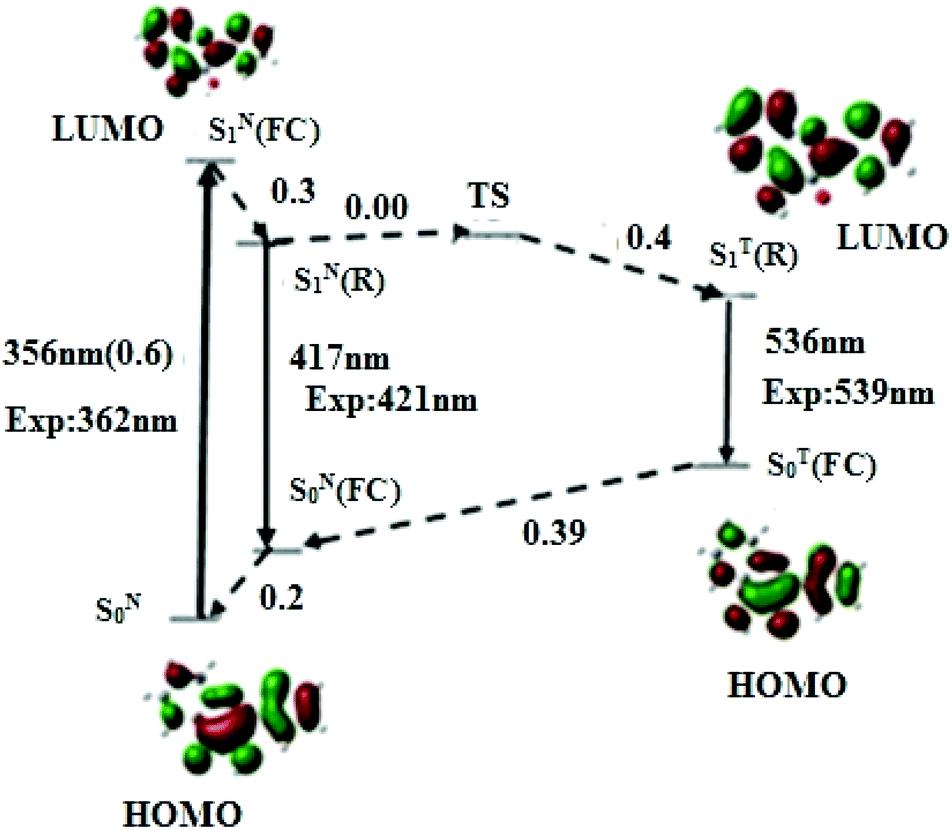 | ||
| Fig. 2 Energy levels and frontier molecular orbitals involved in the photophysics of M in acetonitrile, generated by PBE0-D/TZVP calculations. Upward arrow: absorption; downward arrow: emission; dashed arrow: non radiative relaxation. FC denotes the Franck–Condon state and R the relaxed excited states. The subscripts N and T denote the normal and tautomer forms, respectively. The radiative transitions energies are expressed in nm and non radiative transitions in eV. | ||
The S0 → S1 excitation corresponds to the transition of one electron from HOMO (Highest Occupied Molecular Orbital) to LUMO (Lowest Occupied Molecular Orbital). In accordance with the above mentioned charge delocalisation, this transition is assigned to a ππ* character, featuring an appreciable charge displacement from the thienyl group to the rest of the molecule, as the Mulliken charge of this group obtained by PBE0-D/TZVP calculation varies from −0.49 a.u. in the ground state to +1.42 a.u. in the S1 state. This assignment was further confirmed with M06-2X/TZVP calculation, taken as control method. Concomitantly, the magnitude of the molecular dipole moment varies from 3.94 D in the ground state to 7 D in the S1 excited state. A charge displacement from an aryl group is common for ESIPT dyes,42,43 and is responsible of the strongly increased dipole moment of N* as compared to N.44
To understand the mechanism that governs the ESIPT reaction in acetonitrile, we have calculated the energies of the transition state (TS) and all the other states that likely intervene in the photophysics of M (Fig. 2). From the absorption and fluorescence energies given by the PBE0-D/TZVP calculation, and the 0.2 eV energy gap between SN0 and SN0(FC) obtained from a single point energy calculation, the energy gap between SN1(FC) and SN1(R) was estimated to be 0.30 eV. Moreover, the energy gap between SN1(R) and TS states, corresponding to the activation barrier to the ESIPT reaction, was estimated to be 0.00 eV. By using the TDDFT data on the T* form, an energy difference of 0.4 eV between SN1(R) and ST1(R) was obtained, indicating that ST1(R) is energetically favourable. Thus, the T* state is expected to be predominantly populated, in line with the experimental dominant contribution of the T* band to the emission spectrum (IN*/IT* = 0.13).11 Taken together, our data suggest a very fast ESIPT reaction, facilitated by the absence of activation barrier, the relative low energy of the ST1(R) state with respect to SN1(R), and the pre-existing intramolecular H-bond.
The ESIPT reaction is an environment sensitive process, which is influenced by specific solute–solvent interactions like H bonding.45,46 To examine how H-bonds with solvent affect the ESIPT reaction, we considered M in water. Only the first shell of solvent molecules was taken into account. The number of water molecules H-bonded to M was estimated from 100 ns of MD simulations. In this respect, the pair distribution function g(r) between the M carbonyl oxygen (Oa) and water oxygen (O) was plotted for the ground state (Fig. S1A†). g(r) measures the probability of finding O at a distance r from Oa, relative to that for an ideal gas. The g(r) curve presents a profile similar to that obtained experimentally for the O–O radial distribution of water oxygen atoms47 (Fig. S1†), featuring strong peaks around 3 and 5 Å, attributed to the first and second solvent shells. The number n of water molecules in the vicinity of M in the first solvent shell was obtained from the area under the first peak of g(r) at 3.45 Å.47 A value of 3.62 was obtained for n at the first minimum, indicating that about 3 water molecules are in the first solvent shell.
Consequently, a superstructure of M in complex with three water molecules was considered and optimised to obtain the most stable conformation of NH, the hydrated N form in the ground state, by using the DFT at PBE0-D/TZVP level. The geometry of NH retrieved from these calculations was further used as a starting structure in TDDFT calculations to obtain the geometry of the excited-state N*H and T*H complexes (Fig. 3B and D). Comparison of NH and N*H complexes (Fig. 3A and B) reveals that the conversion from NH to N*H is accompanied by a shortening of the intermolecular H-bond. Moreover, the transition state (Fig. 3C) is characterised by an appreciable lengthening of the distance between the transferring proton and the closest water molecule that increases from 1.50 to 2.493 Å. This result is consistent with the weakening or disruption of the intermolecular H-bond prior to the ESIPT reaction.30,31
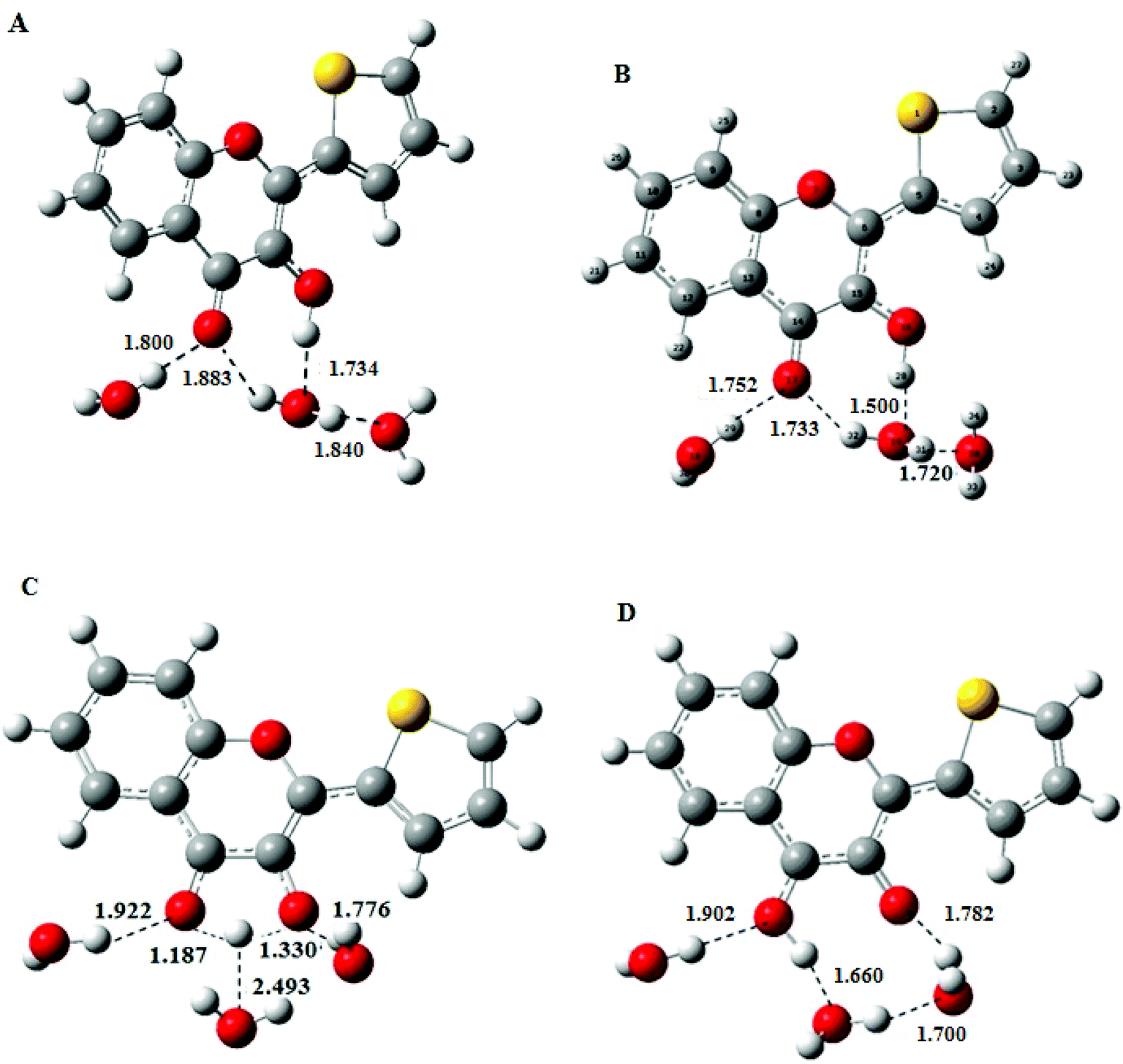 | ||
| Fig. 3 Structure of the M-water complexes for (A) the normal NH form, (B) the relaxed N*H form, (C) the transition state and (D) the tautomer T*H form. The possible H-bonds and the interatomic distances (in Å) obtained from PCM-DFT PBE0-D/TZVP calculations are shown. | ||
From the optimised geometries of NH and N*H and T*H complexes, the electronic transitions of M in water were calculated. As in acetonitrile, the PBE0-D functional along with the TZVP atomic basis set (Fig. 4) positions the S0 → S1 absorption maximum of NH in water at 368 nm (f = 0.7), and the N*H and T*H emission at 441 and 519 nm, close to the experimental values.12 Our calculations further predict that the S0 → S1 transition arises from an electron promotion between HOMO and LUMO. Moreover, a comparison of M with the popular 2AP shows that the oscillator strength of S0 → S1 transition in M (0.7) is about five times that of 2AP (0.127), thus explaining the superior absorptivity of M in solution.11
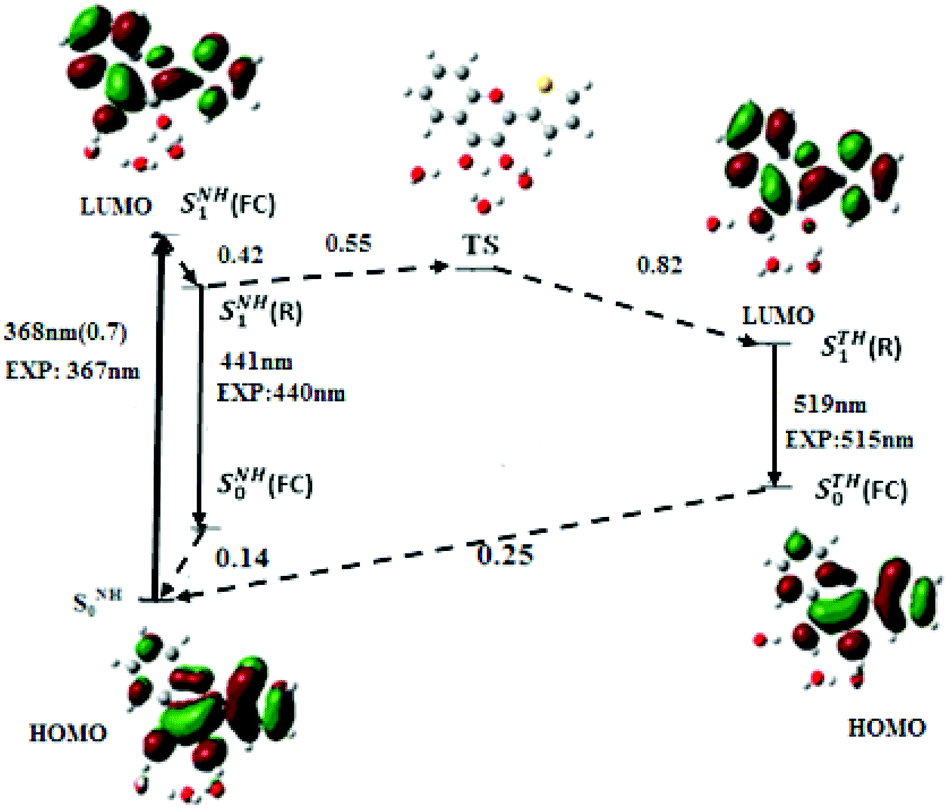 | ||
| Fig. 4 Energy levels and frontier molecular orbitals involved in the photophysics of M in water, generated by PBE0-D/TZVP calculations. Upward arrow: absorption; downward arrow: emission; dashed arrow: non radiative transition. (FC) and (R) denote the Franck–Condon and relaxed excited states, respectively. The superscripts NH and TH denote respectively the hydrated normal and tautomer forms. The radiative transitions energies are expressed in nm and non radiative transitions in eV. | ||
The solvatochromism observed on going from acetonitrile to water suggests that H-bonds and the dielectric constant play a major role in M photophysics. To distinguish the general solvent effects from specific solute–solvent effects, the electronic transitions of M were also calculated in bulk water. The positions of the lowest energy absorption band, and of the N* and T* emission bands obtained by PBE0-D/TZVP method are at 354, 424 and 540 nm, respectively. This shows that, the dielectric constant alone has a negligible effect on the absorption position maximum but dramatically affects the quality of the predictions for the positions of the emission bands as evidenced by the large shifts with respect to the experimental data (16 and 25 nm for the N* and T* bands, respectively). These results highlight the key importance of specific intermolecular H-bonding with water molecules to account for the photophysics of M in aqueous media, a behaviour already observed with other 3HC derivatives.42
To get insight into the kinetics of the ESIPT reaction in water, the conformation and energy of the transition state (TS) were calculated. The energy of the TS state was found 0.55 eV above the SNH1(R) state, so that the activation barrier to the ESIPT reaction is very high. Nevertheless, ESIPT is thermodynamically favoured, as the emissive state of STH1(R) is 0.27 eV below SNH1(R). From the energy diagram of M in water (Fig. 4), it may be inferred that after excitation from the SNH0 ground state, the SNH1(FC) relaxes to SNH1(R) and returns to the ground state by fluorescence emission or non-radiative process. However, a small population of M likely crosses the activation barrier and undergoes ESIPT to reach the STH1(R) state, explaining the observation of the low energy emission band and the high value (1.72) of the experimental IN*/IT* ratio.7
Geometries of M-labelled DNA duplexes with an abasic site
To characterize M inserted in ODN duplexes, 100 ns of MD simulations were performed in the ground state. The nucleoside analogue, M connected to 2′-deoxyribose was analysed and geometrically optimized with the DFT. Starting from the main conformation of M in the duplex,15 the three other possible conformations differing by the torsion angles of the glycosidic bond and the thienyl group with respect to the chromone moiety were built on. The calculated energies of the four conformations (Fig. S2†) show that the syn–anti conformation is the most stable. Noticeably, this conformation was similar to the one adopted by M in the resolved NMR structure.15 To monitor the geometry convergence, the root-mean-square deviation (RMSD) of the duplexes with respect to the average structure along the MD trajectories was followed. In Fig. S3,† the RMSD of duplexes bearing M in its syn–anti conformation indicates that their structures were stable during the simulation, with a mean RMSD of 6, 5.5 and 5.2 Å respectively for AMA + TAbT, TMT + AAbA and CMC + GAbG duplexes. The average structures along the 100 ns MD simulations of these duplexes are presented in Fig. S4.† The obtained structures and structural parameters are very close to those of the normal B-form DNA (Tables S3 and S4†),48 explaining the limited decrease of the thermodynamic stability of the M labelled duplexes. Finally, the radial distribution function g(r) (Fig. S5†) showed no significant peak, indicating that M is screened from water molecules in the ground state.Next, the photophysics of M in each of the three duplexes was investigated using ten structures extracted each 10 ns of the 100 ns simulation. To calculate the behaviour of M in the excited state, we replaced the ground-state structure of M in each of the selected MD frame by the SN1(R), TS and ST1(R) structures obtained from DFT/TDDFT geometry optimisation of the free probe in water. Then, the XMX + YAbY trimers were excised from the duplexes obtained by MD calculations.
Electronic properties and transitions in DNA duplexes containing M and an abasic site
The average absorption electronic transitions wavelength calculated on the trimers extracted from MD simulations are reported in Table 2.| Sequence | Abs. (nm) | f | Transitions | Sequence | Abs. (nm) | f | Transitions | Sequence | Abs. (nm) | f | Transition |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AMA + TAbT-1 | 366 | 0.37 | H → L | TMT + AAbA-1 | 366 | 0.33 | H → L | CMC + GAbG-1 | 409 | 0.016 | H → L |
| 395 | 0.012 | H−1 → L | |||||||||
| 376 | 0.34 | H−2 → L | |||||||||
| AMA + TAbT-2 | 371 | 0.35 | H → L | TMT + AAbA-2 | 369 | 0.36 | H → L | CMC + GAbG-2 | 404 | 0.012 | H → L |
| 394 | 0.016 | H−1 → L | |||||||||
| 370 | 0.33 | H−2 → L | |||||||||
| AMA + TAbT-3 | 367 | 0.38 | H → L | TMT + AAbA-3 | 374 | 0.35 | H → L | CMC + GAbG-3 | 419 | 0.004 | H → L |
| 389 | 0.04 | H−1 → L | |||||||||
| 368 | 0.39 | H−2 → L | |||||||||
| AMA + TAbT-4 | 383 | 0.31 | H → L | TMT + AAbA-4 | 383 | 0.41 | H → L | CMC + GAbG-4 | 407 | 0.007 | H → L |
| 388 | 0.022 | H−2 → L | |||||||||
| 367 | 0.37 | H−1 → L | |||||||||
| AMA + TAbT-5 | 386 | 0.27 | H → L | TMT + AAbA-5 | 371 | 0.37 | H → L | CMC + GAbG-5 | 412 | 0.03 | H → L |
| 396 | 0.054 | H−1 → L | |||||||||
| 356 | 0.33 | H−2 → L | |||||||||
| AMA + TAbT-6 | 381 | 0.34 | H → L | TMT + AAbA-6 | 377 | 0.33 | H → L | CMC + GAbG-6 | 421 | 0.04 | H → L |
| 413 | 0.01 | H−1 → L | |||||||||
| 357 | 0.35 | H−2 → L | |||||||||
| AMA + TAbT-7 | 373 | 0.36 | H → L | TMT + AAbA-7 | 387 | 0.24 | H → L | CMC + GAbG-7 | 417 | 0.006 | H → L |
| 390 | 0.004 | H−1 → L | |||||||||
| 367 | 0.40 | H−2 → L | |||||||||
| AMA + TAbT-8 | 370 | 0.40 | H → L | TMT + AAbA-8 | 382 | 0.3 | H → L | CMC + GAbG-8 | 402 | 0.024 | H → L |
| 383 | 0.01 | H−1 → L | |||||||||
| 346 | 0.14 | H−2 → L | |||||||||
| AMA + TAbT-9 | 375 | 0.37 | H → L | TMT + AAbA-9 | 382 | 0.40 | H → L | CMC + GAbG-9 | 407 | 0.024 | H → L |
| 400 | 0.014 | H−1 → L | |||||||||
| 358 | 0.33 | H−2 → L | |||||||||
| AMA + TAbT-10 | 373 | 0.32 | H → L | TMT + AAbA-10 | 370 | 0.25 | H → L | CMC + GAbG-10 | 405 | 0.01 | H → L |
| 390 | 0.0006 | H−1 → L | |||||||||
| 363 | 0.370 | H−2 → L | |||||||||
| Average value | 375 | 0.35 | H → L | 376 | 0.31 | H → L | 410 | 0.018 | H → L | ||
| 392 | 0.018 | H−1 → L | |||||||||
| 363 | 0.33 | H−2 → L |
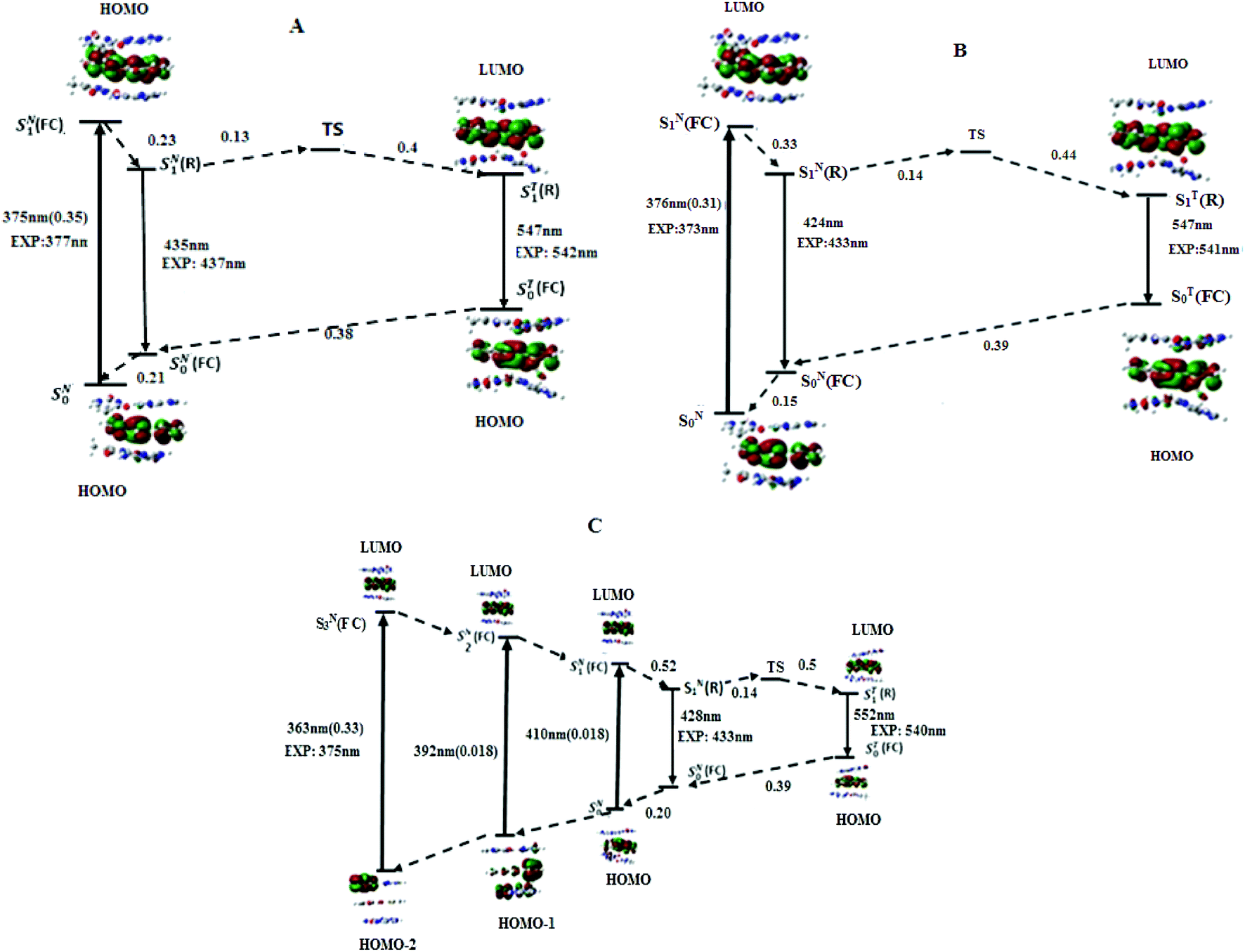 | ||
| Fig. 5 Energy diagrams and frontier molecular orbitals involved in the photophysics of M in (A) AMA + TAbT, (B) TMT + AAbT and (C) CMC + GAbG trimers, as generated by PBE0-D/TZVP calculations. Upward arrow: absorption; downward arrow: emission; dashed arrow: non-radiative transition. (FC) and (R) denote the Franck–Condon and relaxed excited states, respectively. The superscripts N and T denote respectively the normal and tautomer forms. The radiative transitions energies are expressed in nm and non radiative transitions in eV. | ||
According to the energy diagram (Fig. 5B), ST1(R) was 0.30 eV below SN1(R) and the transition state TS was 0.14 eV above SN1(R). As for the AMA + TAbT trimer, the ESIPT reaction is thus expected to be fast and lead to an efficient accumulation of T*, in line with the low experimental value (0.08) of the IN*/IT* ratio.
To get a deeper insight on the origin of the quenching of M when sandwiched by CG base pairs, its ionization potential IP (eV) and electronic affinity EA (eV) as well as the energies of HOMO and LUMO were calculated at the DFT/PBE0-D/TZVP level, using the adiabatic approach.51 2AP as well as A, C, G, T were used as references for comparison (Table 3). The G HOMO appears higher in energy (0.05 eV, 4.8 kJ mol−1) than the M and 2AP ones. As a result, the supermolecule HOMO is highly localized on the G.10 Moreover the IP of G (5.85 eV) is comparatively small as compared to M (5.93 eV), which may facilitate the hole transfer to this nucleobase during the excitation process. Another remarkable result is the very low energy level of the LUMO in M (−2.28 eV), as compared to 2AP (−1.1 eV) and other natural bases. This probably explains the localization of the LUMO on M in the CMC-GAbG trimer, and the photo-induced electron transfer (PET) from G to M in the excited state, further contributing to M quenching.
| Bases | IP (eV) | AE (eV) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|
| Adenine | 6.28 | −1.17 | −6.55 | −0.71 |
| Cytosine | 6.71 | −1.46 | −6.96 | −0.96 |
| Guanine | 5.85 | −0.93 | −6.23 | −0.35 |
| Thymine | 6.70 | −1.68 | −6.99 | −1.2 |
| M | 5.93 | −2.64 | −6.28 | −2.28 |
| 2AP | 6.05 | −1.52 | −6.28 | −1.1 |
Our calculations can also rationalize the observed differences in the spectroscopic properties of 2AP and M in the same ODNs. In the T2APT trimer, the oscillator strength of the S0 → S1 transition is only 0.012 (ref. 9) and thus, 40-fold less than in the TMT trimer. In addition, the close energy level of the LUMOs in 2AP (−1.1 eV), T (−1.2 eV) and A (−0.71 eV) likely favours CT from 2AP to T or A, whereas in M the gap between the LUMOs of M (−2.28 eV) and T (−1.2 eV) or A (−0.71 eV) is higher than 1 eV, thus precluding CT to T. Altogether the superior oscillator strength in absorption and the unfavourable CT mechanism are consistent with the superior brightness of M when it is intercalated between A and T nucleobases in duplexes. In contrast, the higher ionisation potentials of M (5.93 eV) and 2AP (6.05 eV) as compared to G (5.85 eV) well explain the pronounced CT of both 2AP and M to G in the ground state, and thus, the low quantum yield of both probes when close to G.
Influence of thienyl torsional motion on M electronic transitions
To explain the high quantum yield of M when flanked by AT and TA base pairs in DNA duplexes as compared to the free probe in solution,7 one likely hypothesis is that the DNA context restricts the rotation of the thienyl and chromone moieties of M around the C2–C6 single bond, thus favouring the radiative deactivation channel.13 To check this hypothesis, we examined the torsional motion of the two moieties by monitoring the dihedral angle formed by the C3, C2, C6 and C11 atoms during 10 ns of molecular dynamics in the ground state, both for the free probe and the probe in DNA. For the free probe, the rotational motion of the thienyl group around the C2–C6 bond is symmetrical and can be described by a Gaussian distribution with a full width at half maximum (FWHM) value of 42°. In the DNA context, the FWHM decreases to 27.2° and 26.6° for AMA + TAbT and TMT + AAbA, respectively (see Fig. 6). This restricted motion of the thienyl group favours a more planar conformation of M in the DNA, which increases the conjugation of the π electrons of the thienyl group with the rest of the molecule. This in turn is thought to increase the oscillator strength of the transitions localised on M and presenting a CT from thienyl. This increase in the oscillator strength accompanied by a concomitant red shift of the S0 → S1 wavelength position with a maximum for a fully planar conformation (dihedral angle of 0°) was clearly confirmed by our calculations (Fig. 7).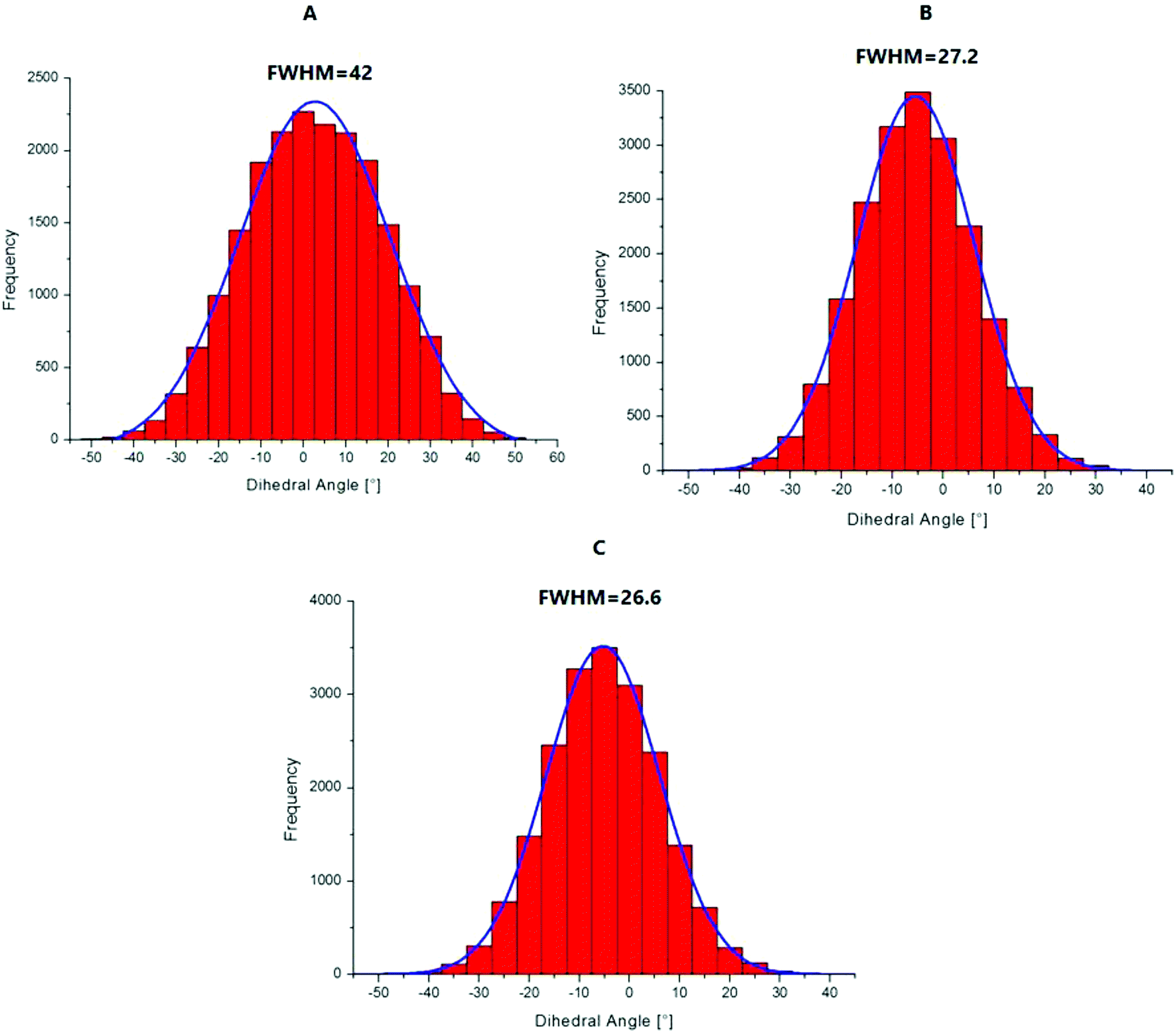 | ||
| Fig. 6 Histograms of the distribution of the dihedral angle C3–C2–C6–C11 describing the torsional motion of the thienyl group with respect to the chromone moiety in the ground state for the free probe in solution (A), and for M in AMA-TAbT (B) and TMT-AAbA (C). | ||
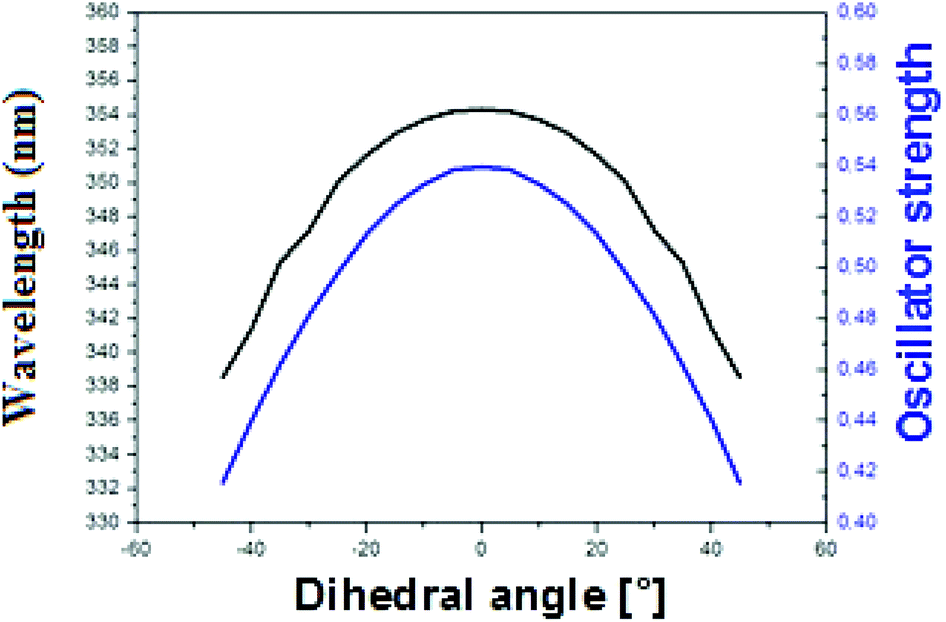 | ||
| Fig. 7 Variation of the S0 → S1 wavelength position (black) and oscillator strength (blue) with the value of the dihedral angle C3–C2–C6–C11 in acetonitrile. | ||
Conclusion
In the present study, the electronic transitions and the energy of the states involved in the absorption and emission processes of M free in solution and inserted in DNA were investigated. The most stable conformations of M were retrieved from DFT calculations for the free probe in solution and MD simulations for M in the DNA context, and further used to calculate the electronic transitions. The accessibility of the transferring proton to the surrounding water molecules was monitored by considering the pair radial distribution function in the ground and excited states. Taken together, our calculations show that ESIPT is slow in buffer because of a high energy barrier that results from the intermolecular H-bonding of both the transferring proton and acceptor oxygen with surrounding water molecules. This consequently disrupts the pre-existing intramolecular H-bond, which favours ESIPT. In the DNA context, the transferring proton of M is screened from H-bonding with water molecules by its flanking nucleobases in the ground state. The electronic properties of M were described using the trimer supermolecule approach. In AMA + TAbT and TMT + AAbA trimers, the electronic density in the frontier MOs was localised on M. In addition, the screening of M from water molecules and the prevention of free rotation between the thienyl and chromone groups probably decrease the non radiative pathways and hence increase the fluorescence quantum yield. The small energy barrier for the ESIPT process is consistent with an efficient accumulation of the T* state, in line with experimental fast ESIPT kinetics and low intensity ratio (IN*/IT*) of M. Differently, in the CMC + GAbG trimer, the electronic density was shifted to the neighbouring G in the ground state and localized to M in the excited state. The very low quantum yield observed for M in this trimer is probably the consequence of the mixed ground state configuration favouring hole transfer to G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. A. K. acknowledges the Abdus Salam Inter-national Centre for Theoretical Physics (ICTP) for its support to CEPAMOQ through the OEA-AC-71 project and the High Performance Computing Center of the University of Strasbourg for supporting this work by providing scientific support and access to computing resources. Part of the computing resources was funded by the Equipex Equip@Meso project (Programme Investissements d'Avenir). YM and AB thank ANR Fluometadn, ANR Pico2 and the FRM for financial support. YM is grateful to the Institut Universitaire de France (IUF) for support and providing additional time to be dedicated to research.References
- A. S. Klymchenko, V. V. Shvadchak, D. A. Yushchenko, N. Jain and Y. Mély, J. Phys. Chem. B, 2008, 112, 12050 CrossRef CAS PubMed.
- V. V. Shvadchak, A. S. Klymchenko, H. de Rocquigny and Y. Mély, Nucleic Acids Res., 2009, 37, e25 CrossRef PubMed.
- S. V. Avilov, E. Piémont, V. Shvadchak, H. de Rocquigny and Y. Mély, Nucleic Acids Res., 2008, 36, 885 CrossRef CAS PubMed.
- J. Godet, C. Kenfack, F. Przybilla, L. Richert, G. Duportail and Y. Mély, Nucleic Acids Res., 2013, 41, 5036 CrossRef CAS PubMed.
- C. A. Kenfack, E. Piémont, N. Ben Gaied, A. Burger and Y. Mély, J. Phys. Chem. B, 2008, 112, 9736 CrossRef CAS PubMed.
- A. A. Kuznetsova, N. A. Kuznetsov, Y. N. Vorobjev, N. P. F. Barthes, B. Y. Michel, A. Burger and O. S. Fedorova, PLoS One, 2014, 9, e10007 CrossRef PubMed.
- D. Dziuba, V. Y. Postupalenko, M. Spadafora, A. S. Klymchenko, V. Guérineau, Y. Mély, R. Benhida and A. Burger, J. Am. Chem. Soc., 2012, 134, 10209 CrossRef CAS PubMed.
- W. Xu, K. M. Chan and E. T. Kool, Nat. Chem., 2017, 9, 1043 CrossRef CAS PubMed.
- N. Ben Gaied, N. Glasser, N. Ramalanjaona, H. Beltz, P. Wolff, R. Marquet, A. Burger and Y. Mély, Nucleic Acids Res., 2005, 33, 1031 CrossRef CAS PubMed.
- J. M. Jean and K. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 37 CrossRef CAS.
- J. M. Jean and K. B. Hall, Biochemistry, 2002, 41, 13152 CrossRef CAS PubMed.
- M. Spadafora, V. Y. Postupalenko, V. V. Shvadchak, A. S. Klymchenko, A. Burger and Y. Mély, Tetrahedron, 2009, 65, 7809 CrossRef CAS.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579 CrossRef CAS PubMed.
- L. M. Wilhelmsson, Q. Rev. Biophys., 2010, 43, 159 CrossRef CAS PubMed.
- L. Zargarian, A. Ben Imeddourene, K. Gavvala, N. P. F. Barthes, B. Y. Michel, C. A. Kenfack, N. Morellet, B. René, P. Fossé, A. Burger, Y. Mély and O. Mauffret, J. Phys. Chem. B, 2017, 121, 11249 CrossRef CAS PubMed.
- V. Kilin, K. Gavvala, N. P. Barthes, B. Y. Michel, D. Shin, C. Boudier, O. Mauffret, V. Yashchuk, M. Mousli, M. Ruff, F. Granger, S. Eiler, C. Bronner, Y. Tor, A. Burger and Y. Mély, J. Am. Chem. Soc., 2017, 139, 2520 CrossRef CAS PubMed.
- D. Dziuba, J. A. Karpenko, N. P. F. Barthes, B. Y. Michel, A. S. Klymchenko, R. Benhida, A. P. Demchenko, Y. Mély and A. Burger, Chem.–Eur. J., 2014, 20, 1998 CrossRef CAS PubMed.
- (a) K. Gavvala, N. P. F. Barthes, D. Bonhommes, A. S. Dabert-Guy, D. Debayle, B. Y. Michel, A. Burger and Y. Mély, RSC Adv., 2016, 6, 87142 RSC; (b) S. Lobsiger, S. Blaser, R. K. Sinha, H.-M. Frey and S. Leutwyler, Nat. Chem., 2014, 6, 989 CrossRef CAS PubMed.
- T. Liu and J. K. Barton, J. Am. Chem. Soc., 2005, 127, 10160 CrossRef CAS PubMed.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642 CrossRef CAS PubMed.
- T. Very, S. Despax, P. Hebraud, A. Monari and X. Assfield, Phys. Chem. Chem. Phys., 2012, 14, 12496 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- B. Santra, A. Michaelides and M. Scheffler, J. Chem. Phys., 2007, 127, 184104 CrossRef PubMed.
- B. Wang, W. Jiang and R.-Q. Zhang, Sci. Rep., 2016, 6, 22099 CrossRef CAS PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpete and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420 CrossRef CAS PubMed.
- D. Jacquemin, J. Preat, M. Charlot, V. Wathelet, J. M. Andre and E. A. Perpete, J. Chem. Phys., 2004, 121, 1736 CrossRef CAS PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (b) J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- P. Y. Ayala and H. B. Schlegel, J. Chem. Phys., 1997, 107, 375 CrossRef CAS.
- S. Yamazaki and T. Taketsugu, Phys. Chem. Chem. Phys., 2012, 14, 8866 RSC.
- R. Das, A. S. Klymchenko, G. Duportail and Y. Mély, Photochem. Photobiol. Sci., 2009, 8, 1583 RSC.
- A. J. G. Strandjord and P. F. Barbara, J. Phys. Chem., 1985, 89, 2355 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 16, Gaussian, Inc., Pittsburgh, PA, 2016, licence g16 Search PubMed.
- K. E. Furse and S. A. Corcelli, J. Chem. Theory Comput., 2009, 5, 1959 CrossRef CAS PubMed.
- D. A. Pearlman, D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. De Bolt, D. Fergurson, G. Seibel and P. A. Kollman, Comput. Phys. Commun., 1995, 91, 1 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269 CrossRef CAS.
- P. Cieplak, W. D. Cornell, C. I. Bayly and P. A. Kollman, J. Comput. Chem., 1995, 16, 1357 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly and P. A. Kollman, J. Am. Chem. Soc., 1993, 115, 9620 CrossRef CAS.
- G. Zheng, X.-J. Lu and W. K. Olson, Nucleic Acids Res., 2009, 37, 240 CrossRef PubMed.
- A. Onufriev, D. Bashford and D. A. Case, Proteins: Struct., Funct., Genet., 2004, 55, 383 CrossRef CAS PubMed.
- F. Fogolari, A. Brigo and H. Molinari, J. Mol. Recognit., 2002, 15, 377 CrossRef CAS PubMed.
- G. Ricciardi, A. Rosa, S. J. A. Van Gisbergen and E. J. Baerends, J. Phys. Chem. A, 2000, 104, 635 CrossRef CAS.
- C. A. Kenfack, A. S. Klymchenko, G. Duportail, A. Burger and Y. Mély, Phys. Chem. Chem. Phys., 2012, 14, 8910 RSC.
- A. D. Roshal, J. A. Organero and A. Douhal, Chem. Phys. Lett., 2003, 379, 53 CrossRef CAS.
- N. A. Nemkovich, W. Baumann and V. G. Pivovarenko, J. Photochem. Photobiol., A, 2002, 153, 19–24 CrossRef CAS.
- A. P. Demchenko, K. C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC.
- A. S. Klymchenko, C. Kenfack, G. Duportail and Y. Mély, J. Chem. Sci., 2007, 119, 83 CrossRef CAS.
- G. N. I. Clark, C. D. Cappa, J. D. Smith, R. J. Saykally and T. Head-Gordon, Mol. Phys., 2010, 108, 1415 CrossRef CAS.
- (a) V. A. Bloomfield, D. M. Crothers, and I. Tinoco Jr, Nucleic Acids, Structure, Properties, and Function, University Science Books, California, USA, 2000 Search PubMed; (b) R. E. Dickerson, M. Bansal, C. R. Calladine, S. Diekmann, W. N. Hunter, O. Kennard, E. Von Kitzing, R. Lavery, H. C. M. Nelson and W. K. Olson, J. Mol. Biol., 1989, 205, 781 Search PubMed.
- A. East and E. C. Lim, J. Chem. Phys., 2000, 113, 8981 CrossRef CAS.
- I. Tinoco Jr, J. Am. Chem. Soc., 1960, 82, 4786 CrossRef.
- E. Cauet, D. Dehareng and J. Liévin, J. Phys. Chem. A, 2006, 110, 9200 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10419d |
| This journal is © The Royal Society of Chemistry 2020 |