DOI:
10.1039/C9RA10541G
(Paper)
RSC Adv., 2020,
10, 3734-3744
Tuning catalysis of boronic acids in microgels by in situ reversible structural variations†
Received
15th November 2019
, Accepted 6th January 2020
First published on 22nd January 2020
Abstract
The catalysis of boronic acids immobilized in polymer microgels can be modulated by bubbling with N2/CO2 gas, and in some cases by adding glucose, making their catalytic activity comparable or even superior to that of the corresponding free boronic acid monomers homogeneously dispersed in solutions and, more importantly, making these boronic-acid-containing polymer microgels able to catalyze alternate reactions that may extend the usefulness. This enhanced catalytic function of these boronic-acid-containing microgels as organoboron acid catalysts is plausibly achieved via in situ reversibly structural variations. Kinetic studies have been carried out on the model boronic-acid-catalyzed aza-Michael addition, aldol, amidation, and [4 + 2] cycloaddition reactions in order to better understand the catalytic process.
1. Introduction
The catalysis by organoboron acids is among the most active fields in synthetic chemistry.1–4 Since organocatalysis highlights the formation and breakage of temporary bonds, organoboron acids possessing the unique ability of reversible covalent binding acidic functional groups should display reactivity patterns that are distinct from those of common organocatalysts.5–13 While a broad spectrum of transformations with organoboron acids as reaction catalysts have been documented in the literature, a problem that accompanies this exciting field from its early beginning, however, is the limited knowledge about the possible action modes and, therefore, the obstacle in the design and optimization of the catalysts, making it difficult to ensure a meaningful and popularized use of organoboron acid catalysis.3,4 The challenge of identifying organoboron acid catalysts that can exhibit enhanced catalytic function remains to be solved.
Traditionally, the widely employed strategy for the design and optimization of organoboron acids as organocatalysts involves structural modification followed by laborious screening, prior to performing the catalytic reactions for target chemical transformations.1–13 This approach, by also combining computational modeling, in principle could allow tailoring the molecular factors that impact the catalysis, including the charge, coordination, interatomic distance, bonding and orientation of the catalytically active atoms, and thus should enable empirical modulation of the attractive and repulsive noncovalent interactions that determine the activity and selectivity in the catalyzed processes.14 Such a tailored catalyst does often much better than others, and most of the time is quite selective for one class of reactants, so that each transformation might require its own tailored catalyst, and in some cases co-catalysts, through a substantial process of design and optimization.4 However, in many successful instances, the catalysts and co-catalysts have been found by chance rather than by logical prediction.4,15,16 The catalytic reaction specificity and the availability of suitable organoboron acids as organocatalysts remain primary restrictions for the targets of interest. Naturally, a question arises of how to appropriately introduce an in situ structural variation to tune the catalysis of ready-prepared organoboron acid catalysts, making it able to catalyze alternate reactions that may extend their usefulness.17
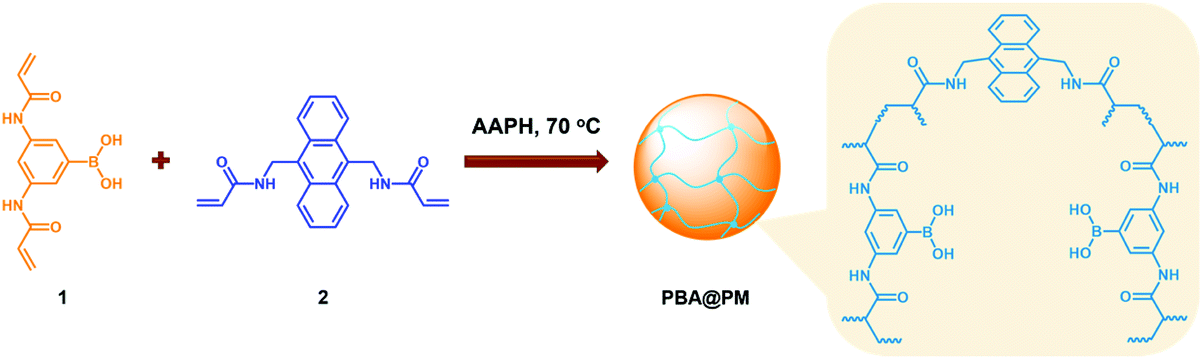
In this paper, as a proof of the concept, we report a new class of organoboron acid catalysts (denoted as PBA@PM) by immobilizing a representative organoboron acid, phenylboronic acid (PBA), in polymer microgels that are dispersible but undissolvable colloids of three-dimensional crosslinked gel network structures internally. To the best of our knowledge, so far, catalysis with PBA-containing polymer microgels acting as organoboron acid catalysts has not been recognized as a viable option, and no example exists in the literature; PBA-containing polymer microgels are better known for molecular recognition, biosensing, and drug delivery.18–24 Herein, our intention in exploiting PBA-containing polymer microgels is based on a combination of recent progresses in microgel synthesis, stimuli-responsive polymers, and supramolecular chemistry: PBA can anchor covalently in microgels and bind cis-diols of saccharides (e.g. glucose) to form boronates,18–24 where tetrahedral species would change into trigonal boronic acids upon CO2 bubbling,25 and recover to the original state without accumulation of by-products by bubbling a suitable gas (e.g., N2) to expel CO2.26,27 With the catalytic, saccharide-responsive, and CO2-sensitive components being integrated into a single object, these organoboron acid catalysts should present prospects for in situ structural variations to tune catalysis by bubbling with N2/CO2 gas.
2. Results and discussion
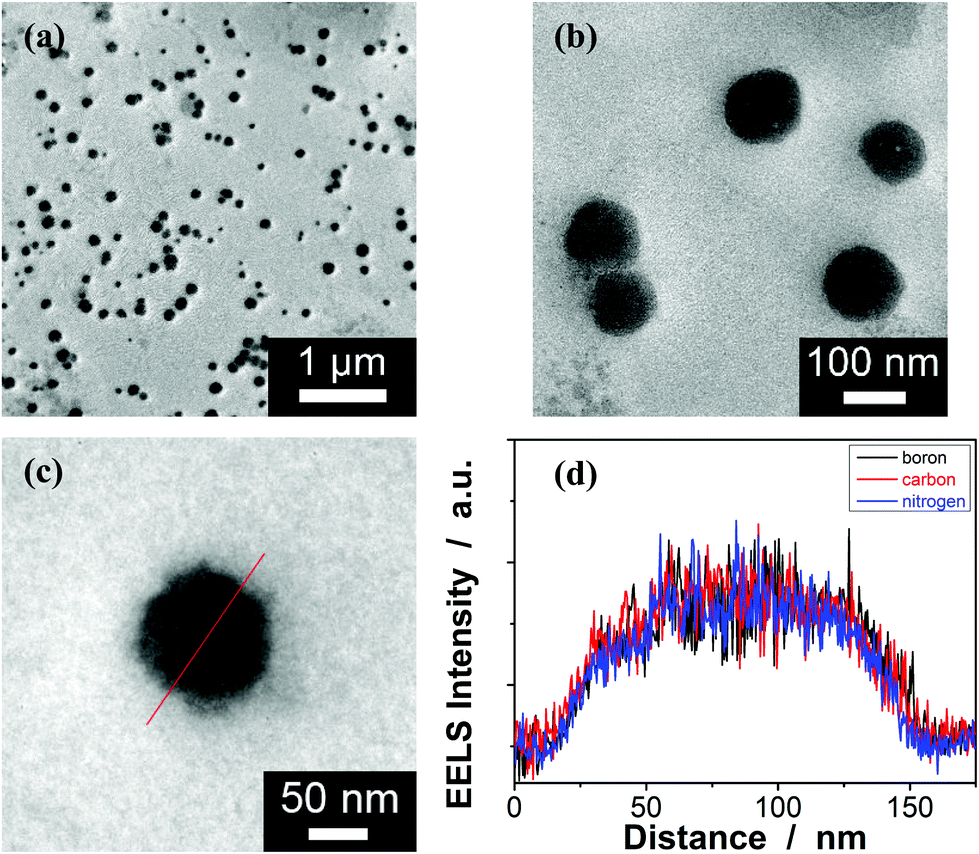
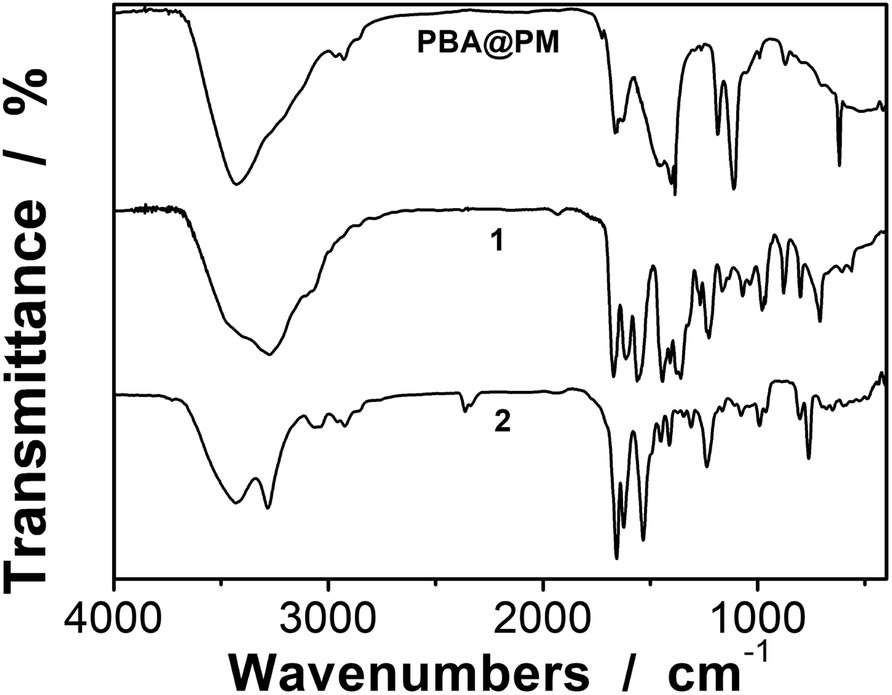
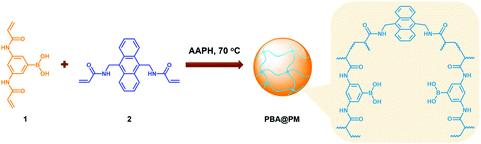
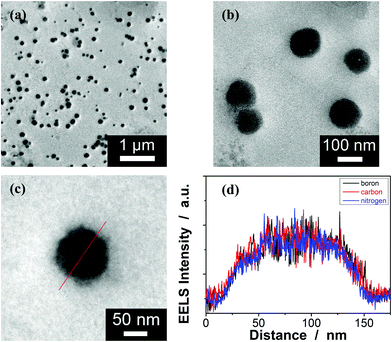
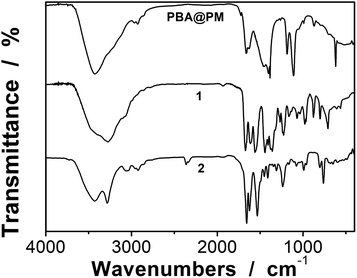
Typically, these PBA-containing polymer microgels were prepared (Scheme 1) according to a reported literature method,23,24 which involves first, the synthesis of vinyl monomers (3,5-diacrylamidophenyl)boronic acid (1; Fig. S1 in ESI†) and N,N′-(anthracene-9,10-diylbis(methylene))diacrylamide (2; Fig. S2 in ESI†), followed by free-radical polymerization of the two vinyl monomers using 2,2′-azobis(2-methylpropionamidine)dihydrochloride (AAPH) as an initiator in a N2 atmosphere at 70.0 °C. The choice of these two vinyl monomers for microgel synthesis is specifically considered to ensure negligible change in the average hydrodynamic diameter (〈Dh〉; Fig. S3 in ESI†) of the ready-prepared PBA@PM in solvents upon heating/cooling.18–24 The feeding molar ratio of 1 and 2 was set to a moderate value of ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.86 to ensure the formation of bis-boronate complexes upon adding glucose in a N2 atmosphere (see below).23,24 TEM images of microgels indicate the formation of a sphere-like morphology (Fig. 1a–c). The distribution of the boronic acid inside the microgels was studied by electron energy loss spectroscopy (EELS; Fig. 1d), which showed that the line scan curves of normalized EELS intensities of carbon, nitrogen and boron elements displayed a similar profile, indicating that those elements and thus PBA groups of 1 and anthracene rings of 2 are homogeneously distributed throughout the whole microgels. IR spectral comparison between 1, 2, and the microgels (Fig. 2) confirmed the incorporation of PBA groups of 1 and anthracene rings of 2 into the microgels, wherein the characteristic bands of B–O stretching vibration at ca. 1384 cm−1 and O–H stretching vibration at ca. 3260 cm−1, and those of aliphatic C–H stretching vibrations at ca. 2850–2960 cm−1 and aromatic = C–H stretching vibrations at ca. 3000–3100 cm−1 were observed. The content of PBA groups immobilized in the microgels was determined (ca. 5.2 × 10−4 mol per (g dried microgels)) by the mannitol-assisted UV-vis spectrophotometric titration analysis,28 from which the molar ratio of 1 and 2 moieties incorporated into the microgels was estimated to be ca. 1:4.79. This is close to the feeding molar ratio (1:4.86) of 1 and 2 for the microgel synthesis, together with the high yield of the microgels (≥96%), indicating a high monomer conversion. These microgels can be reproduced from batch to batch.
:4.86 to ensure the formation of bis-boronate complexes upon adding glucose in a N2 atmosphere (see below).23,24 TEM images of microgels indicate the formation of a sphere-like morphology (Fig. 1a–c). The distribution of the boronic acid inside the microgels was studied by electron energy loss spectroscopy (EELS; Fig. 1d), which showed that the line scan curves of normalized EELS intensities of carbon, nitrogen and boron elements displayed a similar profile, indicating that those elements and thus PBA groups of 1 and anthracene rings of 2 are homogeneously distributed throughout the whole microgels. IR spectral comparison between 1, 2, and the microgels (Fig. 2) confirmed the incorporation of PBA groups of 1 and anthracene rings of 2 into the microgels, wherein the characteristic bands of B–O stretching vibration at ca. 1384 cm−1 and O–H stretching vibration at ca. 3260 cm−1, and those of aliphatic C–H stretching vibrations at ca. 2850–2960 cm−1 and aromatic = C–H stretching vibrations at ca. 3000–3100 cm−1 were observed. The content of PBA groups immobilized in the microgels was determined (ca. 5.2 × 10−4 mol per (g dried microgels)) by the mannitol-assisted UV-vis spectrophotometric titration analysis,28 from which the molar ratio of 1 and 2 moieties incorporated into the microgels was estimated to be ca. 1:4.79. This is close to the feeding molar ratio (1:4.86) of 1 and 2 for the microgel synthesis, together with the high yield of the microgels (≥96%), indicating a high monomer conversion. These microgels can be reproduced from batch to batch.
 |
| | Scheme 1 Synthesis of the proposed PBA@PM. | |
 |
| | Fig. 1 Typical (a–c) TEM images of the PBA@PM. (d) EELS spectra of the PBA@PM along the line shown in the TEM image in (c). | |
 |
| | Fig. 2 Typical IR spectrum of the PBA@PM. IR spectra of 1 and 2 are shown for comparison. | |
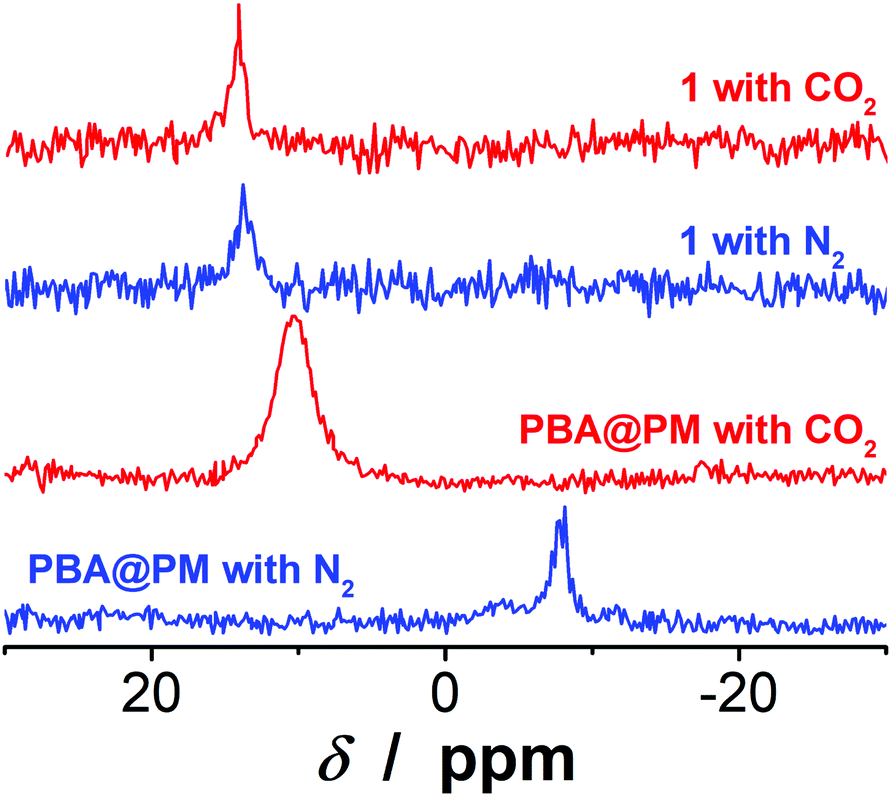
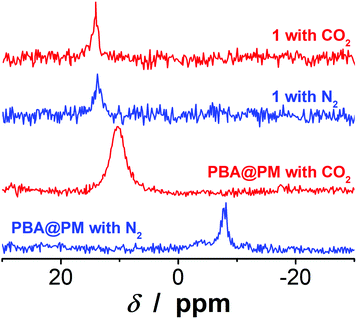
The PBA@PM could be well dispersed in many organic solvents, such as dichloromethane (CH2Cl2), ethanol, and their mixtures, or even in the presence of some water. For instance, in an immiscible mixture of CH2Cl2 and water at a volume ratio of 9:1 (after adding PBA@PM, the mixture would not be stratified, but this may occur if more water was added), or a ternary miscible solvent of CH2Cl2, ethanol and water at a volume ratio of 7:2:1, PBA@PM (the concentration of boronic acids in the final dispersion was ca. 1.2 × 10−5 mol L−1) showed very good stability. No sediment was observed even after 6 months of storage at room temperature in a N2 atmosphere, but there was only a marginal change on the size distribution measured by dynamic light scattering (DLS) (Fig. S4 in ESI†), implying that both 〈Dh〉 and dispersibility of the PBA@PM remained nearly unchanged, and that the degradation/aggregation was negligible. The good dispersibility of the PBA@PM in the presence of water may be due to the partial ionization of PBA groups (Fig. S5 in ESI†) derived from the localization of PBA groups adjacent to the anthracene rings.23,24 It is reported that anthracenes could be used as electron acceptors due to their strong electron affinities.29 This should favor the interaction of anthracene rings with neighboring PBA groups via Nδ+⋯Bδ− interactions,30,31 and possibly π⋯Bδ− interactions as well,23,24 both of which could promote the dissociation equilibrium of boronic acids moving to right, resulting in a decrease in pKa (ca. 7.5 for PBA groups in the PBA@PM, much lower than ca. 8.8 for 1).23,24 Indeed, the 11B NMR spectrum (Fig. 3) of the PBA@PM upon bubbling with N2 gas displayed a single broad peak that centered at ca. −7.9 ppm, which is due to the fast exchange averaging of the two boronic acid species, i.e., tetrahedral and trigonal species; in contrast, the 11B NMR spectrum of 1 displayed a peak at ca. 13.9 ppm, which is consistent with the preponderance of trigonal species.32 Interestingly, if bubbling with 30 min (the same below) of CO2 gas, the solution pH decreased from ca. 7.1 to ca. 5.8; meanwhile, a remarkable increase in the chemical shift for the 11B NMR peak of the PBA@PM (ca. 10.4 ppm) toward for 1 (ca. 14.1 ppm) was observed (Fig. 3), accompanied with an increase in the zeta potential from ca. −13.0 mV to ca. −2.6 mV (Fig. S6 in ESI†) and a decrease in the 〈Dh〉 from ca. 136.1 nm to ca. 66.1 nm (Fig. S3 and S7 in ESI†), deriving from the reduction in the ionization degree of PBA groups along with the decrease in the solution pH.18–20,33 All 11B NMR, solution pH value, surface zeta potential and size changes are completely reversible upon bubbling once again with 30 min (the same below) of N2 gas to expel CO2. Similar changes upon bubbling with N2/CO2 gas were observed on the PBA@PM dispersed in other solvents containing water. These results can not only provide additional proofs for the successful incorporation of both 1 and 2 into the microgels, but also foreshadow a novel class of organoboron acid catalysts that may allow tuning catalysis by simply bubbling with N2/CO2 gas.
 |
| | Fig. 3 Typical 11B NMR spectra of the PBA@PM dispersed in CD2Cl2/D2O (9:1 in volume) upon bubbling with N2 or CO2 gas. Results of 1 are shown for comparison. | |
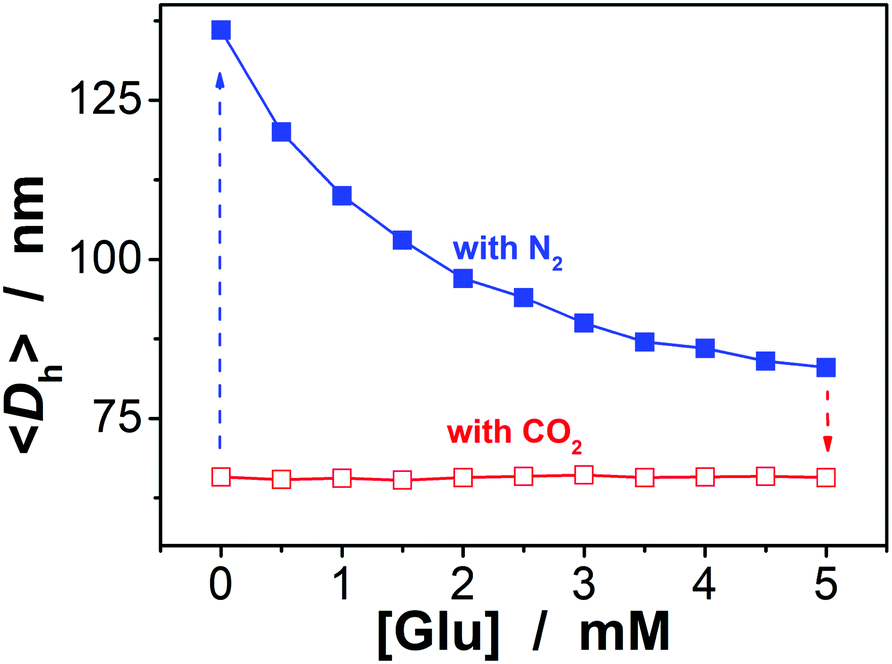
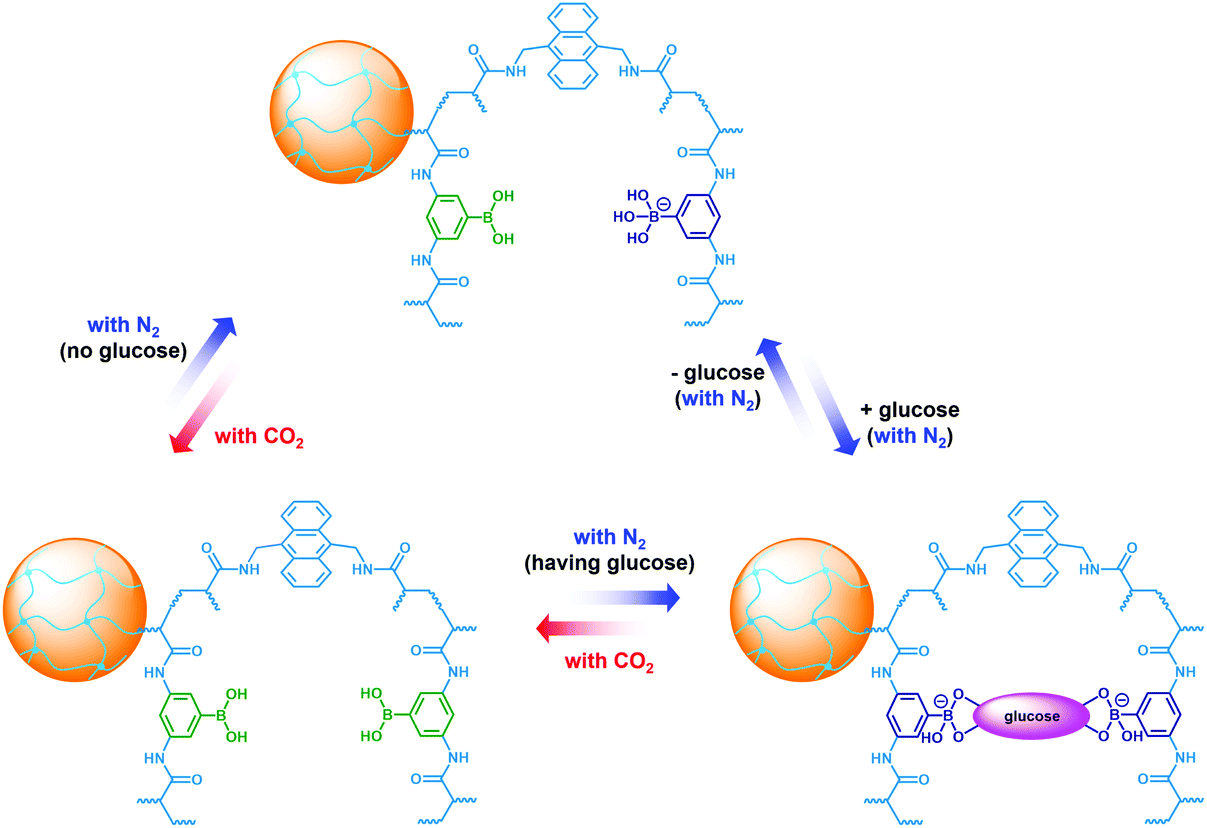
To be an efficient catalyst, the PBA@PM should be so permeable to both the reactants and products that mass-transfer limitations can be avoided to maximize the chemical reaction rates. High porosity of the PBA@PM is then crucial for the catalysis application. Glucose-responsive swelling–deswelling phase transition behaviors, which were quantitatively measured by DLS that is a powerful tool to study volume phase transition behavior in situ of polymer gel particles in solutions,18–26 do confirm the highly porous nature of the PBA@PM. Fig. 4 shows that 〈Dh〉 decreases by ca. 1.64 times when the solution glucose concentration ([Glu]) increases from 0.0 mM to 5.0 mM upon bubbling with N2 gas, verifying the deswelling behavior of the PBA@PM upon addition of glucose. This deswelling behavior of the PBA@PM upon addition of glucose can be interpreted as the occurrence of additional cross-linking junctions due to the formation of 1:2 (i.e., glucose–bis-boronate) complexes, exceeding over the contribution of glucose binding on the ionization of PBA groups.23,24 This is also supported by the inverse, swelling behavior of the PBA@PM upon addition of fructose, galactose or mannose (Fig. S8 in ESI†), which are the natural stereoisomers of glucose, but that can only form 1:1 (i.e., glucose–boronate) rather than 1:2 complexes.34,35 Upon the dissociation of the complexes by removing glucose via dialysis,23,24 the PBA@PM could swell reversibly, with 〈Dh〉 nearly recovered to the original values (Fig. S9 in ESI†). A facile but different way to realize the swelling of the PBA@PM was achieved by bubbling with CO2 gas (Fig. 4), or others containing an appropriate amount of CO2 (Fig. S10 in ESI†). The distinct difference in those phase-transition profiles upon bubbling with N2/CO2 gas was derived from the structural variations in the microgels (Scheme 2). Upon neutralization of alkaline boronate anions after bubbling with CO2 gas,25 the dissociation of the complexes and, thus, a decreased degree of ionization should reduce both the polymer water solubility and Donnan potential,18–20,33 resulting in a decrease in 〈Dh〉 to ca. 66.1 nm in our experimental [Glu] window of 0.0–5.0 mM (Fig. 4), which can be readily recovered upon the subsequent purging with N2 gas to remove CO2.26,27 In the control experiments adding fructose, galactose or mannose, the neutralization would also occur upon bubbling with CO2 gas (Fig. S11 in ESI†), with 〈Dh〉 also dropping to ca. 66.1 nm, and recover by purging with N2 gas (Fig. S12 in ESI†).
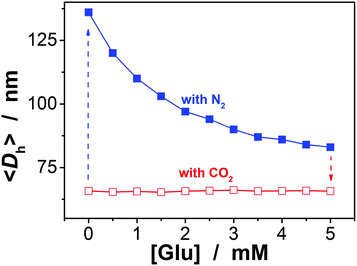 |
| | Fig. 4 Glucose-dependent 〈Dh〉 of the PBA@PM dispersed in CH2Cl2/water (9:1 in volume) upon bubbling with N2 or CO2 gas, measured at 10.0 °C and at a scattering angle of 45°. | |
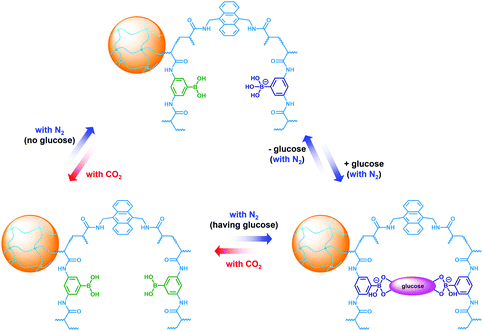 |
| | Scheme 2 In situ structural variations in the PBA@PM upon bubbling with N2/CO2 gas, in the absence or in the presence of glucose. | |
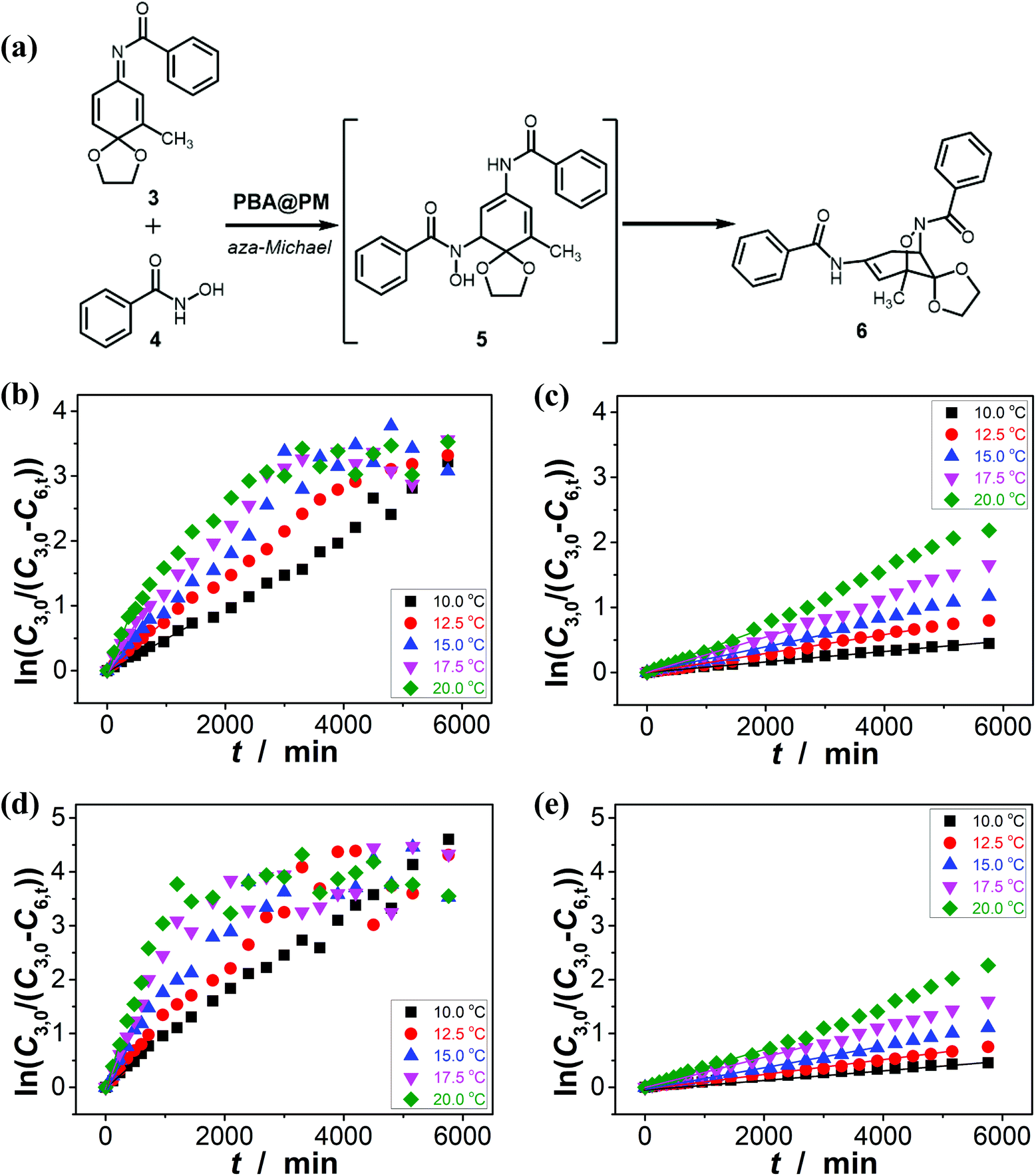
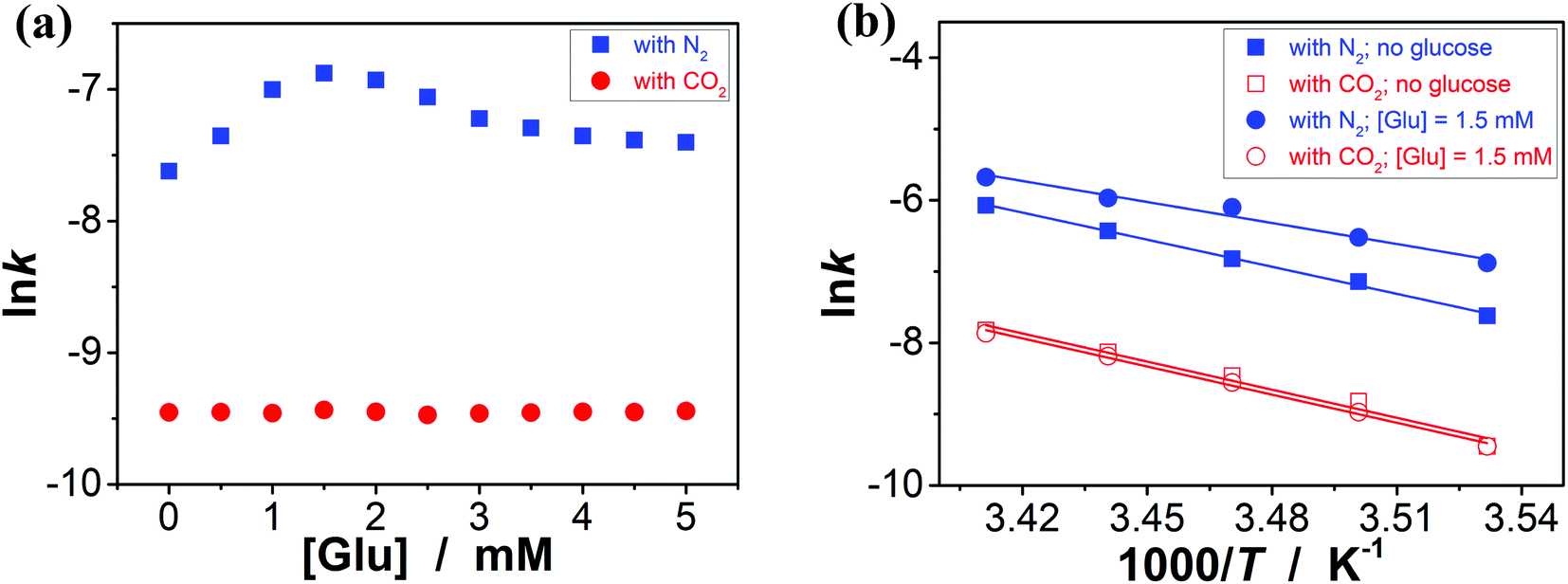
It has been documented that trigonal boronic acids can provide electrophilic activation of an alcohol or a carboxylic acid for a desired nucleophilic substituent or addition; with the formation of tetrahedral neutral or even anionic boronate anions, boronic acids can facilitate the enolization of carbonyl compounds, or nucleophilic activation of hydroxy groups, wherein the reactivity is enabled by an increase in the electronic density of the oxygen atoms, which, in turn, translates into an increase in their nucleophilicity towards potential electrophiles.3,4 Stemmed from these key points reported previously, and motived from our observations on structural variations in the PBA@PM upon bubbling with N2/CO2 gas, our initial venture into organoboron acid catalysis of the PBA@PM (the concentration of boronic acids in the reaction system was ca. 1.2 × 10−5 mol L−1) was commenced by performing a catalytic aza-Michael addition of hydroxamic acid 4 (2.5 × 10−3 mol L−1) to quinone imine ketal 3 (1.0 × 10−4 mol L−1; Fig. S13 in ESI†) to give final product 6 (Fig. S14 in ESI†) in CH2Cl2/water (9:1 in volume; 100.0 mL) at 10.0 °C (Fig. 5a),11 wherein the quinone imine ketal can be activated by hydrogen bonding with boronic acids and attacked by the nucleophilically activated hydroxamic acid,11 which would not progress without any organoboron acid catalysts under otherwise identical reaction conditions (Fig. S15 in ESI†). The glucose (if any) that was added into the catalytic system is stable, which could be separated from the products by chromatography and confirmed simply by spectroscopy (data not shown). Since the uncyclized adduct 5 that may derive from initial aza-Michael addition reported previously11 was not isolated in our cases, and the concentration of 4 exceeds over 25-times than that of 3, the reaction overall may be simplified as pseudo first order.35,36 Fig. 5b–e shows that this is the case for the solution temperature range of 10.0–20.0 °C, as measured by monitoring the yield of 6 (Fig. S16 in ESI†). Linear relationships between ln(C3,0/(C3,0 − C6,t)) and t are obtained at the low conversions (≤60%),37 where C3,0 is the starting concentration of 3, and C6,t is the concentration of 6 at the reaction time t. For present purpose it suffices to discuss the initial rates of the reaction.37 The apparent reaction rate constant k for the reaction catalyzed by the PBA@PM upon bubbling with N2 gas at 10.0 °C thereby is estimated to be ca. 4.90 × 10−4 min−1 in the absence of glucose. As is expected, the reaction can be accelerated in the presence of glucose over the entire range of 0.0 mM < [Glu] ≤ 5.0 mM, whereas instead of a simple linear relationship, the change of the k with the [Glu] can be roughly divided into two regions (Fig. 6a and S17 in ESI†): at low [Glu] of ≤ ca. 1.5 mM, the k increases with the [Glu], but the k decreases gradually when [Glu] ≥ ca. 1.5 mM. This non-linear relationship can be assigned to the formation of glucose–bis-boronate complexes (Fig. 4), consequently the counterbalance of the deswelling of microgels (rendering an inhibiting effect) and the increased amount of boronate anions (rendering a promoting effect here):3,4,38–40 at low [Glu]s that the microgels are in relatively solvophilic and swollen state, the diffusion of chemicals into/out the microgels is slightly or even negligibly affected by the deswelling of microgels, meanwhile the formation of glucose–bis-boronate complexes provides more boronate anions with an increase in the [Glu], leading to an increase in k; when the microgels collapse to some extent with further increase in the [Glu], the cumulative decrease in the porosity would significantly hinder the diffusion (Fig. S18 in ESI†) and surpasses the promoting effect, resulting in a decrease in the k. Nevertheless, in the cases both in the absence and in the presence of glucose, the overall rate for the reaction catalyzed by the PBA@PM upon bubbling with N2 gas is clearly larger than that upon bubbling with CO2 gas. The reaction can be significantly decelerated upon bubbling with CO2 gas, with k decreasing to almost the same value of (7.85 ± 0.13) × 10−5 min−1. After full conversion of the starting 3, the PBA@PM can be simply separated by centrifugation and reused directly for the next cycle. In the more than ten times repeated bubbling with N2 or CO2 gas, no significant erosion of the catalytic activity (the k can return to >92% of original value; Fig. S19 in ESI†) was observed, due to the stability and reversibility of microgels upon bubbling/removing N2/CO2 gas (Fig. 4, S3, S4, S7 and S20 in ESI†). Additional recycling experiments also revealed the robustness of the PBA@PM as organoboron acid catalysts (Fig. S21 in ESI†). The slight decrease in the activities after ten cycles might be due to the occupation of the catalytic site in the PBA@PM by reactants or products, which is confirmed by the nearly unchanged results if the recycled PBA@PM was purified before reuse (Fig. S22 in ESI†).
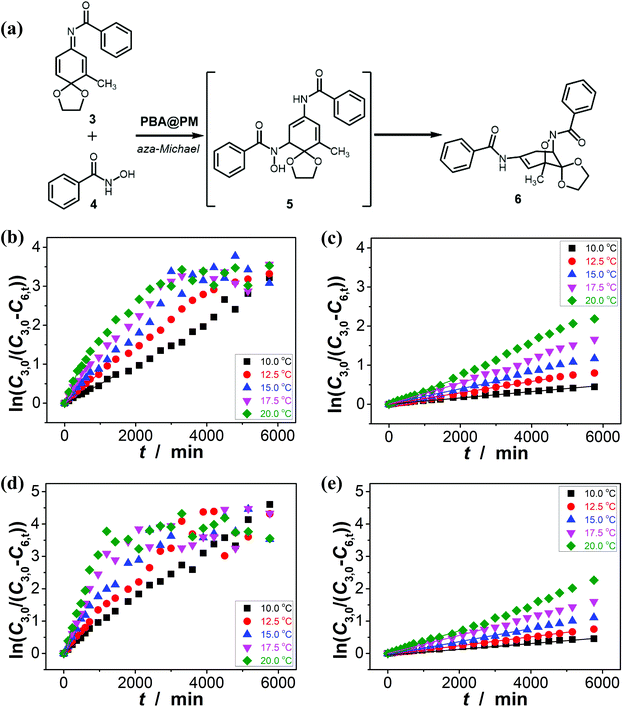 |
| | Fig. 5 (a) The catalytic aza-Michael addition as a model for evaluating the accessibility and the catalytic performance of the PBA@PM. (b–e) Influence of temperature on k, measured in the reaction mixture in the absence (b and c; [Glu] = 0.0 mM) or in the presence (d and e; [Glu] = 1.5 mM) of glucose, upon bubbling with N2 (b and d) or CO2 (c and e) gas, where lines are first-order kinetic fits. | |
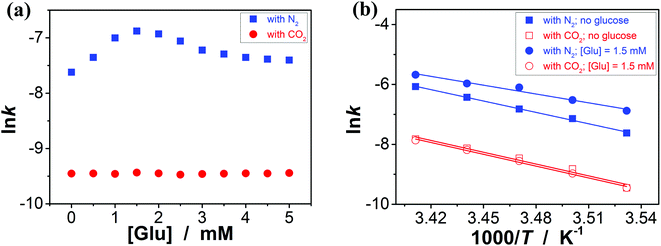 |
| | Fig. 6 (a) Influence of the [Glu] on the k of aza-Michael addition catalyzed by the PBA@PM at 10.0 °C. (b) The lnk–1000/T plots, where the solid lines are linear fitting of the plots. | |
This N2/CO2-modulated catalysis of boronic acids in microgels is observed throughout our experimental temperature window of 10.0–20.0 °C. In a further analysis, both the apparent activation energy (Ea) and the pre-exponential factor (A) can be determined (Table 1) on the basis of the Arrhenius equation and the linear fitting of lnk–T−1 plots (Fig. 6b).37 While the interpretation of the apparent activation parameters is complicated by the fact that these represent the combination of the diffusion and the catalysis processes, for the reaction catalyzed by the PBA@PM upon bubbling with N2 gas, the observation of the smaller Ea (theoretically indicating a lower energy required to reach transition state of reactants, thereby a lower energy required to initiate the reaction) but smaller A (theoretically indicating a smaller total number of collisions per minute) when adding glucose is consistent with a model between diffusional control of the reaction and control by the reaction at the catalytic sites.37,40 In agreement with the observation on the influence of the [Glu] on k (Fig. 6a) discussed above that could be associated with the counterbalance of the two effects, the much denser polymer network (to some extent) of the collapsed microgels imposes a higher diffusional resistance,38–40 and the formation of more boronate anions that is prior to trigonal boronic acids for facilitating the nucleophilic activation should prefer the aza-Michael addition.3,4,11 More importantly, in comparison with the apparent activation parameters obtained for the reaction upon bubbling with N2 gas, both the smaller Ea and A values are obtained for the reaction upon bubbling with CO2 gas; both the collapsed gel and trigonal boronic acids can be envisaged to hinder the reaction to move. These results confirm the feasibility on tuning catalysis of boronic acids in microgels by bubbling with N2/CO2 gas.
Table 1 Apparent activation parameters for the aza-Michael addition catalyzed by the PBA@PM
| [Glu] (mM) |
Bubbling with N2 gas |
Bubbling with CO2 gas |
| A (min−1) |
Ea (kJ mol−1) |
A (min−1) |
Ea (kJ mol−1) |
| 0.0 |
1.3 × 1016 |
101.3 |
1.3 × 1014 |
109.3 |
| 1.5 |
9.1 × 1015 |
85.7 |
1.2 × 1014 |
109.4 |
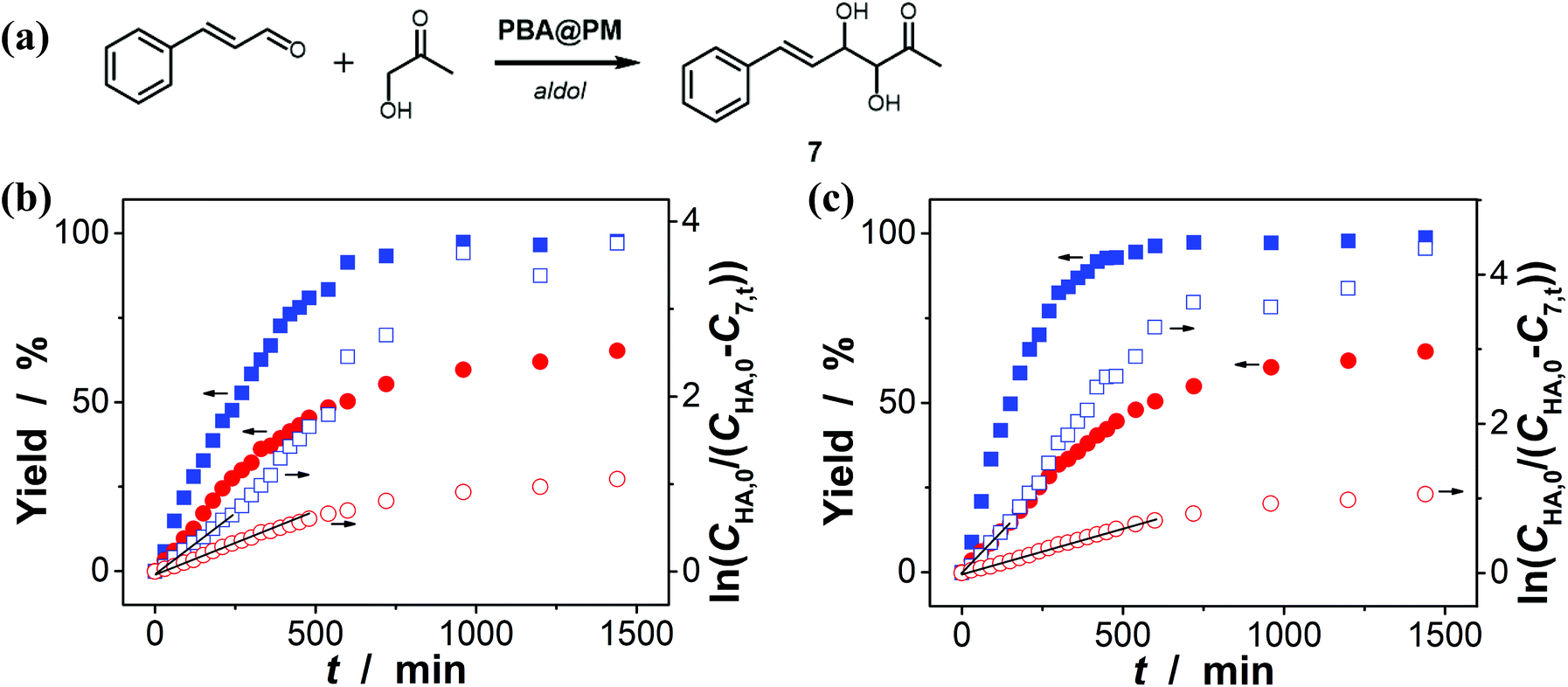
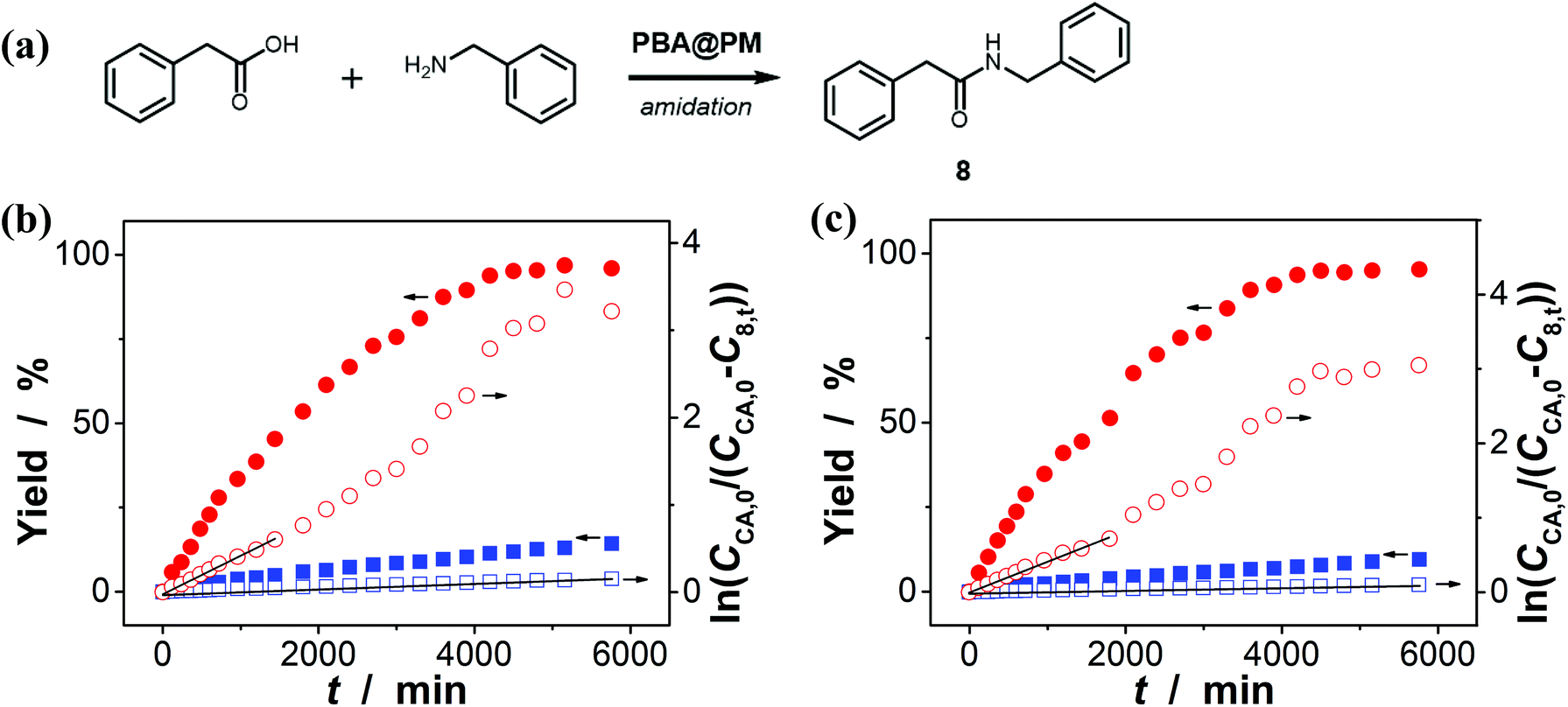
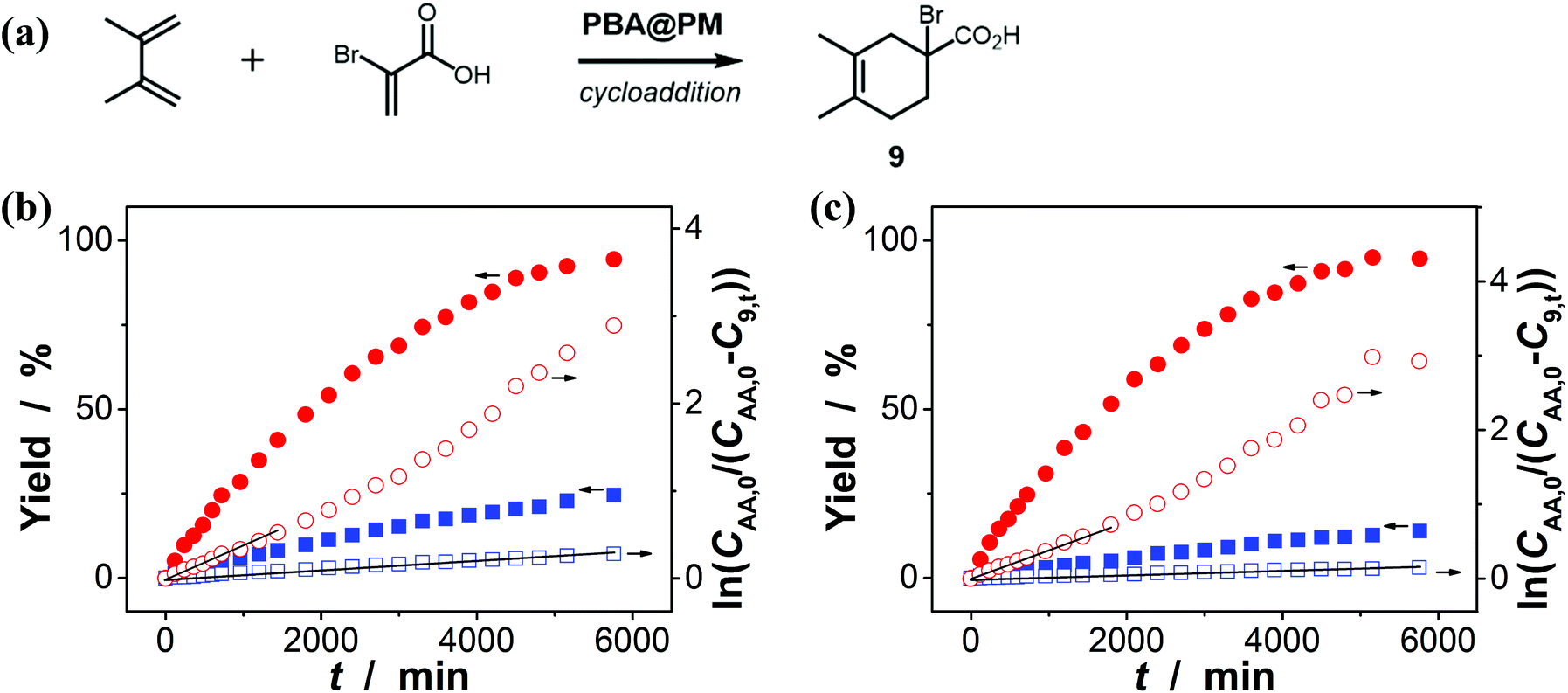
Encouraged by the above-mentioned observation on N2/CO2-modulated catalysis of boronic acids in microgels for the model aza-Michael addition, we tried to extend the use of the PBA@PM (the concentration of boronic acids in reaction systems was ca. 1.2 × 10−5 mol L−1) as organoboron acid catalysts to several other chemical transformations in CH2Cl2/water (9:1 in volume; 100.0 mL) at 10.0 °C, including a model aldol reaction of hydroxyacetone (1.0 × 10−4 mol L−1) and aldehyde (2.5 × 10−3 mol L−1) (Fig. 7 and S23 in ESI†), a model amidation reaction of carboxylic acid (1.0 × 10−4 mol L−1) and amine (2.5 × 10−3 mol L−1) (Fig. 8 and S24 in ESI†), and a model [4 + 2] cycloaddition between acrylic acid (1.0 × 10−4 mol L−1) and diene (2.5 × 10−3 mol L−1) (Fig. 9 and S25 in ESI†). The experimental results showed that for the model aldol reaction, the PBA@PM upon bubbling with N2 gas (k = 2.77 × 10−3 min−1 and 4.72 × 10−3 min−1 for the cases with [Glu] = 0.0 mM and [Glu] = 1.5 mM, respectively) is much more active than that upon bubbling with CO2 gas (correspondingly, k = 1.29 × 10−3 min−1 and 1.25 × 10−3 min−1), similar to the results for the model aza-Michael addition; in both these two reactions reported previously in the literature, organoboron acids are thought to induce nucleophilic activation of carbonyl compounds (herein 4 and hydroxyacetone).4,7,8,11 In contrast, for the model amidation reaction and the model [4 + 2] cycloaddition, organoboron acids are reported to induce electrophilic activation of carboxylic acids.4,9 It is found that the PBA@PM upon bubbling with CO2 gas could provide much faster rates (for amidation and cycloaddition, k = (4.17 ± 0.06) × 10−4 min−1 and (3.56 ± 0.33) × 10−4 min−1, respectively) over upon bubbling with N2 gas (correspondingly, k = 2.61 × 10−5 min−1 at [Glu] = 0.0 mM and 1.68 × 10−5 min−1 at [Glu] = 1.5 mM, and k = 4.89 × 10−5 min−1 at [Glu] = 0.0 mM and 2.63 × 10−5 min−1 at [Glu] = 1.5 mM). These are particularly exciting results as they suggest the possibility of tuning catalysis of the boronic acids in microgels by bubbling with N2/CO2 gas, which can potentially be applied to other chemical transformations, and the ability of the PBA@PM to catalyze alternate reactions that may differ in the functional group and/or the catalytic mechanism involved.
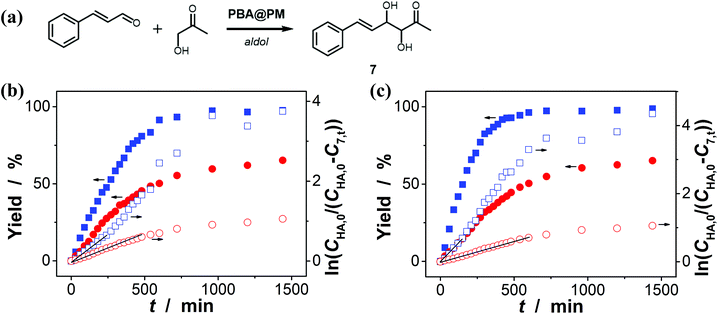 |
| | Fig. 7 (a) A model aldol reaction of hydroxyacetone and aldehyde catalyzed by the PBA@PM. (b and c) Time trace of the yield of aldol adduct 7, measured in the reaction mixture in the absence (b; [Glu] = 0.0 mM) or in the presence (c; [Glu] = 1.5 mM) of glucose, upon bubbling with N2 (■,□) or CO2 (●,○) gas, and at 10.0 °C, where CHA,0 is the starting concentration of hydroxyacetone, C7,t is the concentration of 7 at the reaction time t, and lines are first-order kinetic fits. | |
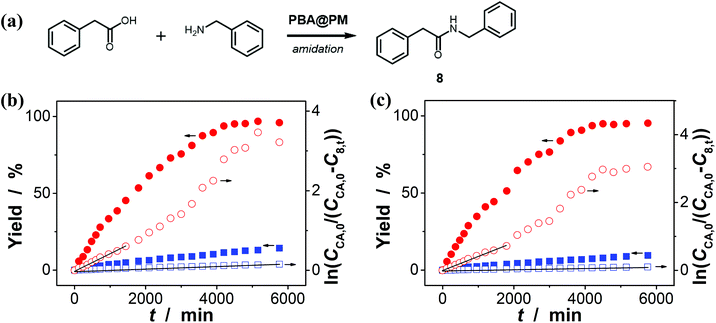 |
| | Fig. 8 (a) A model amidation reaction of carboxylic acid and amine catalyzed by the PBA@PM. (b and c) Time trace of the yield of amide product 8, measured in the reaction mixture in the absence (b; [Glu] = 0.0 mM) or in the presence (c; [Glu] = 1.5 mM) of glucose, upon bubbling with N2 (■,□) or CO2 (●,○) gas, and at 10.0 °C, where CCA,0 is the starting concentration of carboxylic acid, C8,t is the concentration of 8 at the reaction time t, and lines are first-order kinetic fits. | |
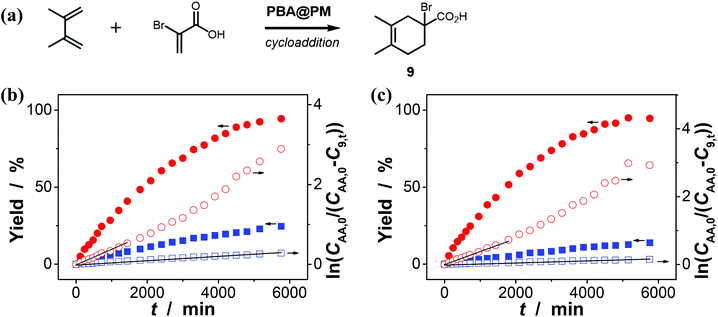 |
| | Fig. 9 (a) A model [4 + 2] cycloaddition of acrylic acid and diene catalyzed by the PBA@PM. (b and c) Time trace of the yield of cycloadduct 9, measured in the reaction mixture in the absence (b; [Glu] = 0.0 mM) or in the presence (c; [Glu] = 1.5 mM) of glucose, upon bubbling with N2 (■,□) or CO2 (●,○) gas, and at 10.0 °C, where CAA,0 is the starting concentration of acrylic acid, C9,t is the concentration of 9 at the reaction time t, and lines are first-order kinetic fits. | |
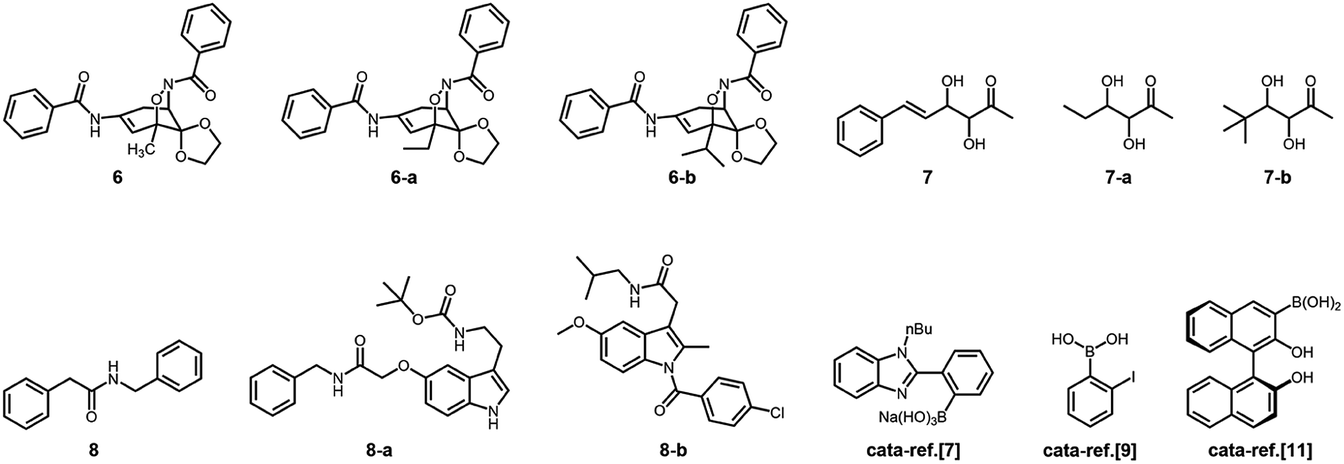
In addition, the control experiments were conducted on the four model reactions with free organoboron acids 1 under otherwise the same reaction conditions, and k was estimated to be (6.58 ± 0.05) × 10−5 min−1, (6.01 ± 0.04) × 10−4 min−1, (5.04 ± 0.07) × 10−6 min−1, and (2.58 ± 0.21) × 10−5 min−1 (Fig. S26 in ESI†), respectively, for those model aza-Michael addition, aldol, amidation, and [4 + 2] cycloaddition reactions; however, no significant difference on k was observed upon bubbling with N2 and CO2 gases, either in the absence or in the presence of 2, due to the negligible change in PBA groups of 1 (Fig. 3). Clearly, the corresponding free boronic acid monomer 1, more or less, displayed smaller k values than the PBA@PM upon bubbling with N2/CO2 gas, indicating a considerable high catalytic activity of the boronic acids in microgels comparable or even superior to that of the corresponding boronic acid monomer homogeneously dispersed in solutions. In particular, while the corresponding free boronic acid monomer 1 might be barely thought to be useful for the model aldol reaction, the PBA@PM can catalyze all four model reactions by tuning catalysis via bubbling with N2/CO2 gas, and in some cases also adding glucose. Further in comparison with those organoboron acid catalysts reported previously in the literature, the PBA@PM still indicates considerable high catalytic activity, as summarized in Table 2. Several reactants were also applied without difficulty to give the desired products, upon properly adjusting the reaction conditions by bubbling with N2/CO2 gas and in some cases adding glucose, making the yields comparable or even superior to the corresponding results with the reactions catalyzed by organoboron acid catalysts reported previously. This enhanced catalytic function of the PBA@PM may extend their usefulness in synthesis.
Table 2 Comparison of results for the model boronic-acid-catalyzed reactions
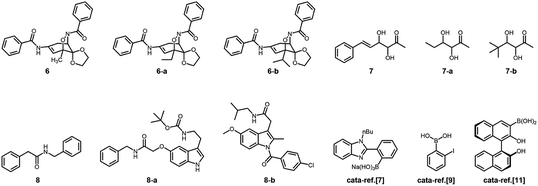
|
| Product |
Catalyst |
Temperature |
Time |
Yield |
|
| Bubbling with N2 gas, [Glu] = 0.0 mM. Bubbling with CO2 gas, [Glu] = 0.0 mM. Bubbling with N2 gas, [Glu] = 1.5 mM. Bubbling with CO2 gas, [Glu] = 1.5 mM. Performed with hydroxamic acid (2.5 × 10−3 mol L−1) and quinone imine ketal (1.0 × 10−4 mol L−1) in CH2Cl2/water (9:1 in volume; 100.0 mL), and the concentration of boronic acids in the reaction system of ca. 1.2 × 10−5 mol L−1. Performed with hydroxamic acid (1.3 × 10−1 mol L−1) and quinone imine ketal (0.1 mol L−1) in CH2Cl2 (1.0 mL); the concentration of boronic acids in the reaction system was ca. 1.0 × 10−2 mol L−1, and o-nitrobenzoic acid (5.0 × 10−2 mol L−1) as a co-catalyst. Performed with hydroxyacetone (1.0 × 10−4 mol L−1) and aldehyde (2.5 × 10−3 mol L−1) in CH2Cl2/water (9:1 in volume; 100.0 mL), and the concentration of boronic acids in the reaction system of ca. 1.2 × 10−5 mol L−1. Performed with hydroxyacetone (5.5 mol L−1) and aldehyde (5.5 × 10−1 mol L−1) in water (2.0 mL), and the concentration of boronic acids in the reaction system of ca. 3.6 × 10−2 mol L−1. Performed with carboxylic acid (1.0 × 10−4 mol L−1) and amine (2.5 × 10−3 mol L−1) in CH2Cl2/water (9:1 in volume; 100.0 mL), and the concentration of boronic acids in the reaction system of ca. 1.2 × 10−5 mol L−1. Performed with carboxylic acid (7.8 × 10−2 mol L−1) and amine (7.1 × 10−2 mol L−1) in CH2Cl2 (7.0 mL), the concentration of boronic acids in the reaction system of ca. 7.1 × 10−3 mol L−1, and adding the activated 4A molecular sieves (1.0 g). |
| 6 |
PBA@PM |
10.0 °C |
16 h |
36% a; 9% b; 62% c; 9% d |
This worke |
| PBA@PM |
12.5 °C |
16 h |
52% a; 11% b; 74% c; 11% d |
This worke |
| PBA@PM |
15.0 °C |
16 h |
58% a; 15% b; 83% c; 15% d |
This worke |
| PBA@PM |
17.5 °C |
16 h |
69% a; 21% b; 91% c; 22% d |
This worke |
| PBA@PM |
20.0 °C |
16 h |
79% a; 27% b; 95% c; 30% d |
This worke |
| Cata-ref. 11 |
−10 °C |
16 h |
89% |
Ref. 11f |
| 6-a |
PBA@PM |
10.0 °C |
16 h |
37% a; 7% b; 61% c; 7% d |
This worke |
| PBA@PM |
20.0 °C |
16 h |
80% a; 25% b; 94% c; 25% d |
This worke |
| cata-ref. 11 |
−10 °C |
16 h |
88% |
Ref. 11f |
| 6-b |
PBA@PM |
10.0 °C |
16 h |
39% a; 7% b; 67% c; 6% d |
This worke |
| PBA@PM |
20.0 °C |
16 h |
82% a; 23% b; 93% c; 24% d |
This worke |
| Cata-ref. 11 |
−10 °C |
24 h |
81% |
Ref. 11f |
| 7 |
PBA@PM |
10.0 °C |
7 h |
76% a; 41% b; 92% c; 40% d |
This workg |
| PBA@PM |
20.0 °C |
7 h |
95% a; 59% b; 97% c; 61% d |
This workg |
| Cata-ref. 7 |
R.T. |
7 h |
64% |
Ref. 7h |
| 7-a |
PBA@PM |
10.0 °C |
7 h |
81% a; 45% b; 93% c; 47% d |
This workg |
| PBA@PM |
20.0 °C |
7 h |
99% a; 61% b; 99% c; 61% d |
This workg |
| Cata-ref. 7 |
R.T. |
7 h |
68% |
Ref. 7h |
| 7-b |
PBA@PM |
10.0 °C |
7 h |
79% a; 39% b; 77% c; 38% d |
This workg |
| PBA@PM |
20.0 °C |
7 h |
96% a; 43% b; 94% c; 44% d |
This workg |
| Cata-ref. 7 |
R.T. |
7 h |
62% |
Ref. 7h |
| 8 |
PBA@PM |
10.0 °C |
48 h |
8% a; 75% b; 5% c; 77% d |
This worki |
| PBA@PM |
20.0 °C |
48 h |
9% b; 97% b; 7% c; 99% d |
This worki |
| Cata-ref. 9 |
R.T. |
48 h |
99% |
Ref. 9j |
| 8-a |
PBA@PM |
10.0 °C |
48 h |
5% b; 64% b; 4% c; 65% d |
This worki |
| PBA@PM |
20.0 °C |
48 h |
6% a; 92% b; 6% c; 91% d |
This worki |
| Cata-ref. 9 |
R.T. |
48 h |
95% |
Ref. 9j |
| 8-b |
PBA@PM |
10.0 °C |
48 h |
3% a; 49% b; 2% c; 55% d |
This worki |
| PBA@PM |
20.0 °C |
48 h |
6% a; 84% b; 5% c; 83% d |
This worki |
| Cata-ref. 9 |
R.T. |
48 h |
73% |
Ref. 9j |
3. Conclusion
We have devised a class of organoboron acid catalysts PBA@PM by immobilizing PBA in microgels. With those boronic-acid-catalyzed aza-Michael addition, aldol, amidation, and [4 + 2] cycloaddition reactions in CH2Cl2/water (9:1 in volume) as the model reactions to evaluate the accessibility and catalytic performance of the PBA@PM, we have demonstrated the feasibility of tuning catalysis of boronic acids in microgels by simply bubbling with N2/CO2 gas, and in some cases adding glucose, making the catalytic activity comparable or even superior to that of the corresponding free boronic acid monomer 1 homogeneously dispersed in solutions and, more importantly, making the PBA@PM able to catalyze alternate reactions that may extend the usefulness. Based on the studies, this enhanced catalytic function of the PBA@PM is plausibly achieved via in situ reversibly structural variations. With significant advances in the development of the stimuli-responsive polymer microgels containing boronic acids, we anticipate that our results may underscore the vast potential of developing high-performance organoboron acid catalysts based on boronic-acid-containing polymer microgels in a wide range of fields.
4. Experimental section
4.1. Materials
The chemicals (3,5-diaminophenyl)boronic acid and 33 wt% HBr acetic acid solution were purchased from 1ClickChemistry Inc. (USA) and Meryer Co., Ltd (China), respectively. Acryloyl chloride, anthracene, paraformaldehyde, hexamethylenetetramine, ethylene bromohydrin, benzoyl chloride, 2-methyl-4-nitrophenol, iodobenzene diacetate (PhI(OAc)2), triethylamine, 2,2′-azobis(2-methylpropionamidine)dihydrochloride (AAPH), sodium dodecyl sulfate (SDS), and other chemicals were purchased from Aldrich. All chemicals were used directly as received without further purification. The water used in all experiments was of Millipore Milli-Q grade.
4.2. Synthesis of 1
(3,5-Diaminophenyl)boronic acid (1.000 g) was dissolved in a 5.0 wt% NaOH solution (20.00 mL) in an ice bath. After 10 min, acryloyl chloride (2.15 mL) was added dropwise into the above-mentioned solution, followed by stirring for 3 h at room temperature. After the reaction, the pH value was adjusted to ca. 1 with a 2 M HCl solution. The yielded 1 was collected, and then washed with water to yield a white solid. 1H NMR (500 MHz, in DMSO-d6; Fig. S1†): δ = 10.10 (2H, NH), 8.22 (1H, ArH), 7.99 (2H, B(OH)2), 7.67 (2H, ArH), 6.47 (2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 6.25 (2H, CHCH2), 5.73 (2H, CHCH2); 13C NMR (500 MHz, in DMSO-d6; Fig. S1†): δ = 113.14, 120.90, 126.53, 132.05, 135.51, 138.42, 163.10; FT-MS (Fig. S1†): m/z 283.09 [M + Na+].
CH2), 6.25 (2H, CHCH2), 5.73 (2H, CHCH2); 13C NMR (500 MHz, in DMSO-d6; Fig. S1†): δ = 113.14, 120.90, 126.53, 132.05, 135.51, 138.42, 163.10; FT-MS (Fig. S1†): m/z 283.09 [M + Na+].
4.3. Synthesis of 2
First, anthracene (10.000 g) and paraformaldehyde (3.400 g) were dissolved in glacial acetic acid (40.00 mL). Then, a 33 wt% HBr acetic acid solution (55.00 mL) was added dropwise (which took ca. 1 h). The mixture was heated to 80 °C and allowed to react overnight. The yielded 9,10-bis(bromomethyl)anthracene was washed with water and further purified by recrystallization in methylbenzene. Second, 9,10-bis(bromomethyl)anthracene (1.000 g) and hexamethylenetetramine (1.000 g) were dissolved in chloroform (150.00 mL) and refluxed for 48 h. After cooling to room temperature, the precipitate was collected, dried in air, and added into ethanol (130.00 mL) and concentrated HCl (25.00 mL), and refluxed for another 48 h. After cooling to ca. 0 °C, the precipitate was collected, washed with cold ethanol, extracted with chloroform/water, and dried to get anthracene-9,10-diyldimethanamine. Finally, anthracene-9,10-diyldimethanamine (0.600 g) was first dissolved in dichloromethane (40.00 mL) in a 100 mL round-bottomed flask, and then anhydrous triethylamine (1.06 mL) was added to the bottle in an ice-bath. Then, acryloyl chloride (0.690 g) was dissolved in anhydrous dichloromethane, and added into the flask dropwise. After that, the reaction was proceeded overnight at room temperature. The yielded 2 was collected, washed with water, and dried in the vacuum. 1H NMR (500 MHz, in DMSO-d6; Fig. S2†): δ = 8.56 (2H, NH), 8.48 (4H, ArH), 7.63 (2H, ArH), 6.21 (4H, ArCH2), 5.59 (2H, CHCH2), 5.40 (4H, CHCH2); FT-MS (Fig. S2†): m/z 367.14 [M + Na+].
4.4. Synthesis of the PBA@PM
SDS (0.017 g), 1 (0.016 g) and 2 (0.103 g) were dissolved in water (50.00 mL) in a 100 mL three-necked bottle, and stirred for 30 min at 70 °C in a nitrogen atmosphere. AAPH (8.0 mg) was dissolved in water (2.94 mL), and added immediately into the bottle dropwise to initiate the polymerization. After proceeding for 5 h, the product was purified by centrifugation (Thermo Electron Co. SORVALL® RC-6 PLUS super speed centrifuge) and redispersed in water, followed by 3 days of dialysis (Spectra/Por® molecular porous membrane tubing, cutoff 12000–14000) against water (replaced water every half day). The concentration of the microgels can be determined by a weighing method via drying a certain volume of the dispersion.
4.5. Catalytic experiments
The aza-Michael addition of 4 to 3, where 3 was synthesized according to a method reported in the literature (1H NMR (in CDCl3): δ = 7.91 (2H), 7.56 (1H), 7.44 (2H), 6.42 (1H), 6.31 (1H), 6.19 (1H), 4.17 (4H), 1.92 (3H); 13C NMR (in CDCl3): δ = 180.3, 155.6, 150.9, 139.4, 133.4, 133.1, 129.6, 128.7, 125.0, 124.1, 100.2, 66.4, 16.9; Fig. S13†),11,41 was chosen as a model catalytic reaction. In a typical run, 3 (1.0 × 10−4 mmol), 4 (2.5 × 10−3 mmol), and the CH2Cl2/water (9:1 in volume; 100.0 mL) mixture were added into a three-necked round flask equipped with a condenser and a magnetic stirrer, and then the temperature was adjusted to 10.0 °C by using a low-temperature constant-temperature stirring reaction bath. After stirring for 30 min, the PBA@PM was added (the concentration of boronic acids in the reaction system was ca. 1.2 × 10−5 mol L−1). This model reaction yielded product 6, i.e., N-((1R,5S)-7-benzoyl-5-methyl-6-oxa-7-azaspiro[bicyclo[3.2.1]octane-8,2′-[1,3]dioxolan]-3-en-3-yl)benzamide (1H NMR (in CDCl3): δ = 7.80–7.74 (4H), 7.66 (1H), 7.48–7.30 (6H), 6.52 (1H), 4.55 (1H), 4.13–4.03 (4H), 3.20 (1H), 3.01 (1H), 1.38 (3H); 13C NMR (in CDCl3): δ = 166.2, 165.6, 136.5, 134.6, 132.8, 131.8, 130.8, 129.2, 128.3, 127.7, 127.1, 112.1, 109.7, 81.6, 66.8, 65.7, 57.7, 34.5, 14.6; Fig. S14†), with a selectivity of nearly 100%. To investigate the yield of product 6, the samples (50.0 μL) were collected at different reaction times and purified for NMR and other analyses.
Those boronic-acid-catalyzed aldol, amidation, and [4 + 2] cycloaddition reactions were also conducted using the PBA@PM (the concentration of boronic acids in reaction systems was ca. 1.2 × 10−5 mol L−1) as an organoboron acid catalyst in CH2Cl2/water (9:1 in volume; 100.0 mL) at 10.0 °C. The model aldol reaction of hydroxyacetone (1-hydroxypropan-2-one; 1.0 × 10−4 mol L−1) and aldehyde (cinnamaldehyde; 2.5 × 10−3 mol L−1) yielded aldol adduct 7, i.e., (E)-3,4-dihydroxy-6-phenylhex-5-en-2-one (1H NMR (in CDCl3): δ = 7.33–7.15 (5H), 6.62 (1H), 6.26 (1H), 4.61 (1H), 4.18 (1H), 3.74 (1H), 2.47 (1H), 2.25 (3H); 13C NMR (in CDCl3): δ = 207.5, 136.2, 132.5, 128.5, 128.1, 127.6, 126.9, 80.2, 72.8, 26.2; Fig. S23†).7,42 The model amidation reaction of carboxylic acid (2-phenylacetic acid; 1.0 × 10−4 mol L−1) and amine (phenylmethanamine; 2.5 × 10−3 mol L−1) yielded amide product 8, i.e., N-benzyl-2-phenylacetamide (1H NMR (in CDCl3): δ = 7.33–7.15 (10H), 6.02 (1H), 4.37 (2H), 3.59 (2H); 13C NMR (in CDCl3): δ = 169.8, 137.2, 134.6, 129.3, 128.9, 128.5, 127.4, 127.3, 127.2, 43.6, 43.4; Fig. S24†).9,43 The model [4 + 2] cycloaddition between acrylic acid (2-bromoacrylic acid; 1.0 × 10−4 mol L−1) and diene (2,3-dimethylbuta-1,3-diene; 2.5 × 10−3 mol L−1) yielded cycloadduct 9, i.e., 1-bromo-3,4-dimethylcyclohex-3-ene-1-carboxylic acid (1H NMR (in CDCl3): δ = 11.62 (1H), 2.85 (1H), 2.68 (1H), 2.27 (2H), 2.18 (2H), 1.63 (6H); 13C NMR (in CDCl3): δ = 177.3, 125.0, 122.7, 59.2, 43.0, 34.1, 30.3, 19.0, 18.6; Fig. S25†).9
The compounds 6-a,11 6-b,11 7-a,7 7-b,7 8-a9 and 8-b9 were prepared using the corresponding procedure described above. The gas bubbling (if any) was continued throughout the reaction, and glucose (if any) was dissolved in the medium before adding the reactants.
4.6. Characterization
TEM images were obtained using a JEOL JEM-1400 transmission electron microscope at an accelerating voltage of 100 kV. Microgel dispersions were dropped on a carbon-coated copper grid, which was quickly put into liquid nitrogen and freeze-dried using a freeze dryer before TEM measurements. IR spectra were recorded using a Thermo Electron Corporation Nicolet 380 Fourier transform infrared spectrometer. NMR spectra were recorded using a Bruker AVIII 500 MHz solution-state NMR spectrometer. The content of PBA groups in the microgels was determined by mannitol-assisted UV-vis spectrophotometric titration, where mannitol was added to convert boronic acids into a relatively strong monobasic acid which was then titrated with 0.2 M NaOH. A change in UV-vis spectra of PBA at 235 nm was used in UV spectrophotometric titration using a Shimadzu UV-2550 UV-vis spectrometer equipped with a temperature controller (±0.1 °C).28 DLS was performed using a standard laser light scattering spectrometer (BI-200SM) equipped with a BI-9000AT digital autocorrelator (Brookhaven Instruments, Inc.). A Mini-L30 diode laser (30 mW, 637 nm) was used as the light source. All samples were passed through Millipore Millex-HV filters with a pore size of 0.80 μm to remove dust before DLS measurements.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21574107, 21774105, 21805164, 21274118, and 91227120), and the National Fund for Fostering Talents of Basic Science (J1310024). The authors thank Xuezhen Lin for the help in the catalytic tests.
References
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107 CrossRef.
- E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945 CrossRef.
- M. S. Taylor, Acc. Chem. Res., 2015, 48, 295 CrossRef CAS.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475 RSC.
- Y. Mori, K. Manabe and S. Kobayashi, Angew. Chem., Int. Ed., 2001, 40, 2815 CrossRef CAS PubMed.
- Y. Mori, J. Kobayashi, K. Manabe and S. Kobayashi, Tetrahedron, 2002, 58, 8263 CrossRef CAS.
- K. Aelvoet, A. S. Batsanov, A. J. Blatch, C. Grosjean, L. G. F. Patrick, C. A. Smethurst and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 768 CrossRef CAS.
- K. Arnold, B. Davies, D. Hérault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673 CrossRef CAS PubMed.
- R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876 CrossRef CAS PubMed.
- D. Lee, S. G. Newman and M. S. Taylor, Org. Lett., 2009, 11, 5486 CrossRef CAS PubMed.
- T. Hashimoto, A. O. Gálvez and K. Maruoka, J. Am. Chem. Soc., 2015, 137, 16016 CrossRef CAS PubMed.
- N. Levi, A. M. Khenkin, B. Hailegnaw and R. Neumann, ACS Sustainable Chem. Eng., 2016, 4, 5799 CrossRef CAS.
- J. W. Liu, H. Q. Yao and C. Wang, ACS Catal., 2018, 8, 9376 CrossRef CAS.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318 CrossRef.
- E. M. Vogl, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 1570 CrossRef CAS PubMed.
- E. Wolf, E. Richmond and J. Moran, Chem. Sci., 2015, 6, 2501 RSC.
- U. T. Bornscheuer and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2004, 43, 6032 CrossRef CAS PubMed.
- Y. J. Zhang, Y. Guan and S. Q. Zhou, Biomacromolecules, 2006, 7, 3196 CrossRef CAS PubMed.
- V. Lapeyre, I. Gosse, S. Chevreux and V. Ravaine, Biomacromolecules, 2006, 7, 3356 CrossRef CAS PubMed.
- T. Hoare and R. Pelton, Macromolecules, 2007, 40, 670 CrossRef CAS.
- Z. Wu, X. Zhang, H. Guo, C. Li and D. Yu, J. Mater. Chem., 2012, 22, 22788 RSC.
- T. Zhou, Y. Guan and Y. J. Zhang, Polym. Chem., 2014, 5, 1782 RSC.
- M. M. Zhou, J. D. Xie, S. T. Yan, X. M. Jiang, T. Ye and W. T. Wu, Macromolecules, 2014, 47, 6055 CrossRef CAS.
- Q. S. Wu, X. Du, A. P. Chang, X. M. Jiang, X. Y. Yan, X. Y. Cao, Z. H. Farooqi and W. T. Wu, Polym. Chem., 2016, 7, 6500 RSC.
- L. Liu, G. J. Ma, M. Zeng, W. Du and J. Y. Yuan, Chem. Commun., 2018, 54, 12475 RSC.
- D. H. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2012, 1, 57 CrossRef CAS.
- D. H. Han, O. Boissière, S. Kumar, X. Tong, L. Tremblay and Y. Zhao, Macromolecules, 2012, 45, 7440 CrossRef CAS.
- S. Soundararajan, M. Badawi, C. M. Kohlrust and J. H. Hageman, Anal. Biochem., 1989, 178, 125 CrossRef CAS PubMed.
- I. A. Fedorov, Y. N. Zhuravleva and V. P. Berveno, Phys. Chem. Chem. Phys., 2011, 13, 5679 RSC.
- I. Hisamitsu, K. Kataoka, T. Okano and Y. Sakurai, Pharm. Res., 1997, 14, 289 CrossRef CAS PubMed.
- M. M. Muscatello, L. E. Stunja and S. A. Asher, Anal. Chem., 2009, 81, 4978 CrossRef.
- J. E. Baldwin, T. D. W. Claridge, A. E. Derome, C. J. Schofield and B. D. Smith, Bioorg. Med. Chem. Lett., 1991, 1, 9 CrossRef CAS.
- J. Boeseken, Carbohydr. Chem., 1947, 4, 189 Search PubMed.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769 CrossRef CAS.
- K. Kondo, Y. Shiomi, M. Saisho, T. Harada and S. Shinkai, Tetrahedron, 1992, 48, 8239 CrossRef CAS.
- S. Lakhdar, M. Baidya and H. Mayr, Chem. Commun., 2012, 48, 4504 RSC.
- K. J. Laidler, The World of Physical Chemistry, Oxford University Press, 1993 Search PubMed.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813 CrossRef CAS.
- S. Carregal-Romero, N. J. Buurma, J. Pérez-Juste, L. M. Liz-Marzán and P. Hervés, Chem. Mater., 2010, 22, 3051 CrossRef CAS.
- R. Roaa, S. Angioletti-Uberti, Y. Lu, J. Dzubiella, F. Piazza and M. Ballauff, Z. Phys. Chem., 2018, 232, 773 Search PubMed.
- T. Hashimoto, H. Nakatsu and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 4617 CrossRef CAS PubMed.
- Y. Zhang and G. A. O'Doherty, Tetrahedron, 2005, 61, 6337 CrossRef CAS.
- W.-K. Chan, C.-M. Ho, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 14796 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10541g |
| ‡ These authors contribute equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Jin Lia,
Ruyue Lana,
Yusong Wangd and
Weitai Wu
c,
Jin Lia,
Ruyue Lana,
Yusong Wangd and
Weitai Wu