
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTurning weak emitters into outstanding luminescent materials using rigid host media†
Umar Sania,
Dmitry Tungulinb,
Claudia Bizzarri b and
Fabio Cucinotta*a
b and
Fabio Cucinotta*a
aSchool of Natural and Environmental Sciences, Newcastle University, King's Road, NE1 7RU Newcastle upon Tyne, UK. E-mail: fabio.cucinotta@newcastle.ac.uk
bInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
First published on 15th January 2020
Abstract
The incorporation into rigid silica host structures leads successfully to a significant luminescence enhancement of two zinc(II) dipyrrins, known to be weak emitters in solution. One of these complexes shows a fluorescence efficiency of 55% and prolonged photo-stability once entrapped in silica, demonstrating high potential for applications in energy conversion.
The search for ever more efficient and sustainable energetic solutions requires the development of new materials that are capable of high conversion rates with the least possible energy losses. Such a target is desirable, and currently tackled, for different types of conversion processes, e.g. light-to-electricity and electricity-to-light, and the primary step typically undertaken is the design of molecular dyes with controlled light absorption and emission bandwidths, alongside high transition efficiencies.1 Among the various classes of dyes studied so far, dipyrromethenes, often called dipyrrins, present particularly attractive advantages over others, such as simple, high-yielding and cheap synthetic routes, wide coordinative versatility towards semi-metals like boron and several metal ions, and finely tuneable optical activity through structural modification on the pyrrole rings by introduction of specific substituents.2–4 Alongside boron dipyrrins, the most extensively studied dipyrrin-based dyes so far, metal dipyrrins have recently gained attention and have been the subject of a growing number of studies on the application of their optical properties for energy conversion.5–8 One of the main drives for studies on metal dipyrrins is the possibility of (i) exploiting the presence of two or three dipyrrin-based chromophores in a complex, as opposed to bodipy dyes bearing only one, thus enhancing the light absorbing capability, and (ii) making use of specific central metal ions to further manipulate the optical properties and the coordination geometries.1,4 Zinc(II) dipyrrins, among other metal complexes, are particularly attractive because of the presence of an earth-abundant central ion and the ability to form supramolecular structures and coordination polymers,9–12 commonly exhibiting a linear geometry, due to the easy functionalization on the meso-position of the dipyrrin. Despite their strong absorption in the blue-green region, though, with molar extinction coefficients up to above 300
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1, they are typically weak emitters because of the strongly competitive non-radiative deactivation pathways, introduced by the distorted tetrahedral coordination and a certain structural flexibility.5
000 M−1 cm−1, they are typically weak emitters because of the strongly competitive non-radiative deactivation pathways, introduced by the distorted tetrahedral coordination and a certain structural flexibility.5
One of the possible solutions that has been recently adopted to improve the emission quantum yields of zinc complexes is the use of bulky coordinating ligands that are able to impose a high structural rigidity to the whole complexes. This strategy involves the design of suitable multi-steps synthetic routes, with the aim of expanding the dipyrrin core from the α- or the β-positions, so to introduce sterically hindering groups.7,13 An alternative solution, which we are presenting in this communication, for increasing the luminescence efficiency without having to introduce lengthy synthetic modifications is to impart the desired rigidity by encapsulation of the complexes into a spatially constraining host structure. This may, in principle, also help circumventing another strict limitation to fluorescence efficiencies that homoleptic bis(dipyrrinato) zinc complexes suffer from: the presence of symmetry-breaking charge transfer excited states, which become the lowest-lying states in polar solvents and, being prone to non-radiative decay, lead to fluorescence quenching.6
Therefore, we chose two among the weakest bis(dipyrrinato) Zn(II) emitters to build host–guest materials, where the role of the host is played by mesoporous silica of COK-12 type.14 The structures and the optical absorption of the two homoleptic complexes, 1 and 2, are shown in Fig. 1. They were first reported, respectively, by Nishihara's and Thompson's groups,6,7 and showed fluorescence quantum yields below 1% in mildly polar solvents, such as chloroform and dichloromethane, and of 2% (complex 1)13 and 16% (complex 2) in cyclohexane, the least polar solvents used in the above studies (see also Table 1).
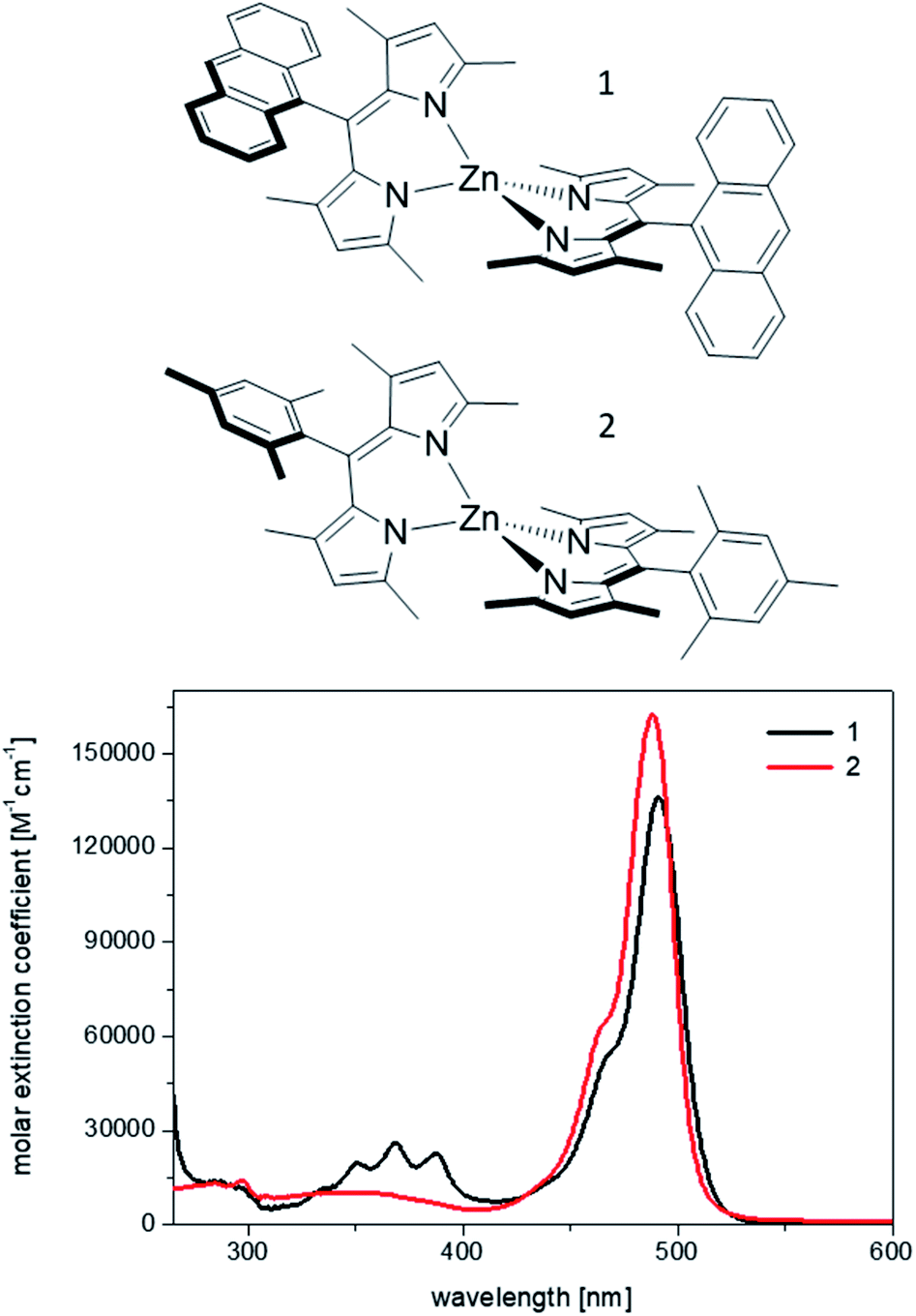 | ||
| Fig. 1 Chemical structures of the complexes 1 and 2, and absorption spectra recorded from 10−6 M dichloromethane solutions. | ||
| Sample | ΦF | τFc/ns | kR/s−1 | kNR/s−1 |
|---|---|---|---|---|
| a From 10−6 M dichloromethane solutions.b From equimolar water suspensions and confirmed by solid state measurements using an integrated sphere.c Laser excitation at 475 nm, pulse width 62 ps, recorded at 530 nm.d In cyclohexane (ref. 13). | ||||
| 1a | 0.003 | 1.75 | 1.71 × 106 | 5.70 × 108 |
| 0.02d | ||||
| 1Sb | 0.035 | 0.43 | 8.14 × 107 | 2.24 × 109 |
| 2a | 0.005 | 1.40 | 3.57 × 106 | 7.11 × 108 |
| 0.16d | 2.4d | 7.5 × 107d | 3.41 × 108d | |
| 2Sb | 0.550 | 1.34 | 4.10 × 108 | 3.36 × 108 |
The experimental approach we used for improving the fluorescence yields further above the values obtained in non-polar solvents takes advantage of a well-established soft-template route: here, amphiphilic surfactants self-assemble in water to form rod-like micelles, around which a silica network grows from the hydrolysis and polycondensation of a suitable precursor.15 Due to the insolubility of the zinc dipyrrins in water, their addition during the self-assembly process leads to their spontaneous diffusion and encapsulation into the solvent-free, rigidified, hydrophobic core of the micelles, which are built using the gemini surfactant Pluronic-P123. In order to enable a homogeneous dispersion of the dyes, minimize intermolecular aggregation and prevent structural disruption of the micelles and, consequently, of the silica particles, we applied a recent strategy established in our group,16 using a low loading of complexes, at a 99:1 molar ratio of Pluronic-P123: complex for each of the host–guest systems, hereafter named 1S and 2S. The final samples were characterized by transition electron microscopy (TEM, Fig. 2) and small-angle X-ray scattering (SAXS, Fig. S7, see ESI†), which confirm that the structural order and the morphology of the dye-doped silica particles are essentially preserved. The SAXS data reported in Fig. S7† show that the most intense peaks for the silica materials occur at q values of 0.64–0.66° and the q ratios of the main peak to the much weaker additional peaks at higher q conform within experimental error to the theoretical q ratios of 1:√3:2:√7 that correspond to the 2D hexagonal reflections of (10), (11), (20) and (21), respectively. The structures are, thus, consistent with literature values for COK-12 silica14 and prove that the dye-doped P-123 micelles maintain the same structural order of plain silica. This can be indeed visualised from the TEM micrographs in Fig. 2, where the silica particles show a highly ordered morphology with well visible parallel channels.
 | ||
| Fig. 2 TEM micrographs of the host–guest systems 1S (top) and 2S (bottom). | ||
Therefore, as the pore channels do not undergo a detectable distortion (i.e. expansion), following encapsulation of the complexes, we can infer that the latter must experience considerably strong spatial constraints once entrapped into the hydrophobic core of the cylindrical micelles. UV-visible spectroscopy studies were then carried out on the samples, to evaluate the effects that such a structural rigidity exerts on the optical properties of the complexes. Because of the strong scattering effects caused by the silica suspensions, no absorption spectra could be recorded. Photoluminescence excitation spectra were measured in dichloromethane (DCM), which has a dielectric constant of 8.93. No changes were observed in the energy of the excitation transitions in comparison to that of the free complexes in solution (Fig. S8, see ESI†). The emission energies of the complexes within the silica particles also remain unaltered, but two striking differences are noted. First, the fluorescence intensities are impressively enhanced, as it can be seen in Fig. 3.
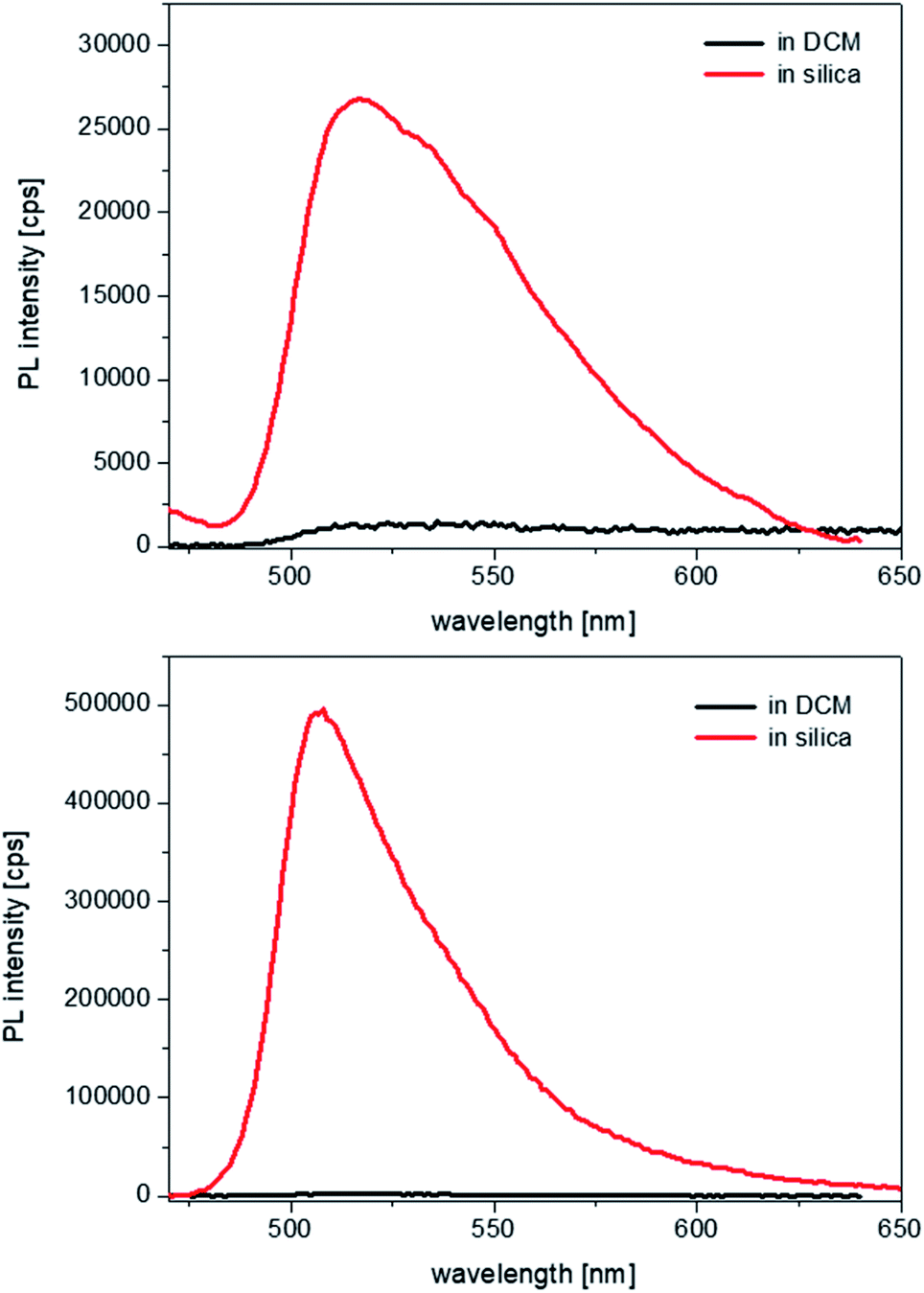 | ||
| Fig. 3 Emission spectra of the complexes 1 (top) and 2 (bottom) from 4 × 10−6 M dichloromethane solutions (black lines), compared with equimolar suspensions of the host–guest silica systems in water. All spectra were recorded at λexc = 450 nm, with both excitation and emission slits set at 5 nm. | ||
Second, the emission bandwidth becomes much narrower in silica than in solution for both complexes, with the full width at half-maxima (FWHM) reducing from values that exceed 4300 cm−1, which are hardly measurable with absolute precision in solution due to the extremely weak and broad emission, to values in silica of ∼2300 cm−1 for 1 and ∼1200 cm−1 for 2. Such an effect is indicative of a restricted vibrational freedom of the complexes within the constrained micellar environment; moreover, considering that the FWHM in the emission band of 2 becomes close to the value in excitation, ∼1100 cm−1, it suggests that the structural rigidification forces the geometries of the ground and the excited state to be more similar to one another.
The fluorescence quantum yields of all samples were recorded and allowed us to make some important observations: a 10-fold increase in the emission of 1 occurs upon incorporation and the enhancement is even 100-fold in the case of 2 (see Table 1, comparing with the values in dichloromethane solution). The high sensitivity of luminescence quantum yields and lifetimes on solvent polarity deserves further clarification here, in light of previous research. As it was demonstrated by Nishihara and Thompson,6,7 homoleptic zinc bis-dipyrrins undergo photoinduced charge transfer from one ligand to the second one, which breaks the symmetry of the excited state. The latter is indeed defined as a symmetry-breaking charge-transfer (SBCT) state and becomes the lowest state even in mildly polar solvents such as dichloromethane. Since SBCT states are typically non-emissive in nature, this leads to a total emission quenching in dichloromethane, with fluorescence yields of 0.3% and 0.5% for 1 and 2, respectively, whereas in relatively apolar cyclohexane the above values increase up to 2% and 16%.13 Such trend reaches its extreme in the solvent-free and apolar environment of the micellar structures within the silica channels: therein, the absence of solvent stabilisation and the rigid environment further reduce the non-radiative deactivation pathways and lead to emission yields of 3.5% and 55%, respectively, for 1 and 2. The excited state lifetimes were also measured and show relatively milder effects from solution to solid state: a reduction by a factor of four for 1, whereas no significant change for 2 (see also the full decay profiles in ESI, Fig. S9†).
More insights on the photophysical dynamics can be gained from the comparison between the radiative and the non-radiative decay rates. For both complexes, the radiative rates increase dramatically, nearly 50-fold for 1 and over 100-fold for 2, and we consider this a further effect of the encapsulation of the complexes. Considering the restricted vibrational and rotational degrees of freedom within the silica host, significant geometrical distortions in the complexes from the ground to the excited state are energetically unfavoured; this may translate in an increased probability of radiative deactivation taking place from an excited state conformations near the initial Franck–Condon form. Such situation has also been demonstrated in previous examples of dipyrromethene complexes, where rigidification of the organic backbones with sterically hindered groups proved to increase the radiative rate.13,17
On the other hand, the non-radiative rate almost quadruples for 1, whereas it halves for 2, showing that 1 is more affected by non-radiative quenching than 2 once it is encapsulated inside the silica template. We ascribe this behaviour to the effect that a partial intermolecular aggregation may have on 1, resulting from the tendency of the anthracene moieties to undergo π-stacking in a more constrained environment than in fluid solution. Such an effect could not be observed for 2, where the sterically hindered mesityl group prevents aggregation. Indeed, the reduced vibrational deactivation pathways in 2 contribute to a much larger increase in emission quantum yield than for 1.
Considering the outstanding fluorescence efficiency of the 2S silica particles, we evaluated their possible exploitation for the design of new luminescent solar concentrators.18,19 For this purpose, thin films were prepared by dispersion of the luminescent silica particles in a transparent polymer, polymethyl methacrylate (PMMA), and the photostability of such films was tested against steady UV light irradiation over the course of twelve hours.
For comparison, the tests were carried out also on polymeric films doped with the free complexes, using the same concentration of complexes present in the films containing the silica particles. The results are shown in Fig. 4, which reports the observed trend in photoluminescence intensity together with photographs of the polymeric films exposed to UV light before and after the stability tests. It is evident that the complex 2, once incorporated into the silica, retains its luminescence intensity throughout the test, whereas the emission from the free complex dispersed in PMMA halves its intensity in less than two hours of irradiation and photobleaches almost completely after twelve hours.
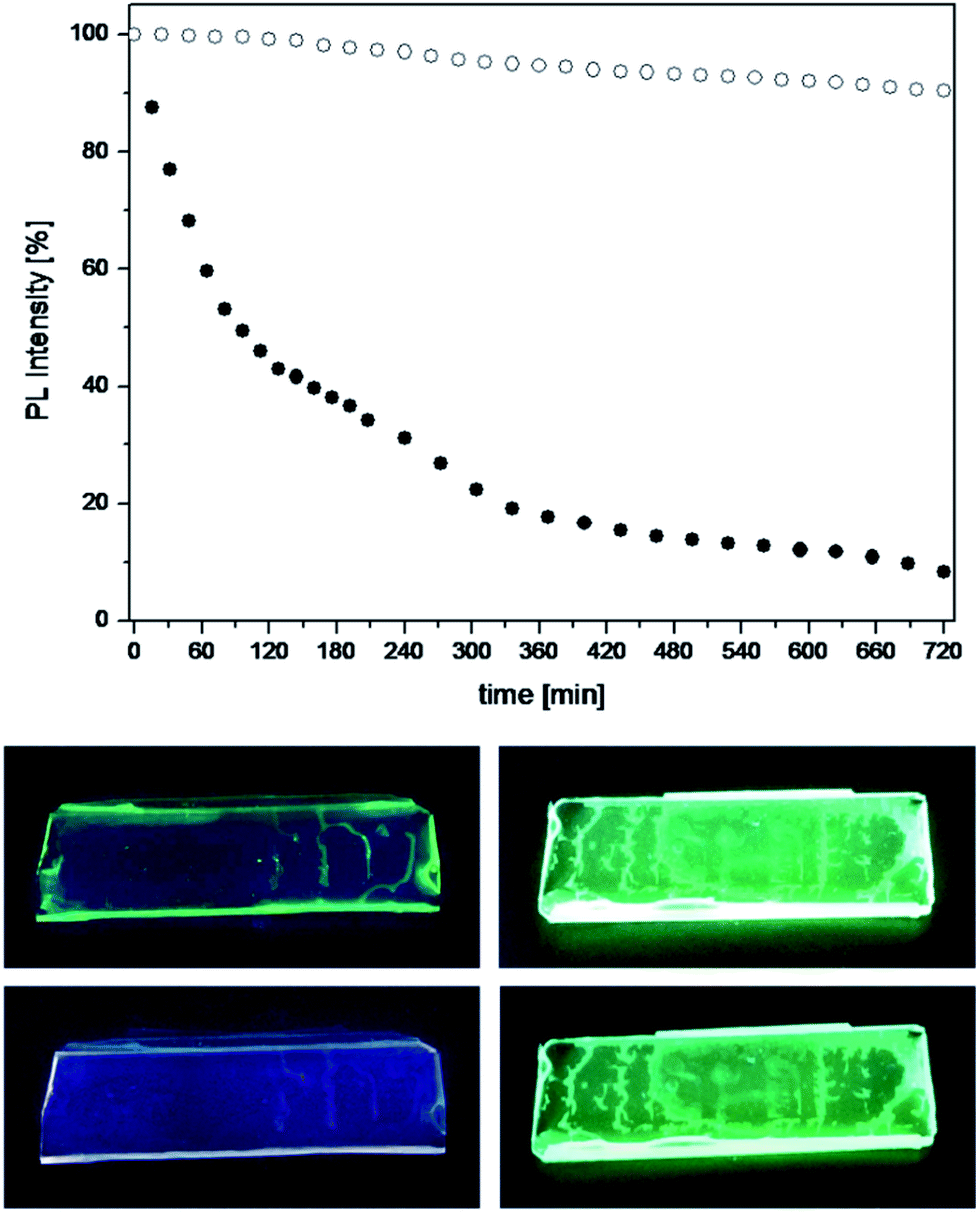 | ||
| Fig. 4 Top: photoluminescence stability tests performed on thin polymer films doped with the free complex 2 (black circles) and with silica particles embedding the complex (white circles); irradiation wavelength = 450 nm; detection wavelength = 520 nm. Bottom: photographs of the 2-doped (left) and the 2S-doped PMMA films (right), under UV light (365 nm), before (top pictures) and after (bottom pictures) the irradiation tests. | ||
It can then be concluded that the assembly strategy used in this study to improve the luminescence properties of notoriously weakly emissive zinc dipyrrins is impressively beneficial and it also serves as a mean to suppress photo-induced degradation of the complexes, demonstrating the potential of the systems presented in this communication for applications in sensitisation and light-emission technologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support by the EPSRC (grant code EP/P015395/1) and the BBSRC (grant code BB/R013942/1). We also thank the Petroleum Technology Development Fund (US), the Toso Montanari Fellowship (DT) and the staff of Newcastle University Electron Microscopy Research Services for their assistance. CB thanks Prof. Bräse (KIT) for laboratory space and fruitful discussion. Data supporting this publication is openly available under an ‘Open Data Commons Open Database License’. Additional metadata are available at: http://dx.doi.org/10.25405/data.ncl.11558826.Notes and references
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342 RSC.
- T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- S. Baudron, Dalton Trans., 2013, 42, 7498 RSC.
- I. V. Sazanovich, C. Kirmaier, E. Hindin, L. Yu, D. F. Bocian, J. S. Lindsey and D. Holten, J. Am. Chem. Soc., 2004, 126, 2664 CrossRef CAS PubMed.
- C. Trinh, K. Kirlikovali, S. Das, M. E. Ener, H. B. Gray, P. Djurovich, S. E. Bradforth and M. E. Thompson, J. Phys. Chem. C, 2014, 118, 21834 CrossRef CAS PubMed.
- R. Sakamoto, T. Iwashima, J. F. Kçgel, S. Kusaka, M. Tsuchiya, Y. Kitagawa and H. Nishihara, J. Am. Chem. Soc., 2016, 138, 5666 CrossRef CAS PubMed.
- R. Sakamoto, T. Iwashima, M. Tsuchiya, R. Toyoda, R. Matsuoka, J. F. Kögel, S. Kusaka, K. Hoshiko, T. Yagi, T. Nagayamam and H. Nishihara, J. Mater. Chem. A, 2015, 3, 15357 RSC.
- R. Sakamoto, K. Hoshiko, Q. Liu, T. Yagi, T. Nagayama, S. Kusaka, M. Tschiya, Y. Kitagawa, W.-Y. Wong and H. Nishihara, Nat. Commun., 2015, 6, 6713 CrossRef CAS PubMed.
- R. Matsuoka, R. Toyoda, R. Sakamoto, M. Tsuchiya, K. Hoshiko, T. Nagayama, Y. Nonoguchi, K. Sugimoto, E. Nishibori, T. Kawai and H. Nishihara, Chem. Sci., 2015, 6, 2853 RSC.
- R. Sakamoto, T. Yagi, K. Hishiko, S. Kusada, R. Matsuoka, H. Maeda, Z. Liu, Q. Liu, W.-Y. Wong and H. Nishihara, Angew. Chem., Int. Ed., 2017, 56, 3526 CrossRef CAS PubMed.
- R. Matsuoka and T. Nabeshima, Front. Chem., 2018, 6, 349 CrossRef PubMed.
- D. Tungulin, J. Leier, A. B. Carter, A. K. Powell, R. Q. Albuquerque, A. N. Unterreiner and C. Bizzarri, Chem.–Eur. J., 2019, 25, 3816 CrossRef CAS PubMed.
- J. Jammaer, A. Aerts, J. D'Haen, J. W. Seo and J. A. Martens, J. Mater. Chem., 2009, 19, 8290 RSC.
- L. Grösch, Y. J. Lee, F. Hoffmann and M. Fröba, Chem.–Eur. J., 2015, 21, 331 CrossRef PubMed.
- A. J. Bagnall, M. Santana Vega, J. Martinelli, K. Djanashvili and F. Cucinotta, Chem.–Eur. J., 2018, 24, 11992 CrossRef CAS PubMed.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433 CrossRef CAS PubMed.
- M. G. Ddebije and P. C. Verbunt, Adv. Energy Mater., 2012, 2, 12 CrossRef.
- F. Meinardi, F. Bruni and S. Brovelli, Nat. Rev., 2017, 2, 17072 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10727d |
| This journal is © The Royal Society of Chemistry 2020 |