
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Newly synthesized 3-(4-chloro-phenyl)-3-hydroxy-2,2-dimethyl-propionic acid methyl ester derivatives selectively inhibit the proliferation of colon cancer cells
Samir M. El Rayes *a,
Ahmed Aboelmagda,
Mohamed S. Gomaab,
Walid Fathallac,
Ibrahim A. I. Alia,
Faheem H. Pottood and
Firdos Alam Khane
*a,
Ahmed Aboelmagda,
Mohamed S. Gomaab,
Walid Fathallac,
Ibrahim A. I. Alia,
Faheem H. Pottood and
Firdos Alam Khane
aDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, Egypt
bDepartment of Pharmaceutical, College of Clinical Pharmacy, Imam Abdulrahman Bin Faisal University, P. O. Box 1982, Dammam 31441, Kingdom of Saudi Arabia
cDepartment of Physics and Math., Faculty of Engineering, Port-Said University, Port-Said, Egypt
dDepartment of Pharmacology, College of Clinical Pharmacy, Imam Abdulrahman Bin Faisal University, P. O. Box 1982, Dammam 31441, Kingdom of Saudi Arabia. E-mail: fhpottoo@iau.edu.sa
eDepartment of Stem Cell Research, Institute of Research and Medical Consultations (IRMC), Imam Abdulrahman Bin Faisal University, P. O. Box 1982, Dammam 31441, Saudi Arabia. E-mail: fakhan@iau.edu.sa
First published on 28th February 2020
Abstract
A series of 24 compounds were synthesized based on structure modification of the model methyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate as potent HDACIs. Saponification and hydrazinolysis of the model ester afforded the corresponding acid and hydrazide, respectively. The model ester was transformed into the corresponding trichloroacetimidate or acetate by the reaction with trichloroacetonitrile and acetic anhydride, respectively. N-Alkyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropan-amides and methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino] alkanoates were obtained by the reaction of corresponding acid or hydrazide with amines and amino acid esters via DCC and azide coupling methods. Methyl-3-aryl-3-(4-chlorophenyl)-2,2-dimethylpropanoates were obtained in good yields and short reaction time from the corresponding trichloroacetimidate or acetate by the reaction with C-active nucleophiles in the presence of TMSOTf (0.1 eq.%) via C–C bond formation. The antiproliferative and apoptotic activity were further studied with molecular docking. The 48 post-treatments showed that out of 24 compounds, 12 compounds showed inhibitory actions on HCT-116 cells, we have calculated the inhibitory action (IC50) of these compounds on HCT-116 and we have found that the IC50 values were in between 0.12 mg mL−1 to 0.81 mg mL−1. The compounds (7a & 7g) showed highest inhibitory activity (0.12 mg mL−1), whereas compound 7d showed the lowest inhibitory activity (0.81 mg mL−1). We have also examined inhibitory action on normal and non-cancerous cells (HEK-293 cells) and confirmed that action of these compounds was specific to cancerous cells. The cancerous cells were also examined for nuclear disintegration through staining with DAPI, (4′,6-diamidino-2-phenylindole) is a blue-fluorescent DNA stain, and we have found that there was loss of DAPI staining in the compound treated cancerous cells. The compounds were found to potentially act through the HSP90 and TRAP1 mediated signaling pathway. Compounds 7a and 7g showed the highest selectivity to TRAP1 which explained its superior activity.
Introduction
Malignancy is one of the significant factors behind loss of life in the developed countries.1–3 Chemotherapy with cytotoxic medications is one of the primary approaches to dealing with established malignancy.4,5 The primary drawbacks of malignancy chemotherapy is the severe poisonous results such as emesis and myelosuppression, as well as the insufficient selectivity of the drugs against cyst tumor cellular material in comparison with normal cellular material.1,6 Hence, searching for newer anticancer drugs is a never-ending job. Key interactions at protein–protein interfaces constitute important targets for small molecule inhibition because of their specific arrangements and biological importance.7Recently, we reported the synthesis of 3-(4-(2-chloroacetamido)phenyl)-3-hydroxy-2,2-dimethylpropanoate and 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate as potent HDACIs.8 We also showed that the antiproliferative activity for these compounds against HeLa cells (IC50; 11–0.69 μM) was excellent in comparison with the standard drug doxorubicin (IC50; 2.29 μM).8 Our compounds are peptide in nature that mimic peptide inhibitors and non-peptide inhibitors, together with being more suitable for pharmaceutical manipulations and development and this work provides integration of our research effort in discovering and modifying new anticancer agents.8,9
Results and discussions
Chemistry
Herein, we report the synthesis of a series of compounds based on structure modification of the model methyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) as promising HDACIs. The model compound 3 was prepared by the reaction of 4-chlorobenzaldehyde 1 and trimethylsilyl ketene acetal 2 in the presence of pyridine-N-oxide and LiCl in DMF at room temperature under nitrogen atmosphere to afford 3 in 69% yield, Scheme 1. | ||
| Scheme 1 Synthesis of methyl-3-hydroxy-2,2-dimethyl-3-(4-chlorophenyl)propanoate (3). | ||
DCC and azide coupling methods are well-recognized in peptide synthesis centered on carboxylic acid-carbonyl group activation to be attacked by nucleophiles as amines or even weaker nucleophiles as amino acids to form a peptide bond.10–13 Structure modification of ester 3 could be achieved by attachment of alkane amine or amino acids to the carbonyl group of 3 via DCC or azide coupling and the formation of peptide bond. Thus, the reaction of methyl 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) with hydrazine hydrate in ethanol under reflux condition for 9 h afforded the corresponding hydrazide 4. The model ester 3 was hydrolyzed using KOH solution in 50% alcohol water mixture at 78 °C for 6 h to afford the corresponding carboxylic acid 6, Scheme 2.
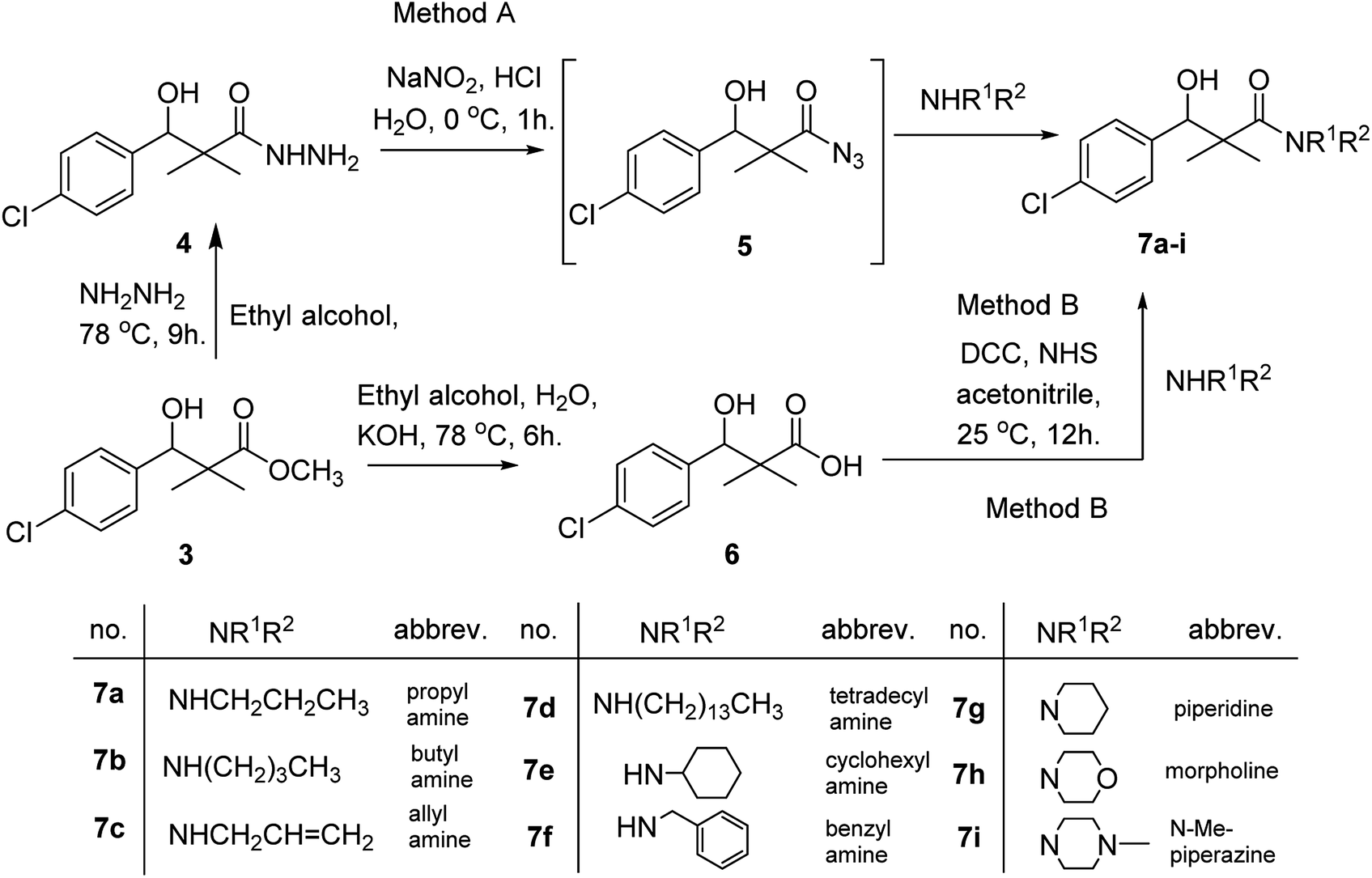 | ||
| Scheme 2 Synthesis of N-alkyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethyl-propanamides 7a–i. | ||
Hydrazide 4 was reacted with NaNO2 and HCl in water at 0 °C for 1 h to afford the corresponding azide 5 and were extracted with ethyl acetate. The in situ generated azide 5 solution was successively added to primary amines; propyl amine, butyl amine, allyl amine, tetradecyl amine, cyclohexyl amine and benzyl amine or secondary amines; piperidine, morpholine and N-methylpiperazine at 0 °C for 12 h to afford the corresponding N-alkyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamides 7a–i following the azide coupling method, Scheme 2.
The reaction of carboxylic acid derivatives 6 with primary amines and secondary amines in the presence of N,N′-dicyclohexylcarbodiimide and N-hydroxylsuccinimide (NHS) in acetonitrile at room temperature for 12 h afforded our products 7a–i as an equivocal method of preparation following HOSu-DCC coupling method, Scheme 2. Comparing both coupling methods leading to 7a–i we found out that azide coupling was more efficient respect to % of yield and simple reaction workup.
The structure assignment of the prepared N-alkyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamides 7a–i is based on 1H and 13C NMR spectral and physicochemical analysis. The 1H NMR spectrum of 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethyl-N-propylpropanamide 7a shows signals at δ 5.72, 4.68, 4.07 and 4.05–4.02 ppm corresponding to NH, OCH, OH and CH2 groups respectively. The 13C NMR spectrum of 7a shows signals at 178.9, 80.9, 60.8 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), (OCH) and (C) groups respectively.
O), (OCH) and (C) groups respectively.
Similarly, methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]alkanoates 8a–g were prepared by the reaction of the in situ generated azide 5 solution in ethyl acetate with amino acid ester hydrochlorides; glycine, β-alanine, γ-amino butyric acid, L-alanine, L-valine, L-leucine and L-methionine in the presence of triethylamine to give 8a–g in excellent yields, Scheme 3. An equivocal synthesis of 8a–g was achieved by the reaction of acid 6 with amino acid ester hydrochlorides in the presence of N,N′-dicyclohexylcarbodiimide and N-hydroxylsuccinimide (NHS) in acetonitrile at room temperature for 12 h afforded our product 8a–g, Scheme 3.
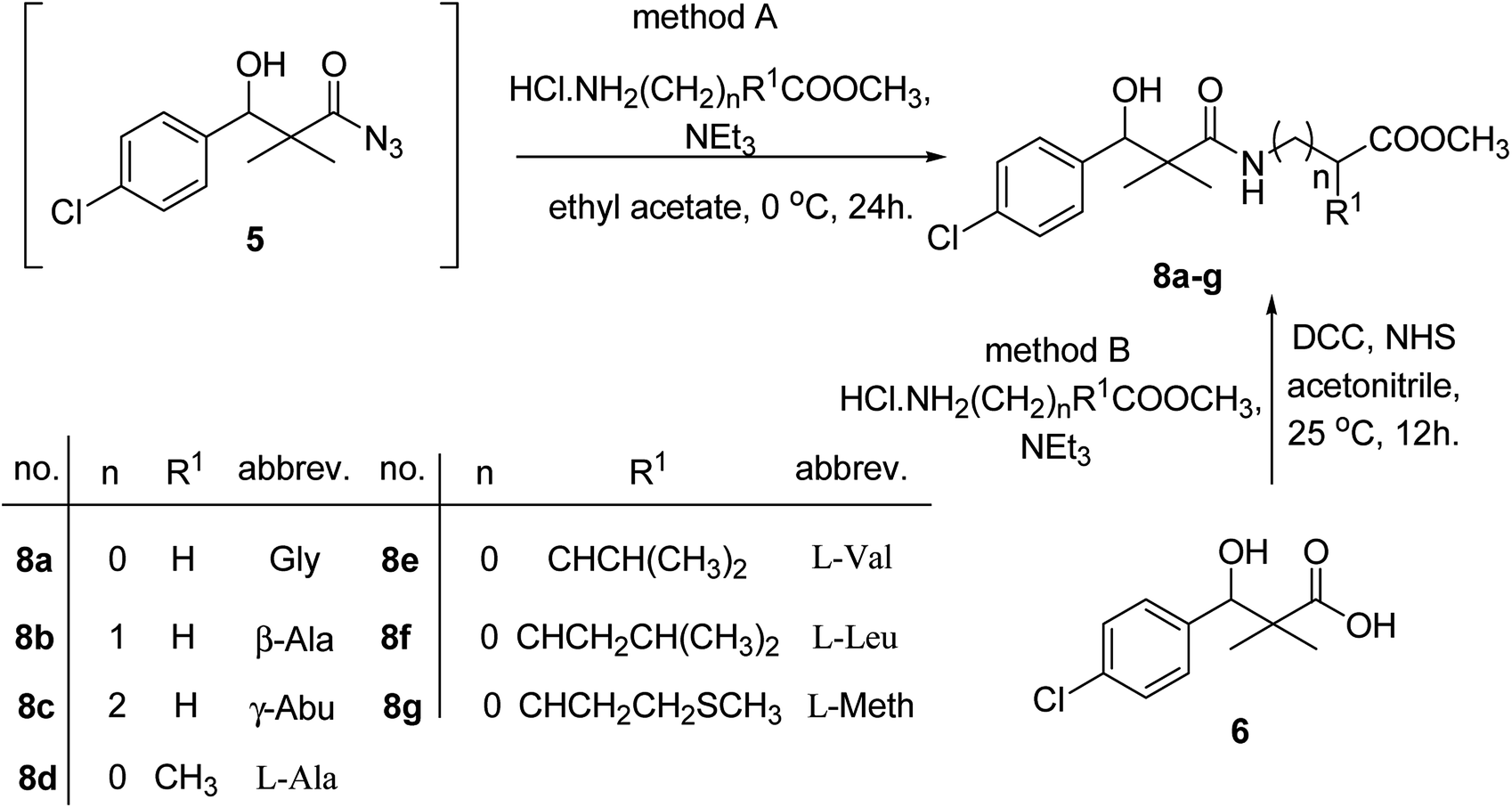 | ||
| Scheme 3 Synthesis of methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]alkanoates 8a–g. | ||
The structure assignment of the prepared methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]alkanoates 8a–g is based on 1H and 13C NMR spectral and physicochemical analysis. The 1H NMR spectrum of methyl 2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]acetate 8a shows signals at δ 6.67, 5.22, 4.63, 3.92 and 3.68 ppm corresponding to NH, OH, CH, NCH2 and OCH3 groups, respectively. The 13C NMR spectrum of 8a shows signals at 177.6, 170.1, 79.7, 51.5, 45.2, 41.4 (CO), (CO), (OCH), (OCH3), (C) and (NCH2) groups, respectively.
Trichloroacetimidate and acetate C–C coupling proved to be excellent methods for structure modifications of alcohols.14–18 These methods are related to transform hydroxyl group substrates into the appropriate trichloroacetimidate or acetate as excellent leaving groups by the reaction with trichloroacetonitrile or acetic anhydride, respectively. The successive addition of C-nucleophiles mainly activated arenes, allyltrimethylsilane and trimethylsiloxyalkenes to the active trichloroacetimidate or acetate intermediates in the presence of Lewis acid gave the desired products through the formation of new C–C bond.14–18 We find it interesting to apply the trichloroacetimidate and acetate coupling methods in the structure modification of methyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) via C–C coupling with methoxybenzene derivatives. Thus, the reaction of the ester 3 with trichloroacetonitrile in presence of DBU in dichloromethane at 25 °C for 2 h afforded the trichloroacetimidate 9. Similarly, the reaction of ester 3 with acetic anhydride in the presence of DMAP (N,N-dimethylaminopyridine) in dichloromethane at 25 °C for 4 h afforded methyl 3-acetoxy-3-(4-chlorophenyl)-2,2-dimethylpropanoate (10). The reaction of trichloroacetimidates 9 or acetate 10 with arene C-nucleophiles; anisole, 1,4-dimethoxybenzene, 1,2-dimethoxybenzene and 1,2,3-trimethoxybenzene in the presence of catalytic amount of trimethylsilyltrifluoro-methanesulfonate (TMSOTf) (0.1 eq.%) at room temperature gave readily the methyl 3-aryl-3-(4-chlorophenyl)-2,2-dimethylpropanoate 11a–e in excellent yields, Scheme 4.
 | ||
| Scheme 4 Synthesis of methyl-3-aryl-3-(4-chlorophenyl)-2,2-dimethylpropanoate 11a–e. | ||
Comparing the efficiency of both C–C coupling methods according to reaction time and percent of yield, results showed that, although, all compounds were prepared in good yields, there was a slight improvement in the percent of yield and in the reaction time (monitored by TLC) using trichloroacetimidate method, Table 1.
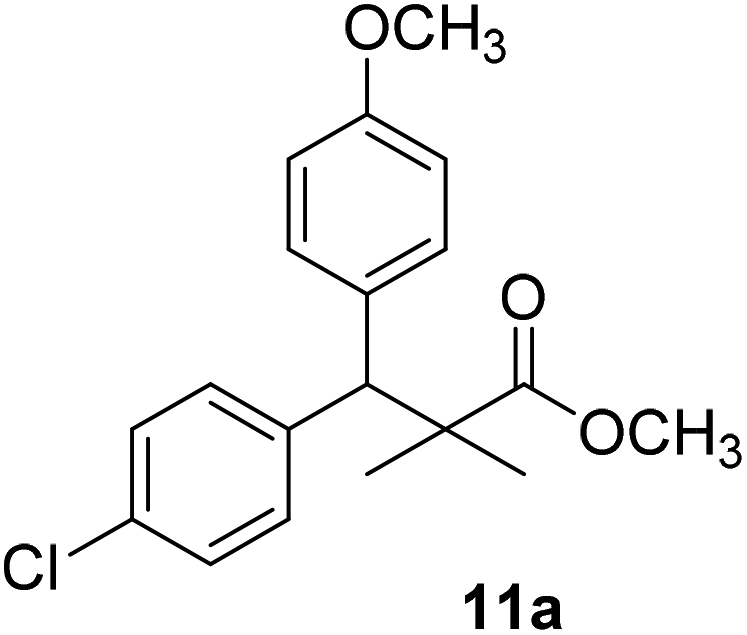
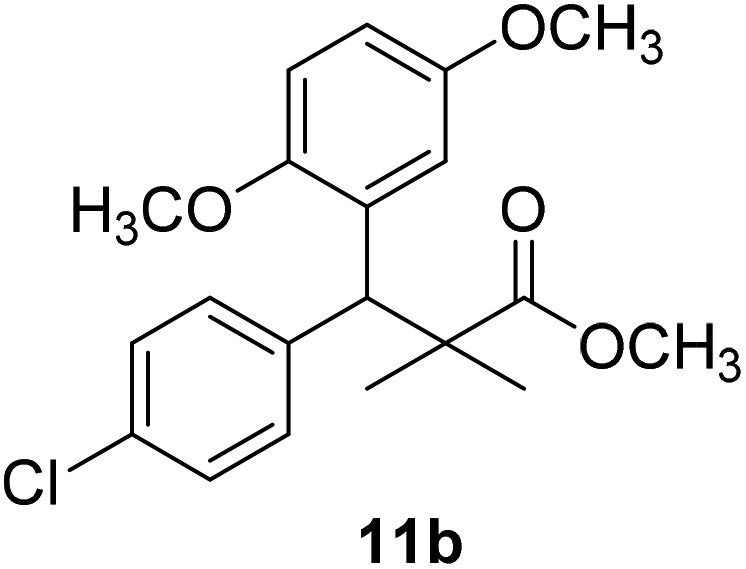

The structure assignment of the prepared methyl 3-aryl-3-(4-chlorophenyl)-2,2-dimethylpropanoate 11a–e is based on 1H and 13C NMR spectral and physicochemical analysis. The 1H NMR spectrum of methyl 3-(4-chlorophenyl)-3-(4-methoxyphenyl)-2,2-dimethylpropanoate (11a) shows signals at δ 4.23, 3.68, 3.44 ppm corresponding to CH, OCH3 and OCH3 groups, respectively. The 13C NMR spectrum of 11a shows signals at δ 176.2, 56.1, 53.2, 48.5, 47.2 ppm corresponding to (CO), (OCH3), (OCH3), (CH) and (C) groups, respectively.
Antiproliferative activities
The cytotoxic impact of compounds on cancer cell's viability was assessed by MTT assay. The 48 post-treatments showed that out of 24 compounds, 12 compounds showed inhibitory actions on HCT-116 cells, whereas 10 compounds did not show any inhibitory actions. We have calculated the inhibitory action (IC50) of these compounds on both HCT-116 and HEK-293 cells and we have found that IC50 were in between is 0.12 mg mL−1 to 0.81 mg mL−1. The compounds (7a, 7g) showed highest inhibitory activity (0.12 mg mL−1), whereas compound 7d showed lowest inhibitory activity (0.81 mg mL−1). We have also examined inhibitory action on normal and non-cancerous cells (HEK-293 cells) to confirm whether these compounds produce any effects on them. We have tested these compounds on normal healthy cells (HEK-293) and MTT results revealed that all these compounds did not produce any inhibitory action on the normal cells which suggest that synthetized compounds are specially targeted to cancerous cells. There are several reports of treatments of biomaterials which showed inhibitory action on different cancerous cells.19–27 The IC50 values for all compounds were given in the Table 2.| Compound number | Structure of compounds | IC50 value HCT-116 | IC50 value HEK-293 |
|---|---|---|---|
| a NA = not active, IC50 value [mg mL−1] = inhibitory concentration (IC) is expressed in mg mL−1. | |||
| 4 | 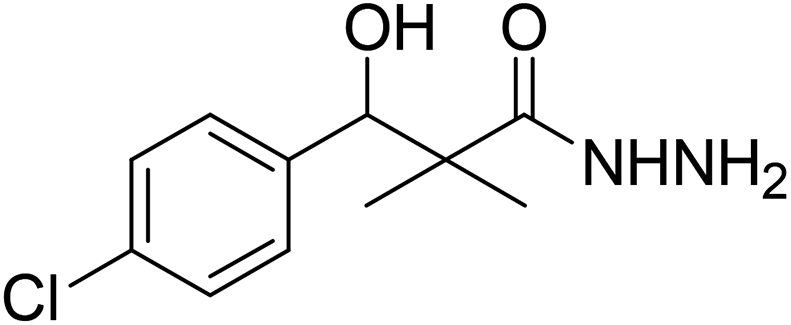 |
0.41 mg mL−1 | NA |
| 6 | 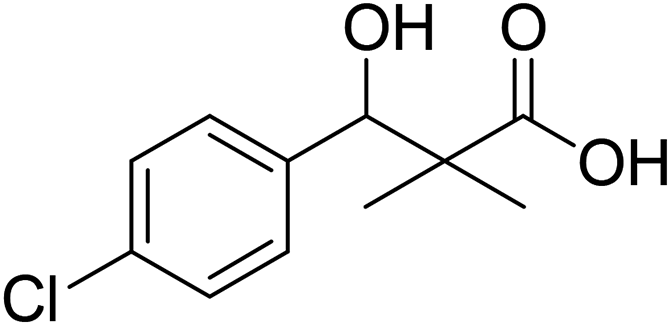 |
0.29 mg mL−1 | NA |
| 7a | 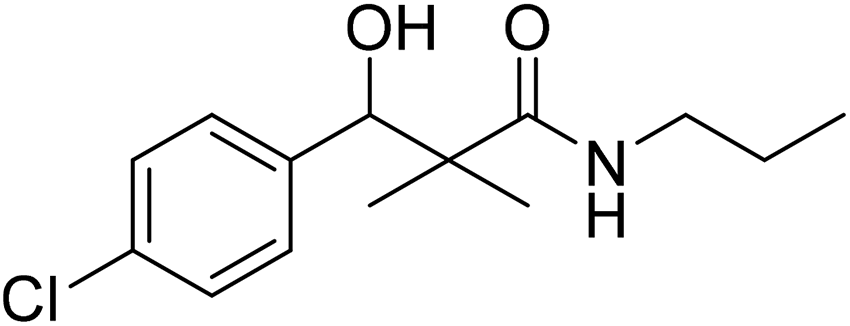 |
0.12 mg mL−1 | NA |
| 7b |  |
NA | NA |
| 7c | 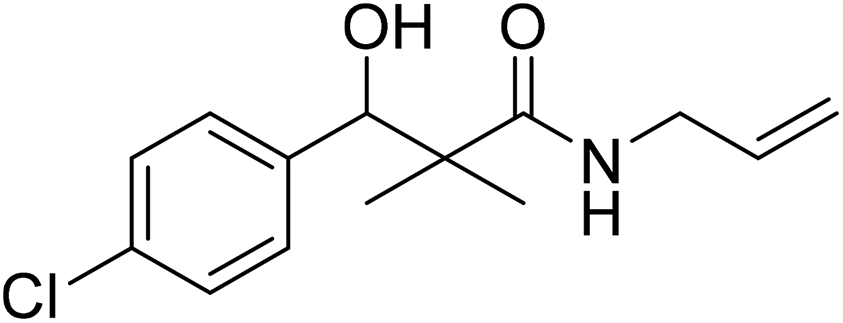 |
0.22 mg mL−1 | NA |
| 7d | 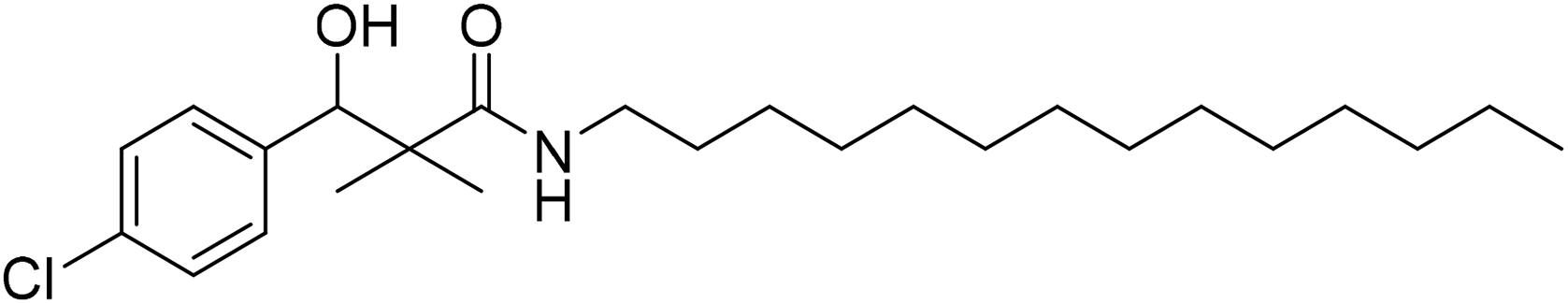 |
0.81 mg mL−1 | NA |
| 7e | 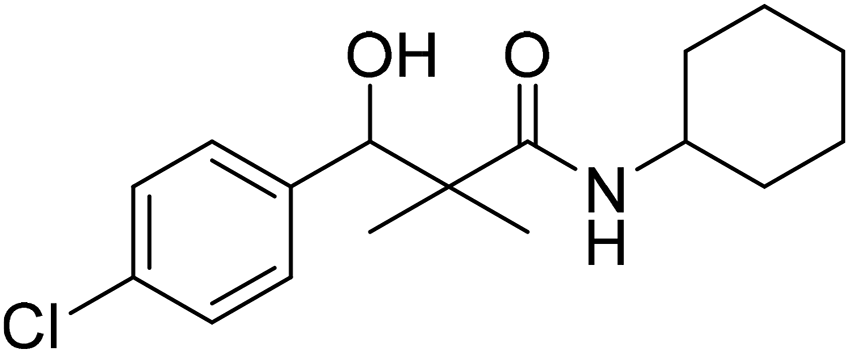 |
NA | NA |
| 7f | 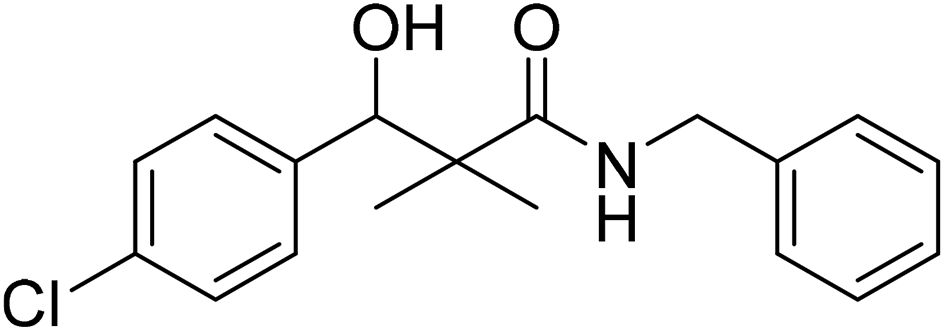 |
0.61 mg mL−1 | NA |
| 7g | 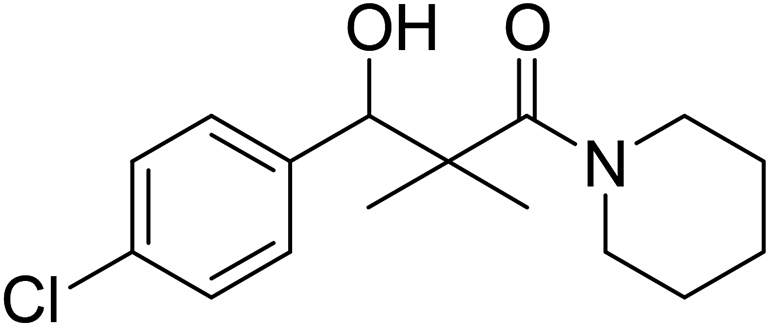 |
0.12 mg mL−1 | NA |
| 7h | 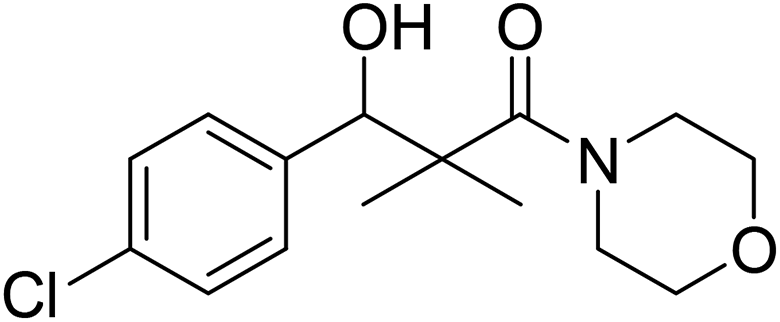 |
0.68 mg mL−1 | NA |
| 7i | 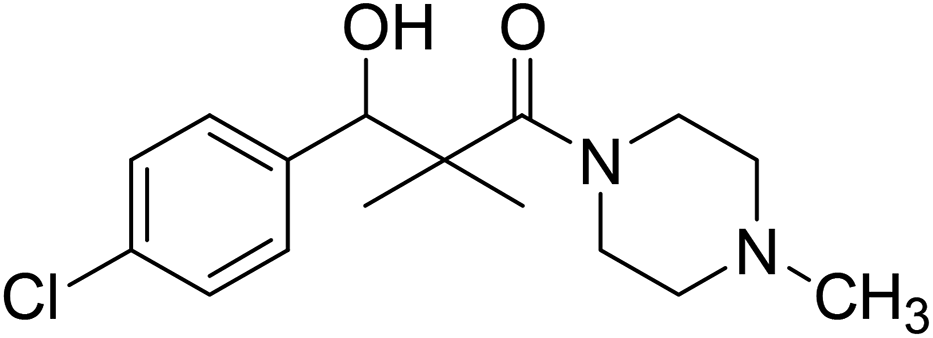 |
0.13 mg mL−1 | NA |
| 8a |  |
0.51 mg mL−1 | NA |
| 8b |  |
0.58 mg mL−1 | NA |
| 8e | 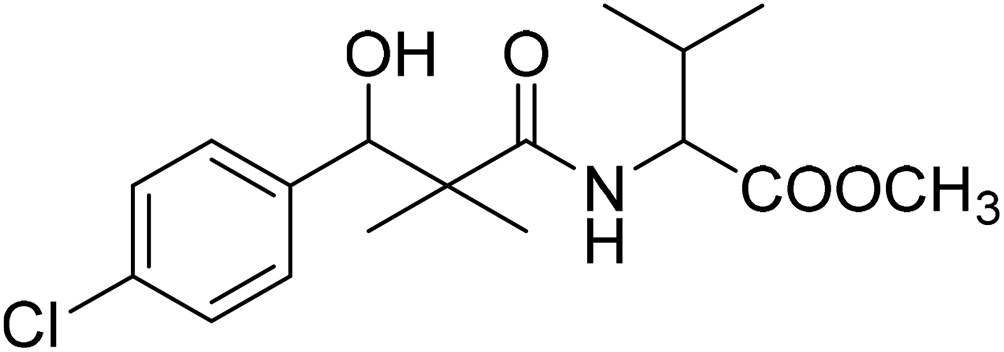 |
NA | NA |
| 8f | 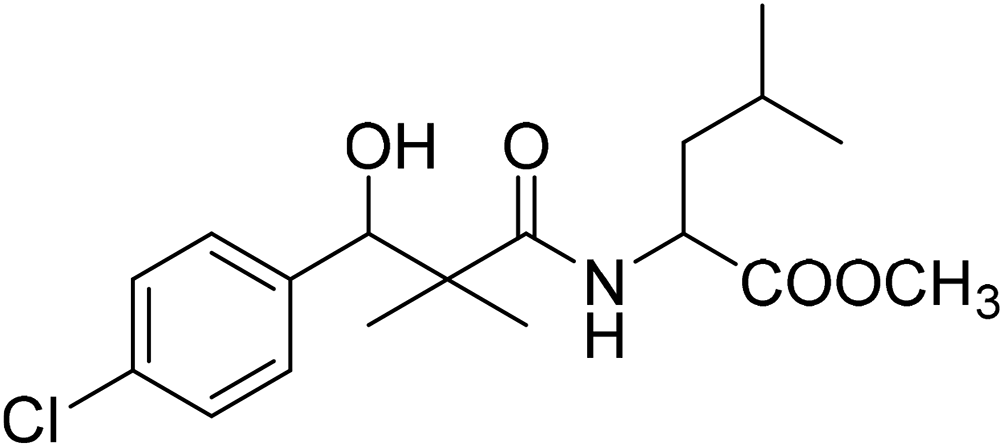 |
0.71 mg mL−1 | NA |
| 8g | 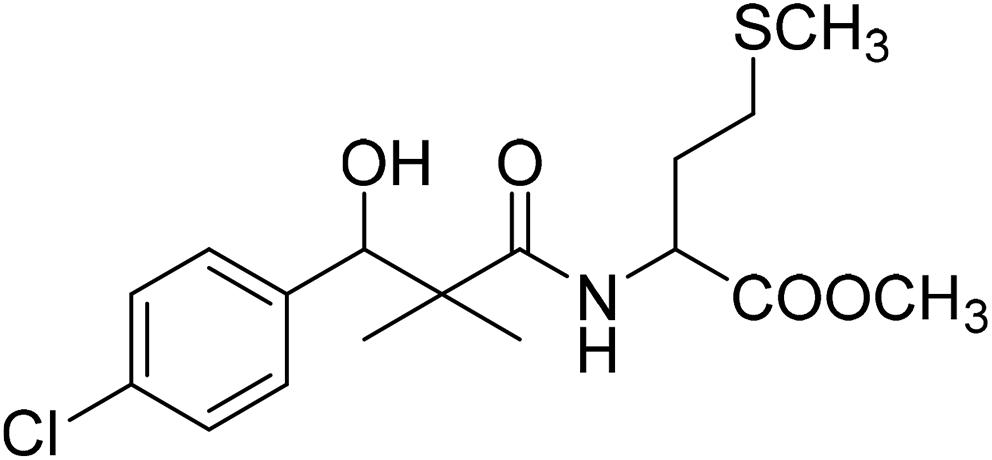 |
NA | NA |
| 10 | 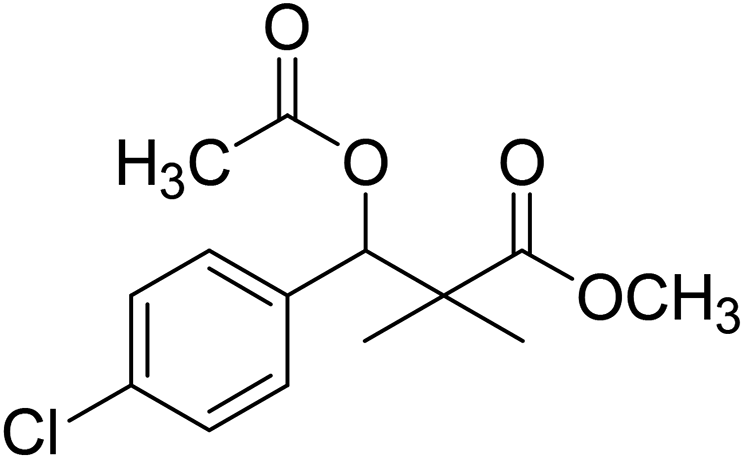 |
NA | NA |
| 11a |  |
NA | NA |
| 11b |  |
NA | NA |
| 11c |  |
NA | NA |
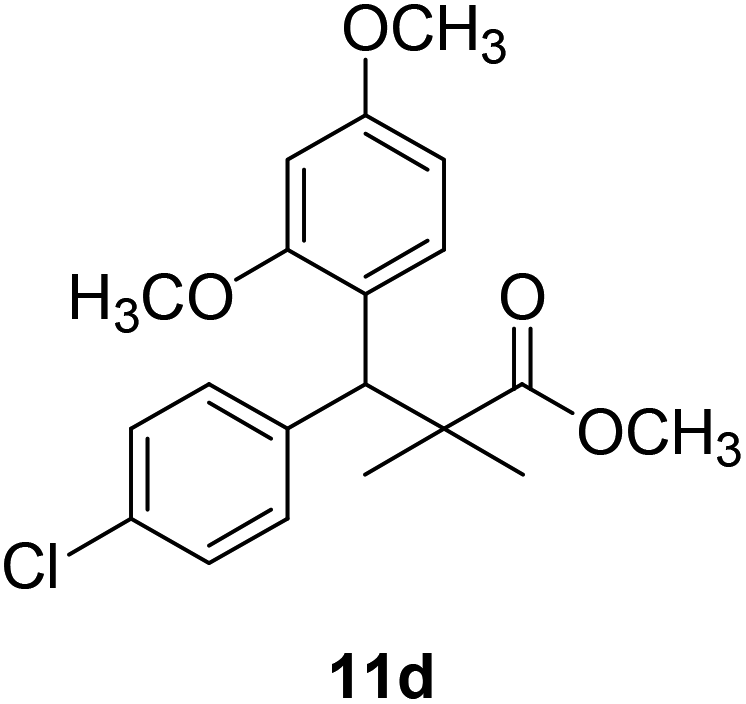
| 11d | 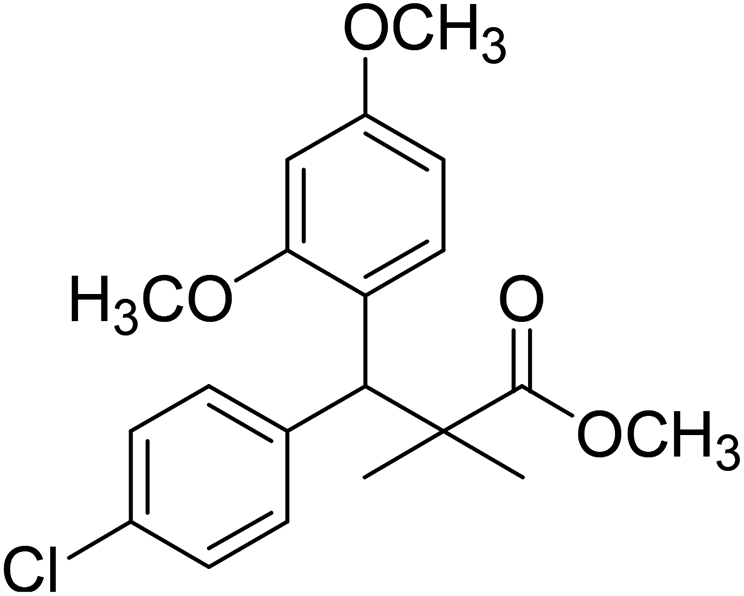 |
NA | NA |
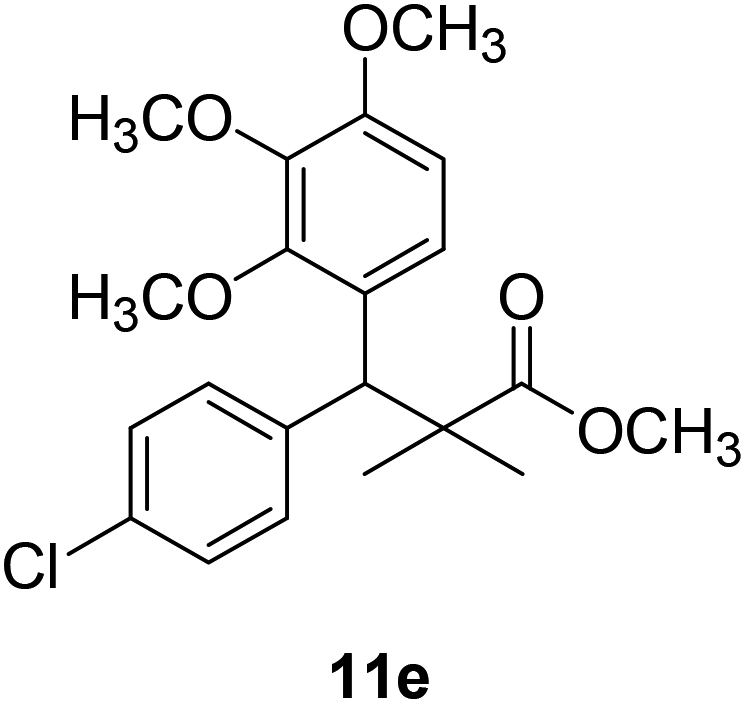
| 11e | 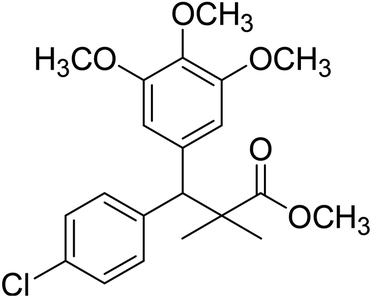 |
NA | NA |
The preliminary structure–activity relationship of the compounds showed that all active compounds contain the free OH on the α carbon (4, 6, 7a, 7c, 7d, 7f, 7g, 7h, 7i, 8a, 8b, 8f), while most of the inactive compounds lack the OH (10, 11a, 11b, 11c, 11d, 11e). All compounds having esters alpha to the isopropyl group are inactive (10, 11a, 11b, 11c, 11d, 11e) while those with an acid or substituted amides are active (4, 6, 7a, 7c, 7d, 7f, 7g, 7h, 7i, 8a, 8b, 8f). This could be partly due to that all the compounds with ester functionality lacks the free OH on the α carbon. Substitution on the amide is crucial; with compounds having bulky branched chain are inactive (8e, 8g). Electron rich amides are more favorable as they can participate better in hydrogen bonding as acceptor (7g and 7i compared to 7h). Therefore, a hydrogen donor group linked to the halo substituted ring is essential. A hydrogen acceptor group such as an acid or alkyl substituted amides are important for activity. Substitution on the amide is detrimental. The amide nitrogen could be a part of a heterocyclic ring however oxygen containing heterocycles are less active due to the higher electronegativity and electron withdrawing properties of the oxygen (7g and 7i compared to 7h).
Nuclear disintegration
In order to understand whether cancer cell death is due to necrosis or apoptosis, we have stained cancer cells with DAPI (4′,6-diamidine-2′-phenylindole dihydrochloride), which is considered as the marker for the apoptosis.29–32 We have observed clear DNA disintegration and fragmentation in the cells treated with compounds (7i and 7g) with compared to untreated or control cells (Fig. 1B and C). In addition, we have observed condensed chromatins and nuclear fragmentations in cancerous cells which is not observed in the untreated (control) cells (Fig. 1A). The nuclear disintegration of cancerous cells is due to programmed cell death and similar results were also observed in other studies.21,22,28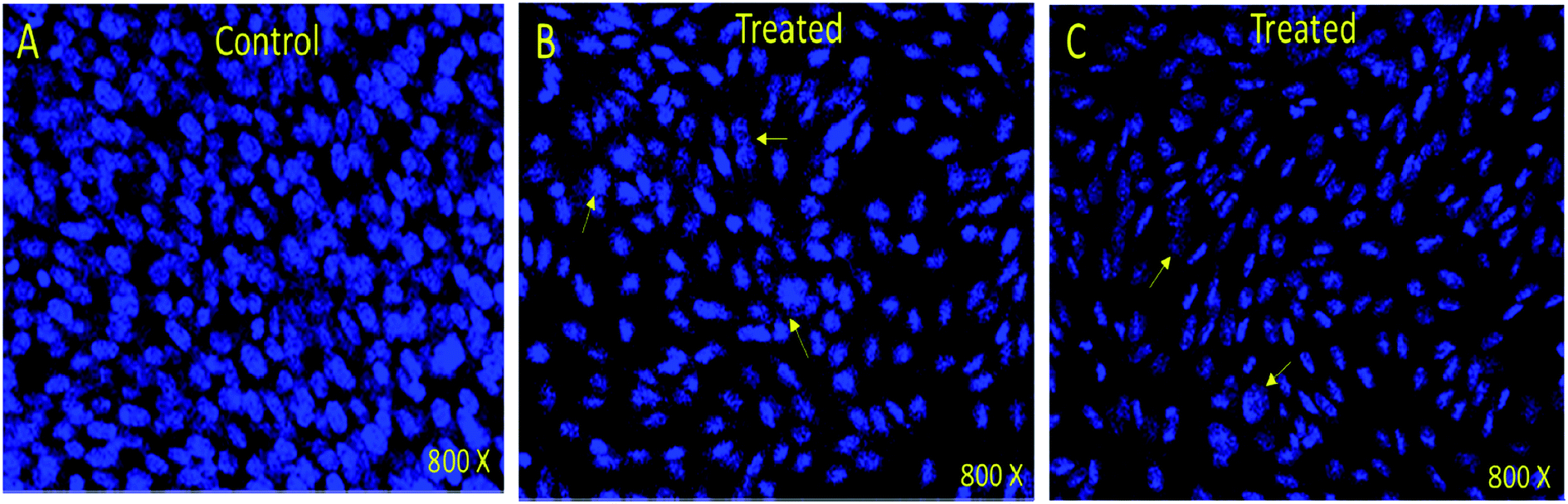 | ||
| Fig. 1 DAPI stained HCT-116 cells. (A) Control (nontreated) (B) treated (7i) compound (0.13 mg mL−1) and (C) treated with (7g) compound (0.12 mg mL−1). Nuclear disintegration, and chromatic condensation are observed (arrows) in the (B) and (C). 800× magnifications. | ||
We have also observed decrease in DAPI staining in the compound-treated cells compared to untreated cells, which suggest that compounds inhibited the growth of the cancer cells as observed in other studies (Fig. 2).33
 | ||
| Fig. 2 DAPI stained HCT-116 cells. (A) Control (nontreated) (B) treated (7i) compound (0.13 mg mL−1) and (C) treated with (7g) compound (0.12 mg mL−1). DAPI stained cells visualized through confocal microscope and (B) and (C) show significant loss (arrows) of staining due to treatment. 200× magnifications. | ||
Molecular modeling
The mechanism of action of the compounds antiproliferative and apoptotic activity were investigated using molecular docking experiments. Heat shock protein 90 (Hsp90) is a molecular chaperone, which plays a key role in signal transduction pathways, whose inhibition has been widely described as one of the potential cancer's treatments.34 Tumor necrosis factor receptor-associated protein 1 (TRAP1), a member of the HSP90 family, is involved in several physiological functions, including cell proliferation, differentiation, and survival. The anti-apoptotic roles of TRAP1 were explored as an important target for anticancer drug development and for tumor control in clinical settings.34 Therefore, the mitochondrial pool of Hsp90 and its mitochondrial paralogue, TRAP1, have been studied as target proteins for anticancer drug development.35Our compounds showed affinity to the heat shock proteins TRAP1 in silico and this could be a potential mode of action that explain the compounds antiproliferative activities. Hsp90 inhibitors anticancer activity has been found modest compared to their TRAP1 inhibitors counterpart.36 Therefore, inhibitors with strong TRAP1-binding but weak Hsp90-binding, activities would have better in vivo activities and clinical applications. Interestingly, the most active compounds 7a, 7g docked well in the active site of TRAP1 while compounds with moderate activity or inactive were docked far away from the active site. The most active compounds showed also lower binding affinity to HSP90. They lacked the hydrogen bonding interactions with Asp93 and Thr184 that was involved in a network of hydrogen bonding in case of 8a and the crystallized ligand. This when added to their high affinity to TRAP1 could give an indication that this is a potential mode of action for further research. The main difference in the TRAP1 active site structure compared to HSP90 were reported to be in the active site lid (Leu172–Phe201) and its flanking conserved residues, Asn171 and Gly202, that have different configurations in TRAP1 compared with those in Hsp90. Only compounds 7a and 7g, the most active ones, were found in the ATP binding site of TRAP1 (Fig. 3a). Comparative docking revealed that those compounds showed docking poses that is almost identical to the co-crystallized inhibitor (Fig. 3b). The surface representation showed that they have their side chain with amide and alcohol functional groups buried deeply in the hydrophobic ATP pocket and their aromatic ring directed toward the solvent exposed surface (Fig. 3b).
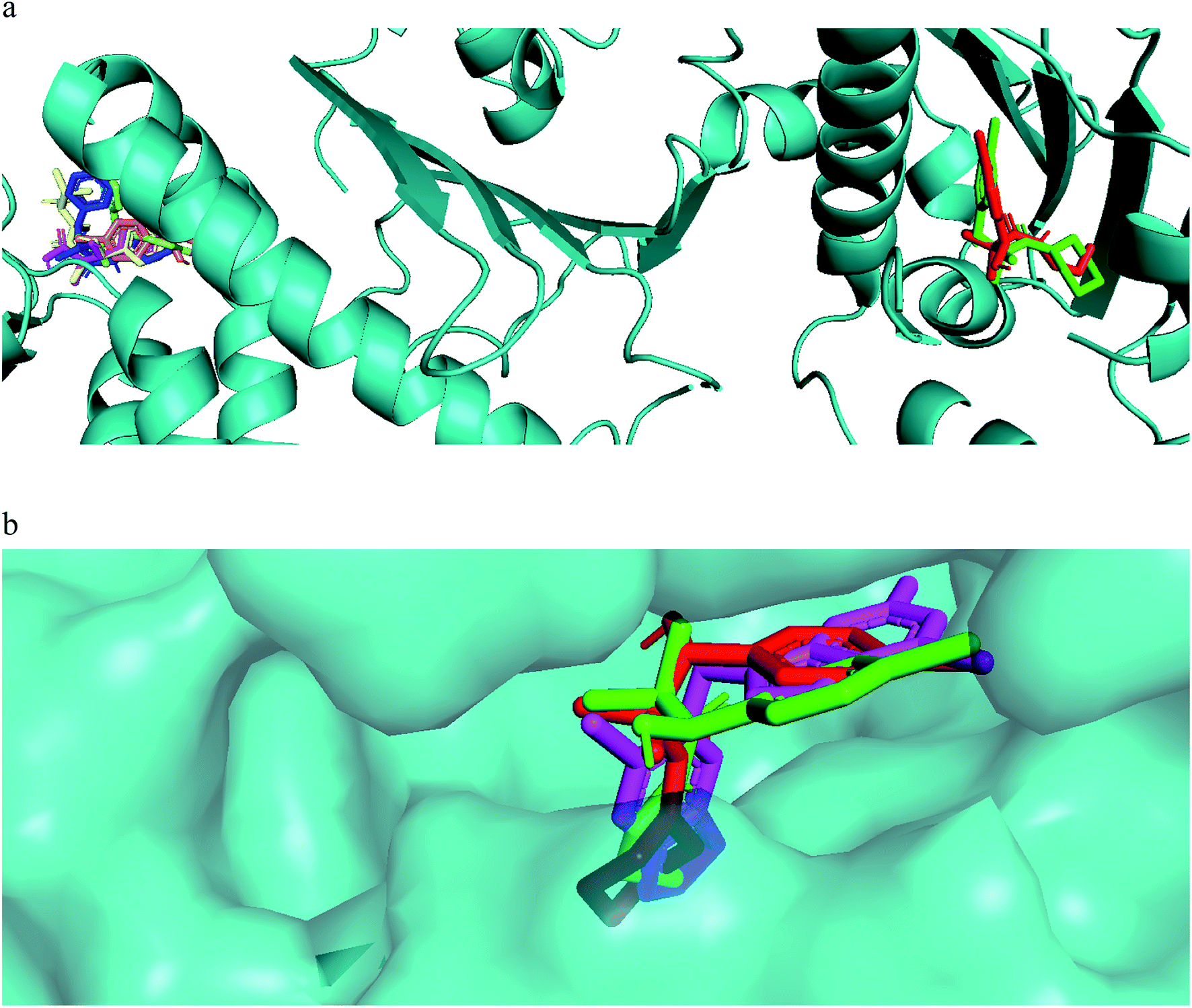 | ||
| Fig. 3 (a) Compound 7a (red) and 7g (green) docked in the ATP binding site of TRAP1, while less active compounds 6 (magenta), 7e (blue), 8a (brown), 8e (yellow) docked in a pocket far away from the binding site. (b) Molecular surface representation of compound 7a (red), compound 7b (green), and co crystallized ligand (magenta) in TRAP1 active site (front view). | ||
Structural analyses of the docking poses revealed that both 4-chlorophenyl ring of 7a and 7b are relatively close to the flanking regions of the disordered TRAP1 active site lid and established additional π–π stacking interactions with the conserved Phe201 residue in TRAP1 (Fig. 4).
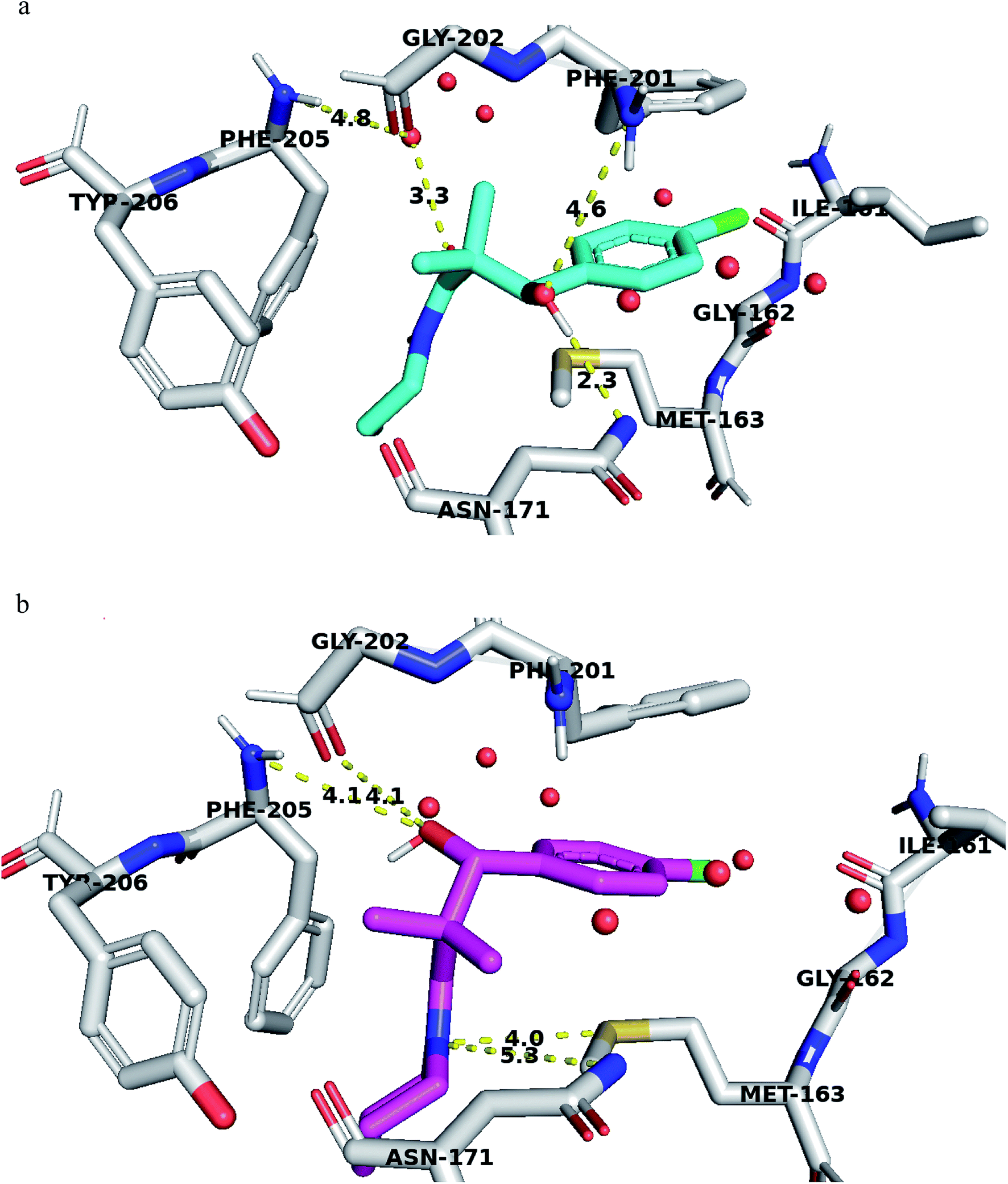 | ||
| Fig. 4 (a) Compound 7a docked in TRAP1 ATP binding site and performing interaction with key residues. (b) Compound 7g docked in TRAP1 ATP binding site and performing interaction with key residues. Distances are represented as yellow dotted lines in angstrom. | ||
Furthermore, 7a exhibited superior binding affinity to TRAP1 through hydrogen bonding between the side chain OH and the Asn171 side chain amide and another potential hydrogen bond with Phe201 (Fig. 4a). An additional water-mediated hydrogen bond was also noted with Phe205 side chain.
Compound 7g showed potential hydrogen bonds of its side chain amide nitrogen with both Asn171 side chain amide and Met163 side chain. The compound side chain OH exhibited potential hydrogen bonds with Gly202 and Phe205 (Fig. 4b).
Thus, the TRAP1 selective binding of 7a and 7g was explained based on their superior and characteristic interactions with Asn171 and Phe201, located on the flanked residues of the disordered TRAP1 structure.
The binding affinity of these compounds was found consistent with their superior TRAP1 activity. Binding affinity calculation showed that compounds 7a and 7g had the best affinity to TRAP1 among all other compounds with values of −7.6 and −7.5 kcal mol−1 for 7a and 7g respectively.
Insufficient mitochondrial penetration of Hsp90 inhibitors is potentially due to a strong binding to abundant (∼2% of total cellular proteins) cytoplasmic Hsp90 proteins.31
To further test the activity of the less active compounds they were docked in the cytoplasmic HSP90. Inhibitors of HSP90 are known to bind to an ATP binding pocket in the N-terminal domain of the protein. Compound 8a displayed a binding mode similar to other known Hsp90 inhibitors. The side chain occupied the adenosine pocket and was involved in a network of hydrogen bonding. The terminal ester carbonyl oxygen made key interactions with Asp93 both through direct hydrogen bonding and through a conserved water molecule while the amide oxygen formed a water-mediated hydrogen bond with Asp93. The side chain amide oxygen also established hydrogen bond with Thr184. The 4-chlorophenyl group was stacked between the side chain of Leu107 and Phe138 and was held tightly in the hydrophobic pocket adjacent to the adenosine binding site through a bidentate hydrogen bond interaction with Leu107 and Met98 (Fig. 5).
 | ||
| Fig. 5 Compound 8a docked in HSP90 ATP binding site and performing interaction with key residues. Distances are represented as yellow dotted lines in angstrom. | ||
TRAP1 and HSP90 have been found to be elevated in cancer cells where these proteins suppress cell death. This finding could partly explain the selectivity shown by those compounds and their inactivity on normal cells. Therefore, the herein described compounds being working on more selective targets have higher chances to be developed to clinically effective agents after the proper pharmacodynamic and pharmacokinetic optimization.
Experimental
General procedures
Solvent were purified and dried in the usual way. The boiling range of the petroleum ether used was 40–60 °C. Thin layer chromatography (TLC): silica gel 60 F254 plastic plates (E. Merck, layer thickness 0.2 mm) detected by UV absorption. Melting points were determined on a Buchi 510 melting-point apparatus and the values are uncorrected. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively (Bruker AC 400) in CDCl3 and DMSO solution with tetramethylsilane as an internal standard. The NMR analyses were performed at Faculty of Science, Sohag University. The mass spectra were measured with a KRATOS analytical compact; on MALDI-MS the spectrometer was using 2,5-dihydroxy benzoic acid (DHB) as matrix. The starting compound 3 were obtained as described in literature.37,38Preparation of 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoic acid hydrazide (4)
To a solution of methyl 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) (0.24 g, 1.0 mmol) in ethyl alcohol (10 mL), hydrazine hydrate (80%) (4.0 mmol) was added and the reaction mixture was heated under reflux for 9 h. The reaction mixture was cooled and evaporated under reduced pressure till dried. The precipitated residue was crystallized from ethyl alcohol. White crystals (89%), mp: 96–98 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 8.70 (1H, bs, NH), 7.34 (2H, t, J = 8.0 Hz, ArH), 7.25 (2H, t, J = 8.0 Hz, ArH), 5.56 (1H, d, J = 4.0 Hz, CH), 4.77 (1H, d, J = 4.0 Hz, OH), 4.17 (2H, bs, NH2), 1.03 (3H, s, CH3), 0.90 (3H, s, CH3). (MALDI, positive mode, matrix DHB): m/z = 265 (M + Na)+.Preparation of 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoic acid (6)
To a solution of methyl 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) (0.24 g, 1.0 mmol) in 70% ethyl alcohol (10 mL), KOH (0.084, 1.5 mmol) and 2 mL H2O were added and the reaction mixture was heated under reflux for 6 h. The reaction mixture was cooled and acidification by dilHCl. The precipitated residue was crystallized from ethyl alcohol. White crystals (74%), mp: 171–173 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 11.98 (1H, bs, OH), 7.37–7.31 (4H, m, ArH), 4.83 (1H, s, CH), 3.29 (1H, bs, OH), 1.03 (3H, s, CH3), 0.89 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 182.9 (CO), 146.3, 136.8, 134.5, 132.5, 130.1, 127.4 (C-Ar), 81.2 (CH), 61.3 (C), 26.4 (CH3), 25.1 (CH3). (MALDI, positive mode, matrix DHB): m/z = 251 (M + Na)+.
Preparation of N-alkyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamides 7a–i
Method A: to a cold solution (−5 °C) of 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoic acid hydrazide (4) (0.24 g, 1.0 mmol) in 1 N HCl solution (5 mL) and acetic acid 96% (2 mL), 5 mL water solution of NaNO2 (0.11 g, 1.5 mmol) was added. The reaction mixture was stirred at 0 °C for 30 min. The reaction mixture was extracted three times with 30 mL ethyl acetate. The combined yellow syrup extract was washed several times with 3% solution of NaHCO3 and water till it became neutral and was finally dried over Na2SO4. The in situ generated azide 5 was used without further isolation and purification. To the previously prepared cold ethyl acetate solution of azide 5, a solution of appropriate amines (1.0 mmol) in ethyl acetate (10 mL) was added dropwise with stirring. The reaction mixture was kept at −5 °C for 12 h, then at room temperature for another 12 h. The reaction mixture was washed with 0.5 N HCl, water and 3% solution of NaHCO3 and dried over Na2SO4. The solution was evaporated to dryness and the residue was recrystallized from ethyl acetate/petroleum ether to give the desired product 7a–i.Method B: to a cold solution (−5 °C) of amines (1.0 mmol) and triethylamine (0.10 mL, 1.0 mmol) in acetonitrile (6 mL) was added 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoic acid (6) (0.23 g, 1.0 mmol), dicyclohexylcarbodiimide (DCC) (0.21 g, 1.0 mmol) and N-hydroxylsuccinimide (NHS) (0.10 g, 1.0 mmol), successively. The reaction mixture was stirred at 0 °C for one hour, at 5 °C for one hour and at room temperature for 12 h. The reaction mixture was set aside overnight. The precipitated dicyclohexyl urea was filtered off and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel column chromatography eluted with EtOAc/petroleum ether 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to give pure 7a–i.
:4 to give pure 7a–i.
3-(4-Chlorophenyl)-3-hydroxy-2,2-dimethyl-N-propylpropanamide (7a)
White crystals (81%), mp: 186–188 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.27–7.20 (4H, m, ArH), 5.72 (1H, bs, NH), 4.68 (1H, s, CH), 4.07 (1H, bs, OH), 4.05–4.02 (2H, m, CH2), 1.39–1.20 (2H, m, CH2), 1.13 (3H, s, CH3), 1.09 (3H, t, J = 4.0 Hz, CH3), 0.90 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 178.9 (CO), 136.8, 133.4, 132.9, 130.5, 128.3, 127.1 (C-Ar), 80.9 (CH), 60.8 (C), 34.5 (CH2), 23.6 (CH2), 23.4 (CH3), 22.2 (CH3), 12.1 (CH3). (MALDI, positive mode, matrix DHB): m/z = 292 (M + Na)+.
Method B, from 6 and propylamine (74%).
N-Butyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (7b)
White crystals (78%), mp: 75–76 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 8.04–8.02 (1H, m, ArH), 7.93–7.90 (1H, m, ArH), 7.76–7.55 (3H, m, ArH, NH), 5.97 (1H, bs, OH), 4.62–4.57 (1H, m, CH), 3.53–3.47 (2H, m, CH2), 2.68–2.64 (2H, m, CH2), 1.58–1.41 (2H, m, CH2), 0.86 (3H, s, CH3), 0.84–0.81 (6H, m, 2CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 175.2 (CO), 139.9, 131.5, 128.4, 128.3, 127.8, 127.7 (C-Ar), 80.1 (CH), 54.2 (C), 39.8 (CH2), 33.5 (CH2), 21.3 (CH3), 20.1 (CH3), 19.2 (CH2), 13.8 (CH3). (MALDI, positive mode, matrix DHB): m/z = 306 (M + Na)+.
Method B, from 6 and butylamine (58%).
N-Allyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (7c)
White crystals (79%), mp: 127–129 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.23–7.14 (5H, m, ArH, NH), 5.92 (1H, bs, OH), 5.76–5.69 (1H, m, CH), 5.08 (1H, s, CH), 4.64 (2H, m, CH2), 3.86–3.60 (2H, m, CH2), 1.12 (3H, s, CH3), 1.01 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 179.2 (CO), 148.4, 137.8, 135.4, 134.5 (CH), 132.9, 130.6, 127.4 (C-Ar), 117.3 (CH2), 81.2 (CH), 61.4 (C), 44.2 (CH2), 20.2 (CH3), 19.4 (CH3). (MALDI, positive mode, matrix DHB): m/z = 290 (M + Na)+.
Method B, from 6 and allylamine (65%).
3-(4-Chloro-phenyl)-3-hydroxy-2,2-dimethyl-N-tetradecyl-propanamide (7d)
White crystals (64%), mp: 89–91 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.39–7.23 (4H, m, ArH), 6.07 (1H, bs, OH), 4.64 (1H, s, CH), 3.39–3.32 (2H, m, NCH2), 1.62–1.57 (4H, m, 2CH2), 1.29–1.19 (16H, m, 8CH2), 1.24–1.22 (2H, m, CH2), 1.04 (3H, s, CH3), 0.99–0.97 (3H, m, CH3), 0.91 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 178.6 (CO), 139.3, 134.5, 129.4, 128.7, 127.3, 127.0 (C-Ar), 79.6 (CH), 45.9 (NCH2), 44.6 (C), 34.2 (CH2), 26.4 (CH2), 26.0 (CH2), 24.3 (CH2), 20.5 (CH3), 19.5 (CH3), 17.7 (CH3). Method B, from 4 and tetradecylamine (51%). (MALDI, positive mode, matrix DHB): m/z = 446 (M + Na)+.
3-(4-Chlorophenyl)-N-cyclohexyl-3-hydroxy-2,2-dimethylpropanamide (7e)
White crystals (81%), mp: 105–107 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.27–7.21 (5H, m, ArH, NH), 5.73 (1H, bs, OH), 4.59 (1H, s, CH), 3.77–3.75 (1H, m, CH), 1.85–1.81 (4H, m, 2CH2), 1.67–1.61 (4H, m, 2CH2), 1.38–1.32 (2H, m, CH2), 1.22 (3H, s, CH3), 1.03 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.3 (CO), 138.8, 134.1, 129.1, 128.8, 126.2, 126.0 (C-Ar), 79.1 (CH), 60.3 (C), 51.2 (CH), 33.2 (CH2), 33.0 (CH2), 25.3 (CH2), 24.1 (CH2), 23.2 (CH2), 22.4 (CH3), 19.8 (CH3). (MALDI, positive mode, matrix DHB): m/z = 332 (M + Na)+.
Method B, from 6 and cyclohexylamine (72%).
N-Benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (7f)
White crystals (83%), mp: 125–126 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.28–7.10 (9H, m, ArH), 6.08 (1H, bs, NH), 4.60 (1H, m, CH), 4.37 (1H, d, J = 8.0 Hz, CH2Ph), 4.33 (1H, d, J = 8.0 Hz, CH2Ph), 3.09 (1H, bs, OH), 1.18 (3H, s, CH3), 1.02 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 177.3 (CO), 139.3, 137.8, 133.5, 132.4, 131.7, 129.0, 128.8, 128.3 (2C), 128.0 (2C), 127.7 (C-Ar), 79.4 (CH), 60.3 (C), 43.6 (CH2), 22.2 (CH3), 20.7 (CH3). (MALDI, positive mode, matrix DHB): m/z = 340 (M + Na)+.
Method B, from 6 and benzylamine (76%).
3-(4-Chlorophenyl)-3-hydroxy-2,2-dimethyl-1-(piperidin-1-yl)propan-1-one (7g)
White crystals (68%), mp: 162–164 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.46–7.18 (4H, m, ArH), 5.63 (1H, bs, OH), 4.52–4.50 (1H, m, CH), 3.51–3.47 (4H, m, 2NCH2), 1.58–1.47 (6H, m, 3CH2), 1.09 (3H, s, CH3), 0.89 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 178.3 (CO), 1375, 133.2, 130.0, 129.5, 128.4, 127.9 (C-Ar), 79.4 (CH), 48.2 (NCH2), 48.0 (NCH2), 45.3 (C), 30.3 (CH2), 29.7 (2CH2), 20.7 (CH3), 20.0 (CH3). (MALDI, positive mode, matrix DHB): m/z = 318 (M + Na)+.
Method B, from 6 and piperidin (52%).
3-(4-Chlorophenyl)-3-hydroxy-2,2-dimethyl-1-morpholinopropan-1-one (7h)
White crystals (62%), mp: 134–135 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.22–7.13 (4H, m, ArH), 5.85 (1H, bs, OH), 4.63–4.60 (1H, m, CH), 3.85–3.59 (4H, m, 2CH2), 3.37–3.29 (4H, m, 2CH2), 1.10 (3H, s, CH3), 0.95 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 175.6 (CO), 138.7, 132.1, 129.0, 128.5, 127.1, 126.8 (C-Ar), 80.2 (CH), 65.7 (OCH2), 65.5 (OCH2), 47.7 (NCH2), 45.3 (NCH2), 44.8 (C), 21.3 (CH3), 19.9 (CH3). (MALDI, positive mode, matrix DHB): m/z = 320 (M + Na)+.
Method B, from 6 and morpholine (69%).
3-(4-Chlorophenyl)-3-hydroxy-2,2-dimethyl-1-(4-methylpiperazin-1-yl)propan-1-one (7i)
White crystals (67%), mp: 156–157 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.41–7.35 (4H, m, ArH), 5.83 (1H, bs, OH), 4.71 (1H, s, CH), 3.31–3.29 (4H, m, 2NCH2), 2.35–3.28 (4H, m, 2CH2), 2.16 (3H, s, CH3), 1.09 (3H, s, CH3), 0.89 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.7 (CO), 140.2, 135.4, 132.8, 128.9, 127.8, 127.2, (C-Ar), 79.2 (CH), 50.5 (NCH2), 50.1 (NCH2), 49.3 (CH2), 49.1 (NCH2), 42.4 (C), 41.2 (NCH3), 21.1 (CH3), 20.2 (CH3). (MALDI, positive mode, matrix DHB): m/z = 333 (M + Na)+.
Method B, from 6 and methylpiperazin (58%).
Preparation of methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)-amino]alkanoates 8a–g
To the previously prepared cold ethyl acetate solution of azide 5 (0.24 g, 1.0 mmol hydrazide), a solution of amino acid esters (1.2 mmol) in ethyl acetate (10 mL) was added dropwise with stirring. The reaction mixture was kept at −5 °C for 12 h, then at room temperature for another 12 h. The reaction mixture was washed with 0.5 N HCl, water and 3% solution of NaHCO3 and dried over Na2SO4. The solution was evaporated to dryness, and the residue was recrystallized from ethyl acetate/petroleum ether to give the desired product 8a–g.Methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]acetate (8a)
White crystals (72%), mp: 168–170 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.32–7.15 (4H, m, ArH), 6.67 (1H, t, J = 4.0 Hz, NH), 5.22 (1H, bs, OH), 4.63 (1H, s, CH), 3.92 (2H, t, J = 4.0 Hz, NCH2), 3.68 (3H, s, OCH3), 1.08 (3H, s, CH3), 0.81 (3H, m, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 177.6 (CO), 170.1 (CO), 139.1, 133.2, 129.0, 128.5, 127.6, 127.4 (C-Ar), 79.7 (CH), 51.5 (OCH3), 45.2 (C), 41.4 (NCH2), 20.1 (CH3), 19.8 (CH3). (MALDI, positive mode, matrix DHB): m/z = 322 (M + Na)+.
Method B, from 6 and glycine methyl ester hydrochloride (66%).
Methyl-3-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]propanoate (8b)
White crystals (86%), mp: 139–141 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.10–6.88 (4H, m, ArH), 6.87 (1H, bs, NH), 4.76 (1H, bs, OH), 4.47 (1H, s, CH), 3.48 (3H, s, OCH3), 3.26 (2H, t, J = 4.0 Hz, NCH2), 2.33 (2H, t, J = 4.0 Hz, CH2), 1.19 (3H, s, CH3), 0.87 (3H, m, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 181.0 (CO), 171.2 (CO), 139.6, 132.9, 129.5, 128.8, 128.3, 127.6 (C-Ar), 78.3 (CH), 53.6 (OCH3), 44.5 (C), 42.4 (NCH2), 33.4 (CH2), 20.3 (CH3), 20.0 (CH3). (MALDI, positive mode, matrix DHB): m/z = 336 (M + Na)+.
Method B, from 6 and β-alanine methyl ester hydrochloride (74%).
Methyl-4-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]butanoate (8c)
White crystals (74%), mp: 139–141 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.42–7.21 (4H, m, ArH), 6.63 (1H, bs, NH), 4.82 (1H, bs, OH), 4.61 (1H, s, CH), 3.56 (3H, s, OCH3), 3.20 (2H, m, NCH2), 2.36 (2H, m, CH2), 2.11 (2H, m, CH2), 1.21 (3H, s, CH3), 0.91 (3H, m, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 179.3 (CO), 174.7 (CO), 138.4, 133.6, 130.5, 129.2, 128.8, 128.0 (C-Ar), 79.6 (CH), 55.2 (OCH3), 45.8 (C), 41.2 (NCH2), 30.0 (CH2), 24.2 (CH2), 20.0 (CH3), 19.7 (CH3). (MALDI, positive mode, matrix DHB): m/z = 350 (M + Na)+.
Method B, from 6 and γ-amino butyric methyl ester hydrochloride (61%).
Methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]propanoate (8d)
White crystals (69%), mp: 88–90 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.42–7.21 (4H, m, ArH), 6.54 (1H, bs, NH), 5.53 (1H, bs, OH), 4.54 (1H, s, CH), 4.41 (1H, m, NCH), 3.58 (3H, s, OCH3), 1.34 (3H, s, CH3), 1.17 (3H, s, CH3), 0.99 (3H, m, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.8 (CO), 172.7 (CO), 138.4, 134.0, 130.4, 129.5, 128.7, 127.2 (C-Ar), 80.2 (CH), 54.8 (OCH3), 48.3 (NCH), 45.9 (C), 20.0 (CH3), 19.6 (CH3), 18.5 (CH3). (MALDI, positive mode, matrix DHB): m/z = 336 (M + Na)+.
Method B, from 6 and L-alanine methyl ester hydrochloride (57%).
Methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]3-methyl butanoate (8e)
White crystals (57%), mp: 109–111 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.35–7.19 (4H, m, ArH), 6.86 (1H, bs, NH), 5.27 (1H, bs, OH), 4.87 (1H, s, CH), 4.52–4.45 (1H, m, NCH), 3.71 (3H, s, OCH3), 2.16–2.13 (1H, m, CH), 1.10 (3H, s, CH3), 0.98–0.86 (9H, m, 3CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.8 (CO), 173.2 (CO), 139.5, 133.8, 129.2, 128.9, 126.7, 127.4 (C-Ar), 79.7 (CH), 55.1 (OCH3), 51.2 (NCH), 46.2 (C), 31.2 (CH), 20.2 (CH3), 19.7 (CH3), 18.6 (CH3), 18.4 (CH3). (MALDI, positive mode, matrix DHB): m/z = 364 (M + Na)+.
Method B, from 6 and L-valine methyl ester hydrochloride (55%).
Methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]4-methyl pentanoate (8f)
White crystals (64%), mp: 68–70 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.35–7.19 (4H, m, ArH), 6.86 (1H, bs, NH), 5.27 (1H, bs, OH), 4.87 (1H, s, CH), 4.52–4.45 (1H, m, NCH), 3.71 (3H, s, OCH3), 2.16–2.13 (1H, m, CH), 1.97–1.93 (2H, m, CH2), 1.10 (3H, s, CH3), 0.98–0.86 (9H, m, 3CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.8 (CO), 173.2 (CO), 139.5, 133.8, 129.2, 128.9, 126.7, 127.4 (C-Ar), 79.7 (CH), 55.1 (OCH3), 51.2 (NCH), 46.2 (C), 38.3 (CH2), 31.2 (CH), 20.2 (CH3), 19.7 (CH3), 18.6 (CH3), 18.4 (CH3). (MALDI, positive mode, matrix DHB): m/z = 378 (M + Na)+.
Method B, from 6 and L-leucine methyl ester hydrochloride (53%).
Methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]4-methyl-sulfanylbutanoate (8g)
White crystals (59%), mp: 96–98 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.38–7.15 (4H, m, ArH), 6.73 (1H, bs, NH), 5.35 (1H, bs, OH), 4.76 (1H, s, CH), 4.63–4.57 (1H, m, NCH), 3.61 (3H, s, OCH3), 2.58–2.51 (2H, m, CH2), 2.19–2.14 (2H, m, CH2), 2.10 (3H, s, CH3), 1.14 (3H, s, CH3), 0.87 (3H, m, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.8 (CO), 173.2 (CO), 139.5, 133.8, 129.2, 128.9, 126.7, 127.4 (C-Ar), 79.7 (CH), 55.1 (OCH3), 51.2 (NCH), 46.2 (C), 33.7 (CH2), 32.3 (CH2), 20.6 (CH3), 19.3 (CH3), 18.8 (CH3). (MALDI, positive mode, matrix DHB): m/z = 396 (M + Na)+.
Method B, from 6 and L-methionine methyl ester hydrochloride (48%).
Preparation of methyl-3-(4-chlorophenyl)-2,2-dimethyl-3-(2,2,2-trichloro-1-iminoethoxy)propanoate (9)
A stirred solution of methyl 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) (1.0 mmol) in dry dichloromethane (30 mL) was treated with trichloroacetonitrile (0.3 mL, 2.0 mmol) and DBU (0.1 mL, 0.5 mmol). The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under reduced pressure and the product was purified by column chromatography 5% triethylamine in dichloromethane, to give 9 as whitish oil (82%). 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 8.65 (1H, bs, NH), 7.47–7.18 (4H, m, ArH), 5.21 (1H, s, CH), 3.73 (3H, s, OCH3), 1.11 (3H, s, CH3), 0.90 (3H, m, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 177.3 (CO), 168.7 (CNH), 134.8, 132.5, 129.1, 128.4, 128.0, 127.7 (C-Ar), 88.7 (CCl3), 79.0 (CH), 53.8 (OCH3), 45.9 (C), 20.0 (CH3), 19.6 (CH3). (MALDI, positive mode, matrix DHB): m/z = 408 (M + Na)+.
Preparation of methyl 3-acetoxy-3-(4-chlorophenyl)-2,2-dimethylpropanoate (10)
To a solution of methyl 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (3) (0.24 g, 1.0 mmol) in dichloromethane, acetic anhydride (1.2 mmol) and dimethyl aminopyridine (DMAP) (0.2 mmol) was added. The reaction mixture was stirred at room temperature for 4 h and was evaporated under reduced pressure to give oily residue. The residue was purified by column chromatography 4:1 pet. ether/ethyl-acetate, to give 10 as white powder (82%).
White crystals (89%), mp: 61–63 °C. 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.28–7.19 (4H, m, ArH), 6.01 (1H, s, CH), 3.66 (3H, s, OCH3), 2.05 (3H, s, CH3), 1.22 (3H, s, CH3), 1.09 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.4 (CO), 169.3 (CO), 135.5, 133.9, 131.4, 128.6, 128.1, 127.4 (C-Ar), 78.4 (CH), 51.9 (OCH3), 47.0 (C), 21.9 (CH3), 21.7 (CH3), 20.1 (CH3). (MALDI, positive mode, matrix DHB): m/z = 307 (M + Na)+.
Preparation of methyl-3-aryl-3-(4-chlorophenyl)-2,2-dimethylpropanoate 11a–e
A solution of trichloroacetimidate 9 (method A) or acetate 10 (method B) (1.4 mmol) in dichloromethane (10 mL) was added C-nucleophiles (1.4 mmol); anisole, 1,4-dimethoxybenzene, 1,2-dimethoxybenzene and 1,2,3-trimethoxybenzene and TMSOTf (13 μL, 0.06 mmol). The reaction mixture was stirred at room temperature under dry nitrogen till completion of the reaction (TLC monitored and the time was recorded). The reaction mixture was neutralized with solid sodium bicarbonate, filtered and concentrated in vacuum. The residue was purified by column chromatography 3:1 pet. ether/ethyl-acetate as eluent, to give 11a–e.
Methyl-3-(4-chlorophenyl)-3-(4-methoxyphenyl)-2,2-dimethylpropanoate (11a)
Oil (84%). 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.21–7.08 (6H, m, ArH), 7.06–6.99 (2H, m, ArH), 4.23 (1H, s, CH), 3.68 (3H, s, OCH3), 3.44 (3H, s, OCH3), 1.16 (3H, s, CH3), 1.02 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.2 (CO), 146.8, 140.3, 135.6, 132.5, 129.4, 129.0, 128.1, 127.8, 125.5, 125.2, 127.1, 116.9 (C-Ar), 56.1 (OCH3), 53.2 (OCH3), 48.5 (CH), 47.2 (C), 20.2 (CH3), 19.6 (CH3). (MALDI, positive mode, matrix DHB): m/z = 355 (M + Na)+.
Method B, oil (77%).
Methyl-3-(4-chlorophenyl)-3-(2,5-dimethoxyphenyl)-2,2-dimethylpropanoate (11b)
Oil (78%). 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.21–7.10 (4H, m, ArH), 6.75–6.66 (3H, m, ArH), 4.43 (1H, s, CH), 3.65 (3H, s, OCH3), 3.63 (3H, s, OCH3), 3.59 (3H, s, OCH3), 1.13 (3H, s, CH3), 1.01 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.5 (CO), 148.2, 146.4, 145.2, 145.0, 141.3, 139.5, 133.4, 133.1, 129.2, 126.5, 118.7, 116.2 (C-Ar), 56.2 (OCH3), 55.4 (OCH3), 52.8 (OCH3), 49.8 (C), 44.2 (CH), 21.1 (CH3), 20.3 (CH3). (MALDI, positive mode, matrix DHB): m/z = 385 (M + Na)+.
Method B, oil (65%)
Methyl-3-(4-chlorophenyl)-3-(2,3-dimethoxyphenyl)-2,2-dimethylpropanoate (11c)
Oil (81%). 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.23–7.13 (4H, m, ArH), 6.81–6.80 (3H, m, ArH), 4.51 (1H, s, CH), 3.68 (3H, s, OCH3), 3.64 (6H, s, 2OCH3), 1.22 (3H, s, CH3), 1.03 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.4 (CO), 149.3, 147.8, 140.2, 140.0, 139.5, 135.7, 131.0, 129.8, 129.1, 123.2, 119.4, 116.0 (C-Ar), 57.7 (OCH3), 56.5 (OCH3), 54.2 (OCH3), 47.9 (C), 45.3 (CH), 20.8 (CH3), 19.9 (CH3). (MALDI, positive mode, matrix DHB): m/z = 385 (M + Na)+.
Method B, oil (76%)
Methyl-3-(4-chlorophenyl)-3-(2,4-dimethoxyphenyl)-2,2-dimethylpropanoate (11d)
Oil (80%). 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.39–7.22 (4H, m, ArH), 7.09–6.79 (3H, m, ArH), 4.53 (1H, s, CH), 3.81 (6H, s, 2OCH3), 3.61 (3H, s, OCH3), 1.20 (3H, s, CH3), 1.08 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 174.1 (CO), 153.9, 150.6, 149.3, 138.2, 133.5, 132.1, 130.3, 129.1, 127.5, 119.7, 114.6, 112.0 (C-Ar), 58.2 (OCH3), 57.2 (OCH3), 55.7 (OCH3), 48.2 (C), 47.5 (CH), 21.7 (CH3), 19.2 (CH3). (MALDI, positive mode, matrix DHB): m/z = 385 (M + Na)+.
Method B, oil (69%)
Methyl-3-(4-chlorophenyl)-2,2-dimethyl-3-(3,4,5-trimethoxyphenyl)propanoate (11e)
Oil (88%). 1H NMR spectrum, (400 MHz, CDCl3), δ, ppm (J, Hz): 7.30–7.20 (4H, m, ArH), 7.00–6.60 (2H, m, ArH), 4.62 (1H, s, CH), 3.88 (9H, s, 3OCH3), 3.68 (3H, s, OCH3), 1.22 (3H, s, CH3), 1.10 (3H, s, CH3). 13C NMR spectrum, (100.0 MHz, CDCl3), δ, ppm: 176.1 (CO), 152.4, 151.9, 150.0, 136.0, 131.8, 130.0, 129.9, 129.7, 128.1, 127.3, 113.8, 112.1 (C-Ar), 58.5 (OCH3), 57.2 (OCH3), 56.1 (OCH3), 54.5 (OCH3), 48.4 (C), 47.6 (CH), 21.3 (CH3), 20.5 (CH3). (MALDI, positive mode, matrix DHB): m/z = 415 (M + Na)+.
Method B, oil (74%).
Experimental method
Antiproliferative activity by MTT assay
The antiproliferative activity of both non-cancerous cells (human embryonic kidney cells – HEK-293) and cancerous cells (human colorectal carcinoma cells – HCT-116) was done by MTT assay. The cell lines (human embryonic kidney cells – HEK-293) and cancerous cells (human colorectal carcinoma cells – HCT-116) used in the study were obtained from Dr Khaldoon M. Alsamman, Clinical Laboratory Science, College of Applied Medical Science, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia. The cell culture and MTT assay was done as per previously described method.39 However, we have described the method in brief. Cells were grown in 96-well plates in the culture media containing DMEM, L-glutamine, fetal bovine serum, selenium chloride and penicillin–streptomycin. The cells were treated with different concentrations (0.06 mg mL−1 to 0.7 mg mL−1) of 15 synthetic compounds. In the control group, no synthetic compounds were added. After 48 hours of treatments, MTT (5.0 mg mL−1) was added in each well and were kept incubated for 4 hours. Thereafter, DMSO was added and plates were read at 570 nm wavelength using ELISA Plate Reader (Biotek Instruments, Winooski, USA). The percentage (%) of cell viability (%) was calculated and all statistical analyses were completed with GraphPad Prism 6 (GraphPad Software). The difference between control and compound-treated groups by a one-way analysis of variance (ANOVA), and p-values were calculated by Student's t-test.Nuclear staining
We examined status of nuclear disintegration of cancerous cells after treatments by staining with DAPI (4′,6-diamidino-2-phenylindole). We have selected compounds 2P and 55P which showed highest inhibitory action on the HCT-116 cell for the DAPI staining. The cancerous (HCT-116) cells were treated with (0.1 mg mL−1) concentration. In control group, compounds 2P and 55P were not added. After 48 hours of the treatment, both control and compounds 2P and 55P-treated groups were immersed in 4% paraformaldehyde solution. Thereafter, fixed cells were treated with Triton X-100 + phosphate buffered saline (PBS). Both control and compounds 2P and 55P-treated cells were stained with DAPI. The nuclear staining of both control and MNPs-treated cells was examined under confocal scanning microscope (Zeiss Germany).Molecular modeling
All molecular modeling studies were performed on a Hewlett-Packard Pentium Dual-Core T4300 2.10 GHz running Windows 10 Ultimate using autodock 4.3 for molecular docking simulation and ligand binding energy calculation. The crystal structure of HSP90 (PDB code; 2xab) and TRAP1 (PDB code; 5y3n) co-crystallized with inhibitor was used as receptor in the docking studies. The selected target was used after deleting the co-crystallized inhibitor. Docking calculations were carried out using the AutoDock 4.3 software (La Jolla, CA). First all hydrogens were added to the ligand PDB file and Gasteiger charges were computed and all the torsion angles of the ligand were defined with the autodock-tools program so they could be explored during molecular modeling. A grid box of 51.3260 × 45.8986 × 25.0000 Å and 88.6644 × 111.8783 × 25.0000 Å for HSP90 and TRAP1 respectively to incorporate the whole protein to further evaluate the selectivity of the tested compounds was used to calculate the atom types needed for the calculation. The Lamarckian genetic algorithm was used as a search method with a total of 30 runs (maximum of 20000000 energy evaluations; 27000 generations; initial populations of 150 conformers). The docking results were evaluated using binding energy calculation in autodock and checking ligand binding position visually in Pymol.
Conclusion
A series of 24 compounds were synthesized based on structure modification of the model methyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate as potent HDACIs. Methyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate was transformed into N-alkyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamides and methyl-2-[(3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoyl)amino]alkanoates via DCC and azide by the reaction with amines and amino acid esters, respectively. Methyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate reacted with acetic anhydride or trichloroacetonitrile to afford acetate or trichloroacetimidate, which was further reacted with C-active nucleophiles in the presence of TMSOTf (0.1 eq.%) to afford methyl-3-aryl-3-(4-chlorophenyl)-2,2-dimethylpropanoates via C–C bond formation. With careful design and optimization of those compounds especially those with the amino acid side chain we would be able to develop a potential candidate that has enough selectivity to progress to clinical trials. Optimization of compounds 7a and 7g for enhanced TRAP1 activity represents our future research direction. The 48 post-treatments showed that out of 24 compounds, 12 compounds showed inhibitory actions on HCT-116 cells, we have calculated the inhibitory action (IC50) of these compounds on both HCT-116 and we have found that IC50 were in between is 0.12 mg mL−1 to 0.81 mg mL−1. The compounds (7a & 7g) showed highest inhibitory activity (0.12 mg mL−1), whereas compound 7d showed lowest inhibitory activity (0.81 mg mL−1). The treated cancerous cells were also examined for nuclear disintegration through staining with DAPI, and we have found that there was loss of DAPI staining in the treated cancerous cells. We have also examined inhibitory action on normal and non-cancerous cells (HEK-293 cells) and confirmed that action of these compounds was specific to cancerous cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Science & Technology Development Fund in Egypt STDF Project ID: 22909 for funding this research proposal.References
- H. P. Rang and M. M. Dale, ELBS With Churchill Livingstone Pharmacology, Cancer Chemotherapy Chapter, 2nd edn, 1994, p. 781 Search PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Global Cancer Statistics, Ca-Cancer J. Clin., 2011, 61, 69 CrossRef PubMed.
- S. Antoni, I. Soerjomataram, B. Møller, F. Braya and J. Ferlay, An Assessment of GLOBOCAN Methods for Deriving National Estimates of Cancer Incidence, Bull. W. H. O., 2016, 94, 174 CrossRef PubMed.
- V. T. DeVita and E. Chu, A History of Cancer Chemotherapy, Cancer Res., 2008, 68, 8643 CrossRef CAS PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy, Angew. Chem., Int. Ed., 2014, 53, 3796 CrossRef CAS PubMed.
- T. S. Reddy, V. G. Reddy, H. Kulhari, R. Shukla, A. Kamal and V. Bansal, Synthesis of (Z)-1-(1,3-Diphenyl-1H-Pyrazol-4-yl)-3-(Phenylamino)Prop-2-en-1-one Derivatives As Potential Anticancer and Apoptosis Inducing Agents, Eur. J. Med. Chem., 2016, 117, 157 CrossRef PubMed.
- S. Fletcher and A. D. Hamilton, Targeting Protein–Protein Interactions by Rational Design: Mimicry of Protein Surfaces, J. R. Soc., Interface, 2006, 3, 215 CrossRef CAS PubMed.
- S. El-Rayes, M. S. Gomaa, A. Abouelmagd, W. Fathalla and I. A. I. Ali, Synthesis and antiproliferative assay of triazolyl-2,2-dimethyl-3-phenylpropanoates as potential HDAC inhibitors, RSC Adv., 2019, 9, 13896 RSC.
- S. M. El Rayes, A. AboElmagd, M. S. Gomaa, I. A. I. Ali, W. Fathalla, F. H. Pottoo and F. A. Khan, Convenient synthesis and antiproliferative activity of methyl 2-[3-(3-phenyl-quinoxalin-2-ylsulfanyl)propanamido]alkanoates and N-alkyl 3-((3-phenylquinoxalin-2-yl)sulfanyl)propanamides, ACS Omega, 2019, 4, 18555 CrossRef CAS PubMed.
- S. Y. Han and Y. A. Kim, Recent Development of Peptide Coupling Reagents in Organic Synthesis, Tetrahedron, 2004, 60, 2447 CrossRef CAS.
- I. A. I. Ali, I. A. Al-Masoudi, B. Saeed, N. A. Al-Masoudi and P. La Colla, Amino Acid Derivatives, Part 2: Synthesis, Antiviral, and Antitumor Activity of Simple Protected Amino Acids Functionalized at N Terminus with Naphthalene Side Chain, Heteroat. Chem., 2005, 16, 148 CrossRef CAS.
- M. Megahed, W. Fathalla and A. Elsheikh, Synthesis and antimicrobial activity of methyl (2-(2-(2-arylquinazolin-4-yl)oxy)acetyl)aminoalkanoates, J. Heterocycl. Chem., 2018, 55, 2799 CrossRef CAS.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, Synthesis of N-hydroxysuccinimide esters of acyl peptides by the mixed anhydride method, J. Am. Chem. Soc., 1967, 89, 178 CrossRef CAS PubMed.
- I. A. I. Ali and W. Fathalla, Synthesis of N-substituted-3,4,5,6-tetrachlorophthalimide using trichloroacetimidate C-C bond formation method, ARKIVOC, 2009,(xiii), 193 Search PubMed.
- W. Fathalla, Synthesis of 1-substituted benzhydryl derivatives using trichloroacetimidate C-C bond formation method submitted, Sci. J. Chem., 2014, 2, 38 Search PubMed.
- I. A. I. Ali and W. Fathalla, Synthesis of N-protected Non-proteinogenic α-amino Acid Esters by Using Trichloroacetimidate and Acetate Coupling Methods, Curr. Org. Chem., 2013, 17, 1903 CrossRef CAS.
- W. Fathalla, I. A. I. Ali and P. Pazdera, Efficient synthesis of ether phosphonates using trichloroacetimidate and acetate coupling methods, RSC Adv., 2018, 8, 7385 RSC.
- W. Fathalla, P. Pazdera, S. El-Rayes and I. A. I. Ali, Efficient synthesis of α-substituted-α-arylmethyl phosphonates using trichloroacetimidate C-C coupling, Tetrahedron, 2018, 74, 1681 CrossRef CAS.
- S. M. Asiri, F. A. Khan and A. Bozkurt, Delivery of Conjugated Silicon Dioxide Nanoparticles Show Strong Anti-Proliferative Activities, Appl. Biochem. Biotechnol., 2019, 189, 760 CrossRef CAS PubMed.
- F. A. Khan, S. Akhtar, D. Almohazey, M. Alomari, S. A. Almofty, I. Badr and A. Elaissari, Targeted delivery of poly(methyl methacrylate) particles in colon cancer cells selectively attenuates cancer cell proliferation, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 1533 CrossRef CAS PubMed.
- S. Akhtar, F. A. Khan and A. Buhaimed, Functionalized magnetic nanoparticles attenuate cancer cells proliferation: transmission electron microscopy analysis, Microsc. Res. Tech., 2019, 82, 983 CrossRef CAS PubMed.
- S. M. Asiri, F. A. Khan and A. Bozkurt, Synthesis of chitosan nanoparticles, chitosan-bulk, chitosan nanoparticles conjugated with glutaraldehyde with strong anti-cancer proliferative capabilities, Artif. Cells, Nanomed., Biotechnol., 2018, 46(sup. 3), S1152–S1161 CAS.
- J. Baharara, T. Ramezani, A. Divsalar, M. Mousavi and A. Seyedarabi, Induction of Apoptosis by Green Synthesized Gold Nanoparticles Through Activation of Caspase-3 and 9 in Human Cervical Cancer Cells, Avicenna J. Med. Biotechnol., 2016, 8, 75 Search PubMed.
- J. Mytych, A. Lewinska, J. Zebrowski and M. Wnuk, Gold Nanoparticles Promote Oxidant-Mediated Activation of NF-κB and 53BP1 Recruitment-Based Adaptive Response in Human Astrocytes, BioMed Res. Int., 2015, 304575 Search PubMed.
- W. Lin-Wei, Q. Ai-Ping, L. Wen-Lou, C. Jia-Mei, Y. Jing-Ping, W. Han, Y. Li and L. Juan, Quantum Dots-Based Double Imaging Combined With Organic Dye Imaging to Establish an Automatic Computerized Method for Cancer Ki67 Measurement, Sci. Rep., 2016, 6, 20564 CrossRef PubMed.
- I. Shiva, S. Zhohreh, M. A. Seyed and M. Shahriar, Induction of Growth Arrest in Colorectal Cancer Cells by Cold Plasma and Gold Nanoparticles, Arch. Med. Sci., 2015, 11, 1286 Search PubMed.
- P. Hye-Kyung, J. Hanbin, K. Eunhwa, L. Geumwoo, L. Ji-Eun, L. S. Kwang, L. An-Jung, Y. I. Jin, H. Sung, H. K. Seong, L. J. Hoon, L. Changwook, K. Soosung and H. K. Byoung, Paralog Specificity Determines Subcellular Distribution, Action Mechanism, and Anticancer Activity of TRAP1 Inhibitors, J. Med. Chem., 2017, 60, 7569 CrossRef PubMed.
- S. Akhtar, S. Rehman, M. A. Almessiere, F. A. Khan, Y. Slimani and A. Baykal, Synthesis of Mn(0.5)Zn(0.5)Sm(x)Eu(x)Fe(1.8-2x)O(4) Nanoparticles via the Hydrothermal Approach Induced Anti-Cancer and Anti-Bacterial Activities, Nanomater, 2019, 9, 1635 CrossRef CAS PubMed.
- S. Z. A. Khader, S. Syed Zameer Ahmed, G. M. Ganesan, M. R. Mahboob, M. Vetrivel, M. Sankarappan and P. Manickam, Rhynchosia rufescens AgNPs enhance cytotoxicity by ROS-mediated apoptosis in MCF-7 cell lines, Environ. Sci. Pollut. Res. Int., 2019, 27(2), 2155–2164 CrossRef PubMed.
- J. Chen, L. Chen, S. Du, J. Wu, M. Quan, H. Yin, Y. Wu, X. Ye, X. Liang and H. Jiang, High sensitive detection of circulating tumor cell by multimarker lipid magnetic nanoparticles and clinical verifications, J. Nanobiotechnol., 2019, 17(1), 116, DOI:10.1186/s12951-019-0548-1.
- Y. Wang, Y. Zhang, Y. Guo, J. Lu, V. P. Veeraraghavan, S. K. Mohan, C. Wang and X. Yu, Synthesis of zinc oxide nanoparticles from Marsdenia tenacissima inhibits the cell proliferation and induces apoptosis in laryngeal cancer cells (Hep-2), J. Photochem. Photobiol., B, 2019, 201, 111624, DOI:10.1016/j.jphotobiol.2019.111624.
- M. Mughees, M. Samim, Y. Sharma and S. Wajid, Identification of protein targets and the mechanism of the cytotoxic action of Ipomoea turpethum extract loaded nanoparticles against breast cancer cells, J Mater Chem B, 2019, 7(39), 6048–6063, 10.1039/c9tb00824a.
- M. H. Abdol mohammadi, S. H. Fouladdel, A. Shafiee, G. H. Amin, S. M. Ghaffari and E. Azizi, Anticancer effects and cell cycle analysis on human breast cancer T47D cells treated with extracts of Astrodaucus persicus (Boiss.) Drude in comparison to doxorubicin, Daru, J. Fac. Pharm., Tehran Univ. Med. Sci., 2008, 16(2), 112–118 Search PubMed.
- M. G. Brasca, S. Mantegani, N. Amboldi, S. Bindi, D. Caronni, E. Casale, W. Ceccarelli, N. Colombo, A. De Ponti, D. Donati, A. Ermoli, G. Fachin, E. R. Felder, R. D. Ferguson, C. Fiorelli, M. Guanci, A. Isacchi, E. Pesenti, P. Polucci, L. Riceputi, F. Sola, C. Visco, F. Zuccotto and G. Fogliatto, Discovery of NMS-E973 as novel, selective and potent inhibitor of heat shock protein 90 (Hsp90), Bioorg. Med. Chem., 2013, 21, 7047 CrossRef CAS PubMed.
- C.-N. Im, Past, present, and emerging roles of mitochondrial heat shock protein TRAP1 in the metabolism and regulation of cancer stem cells, Cell Stress Chaperones, 2016, 21, 553 CrossRef CAS PubMed.
- C. Lee, H.-K. Park, H. Jeong, J. Lim, L. An-Jung, K. Y. Cheon, C.-S. Kim, A. P. Thomas, B. Bae, N. D. Kim, S. H. Kim, P.-G. Suh, J.-H. Ryu and B. H. Kang, Development of a Mitochondria-Targeted Hsp90 Inhibitor Based on the Crystal Structures of Human TRAP1, J. Am. Chem. Soc., 2014, 137, 4358 CrossRef PubMed.
- R. H. Kalita, A. J. Borah and P. Phukan, Mukaiyama aldol reaction of trimethylsilyl enolate with aldehyde catalyzed by CuI, Indian J. Chem., 2013, 52B, 289 Search PubMed.
- F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, An E-factor minimized protocol for the preparation of methyl β-hydroxy esters, Green Chem., 2010, 12, 1301 RSC.
- S. M. El Rayes, A. Aboelmagd, M. S. Gomaa, I. A. I. Ali, W. Fathalla, F. H. Pottoo and F. A. Khan, Convenient Synthesis and Anticancer Activity of Methyl 2-[3-(3-Phenyl-quinoxalin-2-ylsulfanyl)propanamido]alkanoates and N-Alkyl 3-((3-Phenyl-quinoxalin-2-yl)sulfanyl)propanamides, ACS Omega, 2019, 4(20), 18555–18566, DOI:10.1021/acsomega.9b02320.
| This journal is © The Royal Society of Chemistry 2020 |