
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmart regioselectivity towards mono 6-hydroxyl α-cyclodextrin amphiphilic derivatives†
Yanli Cui *ab,
Yangyi Maoc,
Jianwei Maoe and
Yongmin Zhang*ad
*ab,
Yangyi Maoc,
Jianwei Maoe and
Yongmin Zhang*ad
aSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, UMR 8232, 4 Place Jussieu, 75005 Paris, France. E-mail: yongmin.zhang@upmc.fr; Tel: +33 144276153
bDepartment of Chemistry, Zhejiang University, 38 Zheda Road, Hangzhou 310027, China. E-mail: cuiyl@zju.edu.cn; Tel: +86 13858036095
cHangzhou Yanqing Biotechnology Co., Ltd, Hangzhou 310052, China
dInstitute for Interdisciplinary Research, Jianghan University, Wuhan Economic and Technological Development Zone, Wuhan 430056, China
eZhejiang University of Science and Technology, Hangzhou 310013, China
First published on 13th March 2020
Abstract
Following the trend of eco-friendly development, a smart regioselective modification is described herein, for mono 6-hydroxyl and penta-alkyl coexistence on the primary face of α-cyclodextrins with no additional catalysis or no enzyme process, just via the adjustment of the ratio of alkali to alkylation agent, with good yields. The novel procedure minimized the tedious protection, deprotection steps and provided useful intermediates for further cutting edge research. Thus, the scope of green and economical access is extended from penta-pentenyl substitution to C4–C6 alkyl group substitution. It was speculated that the mechanism might be controlled by the concentration of alkali in the system and the steric effects of the electrophilic reagent RBr.
1. Introduction
Cyclodextrins (CDs) are a family of cyclic oligosaccharides derived from starch enzymolysis that possess molecule cavities with topological structures. CDs and their cyclodextrin derivatives (CDDs) have attracted widespread attention for their increasingly expanding applications from traditional to cutting edge research in fields such as: cosmetics, food additives, drug carriers, analysis and catalysis technologies, and recently in stimuli-responsive carriers, cyclodextrin–graphene, supramolecular systems and other CD-based biomolecules.1–7 The monosubstituted CDs are synthesized by introducing a desired functional group or biomolecule to the single hydroxyl, or the mono hydroxyl on cyclodextrins connected with another biological macromolecule or polymer. Monosubstituted derivatives were also utilized in the synthesis of defined CD dimers and oligomers, which exhibit stronger binding constants towards guest molecules than the corresponding monomers due to the cooperative inclusion.8The CD molecule has numerous hydroxyl groups in three different positions that make tiny differences to its reaction activity. The selective derivatization is often a tough task, involving elaborate multistep protection-deprotections, with a low overall yield. In 1998, D'Souza reviewed strategies for selective modification of CDs, including regioselective monomodifications, but nevertheless, the methods used were traditional.9 Traditionally the most popular method for monomodifications at the 6-position of CDs is via nucleophilic attack from a reagent containing the appropriate group on a mono-6-sulfonylcyclodextrin. These monosulfonates are prepared by reacting 1 equiv. of p-toluenesulfonyl chloride with CD in pyridine. However, monotosylation of CD is often a nonselective process and produces a mixture of primary as well as secondary side tosylated products together with di- or tri-tosylated derivatives.9 Alternatively, from the opposite point of view, an approach has been developed over the years, which involves the deprotection of the protected CDs. The methodology uses diisobutylaluminium hydride (DIBAL-H) as a dealkylating reagent and has been successfully applied to benzylated CDs,15 allowing access to polyhetero-functionalized CDs containing two, three or even six different functionalities.16 For example, when perbenzylated α-CD was treated with excess DIBAL-H at room temperature, the mono-debenzylated product was formed in 64% yield, the hydroxyl group being on the primary face.15 The mono-demethylation could also occur on the primary face of hexakis(2,3-di-O-benzyl)-6-hexa-O-methyl-α-CD, but with a low yield (30%).17 Recently Ghosh et al. applied this methodology to per-6-O-tert-butyldimethylsilylated CDs to produce mono- and di-O-desilylated CDs,18 for example, the mono-O-desilylated-α-CD product was isolated in 71% yield.
Herein a one-pot approach is reported for the preparation of a novel series of mono 6-hydroxyl α-CDs with a variety of alkyl groups on the primary face using no additional catalysis or no enzyme process. However, the efficient method and unique structure of the substrates have not been reported as far as is known. Among the linkages (e.g., sulfide,10,11 amine,12 ester,13 ether,14 etc.) available for the attachment of functional groups to CDs, ether bonds are the most desirable, because they are less susceptible to degradation by oxidation or hydrolysis and obtained by a single nucleophilic reaction of a hydroxyl group of the CDs with an appropriate electrophile. The CD ethers either serve as amphiphilic sections, ene-end CDDs for further functionalization, or act as reaction intermediates of click reactions, or are converted into artificial enzymes, and others. In other words, the published substrates can provide versatile variation for fast CD-based research.
2. Materials and methods
2.1. General methods
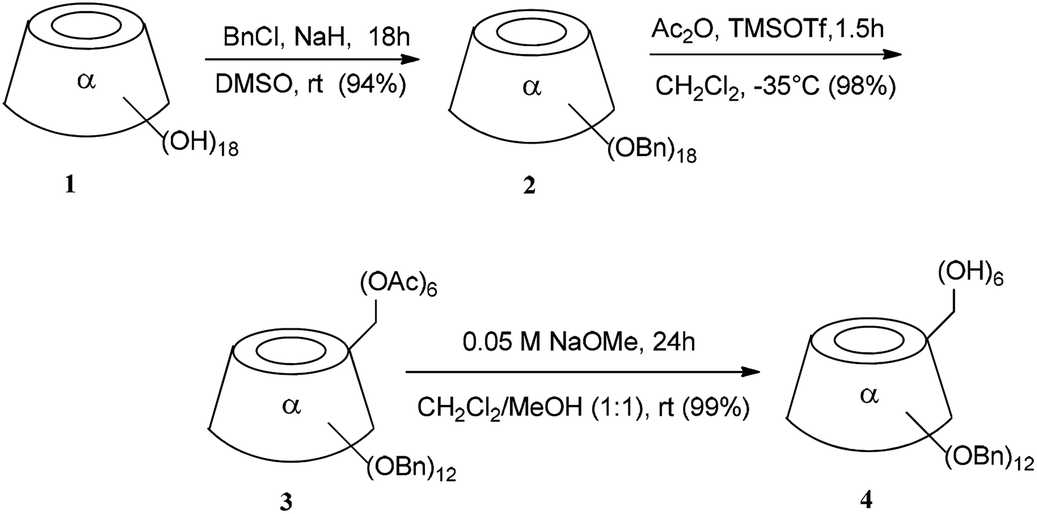
Analytical grade chemicals were used for all reactions and all solvents were dried over standard drying agents and freshly distilled prior to use. Optical rotations were measured at 20 ± 2 °C with a PerkinElmer model 241 digital polarimeter, using a 10 cm, 1 mL cell. Electrospray ionization (ESI)-time-of-flight mass spectra were obtained with a Micromass (UK) LCT ESI-TOF spectrometer. The NMR spectra (1H, 13C) were recorded on a Bruker DRX-400, DMX-500 instrument (Karlsruhe, Germany) at ambient temperature with tetramethylsilane as an internal standard and the coupling constants are represented in Hz. Assignments were aided by carrying out correlated spectroscopy (COSY) experiments. The reactions were monitored by thin-layer chromatography (TCL) on a precoated plate of Merck silica gel 60 F254 (layer thickness: 0.2 mm, Darmstadt, Germany) and detection by charring with sulfuric acid. Flash column chromatography was performed on Merck silica gel 60 (230–400 mesh).2.2. Per-2,3-di-O-benzyl-α-cyclodextrin (4)
A solution of 3 (100 mg, 0.04335 mmol) in 5 mL of 0.05 M NaOMe (dry dichloromethane and dry methanol, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was stirred under argon for 24 h at room temperature. The Amberlite IR 120/H+ resin was then added, and this mixture was stirred until the pH became neutral. The mixture was filtered, and the resin was washed with a mixture of dichloromethane and methanol (1:1). The filtrate was then concentrated. The residue was purified by silica gel chromatography (dichloromethane:methanol, 15:1) to provide the title compound 4 (88.01 mg, 99.00%) as a white powder. The Rf = 0.43 (dichloromethane:methanol, 15:1); [α]D = +50 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.20–7.30 (m, 10H, Ph), 5.16, 4.88 (dd, 2H, J = 11.6 Hz, CH2Ph), 5.08 (d, 1H, J1,2 = 3.52 Hz, H1), 4.88 (1H, OH), 4.65, 4.49 (dd, 2H, J = 12.27 Hz, CH2Ph), 4.17–4.07 (m, 2H, H3, H6a), 3.96–3.90 (m, 1H, H5), 3.78–3.67 (m, 2H, H2, H6b), 3.54 (dd, 1H, J1,2 = 3.52 Hz, J2,3 = 9.81 Hz, H2). 13C-NMR (100 MHz, CDCl3) δ 139.12 (C, Ph), 138.21 (C, Ph), 128.17 (CH, Ph), 127.98 (CH, Ph), 127.68 (CH, Ph), 127.46 (CH, Ph), 127.29 (CH, Ph), 126.95 (CH, Ph), 98.00 (C1), 80.80 (C3), 79.56 (C4), 78.93 (C2), 75.39 (CH2Ph), 73.20 (C5), 73.01 (CH2Ph), 63.00 (C6).
:1) was stirred under argon for 24 h at room temperature. The Amberlite IR 120/H+ resin was then added, and this mixture was stirred until the pH became neutral. The mixture was filtered, and the resin was washed with a mixture of dichloromethane and methanol (1:1). The filtrate was then concentrated. The residue was purified by silica gel chromatography (dichloromethane:methanol, 15:1) to provide the title compound 4 (88.01 mg, 99.00%) as a white powder. The Rf = 0.43 (dichloromethane:methanol, 15:1); [α]D = +50 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.20–7.30 (m, 10H, Ph), 5.16, 4.88 (dd, 2H, J = 11.6 Hz, CH2Ph), 5.08 (d, 1H, J1,2 = 3.52 Hz, H1), 4.88 (1H, OH), 4.65, 4.49 (dd, 2H, J = 12.27 Hz, CH2Ph), 4.17–4.07 (m, 2H, H3, H6a), 3.96–3.90 (m, 1H, H5), 3.78–3.67 (m, 2H, H2, H6b), 3.54 (dd, 1H, J1,2 = 3.52 Hz, J2,3 = 9.81 Hz, H2). 13C-NMR (100 MHz, CDCl3) δ 139.12 (C, Ph), 138.21 (C, Ph), 128.17 (CH, Ph), 127.98 (CH, Ph), 127.68 (CH, Ph), 127.46 (CH, Ph), 127.29 (CH, Ph), 126.95 (CH, Ph), 98.00 (C1), 80.80 (C3), 79.56 (C4), 78.93 (C2), 75.39 (CH2Ph), 73.20 (C5), 73.01 (CH2Ph), 63.00 (C6).
2.3. 6I–O-Hydroxyl-6II–VI-penta-O-pentylenyl-(per-2,3-di-O-benzyl)-α-cyclodextrin (5e) and per-6-O-(pentylenyl-1)-per-2,3-di-O-benzyl-α-cyclodextrin (6e)
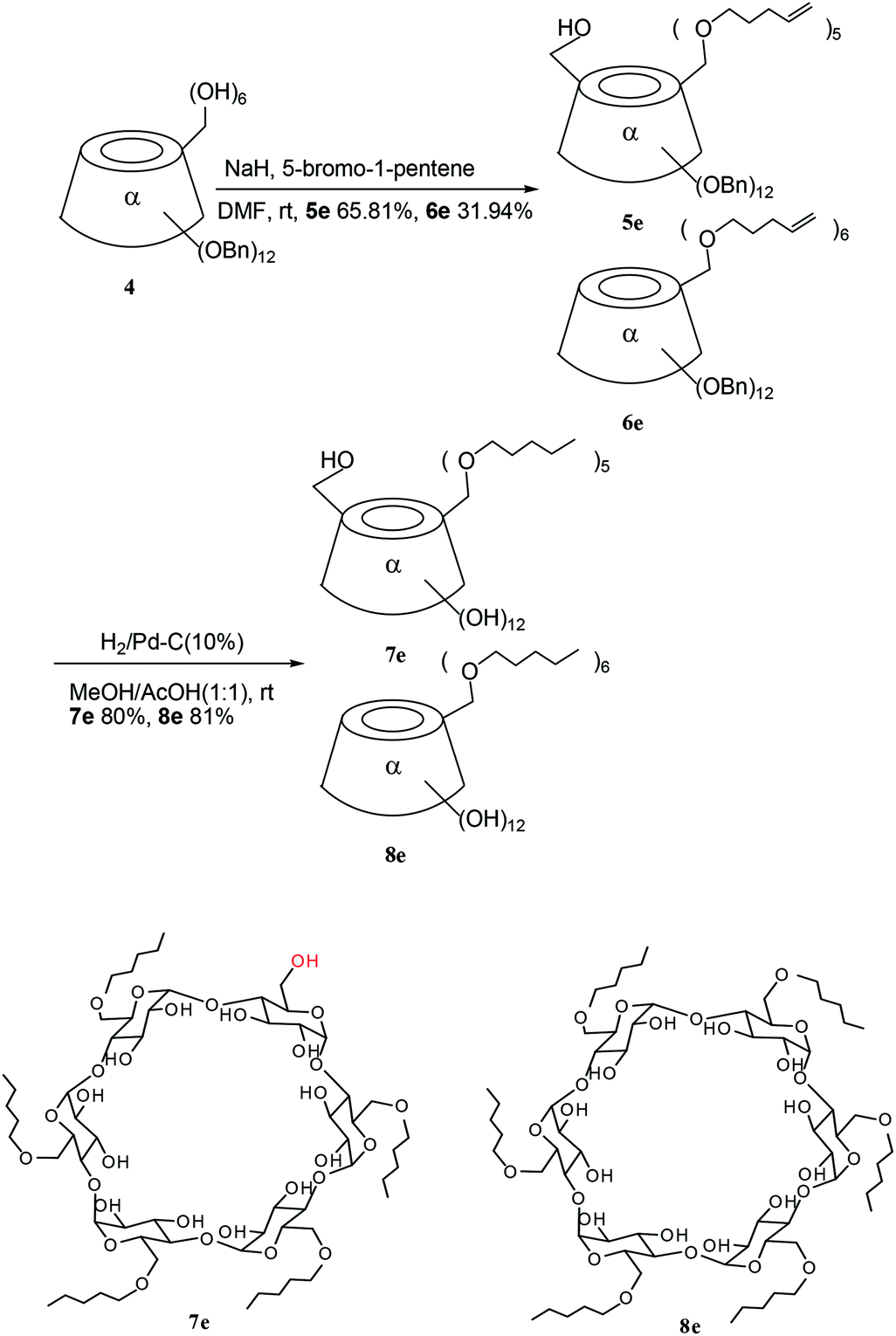
To a solution of 4 (51 mg, 0.02485 mmol) in 7 mL of dry DMF, was added 60% NaH (13.90 mg, 0.3479 mmol) under argon at room temperature, then 5-bromo-1-pentene (88.91 mg, 0.5964 mmol) was added. The mixture was stirred at room temperature until the starting product 4 had disappeared, and was then hydrolyzed by MeOH (7 mL) and evaporated. The residue was dissolved in dichloromethane. The organic phrase was washed with saturated NaCl and water, dried over anhydrous magnesium sulfate, filtered, and then concentrated. The residue was purified by silica gel chromatography (cyclohexane:ethyl acetate, 5:1) to give the title compounds 5e (39.1 mg, 65.81%) and 6e (19.50 mg, 31.94%) as white powders.
:1); [α]D = +27.6 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.40–7.10 (m, 60H, Ph), 5.92–5.76 (m, 5H, 5 × CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.71 (d, 1H, J1,2 = 3.86 Hz, H1), 5.67 (d, 1H, J1,2 = 3.76 Hz, H1′), 5.53, 5.47 (dd, 2H, J = 10.5 Hz, CH2Ph), 5.25 (dd, 2H, J = 10.7 Hz, CH2Ph), 5.16 (d, J1,2 = 3.48 Hz, H1), 5.08–4.97 (m, 2H, CHCH2), 4.57, 4.50 (dd, 2H, J = 12.6 Hz, CH2Ph), 4.22–4.16 (m, 1H, H5), 4.09–4.00 (m, 2H, H4, H6a), 3.98–3.92 (m, 1H, H3), 3.60–3.50 (m, 3H, H2, H6b, H–(O–CH2)), 3.43–3.36
CH2), 5.71 (d, 1H, J1,2 = 3.86 Hz, H1), 5.67 (d, 1H, J1,2 = 3.76 Hz, H1′), 5.53, 5.47 (dd, 2H, J = 10.5 Hz, CH2Ph), 5.25 (dd, 2H, J = 10.7 Hz, CH2Ph), 5.16 (d, J1,2 = 3.48 Hz, H1), 5.08–4.97 (m, 2H, CHCH2), 4.57, 4.50 (dd, 2H, J = 12.6 Hz, CH2Ph), 4.22–4.16 (m, 1H, H5), 4.09–4.00 (m, 2H, H4, H6a), 3.98–3.92 (m, 1H, H3), 3.60–3.50 (m, 3H, H2, H6b, H–(O–CH2)), 3.43–3.36 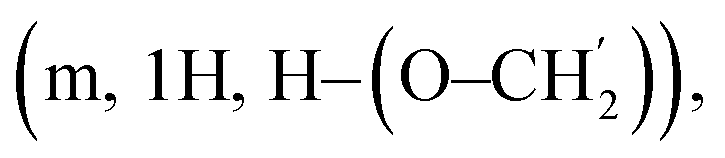 2.20–2.01 (m, 10H, CH2–CHCH2), 1.78–1.63 (m, 10H, CH2CH2–CHCH2). 13C-NMR (100 MHz, CDCl3) δ 139.41, 139.33, 139.29, 139.25, 139.16, 138.63, 138.49, 138.44, 138.22, 138.10, 137.96, (C, Ph), 138.16, 138.02, 137.93, 137.89 (CHCH2), 128.16–126.34 (CH, Ph), 114.88, 114.87, 114.84, 114.78, 114.71 (CHCH2), 98.87, 98.20, 98.14, 98.10, 98.09, 97.73 (C1), 81.60, 81.49, 81.37, 81.19, 81.11, 80.89, 80.84, 80.68, 80.50, 80.20, 79.79, 79.60, 79.17, 79.05, 78.15, 77.69, 75.49, 74.28, 71.94, 71.83, 71.74, 71.41, 71.34, 71.10 (C2, C3, C4, C5), 76.32, 76.17, 76.00, 75.98, 74.15, 74.10, 73.31, 73.22, 73.12, 72.94, 72.35, 72.30, 71.56, 71.21, 71.01, 70.98, 61.05, 61.00 (CH2Ph, O–CH2, C6). 30.28, 30.23, 30.19, 30.18, 30.16 (CH2–CHCH2), 29.01, 28.84, 28.81, 28.60, 28.58 (CH2CH2–CHCH2). HRMS (MALDI-TOF, M + Na+) m/z calcd for C145H172O30Na 2416.1826, found 2416.1944.:ethyl acetate, 5:1); [α]D = +25 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.30–7.20 (m, 10H, Ph), 5.89–5.78 (m, 1H, CHCH2), 5.24, 4.92 (dd, 2H, J = 11.01 Hz, CH2Ph), 5.16 (d, 1H, J1,2 = 3.48 Hz, H1), 5.08–4.97 (m, 2H, CHCH2), 4.57, 4.50 (2d, 2H, J = 12.60 Hz, CH2Ph), 4.19 (dd, 1H, J2,3 = 9.59 Hz, J3,4 = 8.34 Hz, H3), 4.09–4.00 (m, 2H, H4, H6a), 3.97–3.92 (m, 1H, H5), 3.60–3.50 (m, 3H, H2, H6b, one of O–CH2), 3.43–3.36 (m, 1H, one of O–CH2), 2.19–2.03 (m, 2H, CH2–CHCH2), 1.73–1.66 (m, 2H, CH2CH2–CHCH2). 13C-NMR (100 MHz, CDCl3) δ 139.38 (C, Ph), 138.37 (C, Ph), 138.01 (CHCH2), 128.13 (CH, Ph), 127.96 (CH, Ph), 127.74 (CH, Ph), 127.40 (CH, Ph), 127.24 (CH, Ph), 126.89 (CH, Ph), 114.79 (CHCH2), 98.60 (C1), 81.09 (C3), 79.18 (C4), 78.99 (C2), 75.52 (CH2Ph), 72.77 (CH2Ph), 71.39 (C5), 71.08 (O–CH2), 69.56 (C6), 30.25 (CH2–CHCH2), 28.86 (CH2CH2–CHCH2). HRMS (FAB+, M + Na+) m/z calcd for C150H180O30Na 2484.2457, found 2484.2475.
2.20–2.01 (m, 10H, CH2–CHCH2), 1.78–1.63 (m, 10H, CH2CH2–CHCH2). 13C-NMR (100 MHz, CDCl3) δ 139.41, 139.33, 139.29, 139.25, 139.16, 138.63, 138.49, 138.44, 138.22, 138.10, 137.96, (C, Ph), 138.16, 138.02, 137.93, 137.89 (CHCH2), 128.16–126.34 (CH, Ph), 114.88, 114.87, 114.84, 114.78, 114.71 (CHCH2), 98.87, 98.20, 98.14, 98.10, 98.09, 97.73 (C1), 81.60, 81.49, 81.37, 81.19, 81.11, 80.89, 80.84, 80.68, 80.50, 80.20, 79.79, 79.60, 79.17, 79.05, 78.15, 77.69, 75.49, 74.28, 71.94, 71.83, 71.74, 71.41, 71.34, 71.10 (C2, C3, C4, C5), 76.32, 76.17, 76.00, 75.98, 74.15, 74.10, 73.31, 73.22, 73.12, 72.94, 72.35, 72.30, 71.56, 71.21, 71.01, 70.98, 61.05, 61.00 (CH2Ph, O–CH2, C6). 30.28, 30.23, 30.19, 30.18, 30.16 (CH2–CHCH2), 29.01, 28.84, 28.81, 28.60, 28.58 (CH2CH2–CHCH2). HRMS (MALDI-TOF, M + Na+) m/z calcd for C145H172O30Na 2416.1826, found 2416.1944.:ethyl acetate, 5:1); [α]D = +25 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.30–7.20 (m, 10H, Ph), 5.89–5.78 (m, 1H, CHCH2), 5.24, 4.92 (dd, 2H, J = 11.01 Hz, CH2Ph), 5.16 (d, 1H, J1,2 = 3.48 Hz, H1), 5.08–4.97 (m, 2H, CHCH2), 4.57, 4.50 (2d, 2H, J = 12.60 Hz, CH2Ph), 4.19 (dd, 1H, J2,3 = 9.59 Hz, J3,4 = 8.34 Hz, H3), 4.09–4.00 (m, 2H, H4, H6a), 3.97–3.92 (m, 1H, H5), 3.60–3.50 (m, 3H, H2, H6b, one of O–CH2), 3.43–3.36 (m, 1H, one of O–CH2), 2.19–2.03 (m, 2H, CH2–CHCH2), 1.73–1.66 (m, 2H, CH2CH2–CHCH2). 13C-NMR (100 MHz, CDCl3) δ 139.38 (C, Ph), 138.37 (C, Ph), 138.01 (CHCH2), 128.13 (CH, Ph), 127.96 (CH, Ph), 127.74 (CH, Ph), 127.40 (CH, Ph), 127.24 (CH, Ph), 126.89 (CH, Ph), 114.79 (CHCH2), 98.60 (C1), 81.09 (C3), 79.18 (C4), 78.99 (C2), 75.52 (CH2Ph), 72.77 (CH2Ph), 71.39 (C5), 71.08 (O–CH2), 69.56 (C6), 30.25 (CH2–CHCH2), 28.86 (CH2CH2–CHCH2). HRMS (FAB+, M + Na+) m/z calcd for C150H180O30Na 2484.2457, found 2484.2475.2.4. 6I–O-Hydroxyl-6II–VI-penta-O-pentyl-α-cyclodextrin (7e) and per-6-O-pentyl-α-cyclodextrin (8e)
To a solution of 5e (61.01 mg, 0.02476 mmol) in a mixture of methanol/AcOH (1:1, 12 mL) was added Pd–C (10%, 42.10 mg), and the resulting mixture was then was stirred under an atmosphere of H2 for 12 h at room temperature. The results of the TLC (cyclohexane:ethyl acetate, 5:1) indicated that the starting product had disappeared. The catalyst was removed by filtration and was washed with a mixture of dichloromethane and methanol (1:1). The filtrate was then concentrated. The residue was purified by silica gel chromatography (dichloromethane:methanol, 8:1) to give the title compound 7e (27.61 mg, 80.02%) as a white powder. The Rf = 0.54 (dichloromethane:methanol, 6:1); [α]D = +66.7 (c 1, CHCl3/MeOH, 7:3); 1H-NMR (400 MHz, pyridine-d5) δ 5.55 (d, 1H, J1,2 = 3.1 Hz, H1), 5.49 (d, 1H, J1,2 = 3.1 Hz, H1), 5.48–5.43 (m, 4H, 4 × H1), 4.74–4.62 (m, 6H, 6 × H3), 4.52–4.29 (m, 10H, H2, H4, 6 × H5, H6a), 4.23–3.91 (m, 21H, 5 × H2, 5 × H6a, 6 × H6b, 5 × H4), 3.73–3.58 (m, 10H, 5 × OCH2), 1.75–1.58 (m, 10H, 5 × CH2CH2CH2CH3), 1.43–1.23 (m, 10H, 5 × CH2CH3, 5 × CH2CH2CH3), 0.90–0.82 (m, 15H, 5 × CH3). 13C-NMR (100 MHz, pyridine-d5) δ 104.46, 104.28, 104.18 (C1), 83.07, 83.92, 83.83, 83.69 (C2), 75.59, 75.55 (C3), 74.70, 74.62, 74.40, 74.51 (C4), 73.09, 73.03 (C5), 72.47, 72.44, 72.42, 72.37 (O–CH2), 70.91, 70.86, 70.77, 70.74, 70.59, 62.04 (C6), 30.57, 30.56 (CH2CH2CH2CH3), 29.35, 29.31 (CH2CH2CH3), 3.44 (CH2CH3), 14.86 (CH3). HRMS (ESI-TOF, M − H−) m/z calcd for C61H109O30 1321.7004, found 1321.6957.
A similar method to that described for obtaining 7e, was performed to give the title compound 8e (81.10%) as a white powder. The Rf = 0.48 (dichloromethane:methanol, 8:1); [α]D = +81.5 (c 0.7, CHCl3:methanol, 7:3); 1H-NMR (400 MHz, pyridine-d5) δ 5.46 (d, 1H, J1,2 = 3.1 Hz, H1), 5.18–5.02 (br, 2H, 2 × OH), 4.65 (t, 1H, J = 9.14 Hz, H3), 4.44–4.38 (m, 1H, H5), 4.19–4.13 (m, 2H, H4, H6a), 4.11 (dd, 1H, J1,2 = 3.10 Hz, J2,3 = 9.70 Hz, H2), 4.04–3.99 (m, 1H, H6b), 3.72–3.66 (m, 2H, O–CH2), 1.75–1.61 (m, 2H, CH2CH2CH2CH3), 1.44–1.27 (m, 4H, CH2CH3, CH2CH2CH3), 0.87 (t, 1H, J = 7.2 Hz, CH3). 13C-NMR (100 MHz, pyridine-d5) δ 104.26 (C1), 83.90 (C4), 75.52 (C3), 74.55 (C2), 73.01 (C5), 72.43 (O–CH2), 70.79 (C6), 30.55 (CH2CH2CH2CH3), 29.34 (CH2CH2CH3), 23.42 (CH2CH3), 14.85 (CH3). HRMS (ESI, M + Na+) m/z calcd for C66H120O30Na 1415.7762, found: 1415.7760; HRMS (ESI, M + K+) m/z calcd for C66H120O30K 1431.7502, found 1431.7544; HRMS (ESI, M + 2Na+) m/z calcd for C66H119O30Na2 1437.7582, found 1437.7544.
2.5. Per-6-O-ethyl-2,3-di-O-benzyl-α-cyclodextrin (6a)
A similar method to that described for obtaining 6e, was performed to give the title compound 6a (80.50%) as a white powder. The Rf = 0.32 (cyclohexane/ethyl acetate, 5:1.5); [α]D = +20.1 (c 1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.40–7.04 (m, 60H, Ph), 5.20, 4.89 (dd, 12H, J = 10.00 Hz, CH2Ph), 5.04 (d, 6H, J1,2 < 1 Hz, H1), 4.50, 4.42 (2d, 12H, J = 10.00 Hz, CH2Ph), 4.13 (dd, 6H, H3), 3.63–3.56, 3.55–3.38 (m, 42H, H6a, H5, H4, H6b, H2, O–CH2), 1.14 (s, 18H, CH3). 13C-NMR (125 MHz, CDCl3) δ 139.46 (C, Ph), 138.41 (C, Ph), 128.06 (CH, Ph), 127.89 (CH, Ph), 127.79 (CH, Ph), 127.35 (CH, Ph), 127.12 (CH, Ph), 126.79 (CH, Ph), 98.97 (C1), 80.99 (C3), 79.80, 78.98 (C2, C4), 75.45 (CH2Ph), 72.76 (CH2Ph), 71.16 (O–CH2), 69.34 (C5), 66.56 (C6), 15.13 (CH3). HRMS (MALDI/TOF, M + Na+) m/z calcd for C132H156O30Na 2244.0561, found 2244.0574.
2.6. Per-6-O-allyl-per-2,3-di-O-benzyl-α-cyclodextrin (6b)
A similar method to that described for obtaining 6e, was performed to give the title compound 6b (81.70%) as a white powder. The Rf = 0.27 (cyclohexane:ethyl acetate, 5:1); [α]D = +36.6 (c 1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.42–7.05 (m, 10H, Ph), 5.97–5.77 (m, 1H, CHCH2), 5.20, 4.88 (dd, 2H, J = 11.08 Hz, CH2Ph), 5.03 (d, 1H, J1,2 = 2.92 Hz, H1), 5.13–5.11 (m, 2H, CHCH2), 4.47, 4.41 (2d, 2H, J = 10.95 Hz, CH2Ph), 4.17–3.43 (8H, H3, H4, H6a, H5, H6b, O–CH2, H2), 1.26–0.82 (m, 2H, CH2–CHCH2). 13C-NMR (125 MHz, CDCl3) δ 139.44 (C, Ph), 138.37 (C, Ph), 134.87 (CHCH2), 128.10 (CH, Ph), 127.92 (CH, Ph), 127.83 (CH, Ph), 127.39 (CH, Ph), 127.16 (CH, Ph), 126.84 (CH, Ph), 117.85 (CHCH2), 99.00 (C1), 80.91 (C3), 79.75 (C4), 78.91 (C2), 75.49 (CH2Ph), 72.74 (CH2Ph), 72.30 (O–CH2), 71.29 (C5), 69.05 (C6). HRMS (MALDI/DHB, M + Na+) m/z calcd for C138H156O30Na 2316.0574, found 2316.0574.
2.7. 6I–O-Hydroxyl-6II–VI-penta-O-propyl-(per-2,3-di-O-benzyl)-α-cyclodextrin (5c) and per-6-O-propyl-2,3-di-O-benzyl-α-cyclodextrin (6c)
A similar method to that described for obtaining 5e was performed to give the title compound 5c (60.40%) as a white powder. The Rf = 0.15 (cyclohexane:ethyl acetate, 5:1); [α]D = +21.2 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.37–7.05 (m, 60H, Ph), 5.64 (m 1H, H1), 5.67 (m 1H, H1′), 5.48, 5.42 (dd, 2H, J = 9.71 Hz, CH2Ph), 5.23, 5.18 (dd, 2H, J = 11.05 Hz, CH2Ph), 4.95–4.70, 4.58–4.30, 4.28–3.24 (m, 70H, H1, CH2Ph, H5, H4, H6a, H3, H2, H6b, O–CH2), 1.60–1.26 (m, 10H, –CH2–), 0.96–0.81 (m, 15H, –CH3). 13C-NMR (100 MHz, CDCl3) δ 139.47, 139.38, 139.36, 139.34, 139.29, 139.21, 138.71, 138.54, 138.50, 138.25, 138.16, 137.99 (C, Ph), 128.33–126.34 (CH, Ph), 98.97, 98.27, 98.16, 98.11, 98.06, 97.73 (C1), 81.70, 81.51, 81.33, 81.20, 80.95, 80.90, 80.71, 80.64, 80.22, 79.85, 79.67, 79.18, 79.02, 78.15, 77.72, 77.20, 76.35, 76.18, 76.02, 76.00, 75.67, 74.07, 74.04, 73.86, 73.39, 73.32, 73.23, 73.18, 73.11, 72.92, 72.34, 72.29, 71.94, 71.89, 71.75, 71.39, 70.99, 70.49, 69.99, 69.60, 69.43, 60.73 (C2, C3, C4, C5, CH2Ph, O–CH2, C6), 22.96, 22.85, 22.76, 22.65, 22.56 (–OCH2CH3), 10.64–10.38 (–CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C135H162O30Na 2286.1045, found 2286.1043.
A similar method as that described for obtaining 6e, was performed to give the title compound 6c (33.73%) as a white powder. The Rf = 0.44 (cyclohexane:ethyl acetate, 5:1); [α]D = +18.3 (c 1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.36–7.06 (m, 10H, Ph), 5.19, 4.87 (dd, 2H, J = 10.00 Hz, CH2Ph), 5.10 (d, 1H, J1,2 = 3.15 Hz, H1), 4.47, 4.44 (dd, 2H, J = 10.00 Hz, CH2Ph), 4.14 (dd, 1H, J2,3 = 10.10 Hz, J3,4 = 8.56 Hz, H3), 4.03–3.88 (m, 3H, H4, H5 H6a), 3.61–3.27 (m, 4H, H6b, H2, O–CH2), 1.33–1.19 (m, 2H, CH2), 0.92–0.81 (m, 3H, CH3). 13C-NMR (125 MHz, CDCl3) δ 139.43 (C, Ph), 138.38 (C, Ph), 128.06 (CH, Ph), 127.90 (CH, Ph), 127.72 (CH, Ph), 127.32 (CH, Ph), 127.19 (CH, Ph), 126.81 (CH, Ph), 98.75 (C1), 81.05 (C3), 79.49, 79.02 (C4, C2), 75.46 (CH2Ph), 73.21 (CH2Ph), 72.72 (O–CH2), 71.40 (C5), 69.48 (C6), 29.63, (OCH2–CH2), 10.54 (CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C138H168O30Na 2328.1473, found 2328.1513.
2.8. 6I–O-Hydroxyl-6II–VI-penta-O-butyl-(per-2,3-di-O-benzyl)-α-cyclodextrin (5d) and per-6-O-butyl-2,3-di-O-benzyl-α-cyclodextrin (6d)
A similar method as that described for obtaining 5e, was performed to give the title compound 5d (51.88%) as a white powder. The Rf = 0.29 (cyclohexane:ethyl acetate, 5:1); [α]D = +19.5 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.46–6.98 (m, 60H, Ph), 5.66 (d, 1H, J1,2 3.36 Hz, H1), 5.62 (d, 1H, J1,2 = 3.36 Hz, H1′), 5.47, 5.41 (dd, 2H, J = 9.86 Hz, CH2Ph), 5.21, 5.17 (dd, 2H, J = 10.05 Hz, CH2Ph), 4.94–4.64, 4.59–4.27, 4.25–3.32 (m, 70H, H1, CH2Ph, H5, H4, H6a, H3, H2, H6b, O–CH2), 1.65–1.20 (m, 20H, –CH2CH2–), 0.95–0.83 (m, 15H, –CH3). 13C-NMR (100 MHz, CDCl3) δ 139.48, 139.40, 139.36, 139.33, 139.23, 138.69, 138.55, 138.51, 138.28, 138.18, 138.03 (C, Ph), 128.31–126.36 (CH, Ph), 98.90, 98.86, 98.20, 98.10, 97.83, 97.77 (C1), 81.65, 81.55, 81.47, 81.22, 81.11, 80.95, 80.92, 80.75, 80.56, 80.17, 79.88, 79.71, 79.24, 79.12, 78.23, 77.80, 76.29, 76.14, 76.01, 75.96, 75.69, 75.64, 74.35, 74.13, 74.07, 73.30, 73.22, 73.10, 72.90, 72.36, 72.28, 71.95, 71.88, 71.78, 71.61, 71.45, 71.37, 71.33, 71.19, 71.08, 70.54, 69.92, 69.67, 69.50, 69.46, 60.94 (C2, C3, C4, C5, CH2Ph, O–CH2, C6), 31.92–31.49, 29.67, 19.39–19.15 (–OCH2CH2CH2–), 14.03–13.84 (–CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C140H172O30Na 2356.1793, found: 2356.1826.
A similar method to that described for obtaining 6e, was performed to give the title compound 6d (35.37%) as a white powder. The Rf = 0.52 (cyclohexane:ethyl acetate, 5:1); [α]D = +21.6 (c 1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.24–7.11 (m, 10H, Ph), 5.18, 4.87 (dd, 2H, J = 10.97 Hz, CH2Ph), 5.12 (d, 1H, J1,2 = 3.15 Hz, H1), 4.52, 4.45 (2d, 2H, J = 8.93 Hz, CH2Ph), 4.16–4.10 (m, 1H, H3), 4.02–3.97, 3.92–3.88, 3.56–3.44, 3.39–3.32 (m, 7H, H4, H5, H2, H6a, H6b, O–CH2), 1.34–1.18 (m, 4H, CH2–CH2), 0.96–0.78 (m, 3H, CH3). 13C-NMR (125 MHz, CDCl3) δ 139.43 (C, Ph), 138.38 (C, Ph), 128.06 (CH, Ph), 127.90 (CH, Ph), 127.72 (CH, Ph), 127.32 (CH, Ph), 127.19 (CH, Ph), 126.81 (CH, Ph), 98.83 (C1), 81.05 (C3), 79.49, 79.02 (C4, C2), 75.46 (CH2Ph), 73.21 (CH2Ph), 72.72 (O–CH2), 71.40 (C5), 69.48 (C6), 29.63, 22.80 (CH2–CH2), 10.51 (CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C144H180O30Na 2412.2493, found: 2412.2452.
2.9. 6I–O-Hydroxyl-6II–VI-penta-O-pentyl-(per-2,3-di-O-benzyl)-α-cyclodextrin (5f) and per-6-O-pentyl-2,3-di-O-benzyl-α-cyclodextrin (6f)
A similar method to that described for obtaining 5e, was performed to give the title compound 5f (71.68%) as a white powder. The Rf = 0.32 (cyclohexane:ethyl acetate, 5:1); [α]D = +18.2 (c 1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.51–6.86 (m, 60H, Ph), 5.67 (d, 1H, J1,2 = 2.58 Hz, H1), 5.64 (d, 1H, J1,2 = 3.86 Hz, H1′), 5.48, 5.41 (dd, 2H, J = 12.44 Hz, CH2Ph), 5.21, 5.17 (dd, 2H, J = 10.72 Hz, CH2Ph), 4.94–4.64, 4.59–4.27, 4.25–3.32 (m, 70H, H1′′, CH2Ph, H5, H4, H6a, H3, H2, H6b, O–CH2), 1.60–1.20 (m, 30H, –CH2CH2–), 0.95–0.82 (m, 15H, –CH3). 13C-NMR (125 MHz, CDCl3) δ 139.40, 139.36, 139.26, 139.16, 138.64, 138.49, 138.44, 138.22, 138.11, 137.36 (C, Ph), 128.62–126.29 (CH, Ph) 98.75, 98.03, 98.01, 97.96, 97.71, 97.66 (C1), 81.56, 81.42, 81.20, 81.10, 81.06, 80.88, 80.67, 80.47, 80.42, 80.07, 79.92, 79.86, 79.66, 79.15, 79.10, 79.02, 78.94, 78.12, 77.68, 76.32, 76.17, 76.03, 75.97, 75.14, 74.05, 73.99, 73.29, 73.19, 73.07, 72.86, 72.32, 72.21, 71.92, 71.88, 71.82, 71.66, 71.51, 71.37, 71.28, 70.97, 70.48, 69.78, 69.58, 69.43, 60.84 (C2, C3, C4, C5, CH2Ph, O–CH2, C6), 29.63, 28.13, 22.47 (–OCH2–CH2–CH2–CH2), 14.00 (–CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C145H182O30Na 2426.2603, found 2426.2608.
A similar method to that described for obtaining 6e, was performed to give the title compound 6f (24.32%) as a white powder. The Rf = 0.56 (cyclohexane:ethyl acetate, 5:1); [α]D = +16.0 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.25–7.10 (m, 10H, Ph), 5.17, 4.86 (dd, 2H, J = 11.01 Hz, CH2Ph), 5.13 (d, 1H, J1,2 = 3.22 Hz, H1), 4.52, 4.45 (2d, 2H, J = 12.17 Hz, CH2Ph), 4.18–4.09 (m, 1H, H3), 4.04–3.97 (m, 2H, H6a, H4), 3.92–3.87 (m, 1H, H5), 3.56–3.43 (m, 3H, H6b, H2, one of O–CH2), 3.37–3.30 (m, 1H, one of O–CH2), 1.60–1.50 (m, 2H, OCH2–CH2), 1.36–1.19 (m, 4H, –CH2–CH2), 0.90–0.84 (m, 3H, CH3). 13C-NMR (100 MHz, CDCl3) δ 139.38 (C, Ph), 138.39 (C, Ph), 128.10 (CH, Ph), 127.93 (CH, Ph), 127.70 (CH, Ph), 127.35 (CH, Ph), 127.25 (CH, Ph), 126.86 (CH, Ph), 98.35 (C1), 81.13 (C3), 79.05, 78.99 (C4, C2), 75.48 (CH2Ph), 72.73 (CH2Ph), 71.79 (O–CH2), 71.34 (C5), 69.50 (C6), 29.37, 28.22, 22.50 (CH2–CH2–CH2), 14.00 (CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C150H192O30Na 2496.3388, found 2496.3391.
2.10. 6I–O-Hydroxyl-6II–VI-penta-O-hexyl-(per-2,3-di-O-benzyl)-α-cyclodextrin (5g) and per-6-O-butyl-2,3-di-O-benzyl-α-cyclodextrin (6g)
A similar method to that described for obtaining 5e, was performed to give the title compound 5g (45.68%) as a white powder. The Rf = 0.35 (cyclohexane:ethyl acetate, 5:1); [α]D = +22.2 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.40–6.97 (m, 60H, Ph), 5.67 (d, 1H, J1,2 = 4.00 Hz, H1), 5.64 (d, 1H, J1,2 = 4.00 Hz, H1′), 5.48, 5.41 (dd, 2H, J = 12 Hz, J = 8 Hz, CH2Ph), 5.21, 5.17 (dd, 2H, J = 12.00 Hz, CH2Ph), 4.95–4.70, 4.59–4.30, 4.25–3.28 (m, 70H, H1, CH2Ph, H5, H4, H6a, H3, H2, H6b, O–CH2), 1.63–1.47, 1.36–1.20 (m, 40H, –CH2CH2CH2CH2–), 0.92–0.82 (m, 15H, –CH3). 13C-NMR (100 MHz, CDCl3) δ 139.41, 139.30, 139.27, 139.17, 138.64, 138.49, 138.43, 138.22, 138.11, 137.96 (C, Ph), 128.27–126.29 (CH, Ph), 98.74, 98.04–97.96, 97.64 (C1), 81.60, 81.55, 81.41, 81.19, 81.06, 80.88, 80.67, 80.42, 80.06, 79.90, 79.68, 79.14, 79.10, 78.13, 77.70, 77.18, 76.28, 76.16, 76.01, 75.93, 75.16, 74.05, 74.00, 73.29, 73.19, 73.06, 72.86, 72.37, 72.29, 72.22, 71.93, 71.88, 71.91, 71.70, 71.36, 71.30, 70.97, 70.49, 69.81, 69.60, 69.42, 60.84 (C2, C3, C4, C5, CH2Ph, O–CH2, C6), 31.84–31.56, 29.81–29.31, 25.75–25.63, 22.56 (–CH2CH2CH2CH2CH2–), 14.00 (–CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C150H192O30Na 2496.3432, found 2496.3391.
A similar method to that described for obtaining 6e, was performed to give the title compound 6g (34.32%) as a white powder. The Rf = 0.64 (cyclohexane:ethyl acetate, 5:1); [α]D = +30.2 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.27–7.07 (m, 10H, Ph), 5.17, 4.86 (dd, 2H, J = 12.61 Hz, CH2Ph), 5.15 (d, 1H, J1,2 = 3.34 Hz, H1), 4.48 (2d, 2H, J = 12.61 Hz, CH2Ph), 4.13 (t, 1H, J = 9.00 Hz, H3), 4.04–3.95 (m, 2H, H4, H6a), 3.93–3.86 (m, 1H, H5), 3.56–3.41 (m, 3H, H6b, H2, one of O–CH2), 3.37–3.28 (m, 1H, one of O–CH2), 1.58–1.49 (m, 2H, OCH2–CH2), 1.36–1.20 (m, 6H, –CH2–CH2–CH2), 0.89–0.85 (m, 3H, CH3). 13C (100 MHz, CDCl3) δ 139.40 (C, Ph), 138.41 (C, Ph), 128.10 (CH, Ph), 127.94 (CH, Ph), 127.70 (CH, Ph), 127.35 (CH, Ph), 127.25 (CH, Ph), 126.86 (CH, Ph), 98.42 (C1), 81.15 (C3), 79.07, 78.98 (C4, C2), 75.48 (CH2Ph), 72.74 (CH2Ph), 71.85 (O–CH2), 71.34 (C5), 69.52 (C6), 31.73, 29.68, 25.79, 22.61 (OCH2–CH2–CH2–CH2–CH2), 14.05 (CH3). HRMS (MALDI/DHB, M + Na+) m/z calcd for C156H204O30Na 2580.4328, found 2580.4330.
3. Results and discussion
This involved phenomenon caught in our previous research on cyclodextrins.8,20 This study was performed by using compound 4, which was synthesized from native α-CD according to a method the literature, as the starting material (Scheme 3).14 In this nucleophilic substitution, NaH promoted the deprotonation of CD hydroxyl groups for nucleophilic agent formation, or reacts with the bromine ion of RBr. The RBr forms carbon cations for use as electrophiles.Firstly, 5-bromo-1-pentene was chosen for alkylation on the primary face of α-CD derivative 4 because pentenyl has terminal double bond, which can then form cyclodextrin dimers and oligomers or perform click reactions, or be converted into other groups (Scheme 2). When compound 4 was treated with 5-bromo-1-pentene in the presence of NaH at a ratio of 4:NaH:5-bromo-1-pentene = 1:12:10, 6-hexa-O-pentenyl α-CD 6e was obtained as the major product (91%), and 6-hydroxyl-6-penta-O-pentenyl α-CD 5e was isolated as a minor product (6%, Table 1, entry 1). When the amounts of 5-bromo-1-pentene were increased, the yield of mono hydroxyl α-CD derivative 5e increased up to 66% (Table 1, entry 8) whereas the complete alkylation of α-CD 6e decreased. However, only the amount of NaH was increased singly, that not leading to the yield of 5e improved (Table 1, entries 2 and 10). It seemed that a balance between the amount of NaH and 5-bromo-1-pentene existing. The ratio (4:NaH:RBr = 1:14:24) was found from the experiments to be beneficial for obtaining 5e.
| Entry | Reaction ratio (mol) | Yield (%) | |
|---|---|---|---|
| 4:NaH:5-bromo-1-pentene |
5e | 6e | |
| 1 | 1:12:10 |
6.08 | 91.28 |
| 2 | 1:12:12 |
7.00 | 90.00 |
| 3 | 1:12:14 |
20.00 | 73.80 |
| 4 | 1:12:16 |
32.00 | 61.30 |
| 5 | 1:12:18 |
40.00 | 56.78 |
| 6 | 1:12:20 |
56.28 | 40.90 |
| 7 | 1:12:30 |
50.24 | 47.05 |
| 8 | 1:14:24 |
65.81 | 31.94 |
| 9 | 1:14:18 |
62.93 | 30.07 |
| 10 | 1:16:12 |
5.00 | 91.00 |
After accidentally discovering this phenomenon, we were very curious about whether other alkyl bromides caused the same selections. Next a variety of alkyl bromides were used, instead of 5-bromo-1-pentene, to perform a similar alkylation on the primary face of α-CD derivative 4 (Scheme 1).
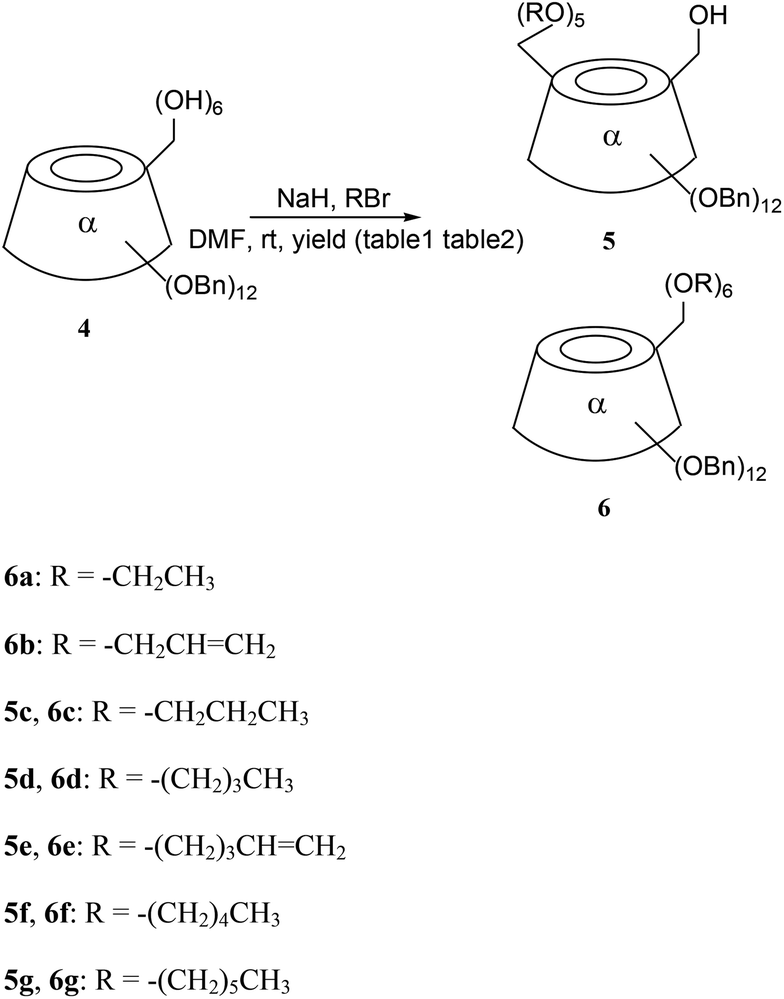 | ||
| Scheme 1 Diverse RBr induced outcomes of 6-substituted α-CDs. | ||
When bromoethane or 3-bromopropene were used, only the complete alkylation of α-CDs 6a, 6b were found even under the conditions of 4:NaH:RBr = 1:14:24 (Table 2, entries 1 and 2). In the other reactions involved, similar phenomena were observed as when 5-bromo-1-pentene was used. In particular when the ratio of 4:NaH:RBr was 1:14:24, some mono hydroxyl α-CD derivatives could be obtained as major compounds, the highest yield (72%) being obtained for 5f (Table 2, entry 6).
| Entry | Yield (%) | ||
|---|---|---|---|
| RBr | 5 | 6 | |
| a Reaction ratio (mol): 4:NaH:RBr = 1:14:24. |
|||
| 1 | –CH2CH3 | 0 | 6a 80.50 |
| 2 | –CH2CHCH2 |
0 | 6b 81.70 |
| 3 | –CH2CH2CH3 | 5c 60.40 | 6c 33.73 |
| 4 | –(CH2)3CH3 | 5d 51.88 | 6d 35.37 |
| 5 | –(CH2)3CHCH2 |
5e 65.81 | 6e 31.94 |
| 6 | –(CH2)4CH3 | 5f 71.68 | 6f 24.32 |
| 7 | –(CH2)5CH3 | 5g 45.68 | 6g 34.32 |
The pentenyl α-CD derivatives 5e and 6e were hydrogenated with hydrogen catalyzed by Pd–C (10%) at 14 kPa to give 7e and 8e for further amphiphilic study (Scheme 2). The hydroxyl group existing in 5e and 7e could also be modified for use in CD applications.
 | ||
| Scheme 2 Reaction of pentenyl α-CD derivatives. | ||
 | ||
| Scheme 3 Synthesis of α-CD derivative 4. | ||
All the new synthetic compounds 5c, 5d, 5f, 5g, 6a, 6b, 6c, 6d, 6f, 6g and 5e, 6e, 7e, 8e were characterized by 1H-NMR and 13C-NMR, as well as HRMS (see ESI†). It can be seen from the NMR spectra that the hydroxyl on the CDDs (5c, 5d, 5e, 5f, 5g, 7e) made the molecules asymmetric (Fig. 1), from their 13C-NMR 6 anomeric carbons were found, whereas the 6-per alkyl CDDs (6a, 6b, 6c, 6d, 6e, 6f, 6g, 8e) were symmetric because the NMR spectra look like those of monosaccharides (Fig. 2). In a paper from 1991, Angibeaud and Utille19, assigned 79.00 to C4, 80.80 to C2. In fact, the 1H–13C correlation spectrum indicated that C2 should be at 79.00, and C4 80.80 (in this case C2 at 78.93 and C4 at 79.56). Moreover, C3 was not assigned at all in ref. 19.
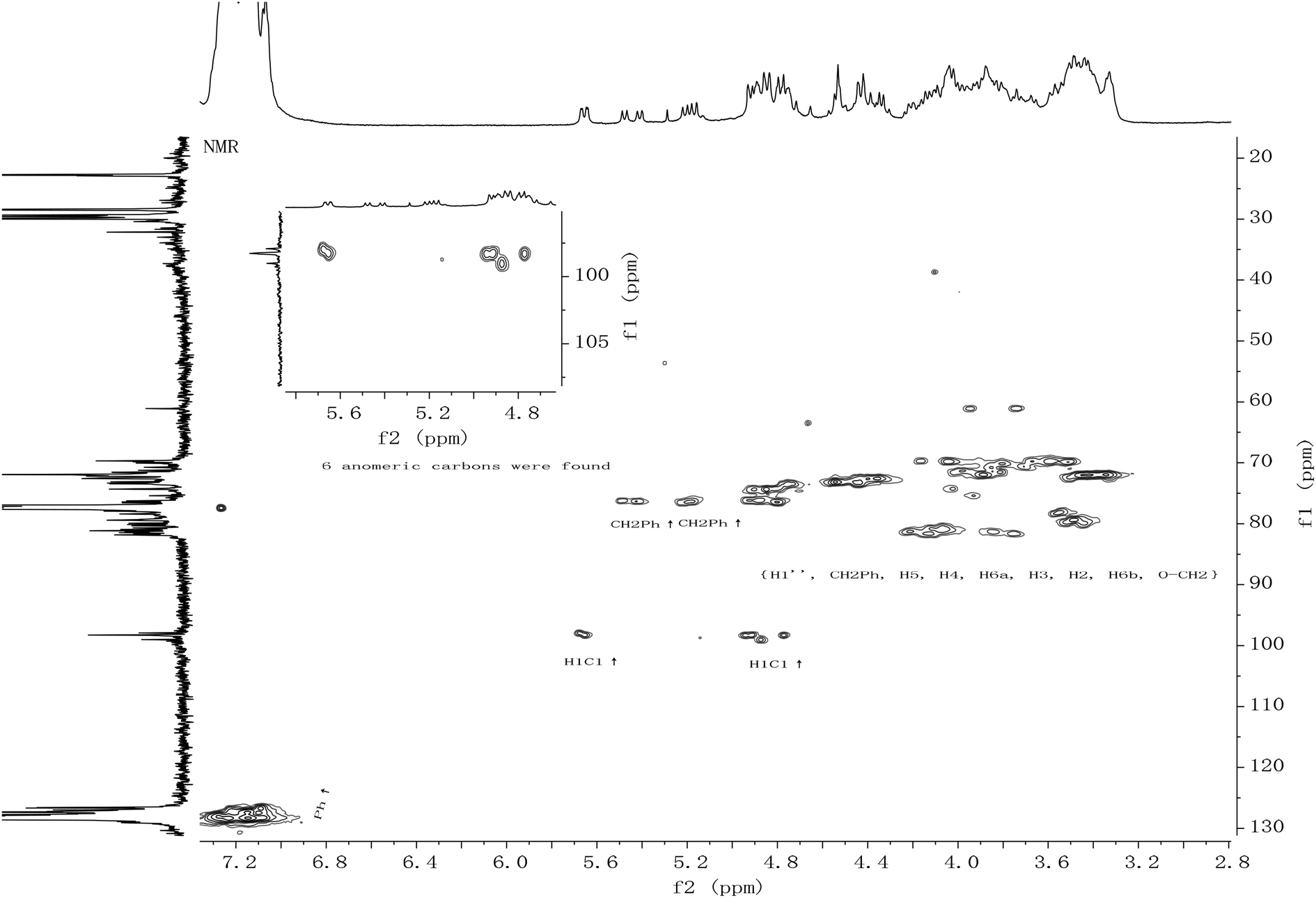 | ||
| Fig. 1 The heteronuclear multiple quantum interference coherence (HMQC) of compound 5f. | ||
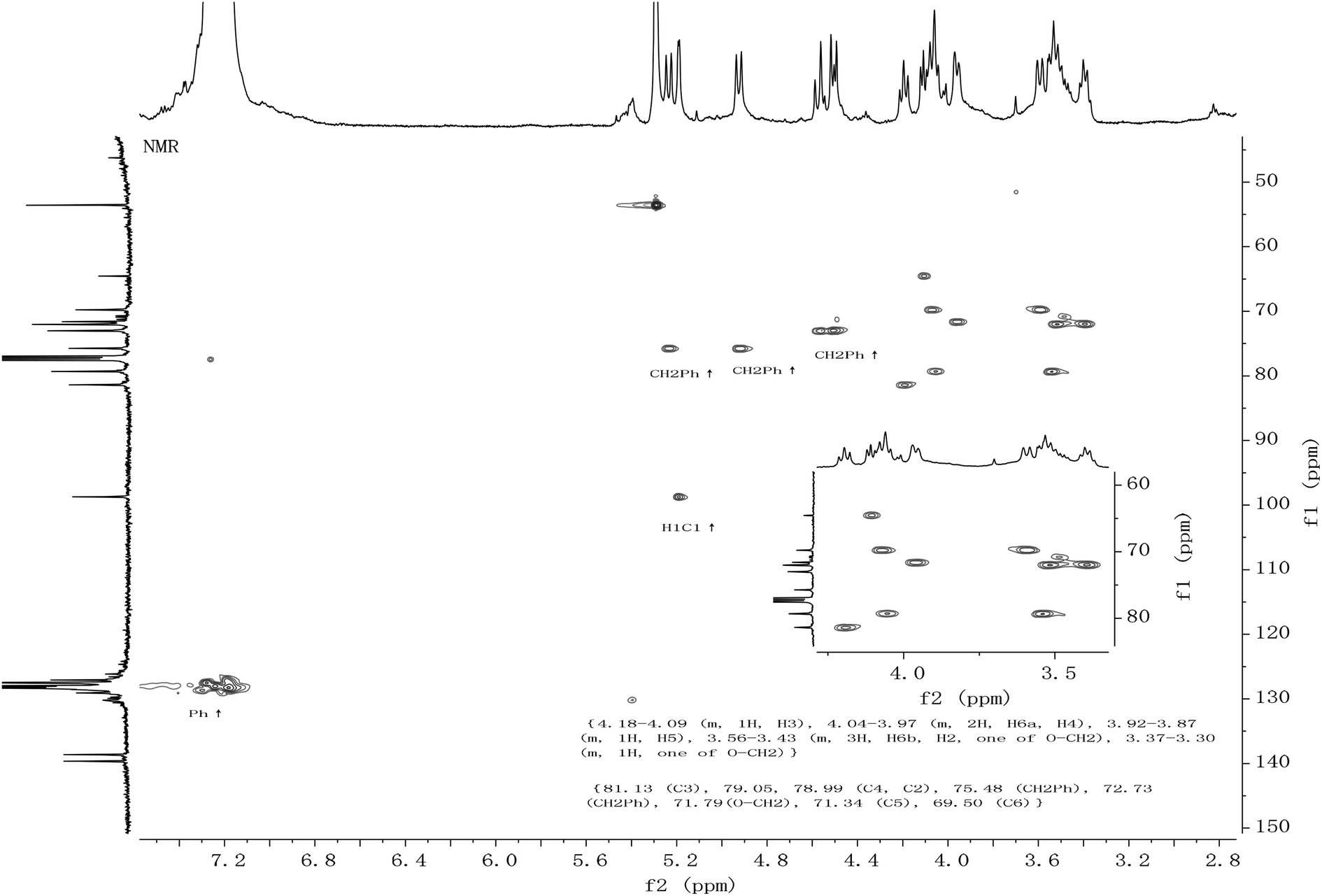 | ||
| Fig. 2 The HMQC of compound 6f. | ||
For the mechanism, it was speculated that there were two interfering factors, the concentration of alkali in the system and the steric effects of the electrophilic reagent RBr. When the RBr amount increased, the NaH concentration decreased and the ratio of the 4:NaH:RBr arrived at a certain value, fewer NaH in the system would lose deprotonation capacity to one of the hydroxyl groups of compound 4 leading to weakening of its nucleophilic attack. Therefore, the regioselective products accounted for a certain yield. The steric hindrance also affected the regioselective reaction. It can be seen from the short chain RBr bromoethane or 3-bromopropene that even if the ratio of 4:NaH:RBr was adjusted no regioselective product was obtained.
4. Conclusion
In summary, we have achieved 6-hydroxyl and penta-alkyl on primary face of α-cyclodextrin in one pot. It seems that the mono 6-hydroxyl α-cyclodextrin derivatives would be difficult to obtain directly through a traditional thought. Even though the exact mechanism is still unclear, the regioselective modifications described previously minimized the reagents required, the labor, the time, and facilitated the availability of a variety of selectively modified α-CDs on the primary face which will be useful for cross-disciplinary researchers. The novel molecule 5e has two reactive points, hydroxyl and pentenyl groups, which can be further used for research in supramolecular chemistry, biological materials, drug delivery, and so on. In our unpublished data, we are exploring the derivatization on two reactive points for further applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China under Grant No. 30870553, the Ministry of Science and Technology of the People's Republic of China under Grant No.2010DFA34370, the Science and Technology Department of Zhejiang Province under Grant No. 2013C14012, the Natural Science Foundation of Zhejiang under Grant No. LQY18B060002 and the Zhejiang Cultural Relic Protection Science and Technology Project under Grant No. 2020011 for this work is greatly appreciated.References
- J. Zhang and P. Ma, Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective, Adv. Drug Delivery Rev., 2013, 65(9), 1215–1233 CrossRef CAS PubMed.
- W. Tang and S. Ng, Monosubstituted positively charged cyclodextrins: synthesis and applications in chiral separation, J. Sep. Sci., 2008, 31(18), 3246–3256 CrossRef CAS PubMed.
- J. Lenik, Cyclodextrins based electrochemical sensors for biomedical and pharmaceutical analysis, Curr. Med. Chem., 2017, 24(22), 2359–2391 CrossRef CAS PubMed.
- R. Liao, P. Lv, Q. Wang, J. Zheng, B. Feng and B. Yang, Cyclodextrin-based biological stimuli-responsive carriers for smart and precision medicine, Biomater. Sci., 2017, 5(9), 1736–1745 RSC.
- N. Erdogar, G. Varan and E. Bilensoy, Amphiphilic cyclodextrin derivatives for targeted drug delivery to Tumors, Curr. Top. Med. Chem., 2017, 17(13), 1521–1528 CrossRef CAS PubMed.
- L. Fan, C. Luo, M. Sun and H. Qiu, Synthesis of graphene oxide decorated with magnetic cyclodextrin for fast chromium removal, J. Mater. Chem., 2012, 22(47), 24577–24583 RSC.
- G. Varan, C. Varan, N. Erdogar and E. Bilensoy, Amphiphilic cyclodextrin nanoparticles, Int. J. Pharm., 2017, 531(2), 457–469 CrossRef CAS PubMed.
- D. Dong, D. Baigl, Y. Cui, P. Sinay, M. Sollogoub and Y. Zhang, Amphiphilic bipolar duplex α-cyclodextrin forming vesicles, Tetrahedron, 2007, 63, 2973–2977 CrossRef CAS.
- A. Khan, P. Forgo, K. Stine and V. D'Souza, Methods for selective modifications of cyclodextrins, Chem. Rev., 1998, 98(5), 1977–1996 CrossRef CAS PubMed.
- F. Sallas, P. Leroy, A. Marsura and A. Nicolas, First selective synthesis of thio-β-cyclodextrin derivatives by a direct Mitsunobu reaction on free β-cyclodextrin, Tetrahedron Lett., 1994, 35(33), 6079–6082 CrossRef CAS.
- G. Nelles, M. Weisser, R. Back, P. Wohlfart, G. Wenz and S. Mittler-Neher, Controlled orientation of cyclodextrin derivatives immobilized on gold surfaces, J. Am. Chem. Soc., 1996, 118(21), 5039–5046 CrossRef CAS.
- R. Petter, J. Salek, C. Sikorski, G. Kumaravel and F. Lin, Cooperative binding by aggregated mono-6-(alkylamino)-beta-cyclodextrins, J. Am. Chem. Soc., 1990, 112(10), 3860–3868 CrossRef CAS.
- A. Croft and R. Bartsch, Synthesis of chemically modified cyclodextrins, Tetrahedron, 1983, 39(9), 1417–1474 CrossRef CAS.
- S. Tian, H. Zhu, P. Forgo and V. D'Souza, Selectively monomodified cyclodextrins: synthetic strategies, J. Org. Chem., 2000, 65(9), 2624–2630 CrossRef CAS PubMed.
- A. Pearce and P. Sinaÿ, Diisobutylaluminium-promoted regioselective de-O-benzylation of perbenzylated cyclodextrins: a powerful new strategy for the preparation of selectively modified cyclodextrins, Angew. Chem. Int. Ed., 2010, 39(20), 3610–3612 CrossRef.
- B. Wang, E. Zaborova, S. Guieu, M. Petrillo, M. Guitet, Y. Blériot, M. Ménand, Y. Zhang and M. Sollogoub, Site-selective hexa-hetero-functionalization of α-cyclodextrin an archetypical C6-symmetric concave cycle, Nat. Commun., 2014, 5, 5354 CrossRef CAS PubMed.
- W. Wang, A. Pearce, Y. Zhang and P. Sinaÿ, Diisobutylaluminium-promoted regioselective de-O-methylation of cyclodextrins: an expeditious entry to selectively modified cyclodextrins, Tetrahedron: Asymmetry, 2001, 12(3), 517–523 CrossRef CAS.
- R. Ghosh, P. Zhang, A. Wang and C. Ling, Diisobutylaluminum hydride mediated regioselective desilylations: access to multisubstituted cyclodextrins, Angew. Chem. Int. Ed., 2012, 51(7), 1548–1552 CrossRef CAS PubMed.
- P. Angibeaud and J. Utille, Cyclodextrin chemistry; part I, application of a regioselective acetolysis method for benzyl ethers, Synthesis, 1991, 9, 737–738 CrossRef.
- Y. Cui, S. Xu and J. Mao, One-pot synthesis of hexias (6-O-acryl) cyclodextrin derivatives at room temperature, J. Inclusion Phenom. Macrocyclic Chem., 2015, 83, 187–191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00299b |
| This journal is © The Royal Society of Chemistry 2020 |