
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectral evidence for generic charge → acceptor interactions in carbamates and esters†
Erode N. Prabhakaran *a,
Shama Tumminakattib,
Kamal Vatsa and
Sudip Ghosha
*a,
Shama Tumminakattib,
Kamal Vatsa and
Sudip Ghosha
aDepartment of Organic Chemistry, Indian Institute of Science, Bangalore, Karnataka, India – 560 012. E-mail: eprabhak@iisc.ac.in
bDOS in Organic Chemistry, University of Mysore, Manasagangotri, Mysore 57006, India
First published on 24th March 2020
Abstract
The correlations of the 1H NMR, 13C NMR and FT-IR spectral data from the R–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in the alkyl carbamates and esters of homologous alcohols reveal R-group-dependent negative charge stabilization at the carbonyl oxygen and their donation to generic acceptors at Cα of even alkyl alcohols (R), which explains several of their apparently anomalous properties.
O groups in the alkyl carbamates and esters of homologous alcohols reveal R-group-dependent negative charge stabilization at the carbonyl oxygen and their donation to generic acceptors at Cα of even alkyl alcohols (R), which explains several of their apparently anomalous properties.
Electronic interactions at the R–O–C
O groups in carbamates and esters are less understood than that at the R–N–CO groups in amides. Carbamates in which both these groups are fused at the carbonyl CO bond, have several ester-like rather than amide-like features1,2 including comparable C–O bond lengths in crystals,3–6 favourable interactions between the dipoles of the CO and O–R bonds in the predominant s-trans rotamers of the R–O–CO groups and unfavourable lone pair repulsion between the two ester oxygens in their trace s-cis rotamers7–10 (Fig. 1a). However, several anomalous properties of carbamates and esters cannot be explained by these dipole and repulsive forces alone. For example, despite the electronic resonance at the O–CO group in esters and additionally at the N–CO group in carbamates (both of which augment the electron density at their carbonyl oxygens), their carbonyls have remarkably lower basicities than the carbonyls of amides11 and even those of ketones, which lack such resonance effects.12 The barriers to the C–N bond rotation for carbamates are 3–4 kcal mol−1 lower than those for analogous amides.13,14 Carbamate and ester carbonyl oxygens also have poor hydrogen bond accepting propensities compared to amides.15–20
 | ||
| Fig. 1 (a) Dipole–dipole, electronic and steric interactions governing the rotamer stabilities at R–O–CO frameworks in carbamates and esters; (b) earlier report of n → π* O⋯Cα interaction in phenyl carbamates; (c) current finding of generic donor → acceptor interactions: (i) charge → HCR* (ii) charge → σ*; (d) list of carbamates (1–8), esters (9–16) and alcohols (17–20) investigated. HCR is hyperconjugative resonance along the Cα–Cβ bond of alcohol groups in carbamates and esters. | ||
Of particular current interest is the apparent anomaly that in the crystals of phenyl carbamates, the phenyl ring plane is oriented perpendicular to the carbamate plane21,22 (Fig. 1b). The presence of an extraordinary n → π* orbital overlap interaction (O⋯Cαphenyl) between the lone-pair electrons on the carbonyl (CO) oxygen and the π* orbital of the phenyl ring has been proposed as the stabilizing force for this conformer based on natural bond orbital (NBO) analysis.22 This is interesting for several reasons: (i) despite a decrease in the stretching frequency of CO in the FT-IR spectrum for this conformer, which indicates a decrease in the bond order and concomitant improvement in the charge density at the carbonyl oxygen, a discussion on the origin and role of such charge on this O⋯Cαalcohol interaction is lacking. Rather, the interaction is assumed to originate from the lone pair on oxygen. (ii) Our investigation of CCDC revealed that the O⋯Cαalcohol distances at the Cα–O–CO groups are quite non-variant (2.62–2.81 Å) for both aliphatic and aromatic carbamates and esters,3–6,23–27 notwithstanding the variations in the structure resolutions. These invoked the following questions: is the O⋯Cα interaction specific to the π* acceptors at Cαalcohol? Can any antibonding orbital at Cαalcohol act as an acceptor of the electrons from carbonyl oxygen? What is the generic nature and role of this O⋯Cα interaction in the Cα–O–CO groups? Here, we present the first spectral evidence for the generic nature of the O⋯Cα interactions even with non-π* orbital acceptors (like σ* and hyperconjugative resonance bonds) at Cα in the Cα–O–CO groups (Fig. 1c) of a variety of model homologous aliphatic carbamates and esters (Fig. 1d). The charge at the carbonyl oxygen is interdependent on the alkoxy groups and forms charge → acceptor O⋯Cα interactions, which influence the rotational states of the O–Cα bonds in the carbamates and esters.
To explore the possibility of the O⋯Cα interactions at Cα–O–CO in aliphatic carbamates and esters, we investigated the 1H NMR, 13C NMR and FT-IR spectra of the secondary and tertiary carbamates (1–4 and 5–8), acetates (9–12) and benzoates (13–16) of homologous aliphatic alcohols (H–CαH2–OH, MeOH; CH3–CαH2–OH, ethanol; (CH3)2–CαH–OH, isopropanol; and (CH3)3–Cα–OH, tert-butanol, 17–20) (Table 1) and correlated their deviations from those of their corresponding alcohols. These are ideal models to investigate the fundamental nature and origin of the O⋯Cα interactions because on increasing the methyl substitution at Cα, there is a systematic increase in the Cα electron density, thus progressively diminishing its electron acceptor propensity. Any O⋯Cα donor–acceptor interactions will thus have least assistance from other local electronic effects. Evidence for the O⋯Cα interactions with such electronically and sterically antagonistic aliphatic Cα would sufficiently reveal the generality of this interaction for any non-π* acceptors. On the other hand, different carbamates and esters account for the generality of carbonyl substituent effects.
| 13C NMR (δ ppm) | ν (cm−1) | 1H NMR (δ ppm) | ||||
|---|---|---|---|---|---|---|
| O–Cα | OCα–Cβ | OC |
CO* |
OCα–Hα | OCα–Cβ–Hβ | |
| a *FT-IR stretching frequencies; values in parentheses are for corresponding alcohols; NA means not applicable; “—” means unambiguous data could not be obtained. | ||||||
| 1 | 52.1 (50.4) | NA (NA) | 155.5 | 1685 | 3.69 (3.68) | NA (NA) |
| 5 | 52.3 | NA | 154.1 | 1705 | 3.78 | NA |
| 9 | 51.6 | NA | 171.5 | 1748 | 3.67 | NA |
| 13 | 51.5 | NA | 165.9 | 1725 | 3.92 | NA |
| 2 | 60.5 (58.3) | 14.6 (18.4) | 155.0 | 1679 | 4.13 (3.72) | 1.26 (1.24) |
| 6 | 61.1 | 14.5 | 153.6 | 1701 | 4.23 | 1.31 |
| 10 | 60.5 | 14.2 | 171.4 | 1740 | 4.12 | 1.25 |
| 14 | 60.8 | 14.3 | 166.5 | 1720 | 4.33 | 1.33 |
| 3 | 67.6 (64.5) | 22.1 (25.1) | 154.7 | 1674 | 4.92 (4.03) | 1.23 (1.20) |
| 7 | 68.7 | 22.1 | 153.7 | — | 5.05 | 1.30 |
| 11 | 67.6 | 21.8 | 170.6 | 1736 | 4.99 | 1.23 |
| 15 | 68.3 | 21.9 | 166.1 | 1716 | 5.24 | 1.35 |
| 4 | 78.7 (69.1) | 28.4 (31.2) | 154.5 | 1683 | NA (NA) | 1.46 (1.27) |
| 8 | 80.4 | 28.3 | 152.7 | 1689 | NA | 1.51 |
| 12 | 80.1 | 28.1 | 170.4 | 1738 | NA | 1.45 |
| 16 | 80.9 | 28.2 | 165.8 | — | NA | 1.58 |
The 13C NMR signals (Table 1) of Cα in homologous alcohols28,29 undergo a large downfield shift (by 18.7 ppm) incrementally as the number of methyl (CβH3) substituents on Cα increases from zero to three (Fig. 2a). There is a simultaneous downfield shift in the 1H NMR signal of the corresponding Hα by 0.55 ppm from methyl to isopropyl alcohol. Such shifts are contrary to what is expected from the increased positive induction on Cα–Hα by the CβH3 substituents. They are rather due to the positive charge at Cα stemming from the polarization of the Cα–O bond, which is stabilized through the hyperconjugation effect (and increasingly so) as the number of methyl substituents on Cα increases. Hence, the Cα–Cβ σ-bonds of alcohols have additional bonding from such hyperconjugative resonance (HCR).
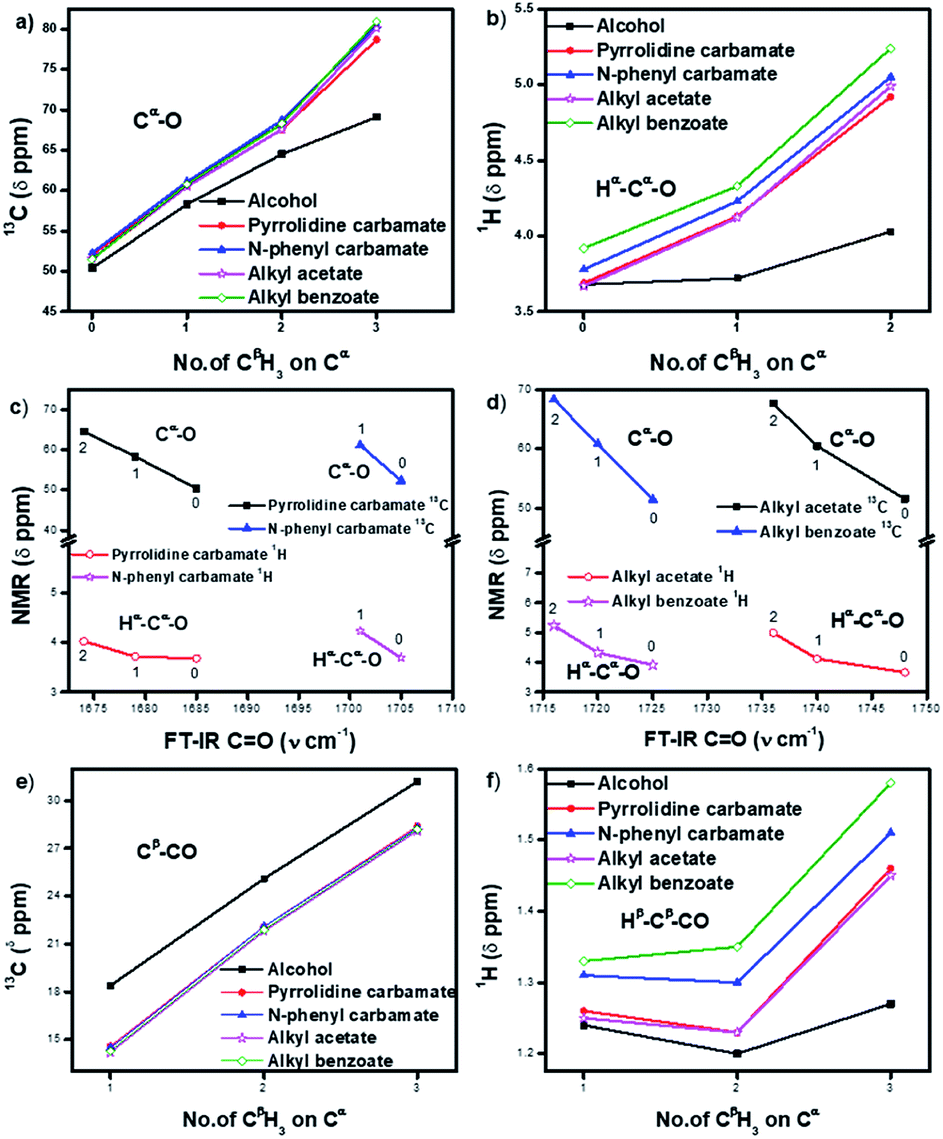 | ||
| Fig. 2 (a and b) Incremental downfield shifts in the 13C and 1H signals of Hα and Cα in carbamates (1–8) and esters (9–16) relative to the corresponding homologous alcohols (17–20), indicating the stabilization of positive charge polarization at Cα by hyperconjugative resonance from the CβH3 groups; (c and d) inverse correlation between 13C and 1H NMR signals of Cα and Hα and FT-IR stretching frequencies at CO in carbamates (1–7) and esters (9–15) with increasing number of methyl (CβH3) substituents (0–2) on Cα (for isopropyl N-phenyl carbamate (7), the FT-IR value is not inserted); (e) upfield shifts in the Cβ signals of carbamates (1–8) and esters (9–16) compared to corresponding alcohols (17–20), indicating electronic back donation towards Cβ; (f) shifts in the Hβ signals of carbamates (1–8) and esters (9–16) compared to corresponding alcohols (17–20), indicating the dominance of charge → HCR* interactions. The lines merely indicate the trends. | ||
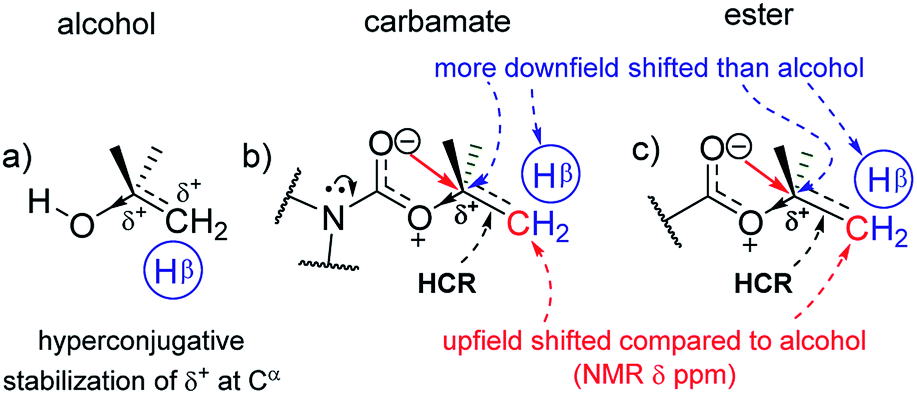
In the corresponding carbamates (1–4, 5–8 (ref. 30–32)) and esters (9–12, 13–16 (ref. 33–35)), there are much larger downfield shifts for Cα (by 26.6 ppm for 1–4; 28.1 ppm for 5–8; 28.5 ppm for 9–12; and 29.4 ppm for 13–16) and Hα (by 1.23 ppm for 1–3; 1.27 ppm for 5–7; 1.32 ppm 9–11; and 1.32 ppm 13–15) compared to that for alcohols28,29 (Fig. 2a and b). These shifts are also incremental on increasing the CβH3 substitution on Cα but show steeper increase compared to that for alcohols (see ESI†). This further substantiates the hyperconjugative stabilization of greater positive polarization at Cα of Cα–O–CO, whose oxygen exists as an oxonium ion in the bipolar resonance form (Cα–O+C–O−) (Fig. 3). Note that if merely the electron-withdrawing induction effect of the carbonyl group was influencing the chemical shifts of Cα and Hα, a constant downfield shift would have been observed on increasing the CβH3 substitution at Cα in either carbamates and esters compared to that for alcohols and not such incremental (steeper) downfield shifts.
 | ||
| Fig. 3 The hyperconjugation effect stabilizing the polarization at Cα of alkoxy groups (a) and the O⋯Cα electronic interactions in the s-trans conformer at the R–O–CO groups in (b) carbamates and (c) esters of homologous alcohols, as observed by the correlation of their 1H NMR, 13C NMR and FT-IR data in the current work. | ||
The remarkable uniformity in the trends of such increments in the Cα and Hα chemical shifts for the R–O–C(R′)O groups (R′ represents primary and secondary amine, R stands for alkyl and aryl groups) of 1–16 reveals that this stabilization of the bipolar R–O+C(R′)–O− intermediates by the hyperconjugative effect from the R group is largely independent of the acyl substituent (R′) effects and is only slightly perturbed by the cross-conjugation from the nitrogen in R′. The longer CO bond lengths in secondary (1.21 ± 0.01 Å)36–40 and tertiary (1.23 ± 0.02 Å)5,6,24,25,41 carbamates compared to that in acetates (1.20 ± 0.02 Å)3,4,23,27 and benzoates (1.21 ± 0.01 Å)42–46 of the homologous alcohols in the solid state (ESI†), which are reflected in their stretching frequencies (1697 ± 8 cm−1, 1679 ± 3 cm−1, 1740.5 ± 7.5 cm−1 and 1723 ± 7 cm−1, respectively) in the FT-IR spectra,30,32,47–50 however, clearly indicate the additional electronic resonance along the N–CO framework. As a result, the 13C nuclear resonance for CO in carbamates shifts upfield compared to that for the esters. However, since the shift is constant and independent of the number of methyl substituents on Cα, the mixing of nitrogen lone pair with the carbonyl π-cloud does not perturb the oxonium charge state or the concomitant positive charge at Cα. The resonance at O–CO is hence quite strong. The shortening of the O–C single bond in the carbamates and esters, noted from the diffraction data, evidences such strong resonance at O–CO. In fact, the lowering of the rotational energy barrier along the C–N bond in carbamates than that in the corresponding amides by 3–4 kcal mol−1,13,14 indicates the overwhelming influence of strong resonance at O–CO in diminishing the resonance at N–CO.
Interestingly, the FT-IR CO stretching frequencies in the carbamates and esters consistently show an inverse correlation with the downfield shifts at their alcohol Cα (Fig. 2c and d), indicating interactive dependence between the negative charge at the carbonyl oxygen and the positive polarization at Cα, an O⋯Cα interaction that remarkably improves on increasing the number of CβH3 substituents on Cα.
Such O⋯Cα interactions are clearly evident in the crystal structures of the carbamates5,6,24,25,36–41 and esters3,4,23,27,42–46 (1–16) (ESI†), especially when there are methyl (CβH3) substituents on Cα: (i) the Cα–Cβ bond is consistently oriented antiperiplanar to CO with an angle of incidence of 160 ± 5 deg for oxygen. Note that in addition to the σ-bond along Cα–Cβ, the current results have established the presence of HCR bonding. Hence, the O⋯Cα charge donation can be either to σ* or HCR* along the Cα–Cβ bond. (ii) Only in methyl carbamates and esters (which lack Cβ), such antiperiplanar orientation of oxygen (159 ± 5 deg) is observed with a Cα–Hα σ-bond. (iii) The O⋯Cα distances are comparable (2.73 ± 0.02 Å) (within errors of estimation). The distances however increase slightly from 2.62 ± 0.01 Å to 2.81 ± 0.00 Å for methyl to t-butyl carbamates and esters due to increased O⋯Cα steric repulsions. (iv) All the atoms of the OC–O–Cα–Cβ group are in plane, indicating lack of distortions due to the steric clashes between the CO and R groups. There is slight deviation of the C–O–Cα–Cβ torsion from planarity (by ∼25 deg) only in the isopropyl carbamates and esters where the carbonyl oxygen is asymmetrically staggered between the small Hα and the bulky CβH3 of the isopropyl group. This slight deviation is reflected in the consistent minor deviations of their NMR and FT-IR data from the trends of their corresponding homologues. (v) The bond angles at the R–O–CO framework are comparable in the carbamates and esters for all the homologous R groups.
The antiperiplanar orientation of CO to the Cα–Cβ bond in the carbamates and esters in the solid state and the O⋯Cα interactions suggested by spectral indicators were consistent with each other. Interestingly, the Cβ signals in the 13C NMR spectra of the carbamates and esters identically shifted upfield on increasing the CβH3 substitution at Cα (Fig. 2e) compared to that for alcohols; this was in contrast to the Cα signals, which shifted down-field. However, the Hβ signals in the 1H NMR spectra showed downfield shifts of decreasing steepness with decrease in the electronic charge at the carbonyl oxygen in the order alkyl benzoates > N-phenyl carbamate > acetates/pyrrolidine carbamates (Fig. 2f). These indicate the predominance of the charge → HCR* (O⋯Cα–Cβ) interaction (Fig. 1c(i)) over the charge → σ* (along Cα–Hα or Cα–Cβ) interaction and slightly greater electronic back donation of the charge from CO to Cα–Cβ in the acetates and pyrrolidine carbamates (Fig. 1c(ii)). There were consistent slight deviations in particularly the δ ppm values of Hβ for all the isopropyl carbamate and ester analogues (3, 7, 11, 15), as observed from the trends obtained for the remaining homologues. This was consistent with the small distortions away from the ideal anti-periplanarity of the carbonyl oxygen and the Cα–Cβ bond observed in their crystal structures, which diminished the charge → HCR* donation and would not be observed unless charge → HCR* predominated over charge → σ* in O⋯Cα.
The generic charge → acceptor O⋯Cα electronic back donation interactions explain the X-ray structures of phenyl carbamates,21 where the inclusive plane of the carbamate group is perpendicular to the plane of the phenyl ring.22 Current data additionally indicate that this interaction is (a) largely charge → π* in nature rather than n → π*; (b) primarily localized between O and Cα of the phenyl ring; and (c) a consequence of the general O⋯Cα charge → acceptor electronic interactions at the R–O–CO groups, which are observed for a variety of Cα acceptors: (1) σ* acceptor in Cα–Hα, when R is a methyl group (Fig. 1c(ii)); (2) HCR* acceptor in Cα–Cβ, when R has a CβH group (Fig. 1c(i)); and (3) π* acceptor, when R is phenyl (Fig. 1b). In other words, the O⋯Cα electronic charge → acceptor back donation interaction is observed at alkyl carbamates and esters as well as they are observed in phenyl carbamate.
Note that only a charge (rather than a lone pair) donor at the carbonyl oxygen, which has a longer coulombic interaction range, is consistent with the reported observations for phenyl carbamates22 that the rest of the π*-orbitals of the phenyl ring (other than at its Cα) that are at distances longer than is conducive for any n → π* orbital overlap interactions also accept electron density from the carbonyl oxygen. Moreover, the constancy (rather than decrease) in the O⋯Cα interactions despite increasing the substitution at Cα and the concomitant upfield shift in the Cβ signals of the current analogues and the O⋯Cα–Cβ periplanarities with little perturbation in the O⋯Cα distances in crystals all substantiate an R group-dependent stabilization of the negative charge on the carbonyl oxygen, which interacts back with Cα of R and influences the rotational states of the O–Cα bond at the R–O–CO groups. Thus, the charge → acceptor O⋯Cα interaction is generic to the Cα–O–CO group and influences the biasing of the rotational states along the O–Cα bond. The incorporation of the corresponding force fields in the computational methods will benefit energy minimization of the rotational states in these molecules. The generic charge → HCR*/σ*/π* interaction model at Cα–O–CO is also consistent with masking the charge at the carbonyl oxygen, hence explaining the low basicities and poor hydrogen bond acceptor propensities of the carbamates and esters.
Finally, it is possible that the O⋯Cα interaction has an electrostatic component as well due to the polarized charge at Cα. Although this may influence the planarity of R–O–CO even when R is a methyl group, the resonance at O–CO would have a major role in such planarity. Moreover, such electrostatic interactions are insufficient by themselves to explain the observed CO⋯Cα–Cβ anti-periplanarities and the relative downfield shifting of Hβ (compared to alcohols). Note that the latter data also discount the possibility of O⋯Hα–Cα or O⋯Hβ–Cβ-type hydrogen bonding interactions.
Conclusions
Correlations between the 1H NMR, 13C NMR and FT-IR spectral data for the Cα–O–CO groups of the model carbamates and esters of homologous alcohols reveal the presence of charge → HCR*/σ* donor/acceptor O⋯Cα interactions along with other structural and electronic features, which explain several of the apparently anomalous properties of carbamates and esters. First, there is positive charge polarization at the Cα carbon attached to the alcohol oxygen, which is stabilized incrementally by hyperconjugative resonance (HCR) with increasing number of CβH3 substituents on it. Interestingly, the resulting dipolar resonance at O–CO is comparable for carbamates and esters and is slightly perturbed by the cross-resonance from the (secondary and tertiary) carbamate nitrogen. Rather, the latter is weakened, explaining the lowering of the transition energy barrier for cis/trans isomerism at the carbamate C–N bonds compared to that for amides. The electronic charge at the carbonyl oxygen of carbamates and esters interacts back with the hyperconjugative resonance (HCR*) along Cα–Cβ or with σ* along Cα–Hα, as substantiated by the NMR and FT-IR spectral shifts (compared to those for the corresponding alcohols). Such an interaction regulates the rotational states along the O–Cα bonds, as seen from the CO⋯Cα–Cβ and CO⋯Cα–Hα anti-periplanarities in the crystal structures of carbamates and esters. Deviations in the anti-periplanarities are directly reflected through spectral shifts. This charge → acceptor O⋯Cα interaction thus occurs with any (HCR*, σ* or π*) acceptor at Cα and explains several unique features of the Cα–O–CO groups: (1) the unusual orthogonality observed between the planes of phenyl and carbamate groups in phenyl carbamates and (2) the diminished basicities and hydrogen bond accepting propensities of the carbamate and ester carbonyl oxygens compared to that for ketones and amides. This is the first insight into the strong interdependence between the alcohol group and the charge at the carbonyl group of the Cα–O–CO groups of carbamates and esters, resulting in charge-stabilizing generic charge → acceptor O⋯Cα interactions that bias the rotational states of the O–Cα bond. Apart from providing a better understanding of the interactions in the R–O–CO groups, the current results will help in chemical biology research51 and drug design,52–54 where carbamates play an important role.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Oki and H. Nakanishi, Bull. Chem. Soc. Jpn., 1971, 44, 3144–3147 CrossRef CAS.
- M. Oki and H. Nakanishi, Bull. Chem. Soc. Jpn., 1970, 43, 2558–2566 CrossRef CAS.
- M. J. Barrow, S. Cradock, E. Ebsworth and D. W. Rankin, J. Chem. Soc., Dalton Trans., 1981, 1988–1993 RSC.
- A. D. Boese, M. Kirchner, G. A. Echeverria and R. Boese, ChemPhysChem, 2013, 14, 799–804 CrossRef CAS PubMed.
- B. Sepehrnia, J. Ruble and G. Jeffrey, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 249–251 CrossRef.
- B. H. Bracher and R. Small, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1967, 23, 410–418 CAS.
- R. Le Fevre and A. Sundaram, J. Chem. Soc., 1962, 3904–3915 RSC.
- J. M. O'Gorman, W. Shand Jr and V. Schomaker, J. Am. Chem. Soc., 1950, 72, 4222–4228 CrossRef.
- J. Wilmshurst, J. Mol. Spectrosc., 1957, 1, 201–215 CrossRef.
- N. Kozak and Y. N. Nizel'skii, J. Struct. Chem., 1987, 28, 310–312 CrossRef.
- H. Rzepa, The Winnower, 2015, 2, 1–4 Search PubMed.
- C. A. Filgueiras and J. E. Huheey, J. Org. Chem., 1976, 41, 49–53 CrossRef CAS.
- C. Cox and T. Lectka, J. Org. Chem., 1998, 63, 2426–2427 CrossRef CAS PubMed.
- M. J. Deetz, C. C. Forbes, M. Jonas, J. P. Malerich, B. D. Smith and O. Wiest, J. Org. Chem., 2002, 67, 3949–3952 CrossRef CAS PubMed.
- H. J. Böhm, S. Brode, U. Hesse and G. Klebe, Chem.–Eur. J., 1996, 2, 1509–1513 CrossRef.
- I. Nobeli, S. Price, J. Lommerse and R. Taylor, J. Comput. Chem., 1997, 18, 2060–2074 CrossRef CAS.
- J.-Y. Le Questel, C. Laurence, A. Lachkar, M. Helbert and M. Berthelot, J. Chem. Soc., Perkin Trans. 2, 1992, 2091–2094 RSC.
- F. Besseau, C. Laurence and M. Berthelot, J. Chem. Soc., Perkin Trans. 2, 1994, 485–489 RSC.
- F. Besseau, M. Luçon, C. Laurence and M. Berthelot, J. Chem. Soc., Perkin Trans. 2, 1998, 101–108 RSC.
- N. Ziao, C. Laurence and J.-Y. Le Questel, CrystEngComm, 2002, 4, 326–335 RSC.
- A. R. Modarresi-Alam, A. Nowroozi, P. Najafi, F. Movahedifar and H. Hajiabadi, J. Mol. Struct., 2014, 1076, 299–307 CrossRef CAS.
- S. K. Singh, K. K. Mishra, N. Sharma and A. Das, Angew. Chem., Int. Ed., 2016, 55, 7801–7805 CrossRef CAS PubMed.
- P. Baillargeon and Y. L. Dory, Cryst. Growth Des., 2009, 9, 3638–3645 CrossRef CAS.
- R. J. Staples and J. A. Gingold, Z. Kristallogr.–New Cryst. Struct., 2009, 224, 121–123 CAS.
- M. Calleri, G. Chiari, A. C. Villa, A. G. Manfredotti, C. Guastini and D. Viterbo, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 479–485 CrossRef.
- M. Bursavich and F. Fronczek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, IUC9700022 CrossRef.
- H. Mouhib, D. Jelisavac, W. Stahl, R. Wang, I. Kalf and U. Englert, ChemPhysChem, 2011, 12, 761–764 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- N. R. Babij, E. O. McCusker, G. T. Whiteker, B. Canturk, N. Choy, L. C. Creemer, C. V. D. Amicis, N. M. Hewlett, P. L. Johnson and J. A. Knobelsdorf, Org. Process Res. Dev., 2016, 20, 661–667 CrossRef CAS.
- A. J. Borah and P. Phukan, Tetrahedron Lett., 2012, 53, 3035–3037 CrossRef CAS.
- R. Ghosh, M. Nethaji and A. G. Samuelson, J. Organomet. Chem., 2005, 690, 1282–1293 CrossRef CAS.
- S. V. Chankeshwara and A. K. Chakraborti, Tetrahedron Lett., 2006, 47, 1087–1091 CrossRef CAS.
- R. J. Abraham, B. Bardsley, M. Mobli and R. J. Smith, Magn. Reson. Chem., 2005, 43, 3–15 CrossRef CAS PubMed.
- S. Pramanik, R. R. Reddy and P. Ghorai, Org. Lett., 2015, 17, 1393–1396 CrossRef CAS PubMed.
- M. Hosseini-Sarvari and E. Sodagar, C. R. Chim., 2013, 16, 229–238 CrossRef CAS.
- R. Laidlaw, Y. Miura, C. Panetta and R. Metzger, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 2009–2013 CrossRef.
- M. Goodman, P. Ganis, G. Avitabile and S. Migdal, J. Am. Chem. Soc., 1971, 93, 3328–3331 CrossRef CAS.
- M. Nethaji and V. Pattabhi, Proceedings of the Indian Academy of Sciences, 1985, 95, 369–374 CAS.
- M. K. Singh, M. Gangwar, D. Kumar, R. Tilak, G. Nath and A. Agarwal, Med. Chem. Res., 2014, 23, 4962–4976 CrossRef CAS.
- X. Zhang, Z. Liu, Y. Gao, F. Li, Y. Tian, C. Li, X. Jia and J. Li, Adv. Synth. Catal., 2018, 360, 272–277 CrossRef CAS.
- M. Bursavich and F. Fronczek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, IUC9700022 CrossRef.
- A. A. Yakovenko, J. H. Gallegos, M. Y. Antipin, A. Masunov and T. V. Timofeeva, Cryst. Growth Des., 2011, 11, 3964–3978 CrossRef CAS.
- Y. Diskin-Posner, S. Dahal and I. Goldberg, Chem. Commun., 2000, 585–586 RSC.
- Y. Diskin-Posner, G. K. Patra and I. Goldberg, Eur. J. Inorg. Chem., 2001, 2515–2523 CrossRef CAS.
- K. Endo, T. Ezuhara, M. Koyanagi, H. Masuda and Y. Aoyama, J. Am. Chem. Soc., 1997, 119, 499–505 CrossRef CAS.
- P. Zou, M. H. Xie, H. Wu, Y. L. Liu and Z. P. Chen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o2806 CrossRef CAS PubMed.
- B. Nolin and R. N. Jones, Can. J. Chem., 1956, 34, 1392–1404 CrossRef CAS.
- N. Sundaraganesan and B. D. Joshua, Spectrochim. Acta, Part A, 2007, 68, 771–777 CrossRef CAS PubMed.
- J. A. Soderquist, I. Rosado, Y. Marrero and C. Burgos, ARKIVOC, 2001, 4, 12–19 Search PubMed.
- X.-S. Jia, H.-L. Wang, Q. Huang, L.-L. Kong and W.-H. Zhang, J. Chem. Res., 2006, 2006, 135–138 Search PubMed.
- Z. Shi and J. H. Griffin, J. Am. Chem. Soc., 1993, 115, 6482–6486 CrossRef CAS.
- W. Ferraz de Souza, N. Kambe and N. Sonoda, J. Phys. Org. Chem., 1996, 9, 179–186 CrossRef CAS.
- H. Takayama, S. Shirakawa, M. Kitajima, N. Aimi, K. Yamaguchi, Y. Hanasaki, T. Ide, K. Katsuura, M. Fujiwara and K. Ijichi, Bioorg. Med. Chem. Lett., 1996, 6, 1993–1996 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00313a |
| This journal is © The Royal Society of Chemistry 2020 |