
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe design, synthesis and catalytic performance of vanadium-incorporated mesoporous silica with 3D mesoporous structure for propene epoxidation†
Agnieszka Held *,
Ewa Janiszewska*,
Justyna Czerepińska and
Jolanta Kowalska-Kuś
*,
Ewa Janiszewska*,
Justyna Czerepińska and
Jolanta Kowalska-Kuś
Adam Mickiewicz University, Faculty of Chemistry, Uniwersytetu Poznańskiego 8, 61614 Poznan, Poland. E-mail: awaclaw@amu.edu.pl; eszym@amu.edu.pl
First published on 10th March 2020
Abstract
V-containing mesoporous silica with 3D structure was prepared by a hydrothermal procedure using NH4VO3 as the vanadium precursor and with varied reaction mixture pH values (pH = 3 and pH = 5). The combined use of DR UV-vis and H2-TPR techniques confirmed the successful incorporation of vanadium into the structure of the mesoporous silica material. The number of acid sites, evidenced by ammonia TPD, strongly correlates with the vanadium content. Propene oxidation with N2O revealed the noticeable activity of the synthesised vanadium-containing mesoporous materials in epoxidation reactions. The activity of the synthesized vanadosilicates is compared with the performance of vanadium-supported catalysts (on mesoporous silica of 3D structures) prepared by wet-impregnation method. On the basis of TOF analysis indicating the activity of particular vanadium ions, it was evidenced that although the presence of isolated V species is crucial in propene epoxidation, the availability of the active species is of paramount importance for proper vanadium utilization.
Introduction
Propylene oxide (PO), the third-largest propene derivative, is recognized as an important bulk chemical intermediate for producing of variety of derivatives, such as polyurethane foams, resins, and propylene glycol.1 The industrial-scale synthesis of PO is mainly performed by the liquid-phase reaction, known as chlorohydrin process (43% of PO production). Other liquid-phase industrial processes of propylene oxide manufacture include the styrene co-product process (33%), the tert-butyl co-product process (15%), the HPPO (hydrogen peroxide-based) process (5%), and the Sumitomo cumene-based process (4%). These methods suffer from substantial drawbacks associated with the production of large amounts of waste salts, plant complexity, or marketing/production inefficiencies.1,2 Compared with the traditional chlorohydrin and organic hydroperoxide processes, direct propene epoxidation in gaseous phase provides a greener, simpler, and more sustainable route.3 The direct synthesis of PO with molecular oxygen is one of the most desirable reactions; however, so far, no economically viable route has yet been found. Most of the studied catalysts show either low selectivity or low PO productivity.4 On the other hand, the use of a combination of H2 and O2 mixture was explored on Au/Ti-based catalysts, which allowed a high selectivity for PO (>90%) but at relatively low conversion (1–2%).5 Therefore, this process still needs to be improved due to the fast catalyst deactivation caused by deposition of carbonaceous species and a broad explosion range of the H2/O2 mixture.6–8Another alternative in the search for a new catalytic system for direct propene epoxidation is the use of N2O as an oxidant. This route has been found to be very promising and has been attempted by using a variety of catalysts. Initially, iron-modified silica supports have been considered as the active catalysts for propene epoxidation.9–11 Apart from Fe-containing catalyst, gold–copper alloy catalysts have also been examined in propene epoxidation.12 Recently, vanadium-supported silica catalysts have been reported as an attractive catalytic system for propene-to-PO oxidation with nitrous oxide.13 The catalytic performance of vanadium-doped materials largely depends on the dispersion of the active phase, the nature of the metal oxide active sites, and the metal-support interaction.14,15 It has been reported that isolated vanadium oxide species are suitable for the formation of partial oxidation products, whereas polymeric or bulk V2O5 favors deep oxidation of propene. Therefore, vanadium species supported on various supports, e.g. SBA-3, MCM-41, SBA-15, MCF, as well as on amorphous SiO2, Al2O3, and TiO2, have been examined in propylene oxide manufacture.13,16 Among the studied catalytic systems, the best results have been reported on V/MCF materials prepared by wet-impregnation method.17 The superior performance of the vanadium-doped mesocellular silica foam (MCF) catalysts in PO synthesis has been attributed to the well-defined 3D mesopore systems leading to favorable conditions for internal mass transfer. The highly porous 3D structure of MCF facilitates easy diffusion, thereby allowing the molecules to access and leave the active sites from all three directions without pore blocking. Nevertheless, vanadium-based catalysts still suffer from relatively low PO yield. Therefore, although the mesoporosity is essential for propene epoxidation, the identification of real active sites is crucial for the design of active catalysts.
The nature of the active metal oxide phase is strongly affected by the preparation procedure as well as by the nature of the support. Most of the studied V-containing mesoporous catalysts are prepared by post-synthesis modification, e.g., the traditional impregnation method. This method usually generates well-dispersed active sites on the surface or in the pores of the support. However, these species may easily aggregate during the reaction. On the other hand, direct hydrothermal method in the preparation of framework-incorporated catalysts could result in the formation of fully isolated and highly dispersed active species firmly anchored in the support structure.18,19 So far, different V-incorporated mesoporous silicas were tested in the selective oxidation of lower alkanes by oxygen.20,21 Recently, vanadosilicates of SBA-3 structure have been successfully explored in propene epoxidation with N2O.22
Based on the high catalytic activity of vanadium-supported MCF materials in propene epoxidation by N2O, in the present study, we report the preparation and catalytic performance of mesoporous vanadosilicates of 3D mesopore systems, namely, KIT-6, SBA-12, and MCF, synthesized by direct one-pot pH adjustment method. MCF, SBA-12, and KIT-6 have a three-dimensional pore architecture which, in principle, enables facile transport of reactant and product molecules and access to active sites. Mesostructured cellular foams (MCFs) are composed of large, uniformly sized spherical cells interconnected by uniform windows to create a continuous 3-D pore system.23 KIT-6 material exhibits a three-dimensional cubic Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d symmetric structure with an interpenetrating bicontinuous network of channels,24 whereas SBA-12 has 3D hexagonal symmetry (P63/mmc).25 For comparison, the samples obtained by wet-impregnation method with the same structures and with comparable V loading were also prepared.
d symmetric structure with an interpenetrating bicontinuous network of channels,24 whereas SBA-12 has 3D hexagonal symmetry (P63/mmc).25 For comparison, the samples obtained by wet-impregnation method with the same structures and with comparable V loading were also prepared.
The main goals of the present study are the estimation of the influence of synthesis method for V-containing silica catalyst with 3D structure, as well as the effect of the mesopore structure and V-aggregation state of V species, on the catalytic efficiency of the systems. The effect of preparation procedure, pH value, and vanadium loading on the structure and physicochemical properties of the catalysts are comprehensively investigated by means of XRD, N2 adsorption/desorption, TEM, SEM, DR UV-vis, XPS, NH3-TPD, and H2-TPR. The catalytic performance of the obtained mesoporous vanadosilicates is studied in direct propene epoxidation with N2O. For comparison, the activity of vanadium-supported mesoporous catalyst prepared by wet impregnation method is explored.
Experimental
Catalyst preparation
VSBA-12 samples were synthesized by a modified procedure based on Kumar et al.26 A typical procedure was as follows: 8 g of Brij-76 (Aldrich) was dissolved in 80 cm3 of 2 M HCl with stirring at 313 K for 3 h. After dissolving, 40 g H2O and another 80 cm3 of 2 M HCl were added. Then, 18.86 cm3 tetraethylorthosilicate (TEOS, Aldrich) and 0.4941 g NH4VO3 (POCh, Poland) were added to the solution. After stirring for the next 20 h at 313 K, the pH of the suspension was increased with aqueous ammonia (25%, StanLab, Poland) up to the value of 3 or 5, respectively. The formed gel was heated at 373 K for 22 h. The obtained precipitate was filtered, washed, dried and calcined at 823 K for 8 h to remove organic matter. The calcined samples were white, but they turned yellow when exposed to atmospheric humidity. The samples were labeled as VSBA-12_x, where x stands for the pH value of the synthesis mixture.VMCF materials were synthesized by a modified procedure based on Piumetti et al.27 The samples were obtained by dissolving 8 g of Pluronic P123 (Aldrich) in the mixture of 60 cm3 H2O and 4.6 cm3 1,3,5-trimethylbenzene (Sigma-Aldrich) with stirring at 313 K for 3 h. Then, 19.3 cm3 TEOS and 0.5031 g NH4VO3 were added. Stirring was continued for the next 20 h at 313 K. Afterwards, the pH of the suspension was adjusted with 0.2 M HCl to the value of 3 or 5, respectively. The formed gel was heated at 373 K for 24 h. The precipitated product was filtered, washed, dried and calcined at 823 K for 8 h to remove surfactant. The calcined samples were white, but they turned yellow when exposed to atmospheric humidity. The samples were labeled as VMCF_x, where x stands for pH value of the synthesis mixture.
VKIT-6 materials were synthesized according to our own procedure. In a typical synthesis, 5.6 g of Pluronic P123 was dissolved in 53.2 cm3 H2O with stirring at 313 K for 2 h. Then, 8.64 cm3 of n-butanol (EuroChem, Poland) was added, and stirring was continued for the next hour. Afterwards, 19.21 cm3 of TEOS and NH4VO3 solution (0.5031 g NH4VO3 dissolved in 159.6 cm3 of H2O) were added. The mixture was stirred for 20 h at 313 K, then the resulting gel was adjusted to pH 3 or 5 by adding aqueous ammonia and aged at 373 K for 48 h. The obtained samples were filtered, washed, dried and calcined at 823 K for 8 h. The samples were labeled as VKIT-6_x, where x stands for the pH value of the synthesis mixture.
For comparison, pure silica SBA-12, MCF and KIT-6 were synthesized using the same procedure in the absence of vanadium precursor and without adjusting the pH. The silica materials were used as the supports for preparation of vanadium catalysts. These were obtained by impregnation of silica supports with NH4VO3 solution, followed by drying at 373 K overnight and calcination at 823 K for 1 h in air. The amount of NH4VO3 was adjusted to obtain a transition metal content in the catalysts equal to vanadium concentration in the synthesized vanadosilicates of the same structure. The samples, labelled as VSBA-12_imp, VMCF_imp, and VKIT-6_imp, contained 1.5, 3.4, and 3.4 wt% V, respectively.
Catalyst characterization
Powder X-ray diffraction patterns of as-prepared non-modified mesoporous matrices and vanadium-containing samples prepared by hydrothermal synthesis and wet-impregnation method were collected on a Philips Bruker D8 Advance diffractometer using Cu Kα radiation (λ = 1.54056 Å) in the range of 2θ equal to 0.3° to 6° (low-angle range) and 6° to 60° (wide-angle range). The textural parameters of the investigated materials were determined by N2 adsorption/desorption isotherms collected at 77 K using a Quantachrome Nova 1000e sorptometer. Prior to the measurements, the samples were outgassed under vacuum at 573 K for 16 h. The specific surface area was determined using the BET equation. The pore volume and diameter were estimated according to the Broekhoff-de Boer method28 for MCF materials and BJH method for SBA-12 and KIT-6 samples. UV-vis diffuse reflectance spectra of vanadium-modified samples were recorded at room temperature on a Cary 100 UV-vis spectrometer (Varian) in the range of 200 to 800 nm. Prior to measurement, the samples were dehydrated by calcination for 1 h at 673 K. The spectra were recorded at room temperature (RT), and BaSO4 was used as a reference material. X-ray photoelectron spectra (XPS) were recorded on an ultrahigh vacuum (UHV) system (Specs, Germany). The examined materials were irradiated with a monochromatic Al Kα radiation (1486.6 eV). The operating pressure in the chamber was close to 2 × 10−9 mbar. Binding energies were referenced to the C 1s peak from the carbon surface deposit at 284.6 eV. The NH3-TPD measurements were performed in a flow reactor. In a typical experiment, about 40 mg of sample was pretreated in He at 773 K for 0.5 h, then cooled down to 393 K and afterwards saturated with ammonia for 0.5 h. The physically adsorbed NH3 was removed by purging with helium flow at 393 K for 1 h, and then, the TPD analysis was carried out. All TPD-NH3 profiles presented in this work were collected in the range of 373 to 873 K, with a heating rate of 10° min−1 and normalized to the same sample weight. Temperature-programmed reduction (H2-TPR) experiments were carried out in a homemade setup equipped with a thermal conductivity detector (TCD). Before the experiment, the sample was pre-treated in helium at 673 K for 60 min to remove adsorbed water. H2-TPR measurements were performed under a flow of 10 vol% H2/Ar (70 cm3 min−1) from 373 K to 1173 K at a constant heating rate (10° min−1). All TPR-H2 profiles presented in this work were normalized to the same sample weight.Transmission electron microscopy (TEM) images were recorded on a JEOL 2000 microscope operating at the accelerating voltage of 80 kV. Scanning electron microscope (SEM) images were recorded on a Hitachi SU3500 microscope. Vanadium content in the calcined samples was determined by means of EDS spectrometer (Thermo Fisher Scientific) with an ultradry silicon drift X-ray detector (SDD).
Catalytic activity measurements
The catalytic tests for propene oxidation were performed in a continuous flow reactor at atmospheric pressure. Catalytic experiments were carried out at 653, 673, and 703 K, with WHSV = 3420 cm3 h−1 gcat−1, related to contact time of 1.1s. The catalysts were pretreated in flowing helium at 723 K for 30 min before each reaction run. The feed composition was C3H6/N2O/He = 1/15/12.5 vol%. The catalytic activity was analyzed at steady-state conditions, and the product composition was analyzed using an online gas chromatograph equipped with FID detector (Varian CP-3800 Gas Chromatograph) with WCOT fused silica capillary column (50 m × 0.53 mm) and TCD detector (SRI 8610C Gas Chromatograph with HayeSep Q packed column). On the grounds of GC analysis, it was found that besides PO, we also recorded propionaldehyde, acetone, acrolein, and carbon oxides (CO and CO2). The catalytic performance is expressed as conversion (%), selectivity (%), and yield (%), calculated as follows:% Propene conversion
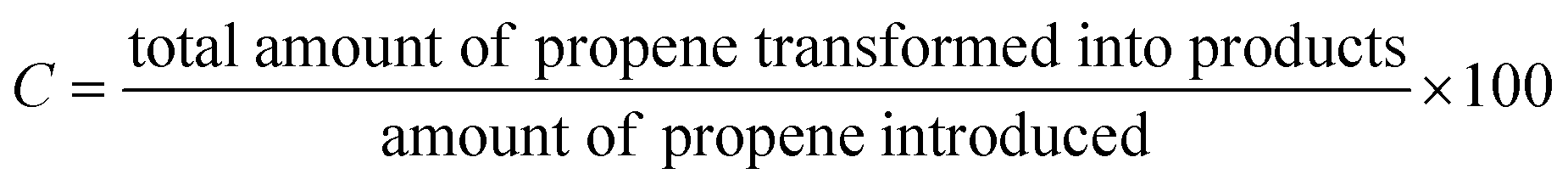 | (1) |
% selectivity
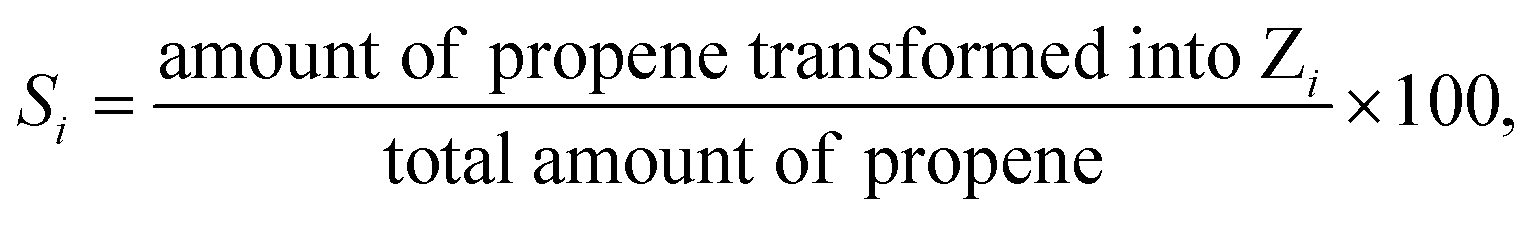 | (2) |
% yield of PO
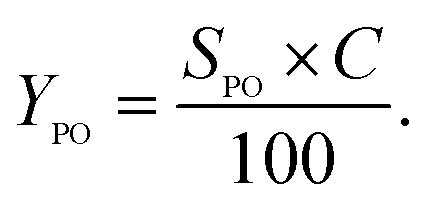 | (3) |
Unless otherwise stated, the catalytic results after 30 min. of the reaction are discussed. Space-time yields (STY) of propene oxide were calculated as g of PO per kg of catalyst per hour. The apparent turnover frequency (TOF) values per V atom were evaluated on the basis of the amount of propene (in moles) transformed to propene oxide related to vanadium present in the catalysts, expressed in moles per second.
Results and discussion
Catalyst characterization
The low-angle XRD patterns of vanadium-containing MCF, KIT-6, and SBA-12 materials synthesized under different pH values or impregnated, as well as those of pure mesoporous matrices, are depicted in Fig. 1. The modification of synthesis mixture pH, facilitating vanadium incorporation in the walls, affects the structure of the 3D vanadosilicates in different ways. All the KIT-6 samples exhibit two well-resolved diffraction peaks, including (211) and (220) at low diffraction angles, which confirm the proper cubic mesostructure. The structure of KIT-6 is slightly affected either by vanadium incorporation, by impregnation, or by the pH of the synthesis. For silica SBA-12 matrix, we observe the presence of three diffraction peaks characteristic for this material. Besides, it is notable that the diffraction peaks shift to lower angles and their intensities decrease with increasing pH value of the synthesis mixture. This can suggest that low-acidity conditions cause some structural irregularities. Similar trends resulting from differences in the synthesis conditions have been reported in literature.29,30 Along with the increase of the reaction mixture pH, some distortions were observed in the long-range order of mesoporous materials. In the case of MCF materials, diffraction peaks at very low angles (<1°) are observed due to their ultra-large pore dimensions, characterized by hexagonal MCF symmetry. Vanadium salt impregnation does not affect the structure, whereas the incorporation of V species during hydrothermal conditions at higher pH causes some disorder in the structure. In order to check the presence of any bulk vanadium oxide species on the surface of the synthesized mesoporous samples, XRD patterns in the wide-angle range were also recorded. The wide-angle XRD patterns of VKIT-6, VSBA-12, and VMCF (synthesized under different pH levels) and samples prepared by impregnation of vanadium salts show a broad peak around 23° due to amorphous SiO2 (Fig. S1, ESI†). Furthermore, no diffraction peaks corresponding to V2O5 crystalline phase are observed on the synthesized VSBA-12 (regardless of pH of the synthesis) and on the impregnated materials. Otherwise, additional diffraction peaks in a wide angle range (located at 2 theta in the range of 15 to 35°) can be observed on the VKIT-6_3 and VMCF_3 samples (comprising the highest V loading, ca. 3 wt% of V), demonstrating the appearance of crystallites of V2O5 species on their surface.19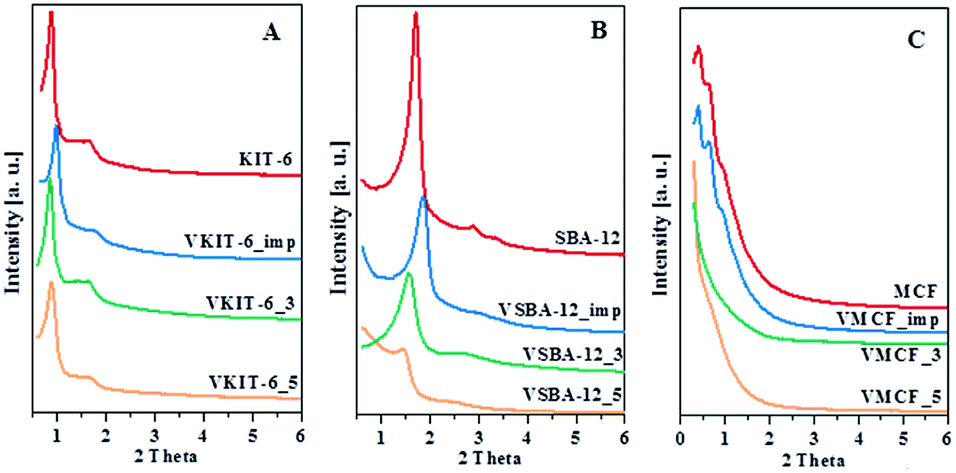 | ||
| Fig. 1 Low-angle XRD patterns of parent and V-containing materials: (A) KIT-6, (B) SBA-12, and (C) MCF. | ||
The N2 adsorption/desorption isotherms of pure silica and V-incorporated mesoporous samples prepared under different pH values, along with those of impregnated samples, are shown in Fig. 2, and Table 1 lists their textural properties.
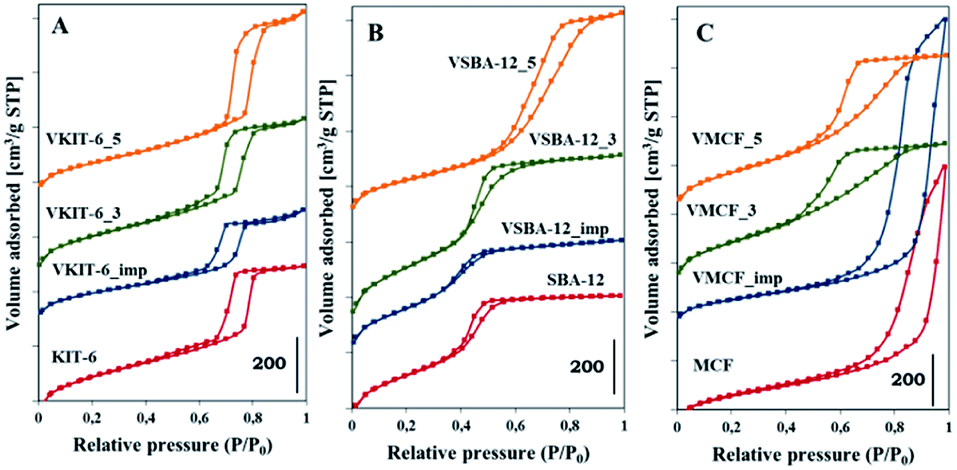 | ||
| Fig. 2 The N2 adsorption/desorption isotherms of the indicated samples with: (A) KIT-6, (B) SBA-12, and (C) MCF structures. | ||
| Sample | pH | Va [wt%] | V-Density [VOx per nm2 | SBETb [m2 g−1] | Vtc [cm3 g−1] | Dd [nm] | NH3-TPD [μmol g−1] |
|---|---|---|---|---|---|---|---|
| a Estimated from the EDS results.b BET specific surface area.c Total pore volume.d The average pore diameter.e Average cell diameter (Dc) determined from the adsorption branches of N2 isotherms.f Average window diameter (Dw) determined from the desorption branches of N2 isotherms. | |||||||
| MCF | — | — | 676 | 1.77 | 20.3e/13.7f | — | |
| VMCF_imp | 3.4 | 0.75 | 533 | 1.69 | 20.0e/12.7f | 1254 | |
| VMCF_3 | 3 | 3.4 | 0.48 | 837 | 0.99 | 3.4e/4.9f | 504 |
| VMCF_5 | 5 | 1.9 | 0.28 | 789 | 1.08 | 5.3e/5.4f | 473 |
| SBA-12 | — | — | 876 | 0.82 | 3.8 | — | |
| VSBA-12_imp | 1.5 | 0.20 | 893 | 0.74 | 3.3 | 916 | |
| VSBA-12_3 | 3 | 1.5 | 0.17 | 1031 | 1.06 | 4.1 | 278 |
| VSBA-12_5 | 5 | 1.3 | 0.25 | 604 | 1.20 | 8.0 | 363 |
| KIT-6 | — | — | 792 | 1.00 | 5.0 | — | |
| VKIT-6_imp | 3.4 | 0.74 | 540 | 0.72 | 5.4 | 1653 | |
| VKIT-6_3 | 3 | 3.4 | 0.59 | 686 | 0.98 | 5.7 | 1045 |
| VKIT-6_5 | 5 | 1.8 | 0.36 | 590 | 1.12 | 7.6 | 602 |
For KIT-6 materials, all the isotherms are of type IV, with a sharp capillary condensation step at high relative pressures and hysteresis loop closing at about 0.65 P/P0, typical for large pores in a narrow range of sizes. The shape of the N2 adsorption/desorption isotherms practically does not change upon vanadium incorporation. These results are consistent with XRD data confirming the preservation of proper KIT-6 structure of the studied catalysts. Data reported in Table 1 show some influence of synthesis pH on the textural parameters of VKIT-6 samples, and a small decrease of the surface area with increasing pH is observed. The decrease of specific surface area (SBET) is even more pronounced for the impregnated sample, which may result from filling of the mesoporous channels.19 SBA-12 structure appears to be less resistant to the modification, and significant changes in the shape of N2 adsorption/desorption isotherms as well as in the textural properties (SBET, the average pore diameter) of hydrothermally synthesized VSBA-12 and VSBA-12_imp samples are observed. The isotherms of VSBA-12_3 and VSBA-12_imp change by a lesser degree; however, for VSBA-12_5, a significant difference is observed, and the hysteresis loop visibly broadens and is shifted to higher P/P0 values. These changes are accompanied by the diversity in textural parameters (Table 1). Except for the VSBA12_3 sample, the surface area decreases, which may result from partial destruction of the structure of VSBA-12_5 sample or filling the pores of the impregnated sample. Significant disorder of SBA-12 structure after V incorporation is supported by XRD analysis (Fig. 1). The most diverse effect of vanadium incorporation on the structure is observed for mesocellular silica foam (MCF). Both XRD patterns and low-temperature N2 adsorption/desorption measurements indicate pronounced changes of the MCF structure during hydrothermal synthesis at higher pH, whereas the impregnation allows preservation of the proper cage-window structure. The shape of N2 adsorption/desorption isotherms of VMCF_3 and VMCF_5 and their textural parameters indicate that the characteristic MCF structure is not formed at the pH values of 3 and 5, although these materials are still mesoporous. Similar conclusions were obtained by Yasyerli et al.30
Pore size distributions of mesoporous silica matrices (KIT-6, SBA-12 and MCF) as well as vanadium-impregnated samples and hydrothermally synthesized vanadosilicates are shown in Fig. 3. Pore size distributions of VSBA-12_3 and VSBA-12_imp were found to be quite similar to the pore size distribution of pure-silica SBA-12. Only for VSBA-12_5 (synthesized at pH = 5) does it broaden and shift to the higher values. These data further support some distortion of SBA-12 mesoporous structure at higher pH. For KIT-6 structure, similar pore size distribution was observed for unmodified as well as all V-doped samples, which confirms their high resistance to the pH of the synthesis mixture and to vanadium introduction. On the other hand, significant differences are observed in the pore size distribution of MCF-based samples. Silica MCF and VMCF_imp materials are characterized by pore size distribution typical of the cage-window structure, whereas the pore size distribution of vanadosilicates of MCF structure prepared at different pH (equal to 3 and 5) are similar to the pore size distributions of SBA-12 and KIT-6 structure. The differences in pore size arrangement of VMCF_3 and VMCF_5 samples indicate a decrease of the long-range order of pores. These results strongly correlate with XRD data confirming disorder of the MCF structure.
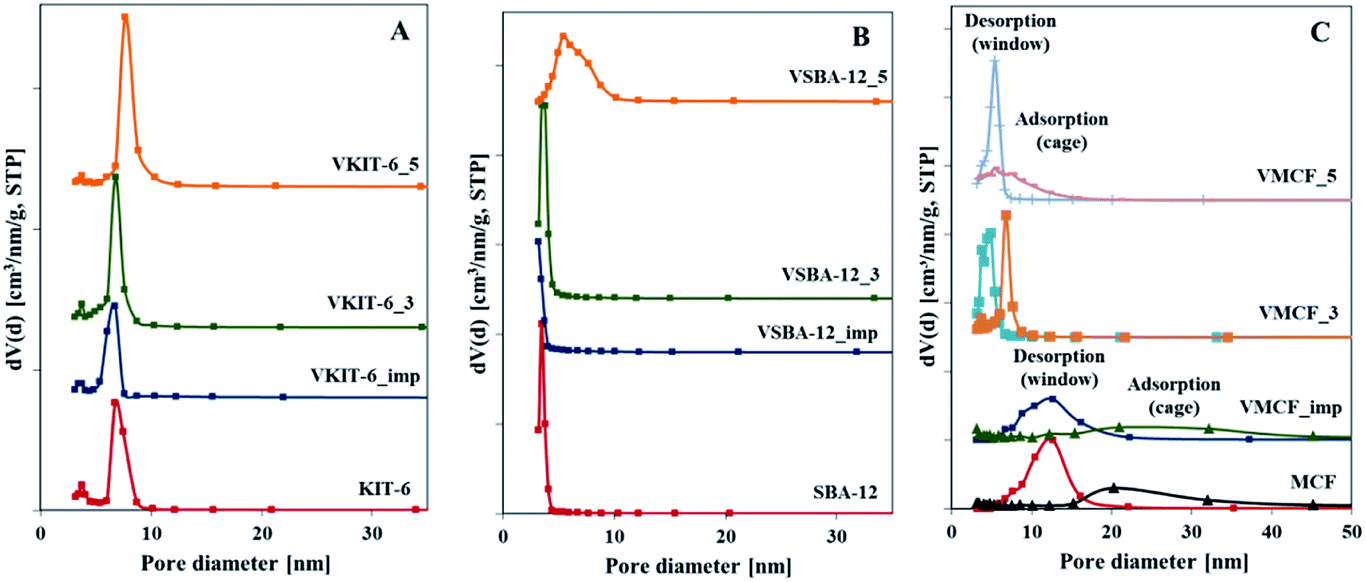 | ||
| Fig. 3 Pore size distributions of the indicated samples with: (A) KIT-6, (B) SBA-12, and (C) MCF structures. | ||
The ordered structure of the synthesized vanadosilicates was examined by transmission electron microscopy (TEM) (Fig. 4). The TEM image of MCF silica support is typical for materials with MCF structure and reveals the presence of cylindrical pores with a disordered arrangement without any channels. The introduction of vanadium during hydrothermal synthesis or by impregnation does not influence the mesostructure, and no crystalline V2O5 phase has been observed. Although, according to XRD and nitrogen adsorption isotherms, the window-cage structure of MCF was significantly distorted in VMCF_3 and VMCF_5, short-range order of the pores was partly conserved. TEM images of KIT-6 catalysts confirm the presence of well-ordered mesopores. This structure is practically conserved for vanadosilicates of KIT-6 structure synthesized at higher pH or prepared by impregnation. TEM analysis also confirmed the long-range, three-dimensional, mesoporous ordering of SBA-12 materials, which is preserved upon vanadium incorporation at higher pH or during impregnation.
 | ||
| Fig. 4 TEM images of the indicated samples. | ||
SEM micrographs of the investigated materials differ significantly depending on the mesoporous structures (Fig. 5). MCF material is characterized by the presence of mostly spherical silica particles of about 1 μm in size. The introduction of a vanadium precursor during synthesis results in significant agglomeration of the particles. A similar effect was observed regarding KIT-6 materials, where after V incorporation, agglomeration was observed. On the other hand, for SBA-12 material, the presence of large hollow spheres reaching 15 μm in size was confirmed. The synthesis of vanadosilicates with SBA-12 structure resulted in the formation of irregular agglomerated particles.
 | ||
| Fig. 5 SEM images of the indicated samples. | ||
Additional information about the reducible nature of vanadium species is provided by the H2-TPR profiles taken at 323–1123 K. The area of the reduction pattern (which corresponds to the amount of consumed hydrogen) is proportional to vanadium concentration. According to previous reports, the reduction peaks with maxima located below 923 K were generally ascribed to monomeric or highly dispersed vanadium species. On the other hand, a high temperature peak (with maximum above 923 K) indicated the presence of polymeric and bulk-like V2O5 species.31,32 No reduction signals were seen in TPR-H2 profiles of the all silica materials (data not shown), which proves that the supports were irreducible. The TPR profiles of the vanadium-modified SBA-12 samples prepared by direct synthesis or by impregnation method exhibit one sharp reduction peak with a maximum at about 748 K (Fig. 6A). For VSBA-12_5 catalysts, a small shoulder appears at ca. 863 K. By analogy with the previous reports concerning reducibility of vanadium-containing catalysts, both peaks are attributed to the reduction of well-dispersed vanadium species.19,27 The appearance of a shoulder at higher temperature may result from vanadium atoms located in the framework position and, therefore, more resistant to reduction, or from the presence of low oligomeric V–O–V groups.19,27,33
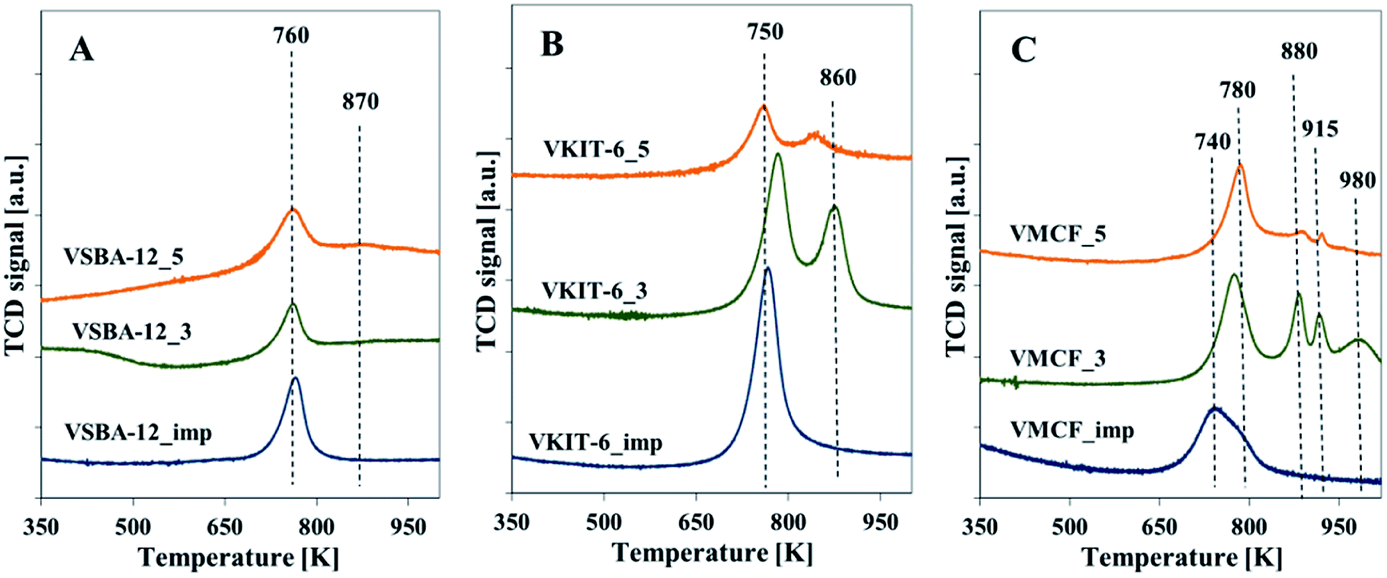 | ||
| Fig. 6 Normalized H2-TPR profiles of the indicated samples with: (A) KIT-6, (B) SBA-12, and (C) MCF structures. | ||
H2-TPR profiles of VKIT-6 samples prepared by hydrothermal synthesis consist of two distinct reduction peaks at ca. 753 and 863 K. Similarly, as for VSBA-12 samples, the coexistence of two different forms of isolated vanadium species is revealed (Fig. 6B). Again, the reduction of vanadium at about 863 K may result from less reducible framework V species. The MCF_imp sample also shows two overlapping reduction maxima at ca. 753 and 783 K (Fig. 6C). In contrast, in the case of VMCF_3 catalyst, four bands at ca. 723, 883, 918, and 983 K are registered. For the sample VMCF synthesized at pH = 5, with lower V content (1.8 wt% of V), the reduction peak at ca. 783 K predominates, whereas the high-temperature peak is much less intense (Fig. 6C). The presence of a few reduction peaks may result from the cage-window structure of the mesopores of MCF-type materials, indicating various localizations of vanadium species characterized with different reducibilities. A similar effect was observed for vanadosilicates of BEA structure.33
The H2-TPR results clearly show that the impregnated samples as well as those synthesized at pH = 5 (comprising lower V content) are characterized by predominance of isolated V species. On the other hand, the VMCF_3 results indicate the coexistence of isolated V species along with low-oligomeric V5+ forms and bulk-like V2O5 crystallites. These results are consistent with XRD analysis confirming the presence of V2O5 crystallites in the selected catalysts (Fig. S1†).
Structural information concerning the coordination environment of VOx species can be additionally obtained from UV-vis diffuse reflectance spectra (Fig. 7, Table S1†). The energy of oxygen → vanadium charge transfer (CT) bands distinguish the coordination of the V5+ centers. Along with the increasing coordination number of V atoms, a shift of the CT band to higher wavelength (lower energy) is observed.34 Fig. 7 shows the DR UV-vis spectra of the dehydrated mesoporous vanadosilicates. A very broad band centered at ca. 250–300 nm is observed for the studied samples. A deconvolution of these spectra helps to define different forms of V species in the studied vanadosilicates. For vanadosilicates of SBA-12 structure, comprising the lowest V loading (ca. 1.2 wt%) among the studied catalysts, the UV-vis bands centered at 250 nm predominate. This confirms the localization of significant portions of V species along the sides of the mesoporous walls, forming a tetrahedral environment.35 These bands overlap with the bands centered >300 nm attributed to the vanadium atoms on the wall surface and in external clusters.36 Additionally, for the samples synthesized at pH = 3 (VSBA-12_3), a shoulder at 380 nm appeared, indicating the presence of polymeric V species in octahedral coordination. In the VSBA-12_imp sample, vanadium species are mainly located on the wall surface (UV-vis bands in the range of 250–300 nm) and in the external clusters (bands >300 nm). In VKIT-6 samples, only a quarter portion of V species are located inside the walls, whereas a predominant number of vanadium atoms exist as external species (bands >300 nm) (Table S2†). On the other hand, in the VKIT-6_imp sample, the introduced vanadium species localized mainly as isolated forms on the wall surface. Similar trend to the VKIT-6_imp catalyst was observed for the VMCF_imp sample, where bands in the range of 250–300 nm (characteristic of isolated V species on the wall surface) predominated. Hydrothermal synthesis of vanadium-containing MCF materials led, in turn, to the formation of extra-framework oligomeric V species (bands >300 nm). The UV-vis results strongly correlate with H2-TPR data and with XRD analysis, indicating the presence of low-oligomeric V5+ forms and bulk-like V2O5 crystallites in the VMCF_3 sample.
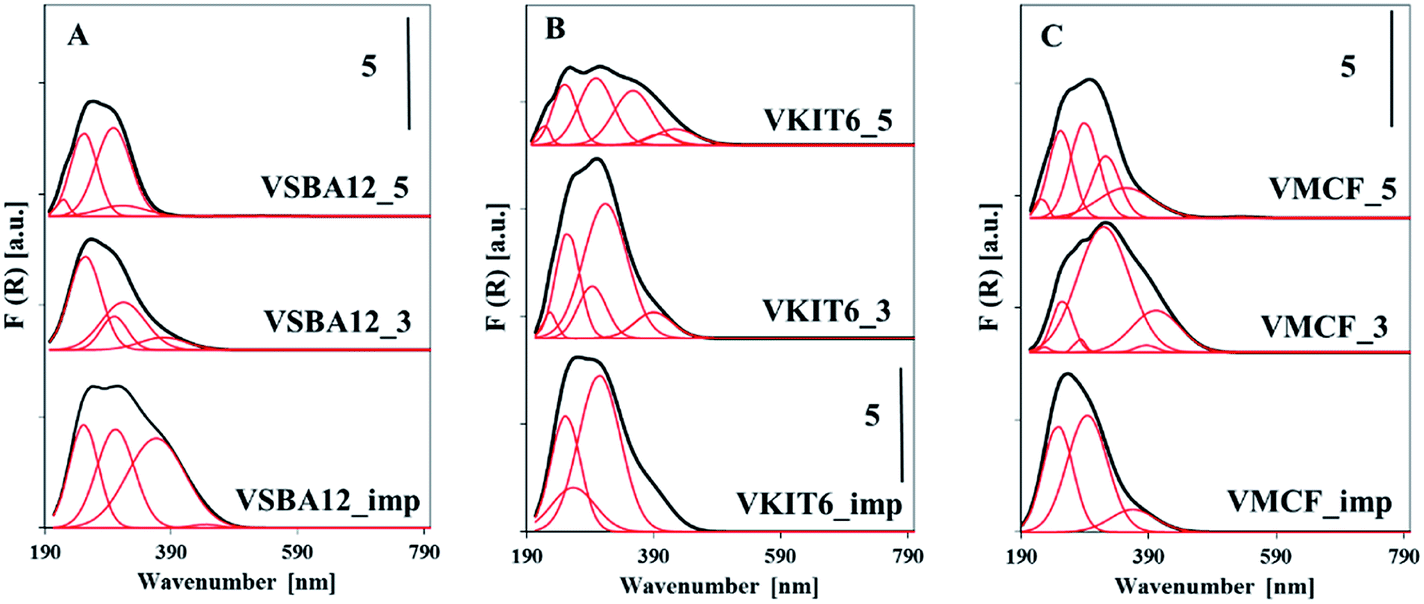 | ||
| Fig. 7 UV-vis reflectance spectra of the indicated dehydrated samples with: (A) KIT-6, (B) SBA-12, and (C) MCF structures. | ||
In order to obtain more details about the nature of vanadium species in the studied samples, X-ray photoelectron spectroscopy measurements were carried out. Vanadium 2p3/2 XPS results of the selected samples (VMCF_imp and VMCF_3) are shown in Fig. S2 ESI.† The areas of the V 2p3/2 peaks of both samples are comparable, which confirms the similar amount of vanadium in the investigated materials. For one-pot synthesized VMCF_3 sample, the vanadium signal was deconvoluted into two binding energies centered at 516.5 eV and 518.1 eV, whereas only one signal at 517.5 eV was registered for VMCF_imp material. According to reports,36,37 the binding energies of V4+ species lie between 516.5 and 517.1 eV, while for V5+, the values are between 517.2 and 518.2 eV. This suggests that in the impregnated sample, only V5+ species are present, whereas in the synthesized sample, V4+ and V5+ species coexist. The presence of V4+ was not detected by UV-vis spectra since the catalysts were thermally dehydrated before collecting the UV-vis spectra by calcination in oxidizing atmosphere for 1 h in order to remove water. On the other hand, the binding energy registered at 517.5 and 518.1 eV in impregnated and directly synthesized samples, respectively, confirms the presence of V5+ in the different coordination environments, in line with UV-vis results.
Total acidity of the synthesized vanadosilicates and the impregnated samples was determined by NH3-TPD. The TPD profiles are characterized by the presence of a broad desorption peak with a maximum in the temperature range of 533–573 K. The number of acid sites was calculated on the basis of the amount of chemisorbed ammonia, and it clearly corresponds to V loading in the studied samples (Fig. 8, Table 1). The highest acidity among the studied catalysts was found in samples prepared by impregnation procedure. This indicates the highly acidic nature of isolated V species located on the external surface of catalysts.
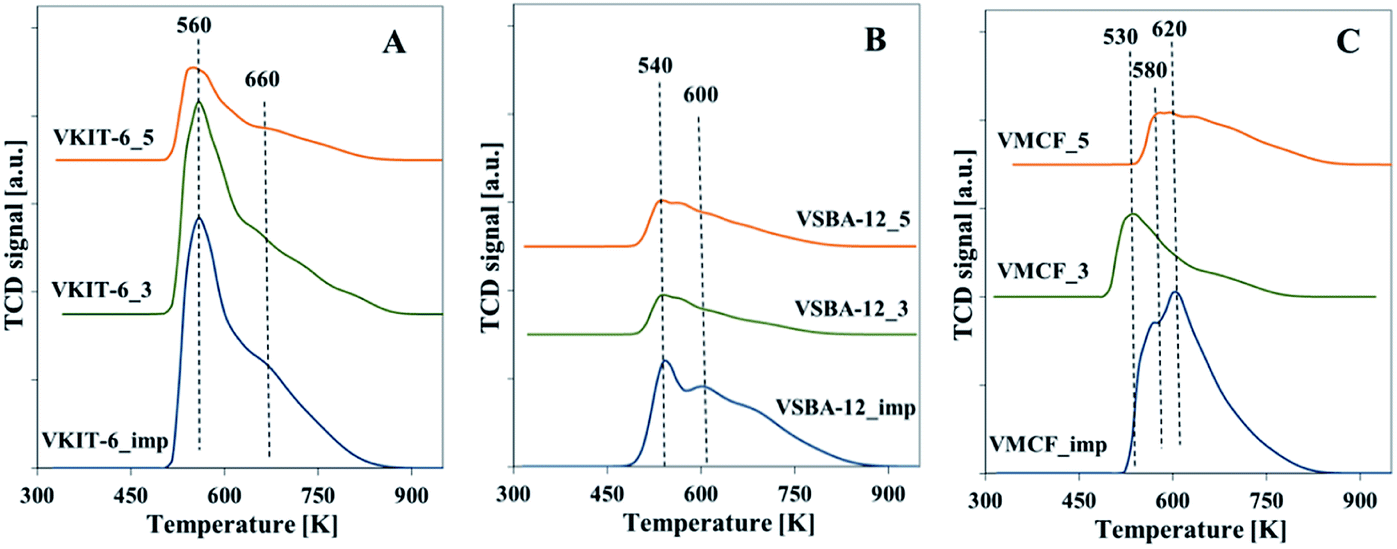 | ||
| Fig. 8 Normalized NH3-TPD profiles of calcined samples with: (A) KIT-6, (B) SBA-12, and (C) MCF structures. | ||
Catalytic performance for propene epoxidation
The activities of the mesoporous vanadium catalysts prepared by direct synthesis and by impregnation method were explored under steady-state conditions at 653–703 K. Fig. 9A presents the propene conversion at different temperatures over vanadosilicates of various structures (KIT-6, SBA-12, and MCF), synthesized at pH = 3 and pH = 5 and containing various V loading. Propene conversion of all catalysts is strongly dependent on the reaction temperature, and it rapidly increases along with increasing reaction temperature (from about 5% at 653 K to about 25% at 703 K). A similar trend has been observed regarding the V content in the catalysts. Samples synthesized at lower pH, characterized by higher V content (Table 1), show higher propene conversion when compared to the samples synthesized at pH = 5.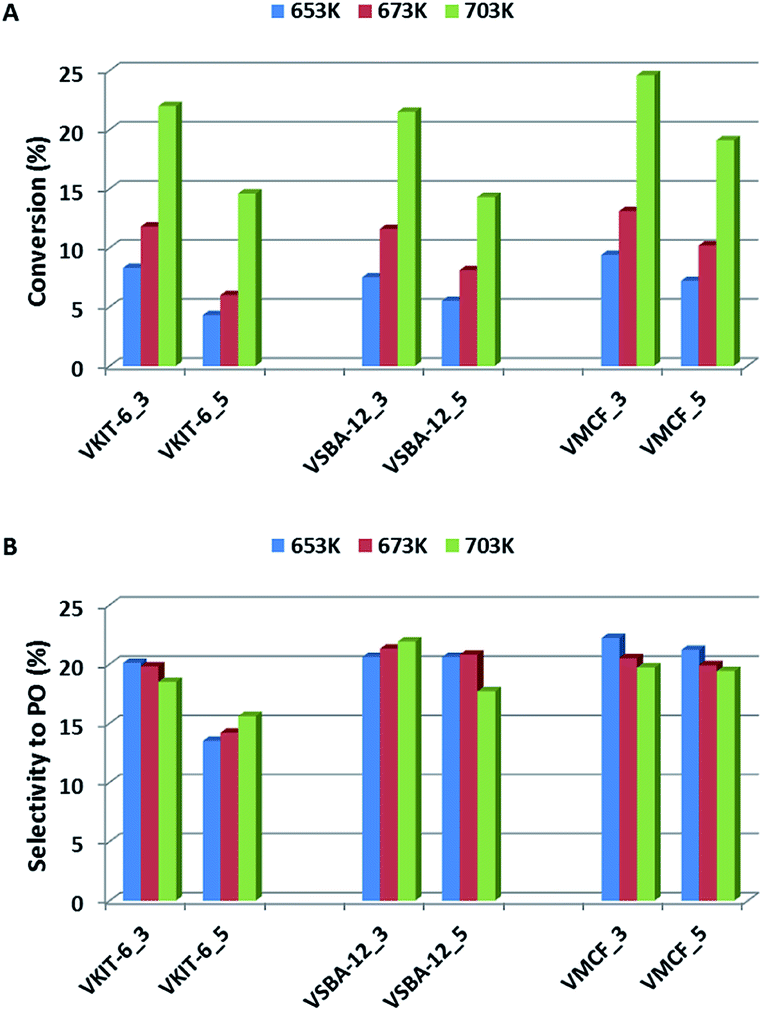 | ||
| Fig. 9 Catalytic activity, expressed in terms of (A) propene conversion and (B) selectivity to propene oxide, of the indicated samples in propene epoxidation at different reaction temperatures. | ||
Catalyst activity evaluation was mainly based on selectivity towards propylene oxide (Fig. 9B). Selectivity towards PO has been slightly affected, either by the reaction temperature or by the mesoporous silica structure, and it did not exceed 23%.
Although selectivity to PO is of paramount importance for the evaluation of catalyst activity in the studied reaction, selectivity for other products is also discussed. Besides PO, other organic oxygen-bearing products, such as propionaldehyde (PA), acetone (ACT), and acrolein (ACR), as well as COx (CO and CO2), were detected in the reaction outlet. Selectivity towards COx strongly correlated with the reaction temperature. Along with the increase of the reaction temperature, selectivity towards COx raised visibly (reaching 20% at 703 K; Fig. 10).
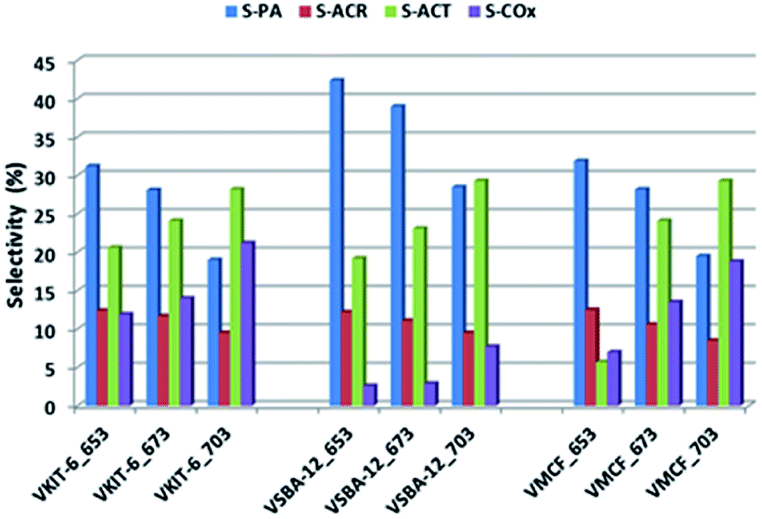 | ||
| Fig. 10 Selectivity towards oxygen-bearing products (PA: propionaldehyde, ACT: acetone, ACR: acrolein, and COx: CO, CO2) of the indicated samples in propene epoxidation at different reaction temperatures. | ||
Among the oxidation products, selectivity towards propionaldehyde (PA) predominated and decreased along with the increase of the reaction temperature. Otherwise, selectivity for acetone (ACT) showed an opposite tendency and increased with growing temperature. Analogous tendency has been reported in literature for V-containing catalysts.38 Selectivity for acrolein (ACR) was affected neither by the reaction temperature nor by the catalyst structure, being about 10%, and only a slight decrease of ACR selectivity with increasing reaction temperature was observed. According to our earlier findings, propene oxide and acetone are mostly primary products of propene oxidation, whereas acrolein is formed over nucleophilic forms of oxygen.39 The lowered selectivity for propionaldehyde at the expense of rising selectivity to acetone indicates the isomerization of PA with increasing reaction temperature. This is consistent with the proposed mechanism of propene oxidation, where the subsequent formation of ACT as a consequence of PA isomerization was reported. The isomerization is further enhanced by catalyst acidity, which was estimated for the studied samples by NH3-TPD measurements (Fig. 8, Table 1).
Fig. S3† presents the formation rates of PO per unit mass of catalyst per unit time, STY, obtained on the mesoporous vanadosilicates of different structures and synthesized under various pH. Higher values of space-time yield are attained over the samples synthesized at lower pH and, as was reported, comprising higher V loading. The activity of vanadosilicates with 3D structure was also compared, taking into account an amount of propene transformed into propene oxide related to the number of vanadium species per second (expressed as turnover frequency (TOF, Fig. S4†)). Since it is very difficult to identify and quantify the active sites because not all the V species are equally active, the “apparent TOF” was used to assess the efficiency of the studied vanadosilicate catalysts. Furthermore, considering the high value of the surface area (reaching 1000 m2 g−1) and relatively low V loading (≤3 wt%), the vanadium surface coverage did not exceed 1 V nm−2 (Table 1). Therefore, according to a previous report,40 we can assume that all V species form monolayer coverage. The apparent TOF values of PO formation are depicted in Fig. S4.† A sharp increase in the rate of PO formation per unit surface area with increasing reaction temperature was observed (Fig. S4A†). When the samples of similar structure but comprising different V loadings were compared, various catalytic effects appeared. The catalysts of SBA-12 and KIT-6 structure, comprising higher vanadium content (synthesized at pH = 3), show higher TOF values when compared with the samples synthesized at pH = 5 (Fig. S4B†). This indicates that in the samples comprising high vanadium loading, a significant amount of vanadium species is active towards mild electrophilic oxygen species generation. These results contradict our previous findings for vanadosilicates of SBA-3 structure.22 The VSBA-3 catalysts showed higher TOF values for low vanadium content (<1 wt%), which suggested that some vanadium species do not show activity in epoxide formation. This could result from the relatively narrow pore system of VSBA-3 causing some diffusion limitation. These obstacles are overcome in the case of mesoporous materials with 3D structure (namely SBA-12 and KIT-6). Otherwise, in the case of MCF material, higher TOF was observed for the VMCF_5 sample (with lower V loading). The observed differences may be explained on the basis of the structure of the applied 3D mesoporous catalysts. SBA-12, as well as KIT-6 materials, are characterized by highly ordered 3D hexagonal mesostructure, whereas MCF material is composed of uniform spherical cells interconnected by windows. Hydrothermal synthesis of V-containing mesoporous materials could result in different locations of V species: inside the cells and on the outer space of the MCF catalysts. The UV-vis spectra and H2-TPR measurements of VMCF_3 material indicate the high contribution of V species located in the extra-framework position, forming oligomeric and even bulk-like V2O5 species. These findings are further supported by the data reported in Table 1, which also confirm the decrease of mesopore size (both Dc and Dw) being the result of some distortions of the structure but also an effect of pore blocking by the external V species. Therefore, although VMCF_3 shows higher STY of PO, a larger number of V species in the catalyst is inactive towards mild electrophilic oxygen species generation.
For selected catalysts, the stability test was performed. The activity of the studied catalysts was evaluated after 30 min on stream. During 2 h of catalytic test, a small decrease in activity was noted. We have already reported that the decrease of the activity of V-containing catalysts with time of stream resulted from coke deposit.13 Therefore, regeneration of the catalysts was performed by heating in air at 753 K. It is clearly seen in Fig. 5S ESI† that the activity was practically restored.
The activity of the presented vanadosilicates was compared with the activity of the samples prepared by wet-impregnation method from the aqueous solution of NH4VO3. Vanadium concentration in the impregnated samples was adjusted to the same values as the corresponding: VSBA-12_3, VKIT-6_3, and VMCF_3 samples (Table 1). In Fig. 11, the activity of synthesized vanadosilicates and impregnated samples is compared. Excluding the VMCF material, vanadosilicates obtained by direct synthesis show slightly higher propene conversion (Fig. 11A). On the other hand, propene conversion over the impregnated MCF material markedly exceeded the activity of the corresponding VMCF_3. Selectivity towards PO was comparable regardless of catalyst structure and preparation method. Selectivity towards other oxygen-bearing products is presented in Fig. 11B. Apart from VMCF materials, VKIT-6 and VSBA-12 samples (both synthesized and impregnated) show very similar selectivities to AP, ACR, and ACT. The impregnated samples are characterized by a slightly higher contribution of COx in the reaction products, compared to one-pot synthesized vanadosilicates. This can result from the larger contribution of acidic sites in the impregnated samples (Table 1) compared to the corresponding vanadosilicates. A similar effect of acidity on the increasing selectivity to COx was reported in the literature.41 On the other hand, significant differences in selectivity were observed for MCF materials. VMCF_imp shows much higher selectivity towards acetone at the expense of selectivity towards PA and COx when compared to VMCF_3 sample. According to the previously proposed pathways of the selective oxidation of propene, COx arises not only from propene but also from consecutive oxidation of oxygenates (PO, PA, ACR, and ACT). Lower selectivity towards COx over VMCF_imp may result from the well-defined 3D, ultra-large mesopore structure of the catalyst leading to favorable conditions for internal mass transfer, and as a consequence, limiting combustion of propene and oxygen-bearing products.
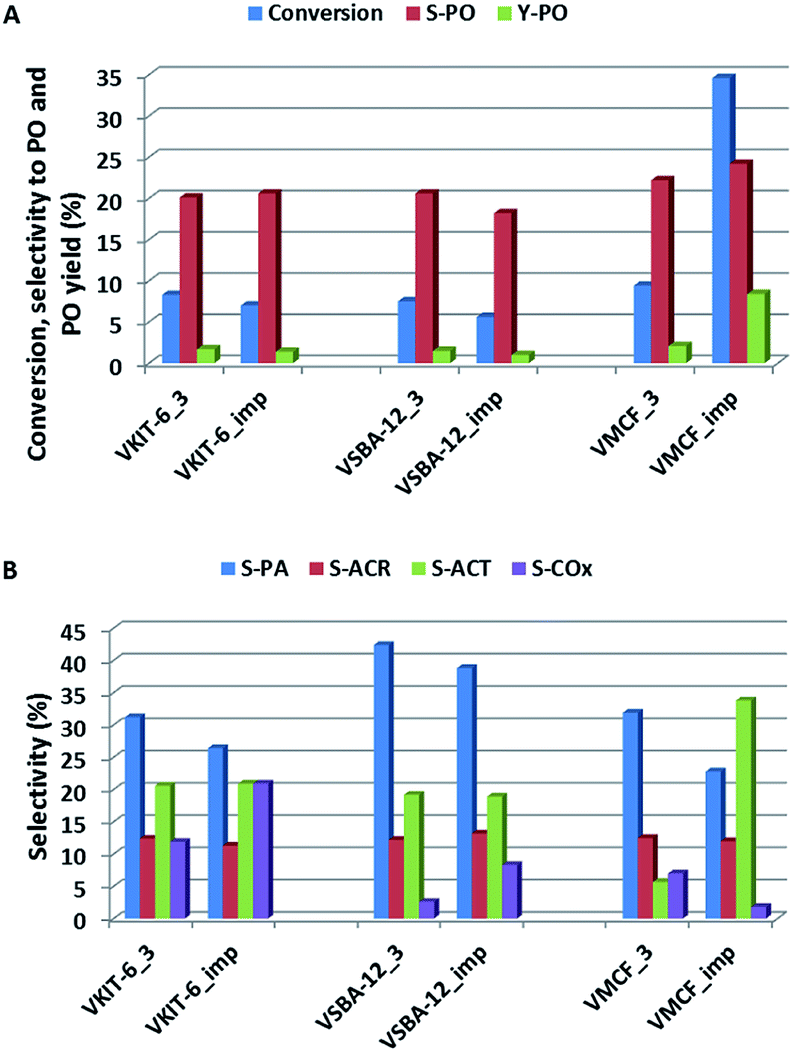 | ||
| Fig. 11 (A) Catalytic activity (propene conversion, selectivity towards PO, and PO yield) and (B) selectivity towards oxygen-bearing products (PA-propionaldehyde, ACR-acrolein, ACT-acetone, and COx) of the indicated catalysts in propene epoxidation at 653 K. | ||
The extended activity of VMCF_imp is also clearly seen when the formation rates of PO and STY are compared (Fig. S6A†). Vanadium-modified mesocellular silica foam prepared by impregnation procedure shows 5 times higher STY value (reaching 25 gPO kgcat−1 h−1) in comparison to the STY of other investigated samples. The catalytic performance of synthesized and impregnated samples was also compared with respect to average catalytic activity of vanadium atoms in the selected catalysts, expressed as TOF (Fig. S6B†). Similarly, as in the case of STY value, the highest TOF was obtained for the VMCF_imp sample.
In summary, these results clearly show that among the parameters influencing the catalytic activity of V-containing samples in propene epoxidation with N2O, a crucial role is played by the elimination of diffusion limitation. Both monomeric VO4 and oligomeric VOx species show activity in propene epoxidation; however, their activity varies depending on the catalyst structure. All the one-pot synthesized vanadosilicates, as well as impregnated samples of SBA-12 and KIT-6 structures, show similar activity, expressed as TOF (in the range of 2.8–8.3 s−1). These values differ significantly for V-containing MCF materials prepared by hydrothermal synthesis and by means of impregnation procedure. The highly disturbed 3D structure of VMCF_3 material results in less porous structure partly blocked by external oligomeric and even bulk-like V2O5 species, causing some diffusion limitation. On the other hand, vanadium sites within VMCF_imp's three-dimensional porous structure helps overcome the problems of limited diffusion of both reactant and product, facilitating high propene oxide productivity.
Conclusions
Vanadium-containing mesoporous silica catalysts with 3D structure, prepared either by one-pot synthesis or by an impregnation method, were characterized, and their catalytic behavior during propene oxidation was examined.The carefully controlled pH of the synthesis gel (pH = 3 or 5) affected both V loading as well as the mesoporous structure. A lower pH value (pH = 3) resulted in higher V loading in the obtained vanadosilicates. The combined use of XRD, N2 adsorption/desorption, and TEM analyses confirmed the formation of a well-ordered mesoporous structure of SBA-12, regardless of the applied synthesis conditions. On the other hand, some distortions were observed for the KIT-6 structure, whereas the cage-window structure of MCF materials appeared to be the least resistant to the pH of the synthesis mixture.
The preparation method of vanadium-containing catalysts, using either a one-pot pH-adjusting method or post-synthesis treatment (impregnation), affected the nature of the vanadium species. The UV-vis results strongly correlate with the H2-TPR data, and they indicate that in the samples synthesized at pH = 5, isolated vanadium species predominated, whereas less acidic conditions (pH = 3) resulted in higher V loading, forming isolated V5+ species as well as low-oligomeric V5+ forms and bulk-like V2O5 crystallites.
Finally, catalytic activity tests in propene epoxidation evidenced that different forms of vanadium, including monomeric VO4 and oligomeric VOx species, show activity in propene epoxidation; however, their activity varies depending on the silica support structure. The crucial parameter, besides the presence of highly dispersed V species, seems to be the highly porous three-dimensional network of the catalyst, which facilitates overcoming the problem of the limited diffusion of both reactants and products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre (grant no. 2016/23/B/ST5/00615).References
- A. H. Tullo and P. L. Short, Chem. Eng. News, 2006, 84, 22–23 CrossRef.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, Ind. Eng. Chem. Res., 2006, 45, 3447–3459 CrossRef CAS.
- F. Cavani, Catal. Today, 2010, 157, 8–15 CrossRef CAS.
- S. J. Khatib and S. T. Oyama, Catal. Rev., 2015, 57, 306–344 CrossRef CAS.
- A. K. Sinha, S. Seelan, M. Okumura, T. Akita, S. Tsubota and M. Haruta, J. Phys. Chem. B, 2005, 109, 3956–3965 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- S. Yao, L. Xu, J. Wang, X. Jing, T. Odoom-Wubah, D. Sun, J. Huang and Q. Li, Mol. Catal., 2018, 448, 144–152 CrossRef CAS.
- J. W. Harris, J. Arvay, G. Mitchell, W. N. Delgass and F. H. Ribeiro, J. Catal., 2018, 365, 105–114 CrossRef CAS.
- V. Duma and D. Hönicke, J. Catal., 2000, 191, 93–104 CrossRef CAS.
- S. Yang, W. Zhu, Q. Zhang and Y. Wang, J. Catal., 2008, 254, 251–262 CrossRef CAS.
- T. Thömmes, S. Zürcher, A. Wix, A. Reitzmann and B. Kraushaar-Czarnetzki, Appl. Catal., A, 2007, 318, 160–169 CrossRef.
- R. J. Chimentão, F. Medina, J. L. G. Fierro, J. Llorca, J. E. Sueiras, Y. Cesteros and P. Salagre, J. Mol. Catal. A: Chem., 2007, 274, 159–168 CrossRef.
- A. Held, J. Kowalska-Kuś and K. Nowińska, Catal. Commun., 2012, 17, 108–113 CrossRef CAS.
- T. Fievez, F. De Proft, P. Geerlings, B. M. Weckhuysen and R. W. A. Havenith, Catal. Today, 2011, 177, 3–11 CrossRef CAS.
- X. Rozanska, E. V. Kondratenko and J. Sauer, J. Catal., 2008, 256, 84–94 CrossRef CAS.
- A. Held, J. Kowalska-Kuś, A. Łapiński and K. Nowińska, J. Catal., 2013, 306, 1–10 CrossRef CAS.
- A. Held, J. Kowalska-Kuś, K. Nowińska and K. Góra-Marek, Catal. Lett., 2018, 48, 2058–2068 CrossRef.
- L. Zhao, Y. Dong, X. Zhan, Y. Cheng, Y. Zhu, F. Yuan and H. Fu, Catal. Lett., 2012, 142, 619–626 CrossRef CAS.
- R. Bulánek, P. Čičmanec, H. Sheng-Yang, P. Knotek, L. Čapek and M. Setnička, Appl. Catal., A, 2012, 415–416, 29–39 CrossRef.
- V. N. Shetti, M. J. Rani, D. Srinivas and P. Ratnasamy, J. Phys. Chem. B, 2006, 110, 677–679 CrossRef CAS PubMed.
- Q. Liu, J. Li, Z. Zhao, M. Gao, L. Kong, J. Liu and Y. Wei, Catal. Sci. Technol., 2016, 6, 5927–5941 RSC.
- E. Janiszewska, A. Held, K. Nowińska and S. Kowalak, RSC Adv., 2019, 9, 4671–4681 RSC.
- P. Schmidt-Winkel, W. W. Lukens, D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1999, 121, 254–255 CrossRef CAS.
- T. W. Kim, F. Kleitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601–7610 CrossRef CAS PubMed.
- Y. Sakamoto, I. Diaz, O. Terasaki, D. Zhao, J. P. Pariente, J. M. Kim and G. D. Stucky, J. Phys. Chem. B, 2002, 106, 3118–3123 CrossRef CAS.
- A. Kumar and D. Srinivas, J. Catal., 2012, 293, 126–140 CrossRef CAS.
- M. Piumetti, B. Bonelli, P. Massiani, S. Dzwigaj, I. Rossetti, S. Casale, L. Gaberova, M. Armandi and E. Garrone, Catal. Today, 2011, 176, 458–464 CrossRef CAS.
- W. W. Lukens, P. Schmidt-Winkel, D. Zhao, J. Feng and G. D. Stucky, Langumir, 1999, 15, 5403–5409 CrossRef CAS.
- O. Aktas, S. Yasyerli, G. Dogu and T. Dogu, Ind. Eng. Chem. Res., 2010, 49, 6790–6802 CrossRef CAS.
- O. Aktas, S. Yasyerli, G. Dogu and T. Dogu, Mater. Chem. Phys., 2011, 131, 151–159 CrossRef CAS.
- F. Arena, N. Giordano and A. Parmaliana, J. Catal., 1997, 167, 66–76 CrossRef CAS.
- E. P. Reddy and S. R. Varma, J. Catal., 2004, 221, 93–101 CrossRef CAS.
- E. Ivanova, K. Hadjiivanov, S. Dzwigaj and M. Che, Microporous Mesoporous Mater., 2006, 89, 69–77 CrossRef CAS.
- Y.-M. Liu, W.-L. Feng, T.-C. Li, H.-Y. He, W.-L. Dai, W. Huang, Y. Cao and K.-N. Fan, J. Catal., 2006, 239, 125–136 CrossRef CAS.
- D. Santhanaraj, C. Suresh, A. Selvamani and K. Shanthi, New J. Chem., 2019, 43, 11554–11563 RSC.
- C. M. Chanquía, A. L. Cánepa, E. L. Winkler, E. Rodríguez-Castellón, S. G. Casuscelli and G. A. Eimer, Mater. Chem. Phys., 2016, 175, 172–179 CrossRef.
- B. Singh and A. Kumar, J. Mater. Chem. A, 2014, 2, 1930–1939 RSC.
- A. Held, J. Kowalska-Kuś, Y. Millot, F. Averseng, C. Calers, L. Valentin and S. Dzwigaj, J. Phys. Chem. C, 2018, 122, 18570–18582 CrossRef CAS.
- A. Held, J. Kowalska-Kuś and K. Nowińska, J. Catal., 2016, 336, 23–32 CrossRef CAS.
- X. Gao and I. E. Wachs, J. Phys. Chem. B, 2000, 104, 1261–1268 CrossRef CAS.
- E. Ananieva and A. Reitzmann, Chem. Eng. Sci., 2004, 59, 5509–5517 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD and UV-vis data. See DOI: 10.1039/d0ra00349b |
| This journal is © The Royal Society of Chemistry 2020 |