
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of surface silicon modification of H-beta zeolites for alkylation of benzene with 1-dodecene
Ruimin Li ,
Shiyong Xing,
Shuai Zhang and
Minghan Han*
,
Shiyong Xing,
Shuai Zhang and
Minghan Han*
Department of Chemical Engineering, Beijing Key Laboratory of Green Reaction Engineering and Technology, Tsinghua University, Beijing 100084, P. R. China. E-mail: hanmh@tsinghua.edu.cn
First published on 10th March 2020
Abstract
H-beta zeolites of 100–200 nm (named BEA-L) and 20–30 nm (named BEA-S) were treated by chemical liquid deposition (CLD) of tetraethyl orthosilicate (TEOS) to improve the selectivity of 2-phenyl linear alkylbenzene (2-LAB) from benzene alkylation with 1-dodecene. The results indicate that H-beta zeolite with a smaller crystal size has a longer lifetime due to shorter channels and less diffusion limitation. The deposited SiO2 layers passivated the external surface acid sites of the zeolite and made the pores narrower. BEA-L lost more external Brønsted acid sites than BEA-S with the same added amount of TEOS, which was due to the severe aggregation of BEA-S grains. This increased passivation gave BEA-L increased 2-LAB selectivity. And when the added amount of SiO2 was 7.20 wt% of the parent zeolite, the selectivity of 2-LAB over BEA-L significantly increased from 41.9% to 54.7% while that of BEA-S only increased by 2%.
1. Introduction
Linear alkylbenzene sulfonate (LAS) is a widely used anionic surfactant.1 Extensive use of detergents affects water quality and causes damage to environment, thus it is necessary to minimize these. Linear alkylbenzenes (LABs) with long chains (C10–C14) produced industrially by the alkylation of benzene with α-olefins are the key intermediates for the synthesis of LAS.2,3 Five isomers of LABs (2-, 3-, 4-, 5-, 6-LAB) can be obtained by the alkylation of benzene with 1-dodecene, via a carbenium ion reaction mechanism in the presence of acidic catalysts, as shown in Fig. 1. 1-LAB is unstable and hard to form on account of the Markovnikov rule. The molecular size of these five isomers is less than 0.529 nm, and 2-LAB is the smallest one.2 He et al. reported that the surface activity and biodegradability of LAS are greatly influenced by the position of the phenyl group in the LAB intermediate used.2 Among all LAB isomers, the 2-phenyl isomer (2-LAB) is the most biodegradable and environmentally favorable.4 Therefore, much research has been devoted to finding a suitable catalyst to attain a high yield of 2-LAB.5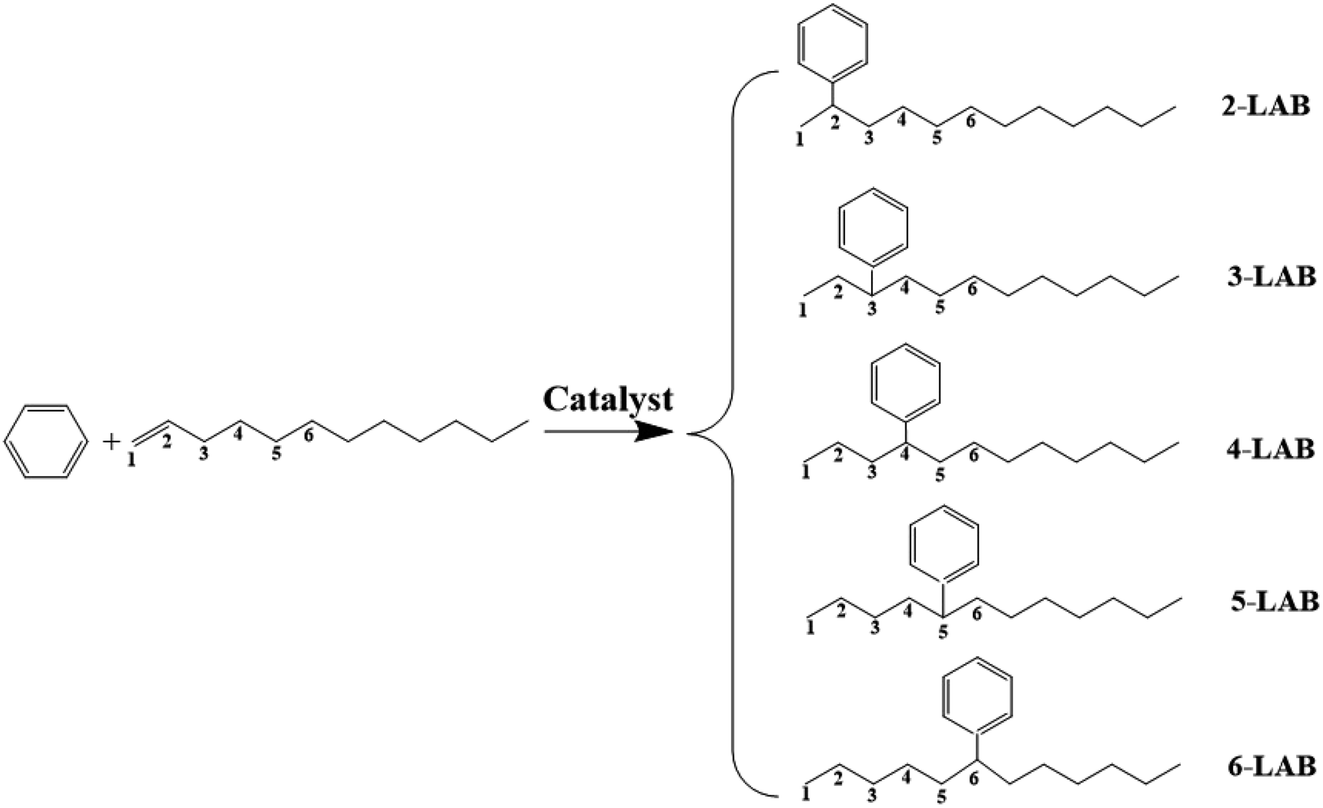 | ||
| Fig. 1 Alkylation reaction of benzene with 1-dodecene. | ||
AlCl3, as the early alkylation catalyst used in industrial development, was gradually replaced by a HF catalyst in 1960s.1 Despite a lot of weakness itself, HF has been the preferred catalyst worldwide for LAB synthesis until now. The weakness includes that (1) the selectivity of 2-LAB in the HF process is less than 20%; (2) the high corrosivity of these homogeneous catalysts causes the corrosion of equipment and environmental pollution; (3) special handling is required not only for the transportation and storage of these catalysts, but also for the separation of homogeneous catalyst, reactants, and products, leading to a high cost.3 Thus, it was a remarkable achievement when U.S. UOP Co. and Spain CEPSA Co. developed noncorrosive fluorided silica-alumina catalysts in 1992. The noncorrosive fluorided silica-alumina catalysts were much better than the homogeneous catalysts, but the selectivity of 2-LAB was only about 30%.1,6 In recent years, more environmentally friendly catalysts, including ionic liquids,7–9 clays,10–12 zirconia,10,11 heteropolyacids13,14 and zeolites,1,5,6,12,15–19 have been extensively studied. Among these catalysts, zeolites have attracted the most attention for the target of attaining a high selectivity of 2-LAB owing to their shape-selectivity catalysis.20 The best zeolites for the alkylation of benzene with long chain olefins contain 12-membered ring (12 MR) channels, and are Y (pore size of 7.4 Å),18 beta (pore size of 6.7 Å × 7.3 Å and 5.6 Å × 5.6 Å)5,19 and MOR (pore size of 6.5 Å × 7.0 Å).4,19,21
However, the rapid deactivation of the zeolite is the primary barrier to industrial application.22 Small crystal size zeolites have been researched to solve the deactivation problem of zeolites. Zeolites with small grains have large specific surface area, short channels and low diffusion limitation, which effectively increase anti-coking capability.23–26 Therefore, small grain zeolite has broad application prospects in the alkylation of benzene with long chain olefins.
In addition, with the stricter environmental protection requirements, increasing the yield of 2-LAB to make detergents more easily degraded is necessary. The acid sites of zeolites are distributed in the channels and external surface. The diffusion limitation of the channels makes it easier for 2-LAB to diffuse out than the other isomers, which facilitates the selectivity to 2-LAB. However, acid sites on the external surface have no diffusion restriction so that they need to be passivated. Chemical silicon deposition is a common method for passivating the acid sites on the external surface of zeolites.27–33 Shang et al. investigated HMCM-22 zeolite silylated with silicon tetrachloride (SiCl4), tetraethoxysilane (TEOS) and 8-hydroxyquinoline (C9H7NO) to passivate the external surface acidity, which sharply increased the selectivity of isobutene.34 Li et al. reported that silanization modification of HZSM-5 zeolites by CLD not only passivated the acidity of the external surface, but also gave narrower pore opening and promoted shape-selectivity.35 Therefore, the passivation of the external surface acid sites of the zeolite is also likely to improve the selectivity of 2-LAB.
H-beta zeolite is potentially the best catalyst for the alkylation of long chain olefins. β Zeolite has a three-dimensional twelve-membered ring (12-MR) ([100] and [010]: 6.7 Å × 7.3 Å, [001]: 5.6 Å × 5.6 Å),21 which is smaller than the size of TEOS (10.3 Å). TEOS cannot enter the internal channels of the zeolite, and can be used to modify the external surface. In this work, different crystal size H-beta zeolites modified by the CLD treatment with TEOS were used as the catalyst for the alkylation of benzene with 1-dodecene. This study focused on the effect of crystal size on the deactivation of catalysts and surface silicon modification on the selectivity of 2-LAB, to devise efficient catalysts for the benzene alkylation with long chain olefins in further studies.
2. Experimental
2.1. Chemicals and reagents
The reagents used were silica sol (40% SiO2, Qingdao Haiyang Chemical Co. Ltd, China), sodium aluminate (NaAlO2, AR, Macklin Chemical Co. Ltd, China), tetraethylammonium hydroxide (TEAOH, 35% in water, Hangzhou FineCHEM, Co. Ltd, China), sodium hydroxide (NaOH, AR, Sinopharm Chemical Reagent Co. Ltd, China), ammonium chloride (NH4Cl, 99.5%, Aladdin Chemical Reagent Co. Ltd, China), n-hexane (C6H14, 97%, Shanghai Titan technology Co. Ltd, China) and tetraethoxysilane (TEOS, Tianjin Heowns technology Co. Ltd, China).2.2. Preparation of catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3:TEAOH:Na2O:H2O = 1:0.03:0.2:0.07:30. The materials used were silica sol, sodium aluminate, TEAOH and sodium hydroxide, which were uniformly mixed in order. The mixture was aged for 2 h under vigorous stirring at room temperature to obtain a homogenous gel and then placed in a 100 ml Teflon-lined stainless steel autoclave. Two kinds of β zeolites with crystal size 100–200 nm and 20–30 nm were obtained by crystallizing at 145 °C for 100 h and 170 °C for 65 h, respectively. After crystallization, the solid product was separated using centrifuge, washed several times with deionized water, dried overnight at 80 °C and calcined in air at 550 °C for 4 h. The H-beta zeolite was obtained after three times ion-exchange with 1.0 mol L−1 NH4Cl aqueous solution at 80 °C for 2 h, drying overnight at 120 °C and calcining in air at 550 °C for 4 h. Smaller crystal size H-beta zeolite was named BEA-S. Another one was named BEA-L.
:Al2O3:TEAOH:Na2O:H2O = 1:0.03:0.2:0.07:30. The materials used were silica sol, sodium aluminate, TEAOH and sodium hydroxide, which were uniformly mixed in order. The mixture was aged for 2 h under vigorous stirring at room temperature to obtain a homogenous gel and then placed in a 100 ml Teflon-lined stainless steel autoclave. Two kinds of β zeolites with crystal size 100–200 nm and 20–30 nm were obtained by crystallizing at 145 °C for 100 h and 170 °C for 65 h, respectively. After crystallization, the solid product was separated using centrifuge, washed several times with deionized water, dried overnight at 80 °C and calcined in air at 550 °C for 4 h. The H-beta zeolite was obtained after three times ion-exchange with 1.0 mol L−1 NH4Cl aqueous solution at 80 °C for 2 h, drying overnight at 120 °C and calcining in air at 550 °C for 4 h. Smaller crystal size H-beta zeolite was named BEA-S. Another one was named BEA-L.The above method was used to modify zeolite BEA-S to obtain the catalysts 1.44% SiO2/g BEA-S, 4.32% SiO2/g BEA-S and 7.20% SiO2/g BEA-S.
2.3. Characterization of catalysts
The crystal structure of the catalysts was determined by a Bruker D8 Advance powder X-ray diffractometer with Cu Kα radiation. XRD patterns were obtained in the 2θ range of 5–50° recorded at 2° min−1. The Si/Al atomic ratio of the catalysts was measured by XRF analysis on ARL PERFORM X instrument from Thermo Fisher. TEM images of the catalysts were recorded by a JEOL JSM-7401F instrument. The scanning transmission electron microscopy (STEM) images were collected using a FEI Titan Cubed Themis G2 300 spherical aberration corrected microscope. The specific surface area and pore size distribution of the catalysts were measured with a Quantachrome Autosorb iQ and AsiQwin instrument using N2 adsorption isotherms at 77 K.The Brønsted and Lewis acid site analysis was carried out by IR spectroscopy with pyridine/2,4,6-trimethylpyridine adsorption between 1350 and 1750 cm−1 using a Nicolet 6700 model Fourier transform infrared spectrometer. The amount of Brønsted and Lewis acid sites was calculated by the following equation: C (pyridine on B or L acid sites) = IA(B,L)·πR2/(IMEC(B,L) × W); IA(B,L) = integrated absorbance of Brønsted acid sites at 1540 cm−1 or Lewis acid sites at 1455 cm−1; R = radius of catalyst disk (cm); IMEC(B,L) = integrated molar extinction coefficient of Brønsted acid sites at 1540 cm−1 (1.67 cm μmol−1) or Lewis acid sites at 1455 cm−1 (2.22 cm μmol−1); W = weight of disk (mg).36 2,4,6-Trimethylpyridine adsorbed on the external Brønsted acid sites gave a band at 1638 cm−1. NH3-TPD (temperature programmed desorption) experiments were performed on an Auto Chem II 2920 to characterize the acid amount of the catalysts. 0.2 g sample was pretreated in a continuous helium flow at 400 °C for 30 min and then cooled to 40 °C. Afterwards, the sample was saturated with NH3 for 45 min. The gas was switched to helium for 30 min to remove excess NH3 and make the baseline stable. The NH3-TPD profiles were recorded from 40 °C to 600 °C in flowing He with a heating rate of 10 °C min−1. The amount of total acid from NH3-TPD was calculated after calibrating the corresponding relation between acid quantity and peak area.
2.4. Catalyst reaction
The alkylation reaction of benzene with 1-dodecene was investigated with a fixed bed microreactor. The catalyst (2 g, 10–20 mesh) was loaded in a reaction tube (i.d. 10 mm, length 50 cm) and pretreated at 300 °C for 3 h under N2 flow. After that, the reactor was cooled down to 136 °C. Benzene and 1-dodecene, as reactants, were mixed in a molar ratio of 15:1 and the mixture was pumped into the reaction tube by a micro flow pump at a WHSV of 4 h−1. The pressure was maintained at 4 MPa. The products collected by a centrifuge tube every hour or two were analyzed by gas chromatography (GC) to get the conversion of 1-dodecene and selectivity of products (2-LAB, 3-LAB, 4-LAB, 5-LAB and 6-LAB). The conversion of 1-dodecene and selectivity of LAB isomers were defined as follows: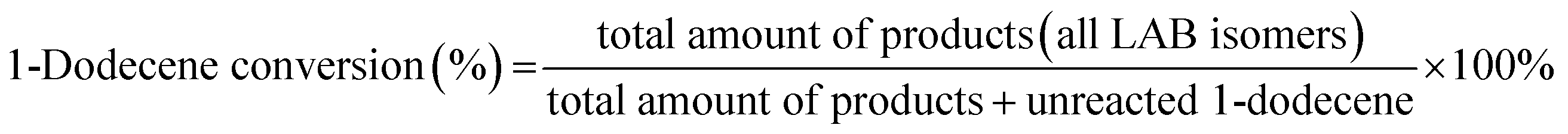
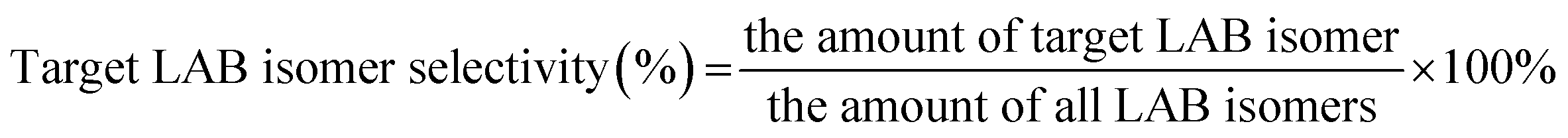
3. Results and discussion
3.1. Basic physicochemical properties characterization
Fig. 2 shows the XRD patterns of the BEA-L, BEA-S and theirs modified H-beta zeolites. Fig. 3 shows the TEM images of the BEA-L, BEA-S and theirs modified H-beta zeolites. The Si/Al atomic ratio of the catalysts analyzed by XRF is given in Table 1. As shown in Fig. 2, all modified zeolites exhibited the characteristic diffraction peaks of H-beta zeolite without the characteristic peaks of SiO2, indicating that the CLD treatment with TEOS did not change the crystal structure of the H-beta zeolite. This also shows that the SiO2 deposited during modification was highly dispersed and present in an amorphous phase. The Si/Al atomic ratio of BEA-L and BEA-S zeolites were 15.7 and 13.8, respectively. For the modified H-beta zeolites, the Si/Al atomic ratio increased significantly with increased SiO2 loading, clearly confirming that SiO2 was effectively deposited on parent zeolites. Moreover, it is worth mentioning that the surface silicon leads to the increase in surface Si/Al atomic ratio without changing in internal Si/Al atomic ratio. TEM images of BEA-L, 7.20% SiO2/g BEA-L, BEA-S and 7.20% SiO2/g BEA-S are presented in Fig. 3. A regular crystal structure and clear pores can be seen in the TEM images (Fig. 3(a) and (c)) of the parent zeolites. The crystal size of the H-beta zeolite named BEA-L was in the range of 100–200 nm while that of BEA-S was in the range of 20–30 nm which showed severe aggregation of the crystals. In the case of Fig. 3(b) and (d) of the modified zeolites, the crystal structure of H-beta zeolite was keep intact although the pore structure became blurred after modification, which was presumably because the external surface of the modified catalysts was covered with a layer of amorphous SiO2.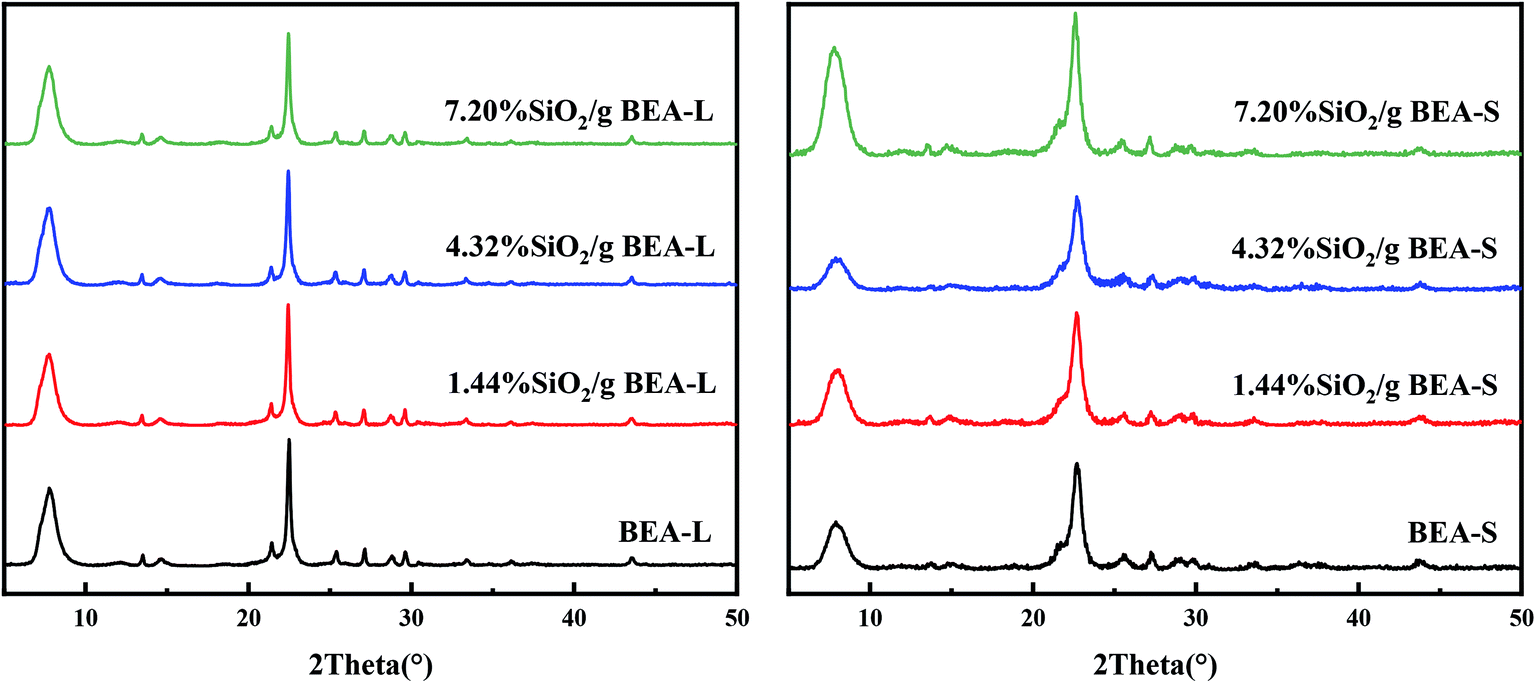 | ||
| Fig. 2 XRD patterns of BEA-L, BEA-S and theirs modified H-beta zeolites. | ||
 | ||
| Fig. 3 TEM images of BEA-L, BEA-S and theirs modified H-beta zeolites. (a) BEA-L, (b) 7.20% SiO2/g BEA-L, (c) BEA-S, (d) 7.20% SiO2/g BEA-S. | ||
| Samples | X% SiO2/g BEA-L | X% SiO2/g BEA-S | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SiO2 loading | 0 | 1.44 | 4.32 | 7.20 | 0 | 1.44 | 4.32 | 7.20 | |
| Si/Al | 15.7 | 16.2 | 16.6 | 16.9 | 13.8 | 14.2 | 14.7 | 15.1 | |
The adsorption isotherms of parent and modified H-beta zeolites are exhibited in Fig. 4. The N2 adsorption isotherms of BEA-L is typical I while that of BEA-S is typical IV, indicating that BEA-L has a mainly microporous structure and BEA-S has more mesoporous structure. Table 2 shows the textural parameters of parent and the modified catalysts. The surface areas of BEA-L and BEA-S were almost the same. As can be seen from Fig. 3, compared with BEA-L, BEA-S exhibit more agglomeration due to the small size of the crystals, which represents more intergranular pores. Therefore, BEA-S contains more mesopores and macropores. When the added amount of modifying SiO2 was 1.44 wt% of the parent zeolites, the micropore volume decreased, implying that silanization can block micropores because of the modified hydroxyl groups located on the external surface. The volume of the macropores was essentially unchanged because the thin SiO2 layer on the external surface of zeolites obtained from the modification did not change the intergranular pore size. As can be seen in Fig. 5,37 with the further increase of silylating reagent, a thicker SiO2 layer was formed, resulting in the decrease of intergranular pore size, which contributed to the increase in micropores. The STEM images of modified H-beta zeolite are shown in Fig. 6, presuming that the silica layer was presented in an amorphous phase and deposited on the external surface and intergranular pores, which is consistent with the above results.
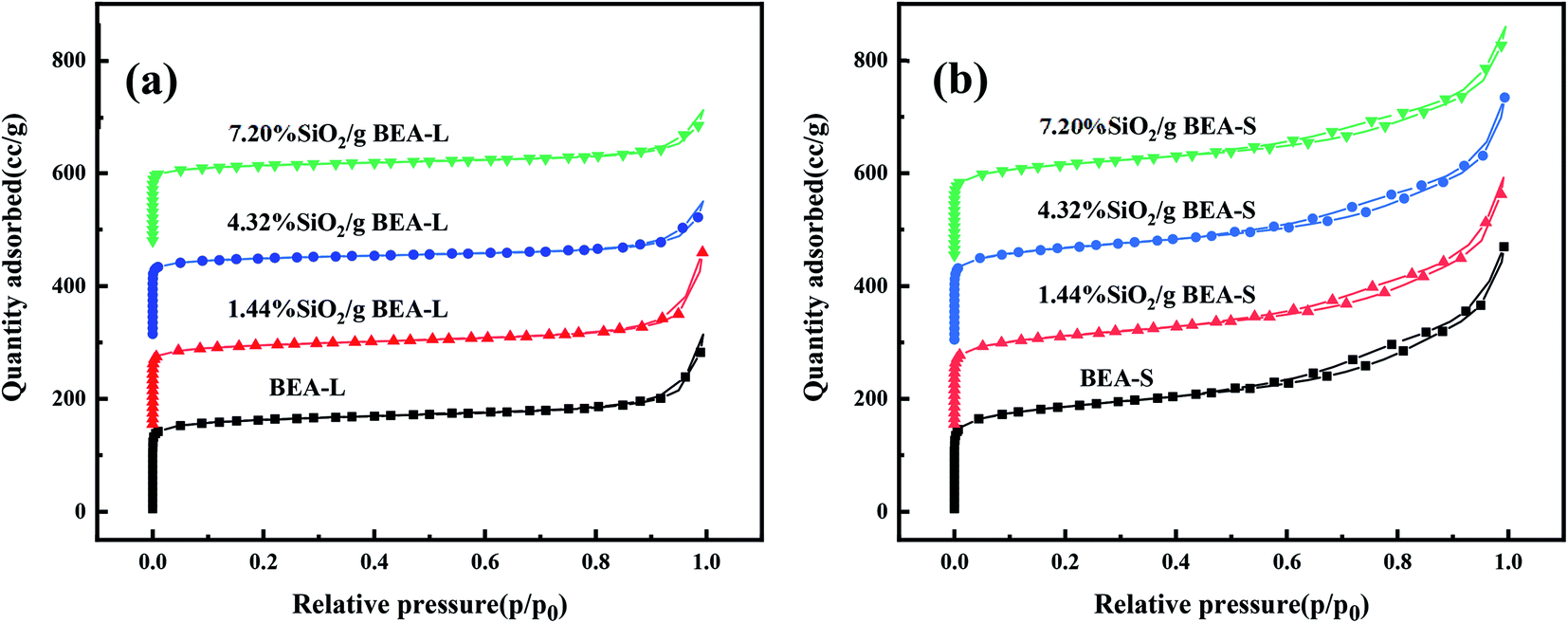 | ||
| Fig. 4 Nitrogen adsorption isotherms of BEA-L, BEA-S and theirs modified H-beta zeolites. (a) BEA-L and modified BEA-L zeolites; (b) BEA-S and modified BEA-S zeolites. | ||
| Samples | SBET (m2 g−1) | Vtol (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Vmacro (cm3 g−1) |
|---|---|---|---|---|---|
| a SBET: surface area by the BET method; Vtol, Vmicro, Vmeso, Vmacro: volume of total pore, micropore, mesopore, macropore by the DFT method. | |||||
| BEA-L | 636 | 0.354 | 0.192 | 0.043 | 0.119 |
| 1.44% SiO2/g BEA-L | 565 | 0.333 | 0.165 | 0.045 | 0.122 |
| 4.32% SiO2/g BEA-L | 547 | 0.265 | 0.160 | 0.039 | 0.066 |
| 7.20% SiO2/g BEA-L | 545 | 0.251 | 0.164 | 0.032 | 0.055 |
| BEA-S | 685 | 0.565 | 0.179 | 0.188 | 0.198 |
| 1.44% SiO2/g BEA-S | 595 | 0.509 | 0.149 | 0.159 | 0.201 |
| 4.32% SiO2/g BEA-S | 620 | 0.506 | 0.161 | 0.160 | 0.185 |
| 7.20% SiO2/g BEA-S | 612 | 0.480 | 0.165 | 0.183 | 0.132 |
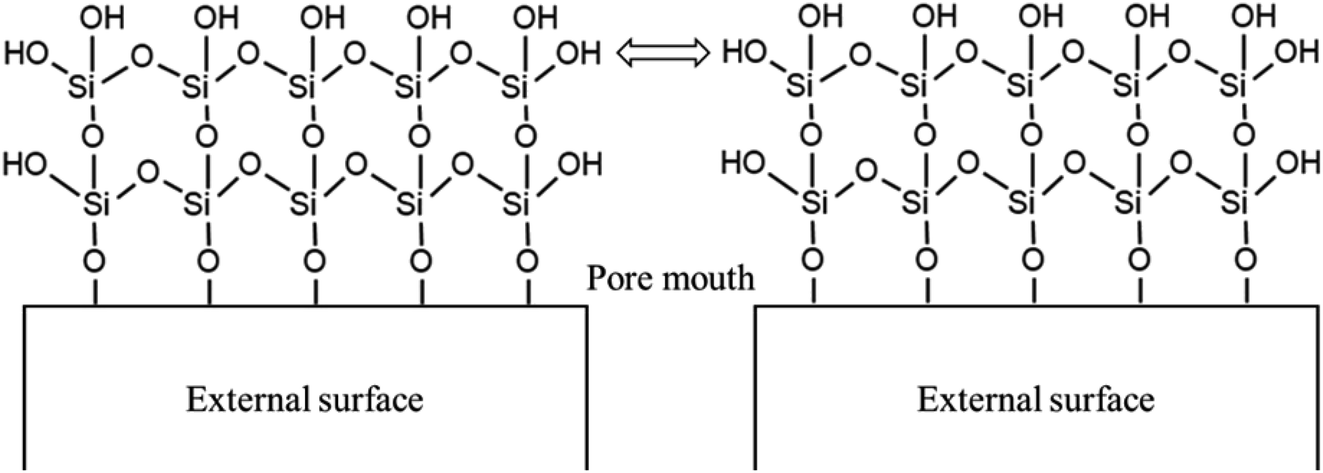 | ||
| Fig. 5 SiO2 layer on the external surface of the catalysts. | ||
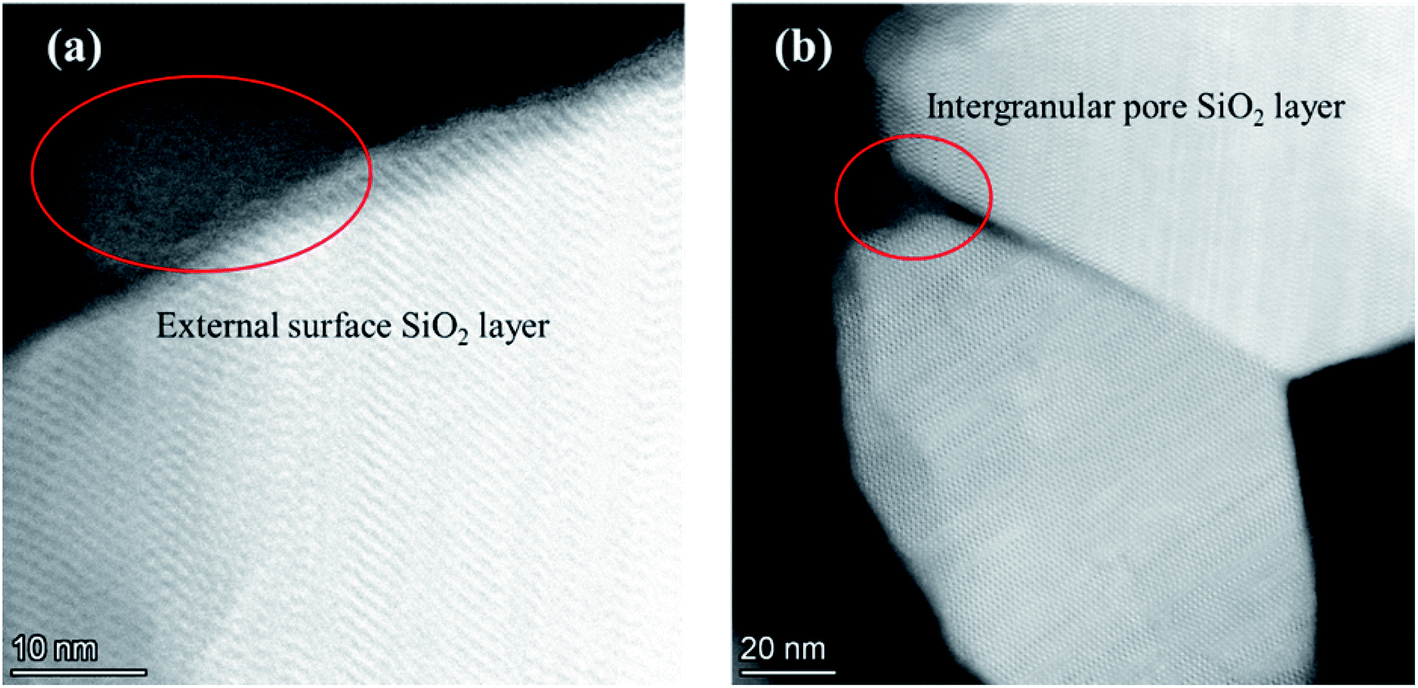 | ||
| Fig. 6 STEM images of modified H-beta zeolite. (a) SiO2 layer on the external surface; (b) SiO2 layer on the intergranular pore. | ||
3.2. Acidity characterization
Fig. 7 and 8 show the ammonia temperature programmed desorption (NH3-TPD) profiles and the FT-IR spectra of pyridine at different desorption temperatures of the BEA-L, BEA-S and theirs modifed zeolites. NH3-TPD is a method to analyze the total acid amount and Py-IR is used to distinguish Brønsted and Lewis acid sites. The amounts of acid determined by these two methods provide a reference to analyze the trend of acid after modification. The amounts of total acid sites, Brønsted and Lewis acid sites are listed in Table 3.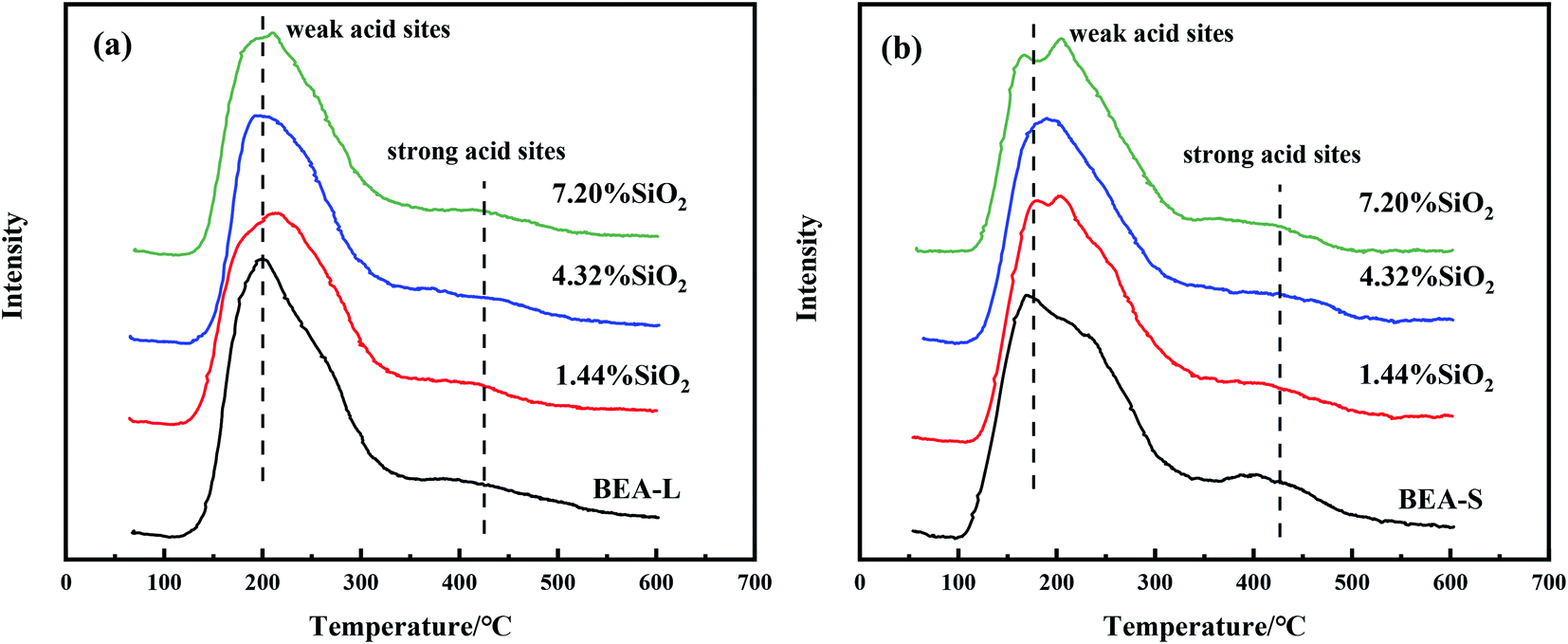 | ||
| Fig. 7 NH3-TPD profiles of BEA-L and modified BEA-L zeolites (a), BEA-S and modified BEA-S zeolites (b). | ||
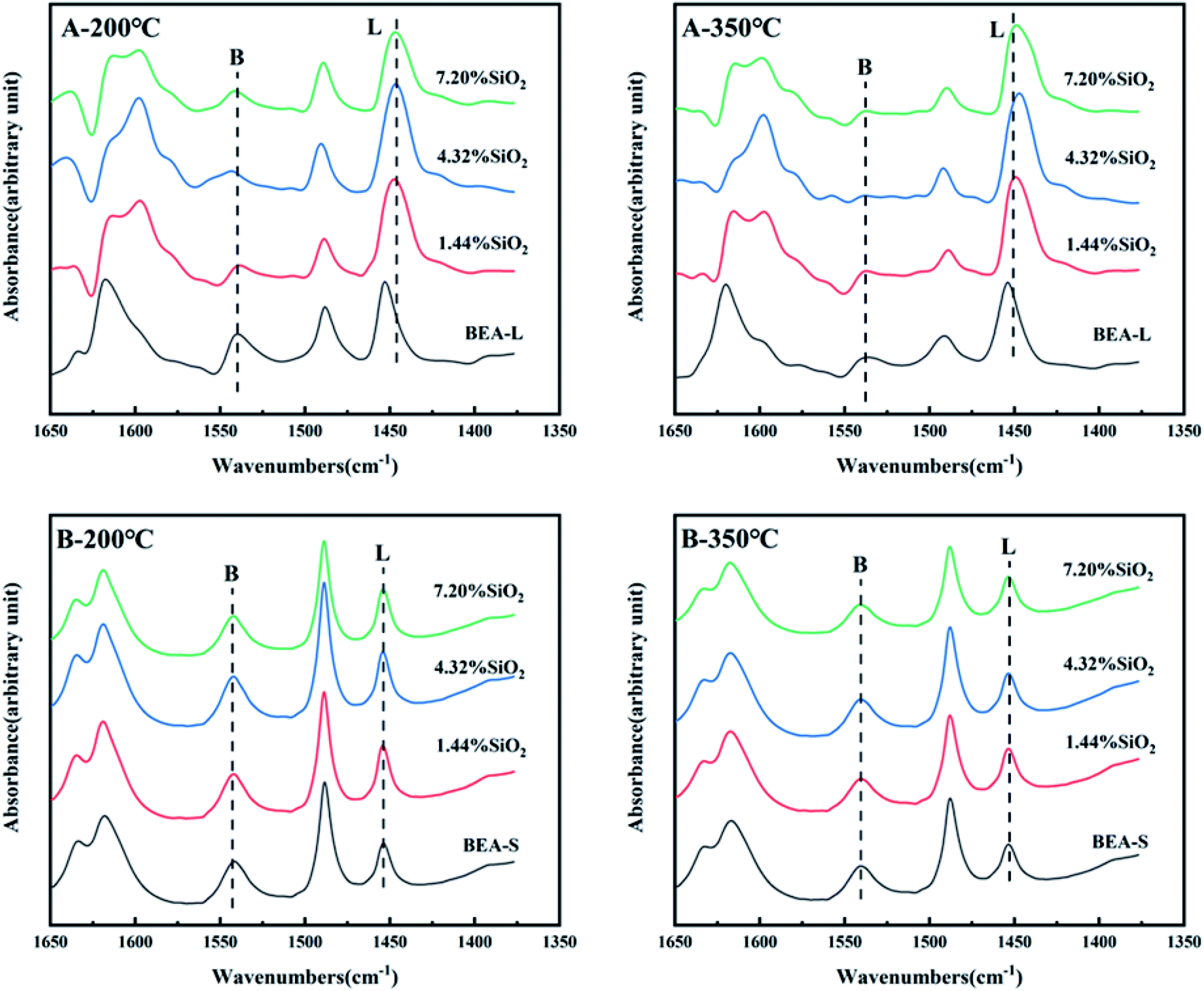 | ||
| Fig. 8 FT-IR spectra of pyridine adsorbed on the BEA-L, BEA-S and theirs modified zeolites under different desorption temperature (200 °C and 350 °C). A-200 °C, A-350 °C: BEA-L and modified BEA-L zeolites; B-200 °C, B-350 °C: BEA-S and modified BEA-S zeolites. | ||
| Samples | Total acid (μmol g−1) | Brønsted acid sites (μmol g−1) | Lewis acid sites (μmol g−1) | ||
|---|---|---|---|---|---|
| 200 °C | 350 °C | 200 °C | 350 °C | ||
| a (): Change in the amount of acid sites. |
|||||
| BEA-L | 197 | 159 | 73 | 204 | 234 |
| 1.44% SiO2/g BEA-L | 170(−14.4%) | 75(−52.8%) | 72(−1.4%) | 255(+25.0%) | 291(+24.3%) |
| 4.32% SiO2/g BEA-L | 167(−16.6%) | 75(−52.8%) | 14(−80.8%) | 304(+49.3%) | 318(+35.9%) |
| 7.20% SiO2/g BEA-L | 164(−16.9%) | 61(−61.0%) | 24(−67.1%) | 212(+3.7%) | 265(+13.2%) |
| BEA-S | 197 | 263 | 211 | 117 | 112 |
| 1.44% SiO2/g BEA-S | 182(−7.6%) | 254(−3.4%) | 183(−13.3%) | 131(+12.0%) | 109(−2.7%) |
| 4.32% SiO2/g BEA-S | 167(−14.8%) | 250(−4.9%) | 179(−15.2%) | 157(+34.2%) | 125(+11.6%) |
| 7.20% SiO2/g BEA-S | 160(−18.7%) | 237(−9.9%) | 172(−18.5%) | 119(+1.7%) | 95(−15.2%) |
As shown in the NH3-TPD curves (Fig. 7), two desorption peaks at 200 °C (weak acid sites) and 400 °C (strong acid sites) were observed on all samples. BEA-S exhibited stronger signal than BEA-L at the strong acid sites. The strong acids sites of BEA-L and BEA-S were both reduced after modification. NH3 can be adsorbed on the internal and external acid sites of zeolites on account of its small size (0.26 nm). Therefore, the adsorption peaks areas correspond to the total amount of acid content. As can be seen in Table 3, the acidity of BEA-L and BEA-S were significantly altered after surface silicon modification. The total acid amount of the parent zeolites decreased with increasing SiO2 loading used in the modification. 16.9% acid sites of BEA-L and 18.7% acid sites of BEA-S were lost when the added amount of modified SiO2 was 7.20 wt% of the parent zeolites. This can be attributed to the passivation of external surface acid sites and the blockage of pores after modification.
The FT-IR spectra of zeolites BEA-L and BEA-S with different loading of SiO2 were investigated in the range of 1650–1400 cm−1 and desorption temperature of pyridine of 200 °C and 350 °C. The adsorption bands at 1540 cm−1 and 1455 cm−1 correspond to Brønsted and Lewis acid sites of H-beta zeolites, respectively. The quantity of Brønsted and Lewis acid sites of the BEA-L, BEA-S and theirs modified zeolites are presented in Table 3. Brønsted acid sites of zeolites BEA-L and BEA-S decreased with increased SiO2 loading. Lewis acid sites of the modified zeolites increased first, then decreased. In the case of silylation modification, TEOS was hydrolyzed which resulted in triethoxy reacting with bridging hydroxyl groups on the zeolites.27 After calcination, new Al–O–Si bonds were formed, leading to the reduction of skeleton aluminum. The mechanism of SiO2 deposit is displayed in Fig. 9.37 As the SiO2 loading increased, more reactions between TEOS with bridging hydroxyl groups were involved, which led to a reduction of Brønsted acid sites. In addition, more Si–O bonds were formed on the surface of the zeolites, resulting in an increase in Lewis acid sites. When the silanization reagent was too much (7.20% SiO2/g BEA-L or BEA-S), blockage caused by reaction around the pore mouth led to the inability of pyridine molecules to enter the pore, resulting in the apparent decrease of Brønsted and Lewis acid sites. The results from the FT-IR spectra Brønsted and Lewis acid sites showed the trend of decreasing Brønsted acid sites, and increasing-decreasing Lewis acid sites. In addition, the results from desorption temperature 200 and 350 °C of Py-IR usually represent total acid and strong acid. As the desorption temperature increases, the BrØnsted and Lewis acid sites of BEA-S obviously decrease. And the conversion of BrØnsted acid sites to strong Lewis acid sites might be the reason for the increase in Lewis acid amount and decrease in BrØnsted acid amount of BEA-L as the desorption temperature increases from 200 °C to 350 °C.38,39
 | ||
| Fig. 9 Mechanism of SiO2 deposition. | ||
The FT-IR spectra with 2,4,6-trimethylpyridine adsorption was used to measure the external surface acid sites of the BEA-L, BEA-S and theirs modified zeolites since it has a larger size than the H-beta zeolite channels. As shown in Fig. 10, 2,4,6-trimethylpyridine adsorbed on external Brønsted acid sites gave the band at 1638 cm−1 while the infrared adsorption peak on Lewis acid sites was not exhibited due to steric constraints.21,40–44 After surface silicon modification, the quantity of external Brønsted acid sites was significantly reduced, indicating that the silylating reagent TEOS has neutralized hydroxyl groups located on the external surface of the zeolites. As can be seen in Fig. 3, BEA-S showed more crystal agglomeration compared with BEA-L. As shown in Table 2, when the added amount of modified SiO2 was 4.32 wt% and 7.20 wt% of the parent zeolites, micropores and mesopores of BEA-S was increased due to the decrease of intergranular pore size. It can be speculated that the silylating reagent TEOS was hard to access the interior of the agglomerated particles of BEA-S. The acid sites located on the external surface of the agglomerated particles were easier to passivate. Almost all external surface acid sites of BEA-L could be passivated by TEOS due to the large size of the crystals. Therefore, more external Brønsted acid sites of BEA-L were lost than that of BEA-S at the same added amount of TEOS, as shown in Fig. 11. Paul J. Kunkeler et al. also proposed that the restriction of surface silicon modification due to extremely rough surface and the destruction of silica layers during calcination were also the reasons for less efficient external acidity passivation of small size crystal zeolite.45
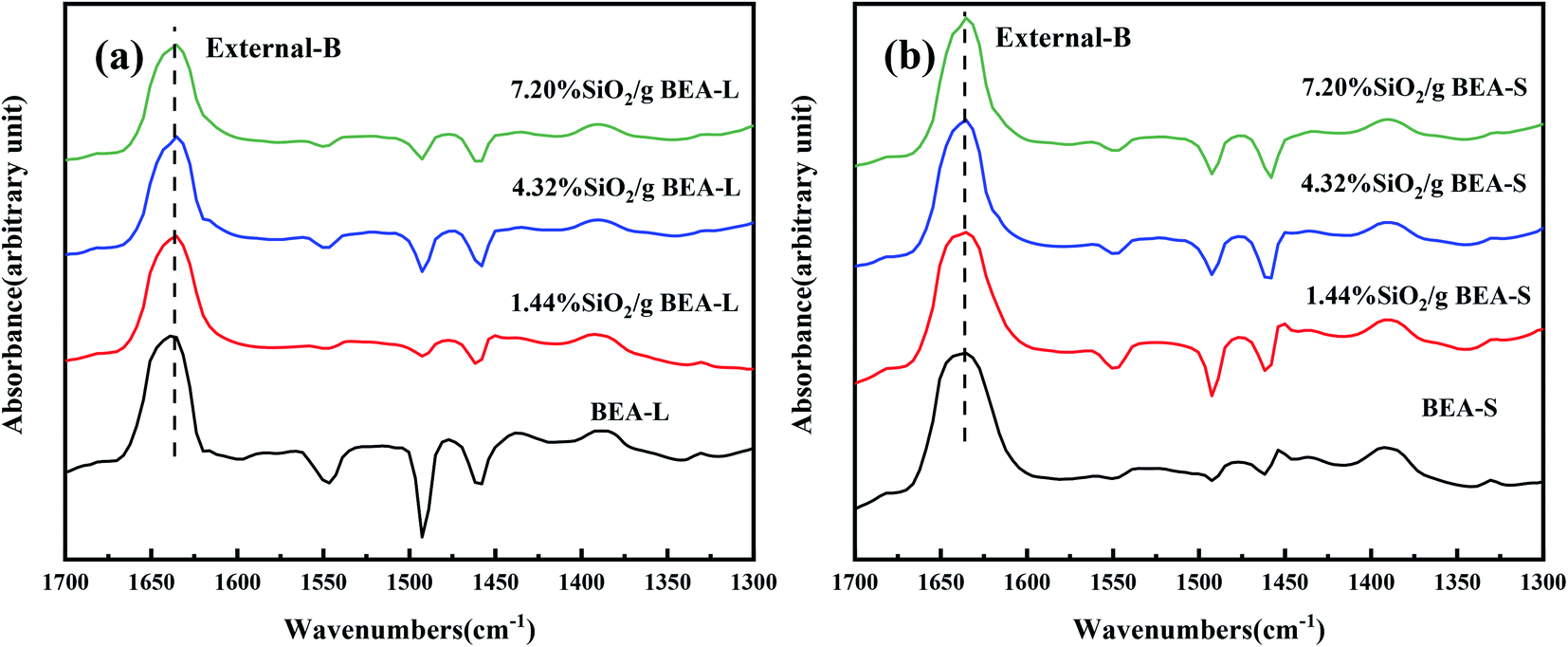 | ||
| Fig. 10 FT-IR spectra of 2,4,6-trimethylpyridine adsorbed on the BEA-L and modified BEA-L zeolites (a), BEA-S and modified BEA-S zeolites (b) at desorption temperature of 200 °C. | ||
 | ||
| Fig. 11 Relative external acid sites of the BEA-L, BEA-S and theirs modified H-beta zeolites with different loading of SiO2. | ||
3.3. Alkylation of benzene with 1-dodecene over H-beta catalysts
The alkylation reaction of benzene with 1-dodecene was studied over the BEA-L, BEA-S and theirs modified H-beta catalysts. Fig. 12 presents the influence of time on stream (TOS) on the conversion of 1-dodecene and selectivity of products. The conversion of 1-dodecene and selectivity of products before the activity of the catalysts declined are displayed in Fig. 13. 1-Dodecene conversion over zeolites BEA-L and BEA-S were ≥98%. As can be seen in Fig. 12, the lifetime of BEA-S could attain 65 h while that of BEA-L was 14 h, which can be attributed to the small crystal size and abundant Brønsted acid sites of BEA-S. Compared to BEA-L, the shorter channels of BEA-S make it easier for reactants and products to diffuse into and out of channels. Moreover, BEA-S was active for a longer time than BEA-L before it started deactivation due to more Brønsted acid sites.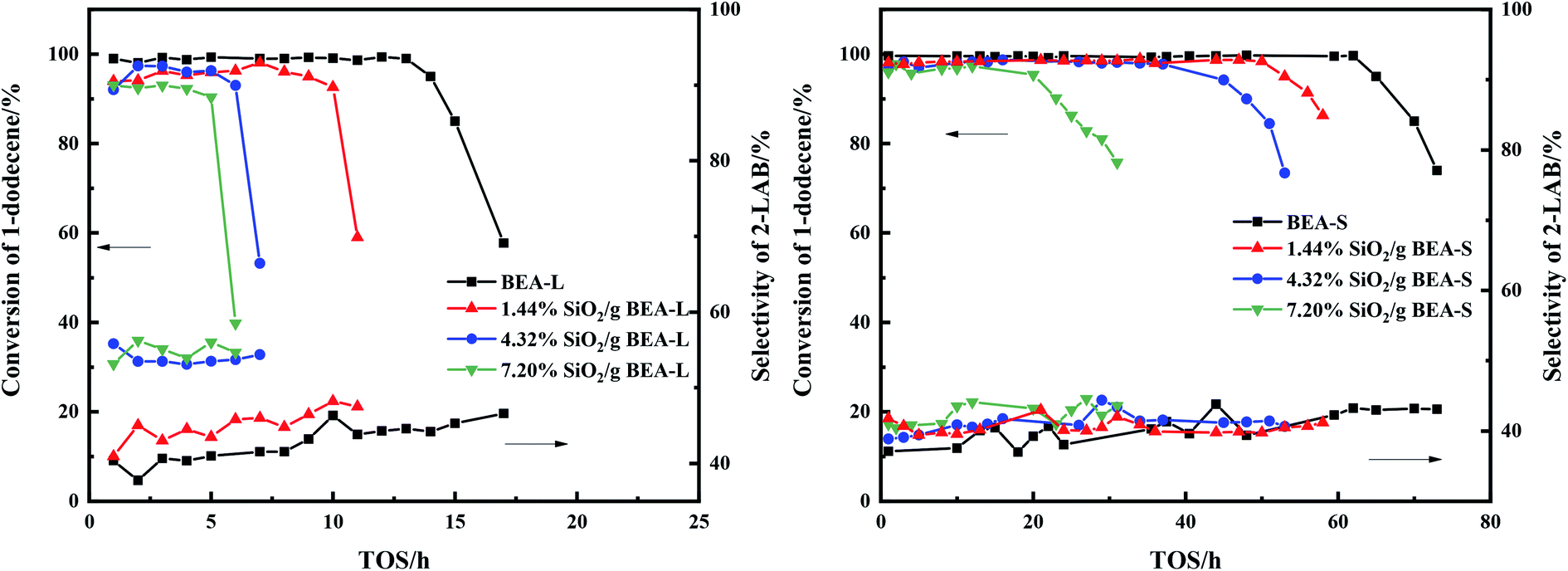 | ||
| Fig. 12 Influence of time on stream (TOS) on the catalytic conversion of 1-dodecene and selectivity of 2-LAB over BEA-L, BEA-S and theirs modified H-beta zeolites. | ||
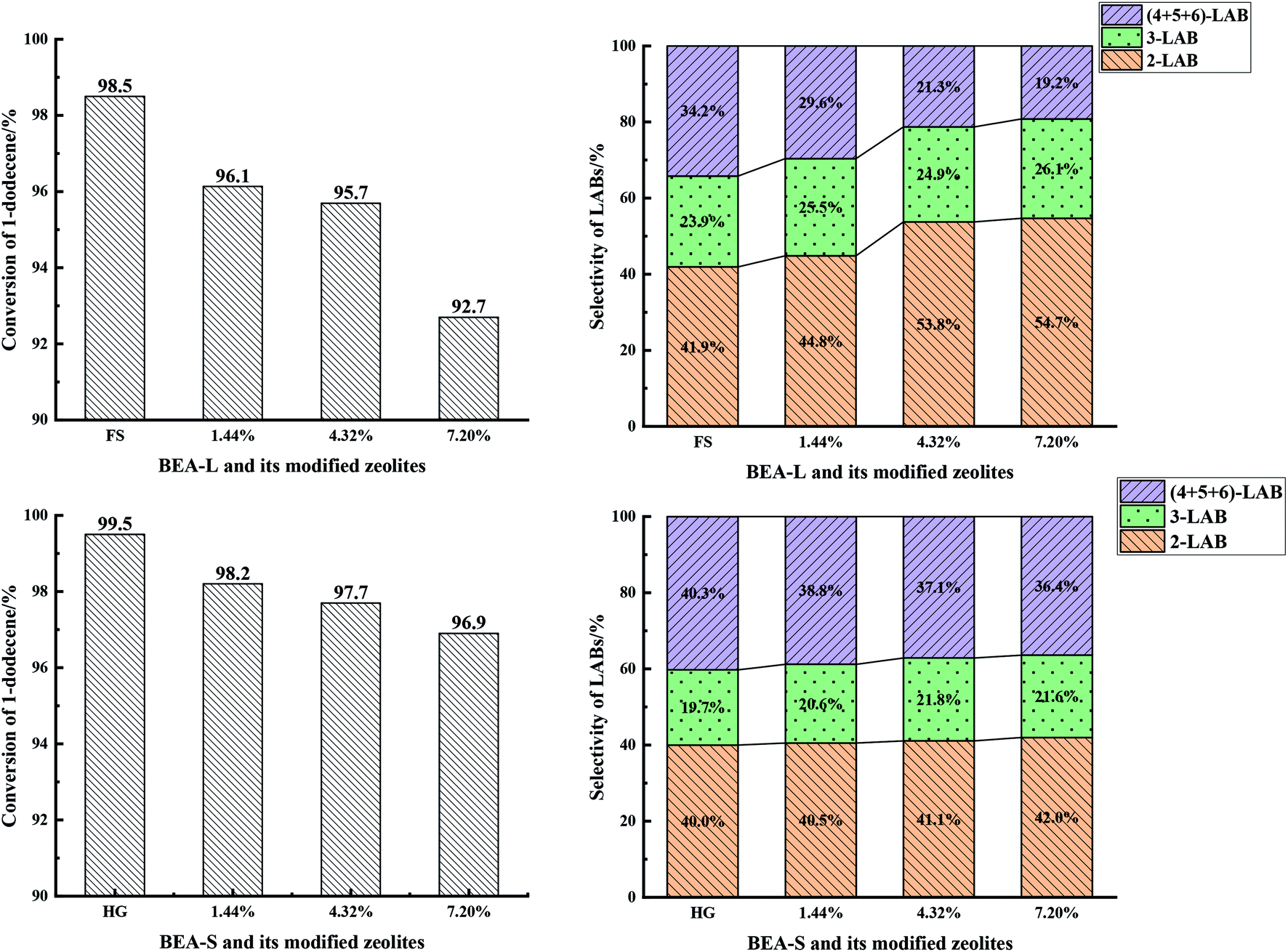 | ||
| Fig. 13 1-Dodecene conversion (left) and selectivity of products (2-LAB, 3-LAB, 4 + 5 + 6-LAB) (right) over BEA-L, BEA-S and theirs modified H-beta zeolites. | ||
With the increase of SiO2 loading, the conversion of 1-dodecene over modified zeolites BEA-L and BEA-S gradually dropped off and eventually reached 92.7% and 96.9% when the added amount of modifying SiO2 was 7.20 wt% of the BEA-L and BEA-S, respectively. Based on the acidity characterization results, the decrease of 1-dodecene conversion was attributed to the passivation of external surface acid sites, which led to the reduction of active sites. In addition, more Brønsted acid sites of BEA-L were lost than that of BEA-S, resulting in the faster decrease of conversion of BEA-L after modification. This also indicated that the alkylation of benzene with 1-dodecene mainly occurred on Brønsted acid sites.
Five dodecylbenzene isomers (except unstable 1-LAB) can be produced during the alkylation of benzene with 1-dodecene. The molecular size of 2-LAB is the smallest while 6-LAB is the largest. Diffusion limitation in the zeolite channels makes it more difficult for larger products to diffuse out. Among the isomers obtained from the alkylation of benzene with 1-dodecene, the selectivity of 2-LAB was the highest. The selectivity of 2-LAB over zeolites BEA-L and BEA-S are similar (41.9% and 40.0%, respectively) which was attributed to the same channel structure of H-beta zeolite. On BEA-L, as the SiO2 loading increased, the selectivity of products 2-LAB and 3-LAB increased, and that of products (4 + 5 + 6)-LAB markedly decreased. The selectivity to 2-LAB increased from 41.9% (BEA-L) to 54.7% (7.20% SiO2/g BEA-L), while that of (4 + 5 + 6)-LAB decreased from 34.2% (BEA-L zeolites) to 19.2% (7.20% SiO2/g BEA-L). However, after modification, the product selectivity of BEA-S was only slightly changed. When the added amount of modified SiO2 was 7.20 wt% of BEA-S, the selectivity of 2-LAB was only increased by 2% and that of (4 + 5 + 6)-LAB decreased by 3.9%.
The active sites of zeolites are located on the internal and external surfaces. External surface acid sites have no shape selective catalysis. The passivation of the external surface area by CLD treatment with TEOS led to a decrease in nonselective acid sites, resulting in the increase of 2-LAB selectivity. The characterization studies showed that more external Brønsted acid sites of BEA-L were lost than that of BEA-S at same added amount of TEOS. However, the main effect was probably that the modification procedure has narrowed the pore mouth and restricted the diffusion of larger products, giving rise to the increase of 2-LAB selectivity. It is hard for the silylating reagent TEOS to access the interior of agglomerated particles of BEA-S. Orifices of the crystals inside agglomerated particles have nearly no change. As a result, the selectivity of 2-LAB from modified of BEA-S increased only slightly. It was easier to make the orifices of BEA-L narrower, leading to the significant increase in selectivity of 2-LAB.
The lifetimes of the zeolites decreased sharply with increased SiO2 loading, as shown in Fig. 12. The lifetime of BEA-L decreased from 14 h (BEA-L) to 5 h (7.20% SiO2/g BEA-L) while that of BEA-S decreased from 65 h (BEA-S) to 20 h (7.20% SiO2/g BEA-S). One reason is that passivation of external surface acidity led to fewer active sites for the alkylation. Another reason is the narrowing of orifice and pores, such that reactants cannot easily enter the pore to react on the active sites, and products are difficult to diffuse out and stay in the channels.
For benzene alkylation with long chain olefins, the rapid deactivation of H-beta zeolites is an obstacle to the industrial application. The main reason for inactivation is that the shape selective catalysis of the zeolites causes the accumulation of macromolecular products in the channels and covers the acid sites.46 From the above studies, it can be seen that the small grain zeolites have short pore channels so that the products are easier to diffused out, which can effectively increase anti-coking capability and obtain longer lifetime. Small grain zeolite is the trend of solid acid catalyst industrialization in the future. Silanization modification, which can passivate external surface acid sites and narrow the pores, is a feasible method to improve 2-LAB selectivity, thus relieving the increasing pressure of environmental protection. However, this method has no obvious effect on small grain zeolite because the silylating reagent cannot access the interior of the agglomerated particles. Silanization reagent can be added during the synthesis process of small grain zeolite, which can modify the external surface acidity and orifice of the crystallites, so as to obtain catalysts with high 2-LAB selectivity. In addition, silanization modification causes the faster deactivation of zeolites, which can be solved by high-temperature benzene washing and coke burning regeneration.46
4. Conclusions
Two H-beta zeolites with different crystal size were modified with different amounts of TEOS by treatment with CLD, and were used as the catalyst for the alkylation of benzene with 1-dodecene. The crystal size of the H-beta zeolite named BEA-L was 100–200 nm while that of BEA-S was 20–30 nm, which led to severe aggregation of the crystallites. H-beta zeolites with a smaller crystal size has a longer lifetime due to the shorter channels and less diffusion limitation. After silanization modification, the external surface acid sites of the H-beta zeolites were passivated and the pore was narrower. When the added amount of modifying SiO2 was 7.20 wt% of the parent zeolites, the selectivity of 2-LAB over BEA-L significantly increased from 41.9% to 54.7% while that of BEA-S only increased by 2%. This is because the silylating reagent TEOS is difficult to access the interior of the agglomerated particles of BEA-S. According to our study, the silanization modification for external surfaces is not a suitable method for improving the 2-LAB selectivity of small-grain β zeolites. It is a feasible method to add silanization reagent during synthesis procedure of β zeolite to passivate the acidity of the external surface of crystallites, which will be our research topic in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the discussion of the experimental results with our group members.References
- J.-J. Wang and T.-C. Tsai, Catal. Today, 2017, 298, 109–116 CrossRef CAS.
- Y. He, X. Zhan, D. Cheng and F. Cheng, Chin. J. Chem. Eng., 2017, 25, 1533–1538 CrossRef CAS.
- N. Toutounchian, A. Ali, M. H. Majid, F. B. Fatemeh and F. Deymeh, Res. Chem. Intermed., 2015, 42, 3283–3301 CrossRef.
- A. Aitani, J. B. Wang, I. Wang, S. Al-Khattaf and T.-C. Tsai, Catal. Surv. Asia, 2014, 18, 1–12 CrossRef CAS.
- M. Horňáček, P. Hudec, A. Smiešková and T. Jakubĺk, React. Kinet., Mech. Catal., 2010, 99, 431–437 Search PubMed.
- L. TIAN, Y. LI and B. CHEN, Chin. J. Catal., 2008, 29, 889–894 CrossRef CAS.
- C. DeCastro, E. Sauvage and M. H. Valkenberg, J. Catal., 2000, 196, 86–94 CrossRef CAS.
- G. P. QI, X. W. Sun and S. Q. ZHAO, Sci. China: Chem., 2010, 53, 1102–1107 CrossRef CAS.
- H. Xin, Q. Wu, D. Wang and Y. Jin, Appl. Catal., A, 2005, 292, 354–361 CrossRef CAS.
- N. S. Doshi, Org. Process Res. Dev., 2002, 6, 263–272 CrossRef.
- S. R. Guerra, L. M. O. C. Merat and R. A. S. San Gil, Catal. Today, 2008, 133–135, 223–230 CrossRef CAS.
- H. Faghihian and M. H. Mohammadi, C. R. Chim., 2012, 15, 962–968 CrossRef CAS.
- J. Zhang, B. Chen, C. Li, Z. Zhu, L. Wen and E. Min, Appl. Catal., A, 2003, 249, 27–34 CrossRef CAS.
- J. Zhang, Z. Zhu, C. Li, L. Wen and E. Min, J. Mol. Catal. A: Chem., 2003, 198, 359–367 CrossRef CAS.
- J.-S. Lin, J.-J. Wang, J. Wang, I. Wang, R. J. Balasamy, A. Aitani and S. Al-Khattaf, J. Catal., 2013, 300, 81–90 CrossRef CAS.
- B. Wang, C. W. Lee and T.-X. Cai, Catal. Lett., 2001, 76, 99–103 CrossRef CAS.
- X.-D. Yuan and J.-N. Park, Korean J. Chem. Eng., 2002, 19, 607–610 CrossRef CAS.
- B. Wang, C. W. Lee and T.-X. Cai, Bull. Korean Chem. Soc., 2001, 22, 1056–1058 CAS.
- W. Aslam, M. A. B. Siddiqui, B. R. Jermy, A. Aitani and J. Cejka, Catal. Today, 2014, 227, 187–197 CrossRef CAS.
- W. Mamo, Y. Awoke and Y. Chebude, Bull. Chem. Soc. Ethiop., 2015, 29, 95–103 CrossRef CAS.
- M. Fan, T. Si and P. Zhang, J. Am. Oil Chem. Soc., 2018, 95, 1357–1365 CrossRef CAS.
- H.-T. Yen, J.-J. Wang, S.-H. Siao, S. H. Cha, S. B. Hong, S. S. Al-Khattaf and I. Wang, Catal. Sci. Technol., 2016, 6, 2715–2724 RSC.
- H. Mochizuki, T. Yokoi, H. Imai, R. Watanabe, S. Namba and J. N. Kondo, Microporous Mesoporous Mater., 2011, 145, 165–171 CrossRef CAS.
- K. Müller, B. Yilmaz, R. M. Jacubinas and U. Müller, J. Am. Chem. Soc., 2011, 133, 5284–5295 CrossRef PubMed.
- N. Taufiqurrahmi, A. R. Mohamed and S. Bhatia, J. Nanopart. Res., 2011, 13, 3177–3189 CrossRef CAS.
- U. Wilkenhöner, G. Langhendries, F. Laar, G. V. Baron, D. W. Gammon and P. A. Jacobs, J. Catal., 2001, 203, 201–212 CrossRef.
- N. Zhang, Y. Sang and D. Yuan, Ind. Catal., 2017, 25, 5–12 Search PubMed.
- J. Qian, G. Xiong, J. Liu and C. Liu, Ind. Eng. Chem. Res., 2019, 58, 9006–9016 CrossRef CAS.
- S. Akiyama, H. Mochizuki, H. Yamazaki, T. Yokoi and T. Tatsumi, Mol. Catal., 2017, 433, 48–54 CrossRef CAS.
- U. Khalil, O. Muraza and A. Al-Amer, Adv. Powder Technol., 2016, 27, 1404–1410 CrossRef CAS.
- E. Kiliç and S. Yilmaz, React. Kinet., Mech. Catal., 2014, 112, 283–294 CrossRef.
- D. Vanvu, M. Miyamoto, Y. Egashira and K. Ueyama, J. Catal., 2006, 243, 389–394 CrossRef.
- C. Zhang, X. Guo, C. Song, S. Zhao and X. Wang, Catal. Today, 2010, 149, 196–201 CrossRef CAS.
- Y. Shang, P. Yang, M. Jia and T. Wu, Catal. Commun., 2008, 9, 907–912 CrossRef CAS.
- J. Li, Y. Wang, W. Jia, Z. Xi, H. Chen and Z. Hu, J. Energy Chem., 2014, 23, 771–780 CrossRef.
- C. A. EMEIS, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- S. G. Hegde, A. A. Behlekar and B. S. Rao, Catal. Today, 1999, 268, 201–209 Search PubMed.
- J. N. Kondo, R. Nishitani, E. Yoda, T. Yokoi, T. Tatsumi and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 11576–11586 RSC.
- X. Liu, S. Chen, T. Zhao, D. Li, X. Bai and L. Li, Ind. Catal., 2015, 23, 817–820 CAS.
- B.-T. L. Bleken, L. Mino, F. Giordanino, P. Beato, S. Svelle and K. P. Lillerud, Phys. Chem. Chem. Phys., 2013, 15, 13363–13370 RSC.
- A. Erigoni, S. H. Newland, G. Paul, L. Marchese and R. Raja, ChemCatChem, 2016, 8, 3161–3169 CrossRef CAS.
- M. S. Holm, S. Svella, F. Joensen, P. Beato, C. H. Christensen and S. Bordiga, Appl. Catal., A, 2009, 356, 23–30 CrossRef CAS.
- S. Inagaki, K. Kamino, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2007, 318, 22–27 CrossRef CAS.
- S. Inagaki, S. Park, H. Yamazaki and J. N. Kondo, Microporous Mesoporous Mater., 2018, 272, 16–23 CrossRef CAS.
- P. J. Kunkeler, D. Moeskops and H. v. Bekkum, Microporous Mater., 1997, 11, 313–323 CrossRef CAS.
- Z. Cui, C. Xu and W. Chen, Appl. Catal., A, 2003, 238, 99–107 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |