
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBroadband emission from zero-dimensional Cs4PbI6 perovskite nanocrystals†
Saikat Bhaumik *ab,
Annalisa Brunoa and
Subodh Mhaisalkarac
*ab,
Annalisa Brunoa and
Subodh Mhaisalkarac
aEnergy Research Institute@NTU (ERI@N), Research Techno Plaza, X-Frontier Block, Level 5, Singapore 637553
bInstitute of Chemical Technology-IndianOil Odisha Campus, Mouza-Samantapuri, Bhubaneswar, Odisha, India 751013. E-mail: s.bhaumik@iocb.ictmumbai.edu.in
cSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798
First published on 15th April 2020
Abstract
To overcome the drawbacks in three-dimensional (3D) perovskites, such as instability, surface hydration, and ion migration, recently researchers have focused on comparatively stable lower-dimensional perovskite derivatives. All-inorganic zero-dimensional (0D) perovskites (e.g., Cs4PbX6; X = Cl−, Br−, I−) can be evolved as a high performing material due to their larger exciton binding energy and better structural stability. The clear understanding of carrier recombination process in 0D perovskites is very important for better exploitation in light-emitting devices. In this work, we comprehensively studied the light emission process in 0D Cs4PbI6 nanocrystals (NCs) and interestingly we observe intense white light emission at low temperatures. According to our experimental observations, we conclude that the white light emission contains an intrinsic exciton emission at 2.95 eV along with a broadband emission covering from 1.77 eV to 2.6 eV. We also confirm that the broadband emission is related to the carrier recombination of both self-trapped excitons (STE) and defect state trapped excitons. Our investigations reveal the carrier recombination processes in Cs4PbI6 NCs and provide experimental guidelines for the potential application of white light generation.
Introduction
In recent years, all inorganic metal-halide perovskites have attracted much attention due to their unique properties, like, high absorption coefficient,1 tunable band-gap,2,3 high photoluminescence quantum yield (PLQY),4 low-threshold stimulated emission,5 the strong nonlinear absorption,6 single photon emission,7 and narrow emission linewidth.2 These perovskite materials are present in various fields of applications, such as solar cells,8–10 light-emitting diodes (LEDs),11 lasing gain materials,5 low-cost nonlinear absorbers,6 and single-photon sources.12 Among these perovskite materials, three-dimensional (3D) all-inorganic lead halide perovskites (ILHP) CsPbX3 (X = Cl−, Br−, or I−) composed of an extended network of corner-sharing [PbX6]4− octahedra with Cs+ ions residing in the cavities of this network, have been extensively explored.1,8–10,13–17 However, under operating conditions, 3D ILHPs suffer from crystal instability, ion migration and surface degradation, hence their reduced-dimensionality counterparts are being hugely investigated.18,19 Recently, zero-dimensional (0D) Cs4PbX6 nanocrystals (NCs) were successfully synthesized by hot-injection colloidal synthesis approach under Cs-rich conditions,20,21 and the resulted nanocrystalline crystal structure formed by isolated octahedra [PbX6]4− separated by Cs+ ions in all three directions.21 This specific structure of 0D perovskites is expected to result in some novel optical and electronic properties. Recent studies on Cs4PbBr6 NCs show some special properties, such as, intrinsic Pb2+ ion emission,22 and small polaron formation on photoexcitation.23 0D Cs4PbI6 perovskite was first reported around two decades ago,24,25 however the photoluminescence (PL) recombination process is still unclear. It's necessary to have further characterizations such that we can unveil some hidden interesting properties.In this work, we have studied the PL recombination processes in 0D Cs4PbI6 NCs synthesized via hot injection method.26 We have observed that the Cs4PbI6 NCs possess a sharp intrinsic exciton absorption peak at 3.38 eV with an excitonic binding energy of around 490 meV. More importantly, we found an extremely broad visible light emission that covers from 1.77 eV to 3.26 eV at low temperature. To get rid of the thermal effect and find out the origin of such broadband emission, we have performed the low-temperature PL measurement excited at variable excitation energies. We observed that the broadband emission of Cs4PbI6 NCs contains three main emission peaks. With comprehensive experiments and careful analysis, we conclude that the emission peak at 2.95 eV originates from the exciton recombination process, while the other two peaks at 2.36 eV and 2.16 eV evolve from recombination processes of self-trapped excitons (STEs) and defect-trapped excitons. The time-resolved PL (TRPL) results reveal that the emission originates from STE process decays faster for shorter wavelength/higher energy photon recombination. This phenomenon indicates that there are a series of emissive STE states and the excited STE states, decay from higher energy states to lower energy states during the emission process.
Results and discussion
Sample preparation and structural characterization
We have synthesized Cs4PbI6 NCs by the hot injection method (see details in the ESI†).26 Firstly, lead oxide (PbO) was dissolved in 1-octadecene (ODE) with oleic acid (OA) and oleylamine (OAm) at 150 °C. Then the solution was cooled to 80 °C, and the tetrabutylammonium iodide (TBAI) was injected. The reaction was kept for ten minutes, and then Cs-oleate precursor, i.e., cesium carbonate (Cs2CO3) dissolved in ODE with OA was injected. The reaction was kept for another five minutes and then stopped by quenching in an ice-water bath. Finally, the NCs were collected through centrifugation.To confirm the crystallinity of Cs4PbI6 NCs, we have performed powder X-ray diffraction (XRD) measurement. More specifically, we prepared a thick film by drop-casting the NCs sample on a glass substrate and dried for the measurement. As shown in Fig. 1a, the experimental XRD pattern matches well with the reported crystal structure as represented in Fig. 1b having lattice parameters of a = b = 14.6019 Å, c = 18.3678 Å.21 The high-resolution transmission electron microscope (HRTEM) image of these NCs is presented in Fig. 1c, which demonstrates the spherical shape with size distribution in the range of 10 ± 3 nm.
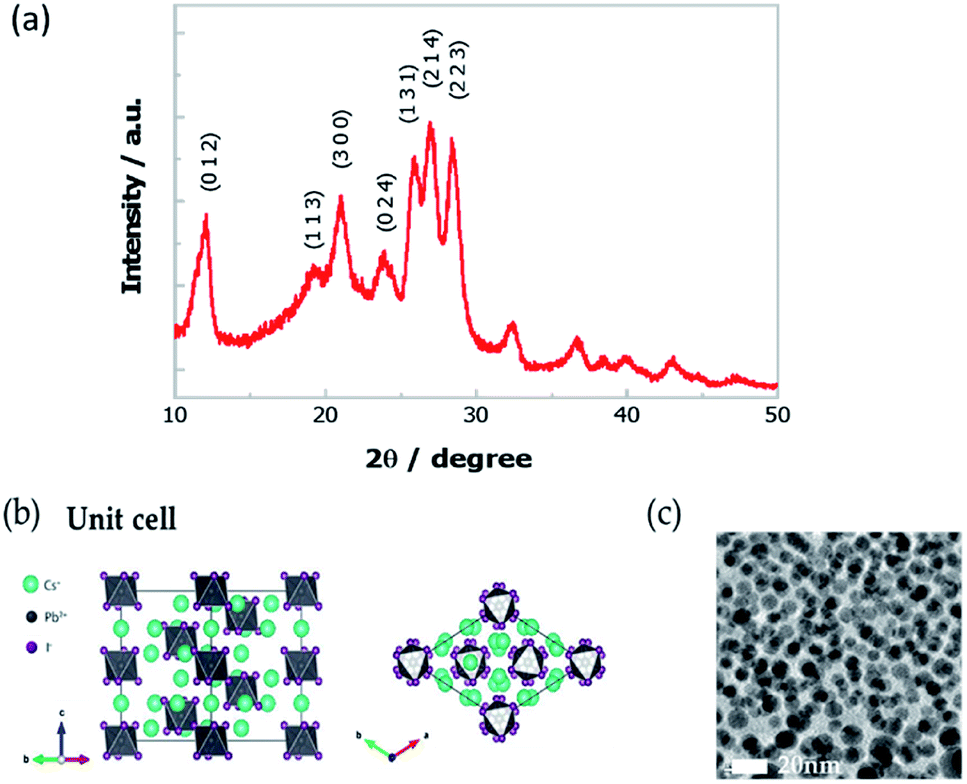 | ||
Fig. 1 (a) XRD pattern of the Cs4PbI6 NCs film. (b) The crystal structure of Cs4PbI6 (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, a = b = 14.6019 Å, c = 18.3678 Å). (c) The HRTEM image of Cs4PbI6 NCs. c, a = b = 14.6019 Å, c = 18.3678 Å). (c) The HRTEM image of Cs4PbI6 NCs. | ||
Low-temperature absorption and PL spectra
The absorption spectra of Cs4PbI6 NCs film at 298 K and 78 K, is displayed in Fig. 2a. The absorption spectrum measured at 298 K shows a strong excitonic absorption peak at 3.36 eV. While decreasing the temperature from 298 K to 78 K, the excitonic absorption peak slightly shifted to 3.38 eV (Fig. S1†) and the spectra become sharper. Two additional higher energy absorption bands (in the range of 4 eV and 4.5 eV) at low temperature is appeared due to reduction of thermal energy. These higher energy bands originate from the crystal-field splitting of the conduction band due to deviation from Oh symmetry of [PbI6]4− octahedra.24 The Tauc plot at 78 K (Fig. S2†) results in the band-gap energy around 3.87 eV and the corresponding exciton binding energy of 0.49 eV. The high exciton binding energy of the NCs indicates the strong confinement effect of excitons that originates from the isolated octahedra.27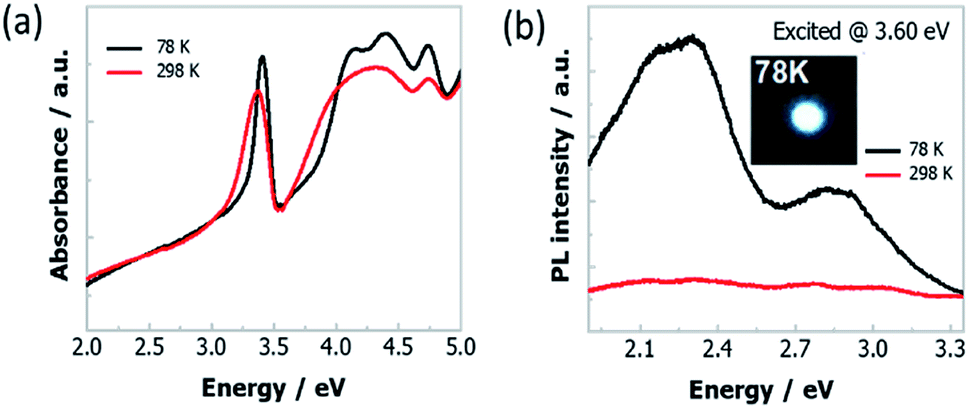 | ||
| Fig. 2 (a) Absorption and (b) PL spectra of Cs4PbI6 NCs films at 298 K and 78 K. The insert of (b) is the optical image of the white light emission from these NCs film at 78 K. | ||
We have also conducted temperature-dependent PL measurement of Cs4PbI6 NCs film that is excited at various excitation energies (2.95 eV, 3.14 eV, 3.38 eV, 3.60 eV) and the corresponding graphical representation is presented in Fig. S3.† Among them, two representative PL spectra excited at 3.60 eV are shown in Fig. 2b. As a comparison of the strong narrow high energy absorption peak of Cs4PbI6 NCs, the PL spectra covering the full visible energy range (1.9 eV to 3.2 eV) compose of two broad bands peaked at 2.27 eV and 2.95 eV, respectively. Moreover, the PL spectrum changes drastically upon cooling and the emission peak intensity increases around thirty times compared to the PL intensity at room temperature. Strong white light emission appears at 78 K is shown in the inset of Fig. 2b and the 1931 CIE chromaticity map is in Fig. S4.† Besides, the relative PL intensity of the emission band at 2.95 eV gradually decreases with increase in temperature and the emission band disappears at around 190 K (Fig. S3†).
The origin of emission bands
To unveil the emission processes of Cs4PbI6 NCs, we firstly performed low-temperature PL measurements at 78 K under different excitation energies of 2.95 eV, 2.36 eV and 2.16 eV (Fig. 3a). It turns out that the PL spectra strongly depend on excitation energies. We fitted the experimental PL spectra by using three peaks as shown in Fig. 3a. We observe that these PL spectra can be deconvoluted as the superposition of three emission bands having peak positions at 2.95 eV, 2.36 eV and 2.16 eV. The distinct PL profiles under different excitation energies are ascribed to the different relative intensities of three emission bands under different excitation energies. Specifically, when the NCs thin-film sample is excited by the energy of 3.6 eV (larger than excitonic band-gap energy of 3.38 eV), the emission intensities at 2.36 eV and 2.16 eV are much stronger than the intensity at 2.95 eV. The emission peak at 2.36 eV becomes most intensive when the sample is excited with the energy of 2.95 eV (smaller than excitonic band-gap energy of 3.38 eV). When the sample is excited with excitonic band-gap energy 3.38 eV, intensities of three emission peaks become comparable.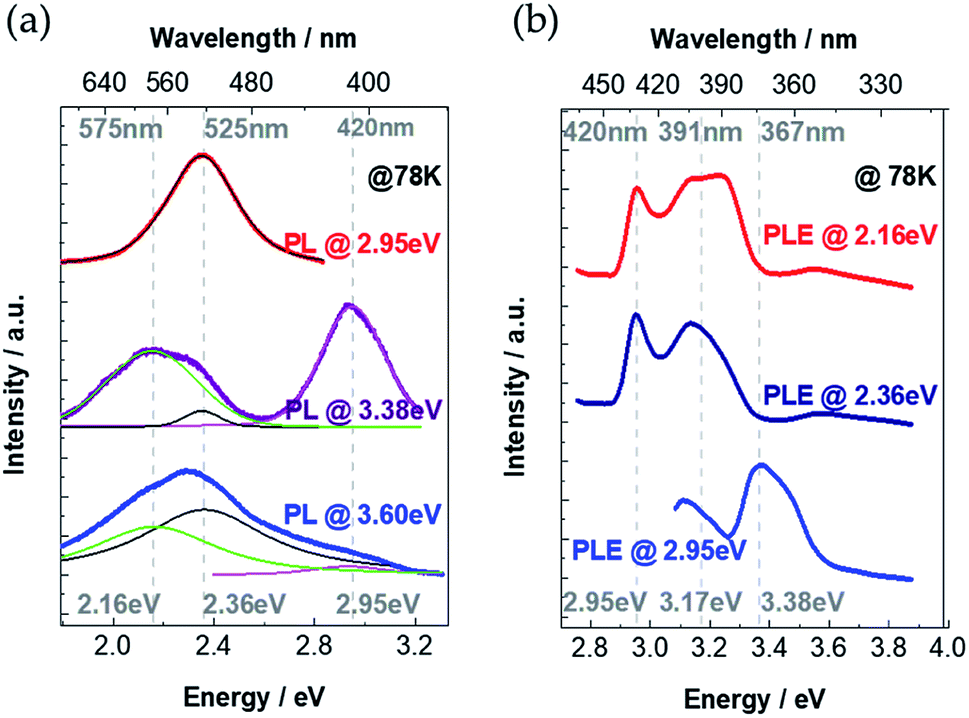 | ||
| Fig. 3 (a) PL spectra of the Cs4PbI6 NCs film excited at 3.6 eV (blue), 3.38 eV (purple) and 2.95 eV (red). The spectra are fitted with three emission peaks (2.95 eV, 2.36 eV, 2.16 eV). The fitting results are shown within the thinner green (2.16 eV), black (2.36 eV) and pink (2.95 eV) lines. (b) PLE spectra detected at 2.95 eV (blue), 2.36 eV (dark blue) and 2.16 eV (red). | ||
To understand the physical origin of these three PL peaks, we continuously change the excitation energy and detect the emission intensity at certain energies of 2.95 eV, 2.36 eV and 2.16 eV. PL excitation (PLE) measurements are shown in Fig. 3b, where the PL emission intensity at 2.95 eV reaches maximum value under excitation energy of 3.38 eV, which coincides with the excitonic absorption band-gap energy as demonstrated in the absorption spectrum (Fig. 2a). Therefore, we attribute that the 2.95 eV PL peak is the intrinsic excitonic emission with a Stoke's shift as large as 0.43 eV. For the PLE spectra detected at 2.36 eV and 2.16 eV, there are two PLE peaks at 2.95 eV and 3.17 eV (Fig. 3b), which indicates that the emissions at 2.36 eV and 2.16 eV originate from the absorption both at 2.95 eV and 3.17 eV. It is noticeable that there is no obvious absorption band below the excitonic absorption band-gap 3.38 eV (Fig. 2a). Considering the extremely large exciton binding energy in Cs4PbI6 NCs, the exciton separation is very unlikely to take place in this material. Thus, the emission at 2.36 eV and 2.16 eV could originate from (a) the self-trapped excitons (self-trapped electrons localizing on Pb2+, self-trapped holes localizing on I−)23,28–32 and/or (b) defect-trapped excitons. To distinguish self-trapped excitons and defect-trapped excitons, we investigate the behaviour of the PL spectra under exciton-power-dependent PL measurements at 78 K.
To understand if any phase transition of perovskites structure causes increase in PL intensity, we have conducted temperature-dependent Raman measurement and XRD diffraction of Cs4PbI6 NCs film from 298 K to 78 K. The corresponding Raman spectra (Fig. S6†) and XRD diffraction pattern (Fig. S7†) of Cs4PbI6 NCs films have been added in the ESI.† In the range of temperature analyzed both Raman and XRD spectra do not show any abrupt shift or change in intensity at a specific temperature, as typically observed when there is a phase transition. We have noticed that 41.53 cm−1 Raman peak at 298 K shifted to 44.71 cm−1 at 78 K, whereas 28.24° XRD peak at 298 K shifted to 27.64° at 98 K. We can just observe a small shift to smaller angles for the XRD peak (with decreasing the temperature the crystal structure shrinks). It concludes that the emission intensity of NCs enhances at low temperature is not related to the phase transition of the perovskite crystal structure.
The mixed STE and defect-assisted emission
In order to find out the origin of the emission peaks at 2.36 eV and 2.16 eV, we have performed the excitation-power-dependent PL measurements. The nature of the charge carrier recombination processes can be determined from the relationship between integral PL intensity and excitation power density. When the sample is excited with a laser energy of 3.6 eV, where the PL profile does not change much even the excitation power density increases two orders of magnitude. We plot the integral PL intensities as a function of excitation power density with logarithmic coordinates as shown in Fig. 4a. By fitting the integral PL intensity with a power law of the form I ∼ Lk (I is the integral PL intensity, L is the power density of excitation laser and k is the fitting parameter), we get k = 0.897 ± 0.005. Since the value of k is very close to 1, we attribute the broad emission to the STEs.2 As a comparison, we also performed power-dependent PL spectra excited with a laser energy of 2.33 eV (Fig. 4b). As shown in the inset of Fig. 4b, we observe a clear saturation behaviour for the PL emission intensity with the value of k = 0.66 ± 0.02. To further confirm this saturation phenomenon, we have conducted power-dependent PL measurements with another laser of 2.71 eV, which was also lower than the excitonic absorption energy. Similarly, saturation behavior is featured with the k value of 0.58 ± 0.03 (Fig. S5†). All these results indicate that emission at 2.36 eV and 2.16 eV originates from a mixed emission process of STEs and defect-trapped excitons.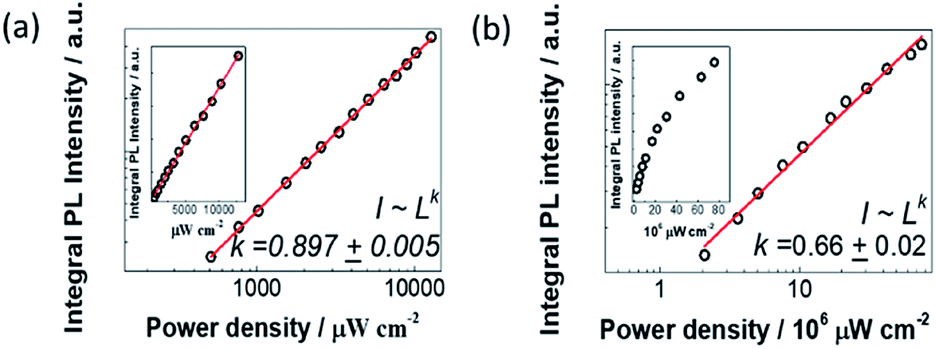 | ||
| Fig. 4 Power-dependent integral PL intensity (IPLI) under different excitation power density of (a) 3.6 eV laser and (b) 2.33 eV laser. Data points are plotted with logarithmic coordinates. The red lines are the linear fitting results of data points. ‘I’ is the integral PL intensity, ‘L’ is the excitation power density. Insets in (a) and (b) are integral PL intensity vs. excitation power density plotted in decimal coordinates. | ||
Emission-wavelength-dependent dynamics of the STEs
To investigate the decay dynamics of STEs, we measured the low temperature (@78 K) TRPL decays of Cs4PbI6 NCs films exciting at 405 nm and selecting the emitted fluorescence at different characteristics wavelengths. The PL decays are strongly wavelength-dependent as shown in Fig. 5a. For all the wavelengths the PL decays are well fitted with a two-exponential decay function,I(t) = I0[a1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(t/τ1) + a2exp(t/τ2)], exp(t/τ1) + a2exp(t/τ2)], |
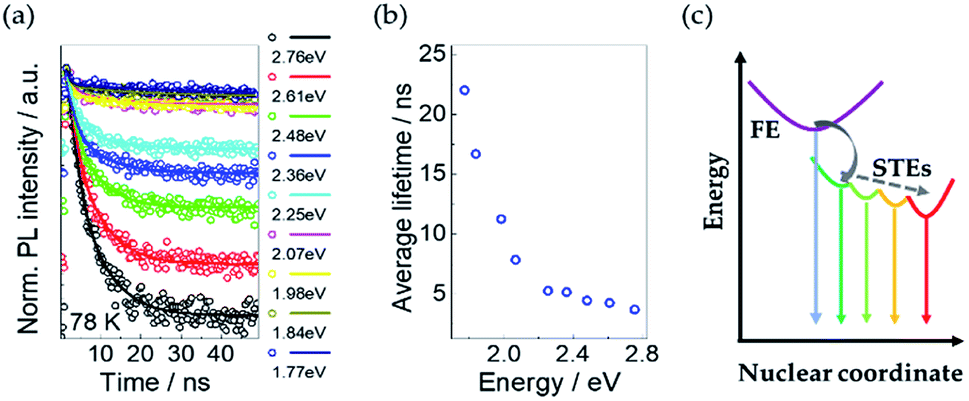 | ||
| Fig. 5 (a) Normalized time-resolved PL spectra of Cs4PbI6 NCs at 78 K excited at 3.38 eV. Open circles are experimental data. Corresponding solid lines are fitting results with the two-exponential function. (b) Average lifetime at different energy obtained from TRPL results in the left panel. (c) The schematic diagram of the decay dynamics of free excitons and STEs. The dashed line indicates the possible nonradiative decay processes of STEs. | ||
The average PL lifetimes according to the equation, τave = Σaiτi2/Σaiτi, are calculated for each decay and plotted in Fig. 5b. It shows that the average PL lifetime increases with a decrease in emission energy of the exciting photons.
This trend of PL lifetimes is consistent with the existence of a series of STE states in Cs4PbI6 NCs as illustrated in the schematic diagram Fig. 5c. Indeed, after excitation, free excitons relax very fast and form STE states through the interaction with the surrounding lattice. The presence of multiple STE states could be induced by different lattice distortions. The excitons from STE states can recombine radiatively and/or nonradiatively. The inhomogeneous distribution of photon energies originated from STE states broaden the resulted PL emission spectra. The deeper self-trapped excitons produce lower-energy PL and take a longer time to recombine, consistently with greater lattice distortions required for lower-energy PL. Besides, a cascade mechanism is also possible, where shallow STE states transfer to deeper STE states, aided by the local heterogeneity of the excited-state potential-energy surface.31 Since the average lifetime of some STE states at specific energies is determined by all of the related radiative and nonradiative transitions. Similar emission-energy-dependent relaxation dynamics have been already reported in case of silicon NCs embedded in SiO2 (ref. 33 and 34) and two dimensional (2D) lead-bromide perovskites31,35,36 and were attributed to the nonradiative quenching of multiple STE states through a phonon-assisted tunnelling mechanism.
To distinguish the PL signal appears from the Cs4PbI6 NCs or the ligands (e.g., oleic acid/oleylamine) we further studied the PLE and lifetime measurements. The optical properties of ligands depend on excitons. The excitonic nature of organics is a result of the weak, electrostatic van der Waals cohesive forces. Hence, the excitons are tightly bound into small Frenkel-like states and results in very high exciton binding energy. However inorganic semiconductors are chemically bound by strong covalent and/or ionic forces, whereby electrons are shared by all the ions forming the crystal lattice and result in comparatively low exciton binding energy than organics. Further, the very long PL lifetime of organic molecules (500 ns to 100 ms) is due to exciton recombination between singlet and triplet energy states,37,38 whereas in inorganic molecules exciton recombines through quantum mechanically allowed singlet energy states and show comparatively short-lived PL lifetime (1–50 ns). Here in Cs4PbI6 NCs, the exciton generation and recombination processes happen within the isolated lead halide octahedra. The PL lifetimes of these NCs is in the range of ns scale. These characteristics confirm that the PL emission contributes to Cs4PbI6 crystal structure.
Excitation dependent emission process
According to our experimental results and careful analysis, we can illustrate the exciton recombination and emission processes within Cs4PbI6 NCs. The schematic diagram of absorption, PL and PLE results are summarized in Fig. 6a. The excitonic absorption peak is situated at 3.38 eV and corresponding emission peak is at 2.95 eV. The trapped exciton emission peaks are located at around 2.36 eV and 2.16 eV. In the following section, we will discuss the exciton recombination process considering two conditions, (a) the excitation energy is higher than excitonic absorption band (3.6 eV) and (b) the excitation energy is smaller than excitonic emission band (2.71 eV and 2.33 eV).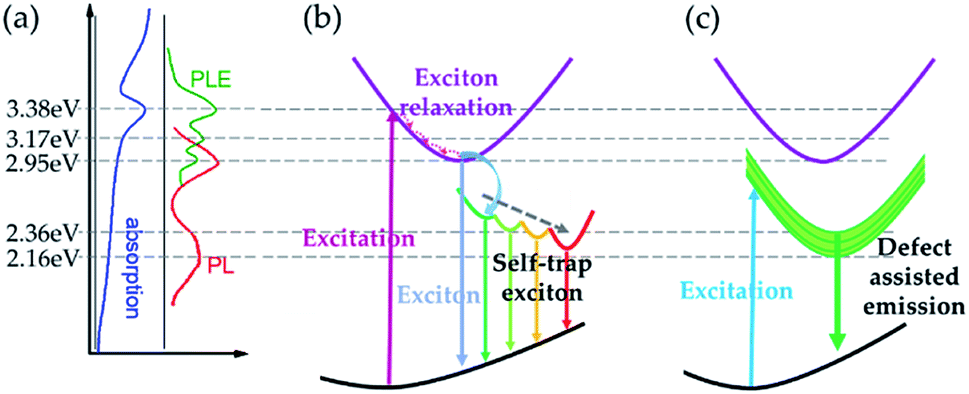 | ||
| Fig. 6 Schematic diagram of PL recombination processes in Cs4PbI6 NC samples. (a) Summary of energy levels in absorption and emission process. (b) Carrier recombination process excited with the energy higher than excitonic absorption band. (c) Carrier recombination process excited with the energy lower than the excitonic emission band. | ||
The emission process for the first case is illustrated in Fig. 6b. In this condition, the excitation energy is high enough to excite free carriers and excitons in Cs4PbI6 NCs. Because of the extremely high exciton binding energy (0.49 eV), there would be little free carriers formed considering the equilibrium between excitons and free carries.39 Then excitons will relax through the interaction with phonons and emit photons with the energy around 2.95 eV, which is much smaller than the excitation energy. During the relaxation process, excitons tend to form STEs with a distribution of energy states. STEs decays through radiative or nonradiative recombination process and results in broad emission spectra. Moreover, there will be a thermal exchange process between intrinsic exciton emission and STEs when the temperature rises, which would result in a variation of the relative intensity of intrinsic exciton emission and STE emission. At low temperatures, the de-trapping rate of STEs is very slow due to the lack of phonons. As a result, emission from STEs would be the dominant emission process at low temperature. Since the de-trapping rate increases with increasing temperature, there would be more free excitons participating in the emission process at higher temperatures. As a result, we observe a much stronger excitonic emission at 78 K than at 298 K as demonstrated in Fig. 6b, and the relative intensity of intrinsic excitonic emission increases gradually with decreasing temperature (Fig. S3†).
For the second condition, the corresponding PL process is represented in Fig. 6c. In this case, the energy of the excitation photon energy is not enough to excite free carriers or excitons. The main excitation species would be the defect states and trapped excitons within the octahedra. Because of the strong confinement from individual octahedron, excited carriers would decay very fast and emit photons with the energy of around 2.36 eV and 2.16 eV. In this case, the defect-assisted emission (from defect-trapped excitons) becomes the dominant contributor.
Conclusions
In summary, we investigate the exciton recombination process in Cs4PbI6 perovskite NCs by absorption, PL, PLE and PL lifetime measurements. We demonstrate that the exciton absorption band peaked at 3.38 eV and the intrinsic emission band peaked at 2.95 eV with a Stoke's shift of 430 meV. Besides, there is a broad emission band from 1.77 eV to 2.6 eV in the PL spectra. Through PLE and power-dependent PL measurements, we conclude that the broad emission band is a mixed emission from the STEs and defect-trapped excitons. Through TRPL experiments, we observe that the PL lifetimes strongly depend on the emission wavelength of STE, which can be attributed to the existence of multiple emissive STE states. When the excitation energy is larger than the excitonic absorption band, self-trapped exciton emission is dominant in the emission because of the strong electron–phonon interaction. When the excitation energy is not enough to excite excitons within the sample, then defects-assisted emission occurs. Our comprehensive studies reveal the properties and mechanism of light emission process in Cs4PbI6 NCs offering a very promising material for white light emission applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation (NRF) Singapore (Program # NRF-CRP14-2014-03). S. B. acknowledges the Department of Science and Technology, India for the financial support through DST-INSPIRE Faculty award (DST/INSPIRE/04/2017/000530). We also acknowledge Qian Chen for his support during experiments.Notes and references
- Q. Zhang and Y. Yin, ACS Cent. Sci., 2018, 4, 668 CrossRef CAS PubMed
.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692 CrossRef CAS PubMed
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635 CrossRef CAS PubMed
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162 CrossRef CAS PubMed
- Y. Wang, X. M. Li, J. Z. Song, L. Xiao, H. B. Zeng and H. D. Sun, Adv. Mater., 2015, 27, 7101 CrossRef CAS PubMed
- Y. Wang, X. M. Li, J. Z. Song, L. Xiao, H. B. Zeng and H. D. Sun, Nano Lett., 2016, 16, 448 CrossRef CAS PubMed
- F. Hu, H. Zhang, C. Sun, C. Yin, B. Lv, C. Zhang, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, ACS Nano, 2015, 9, 12410 CrossRef CAS PubMed
- C. Liu, W. Li, C. Zhang, Y. Ma, J. Fan and Y. Mai, J. Am. Chem. Soc., 2018, 140, 3825 CrossRef CAS PubMed
- H. Yuan, Y. Zhao, J. Duan, Y. Wang, X. Yang and Q. Tang, J. Mater. Chem. A, 2018, 6, 24324 RSC
- L. Yan, Q. Xue, M. Liu, Z. Zhu, J. Tian, Z. Li, Z. Chen, Z. Chen, H. Yan, H. L. Yip and Y. Cao, Adv. Mater., 2018, 30, 1802509 CrossRef PubMed
- N. Yantara, S. Bhaumik, F. Yan, D. Sabba, H. A. Dewi, N. Mathews, P. P. Boix, H. V. Demir and S. Mhaisalkar, J. Phys. Chem. Lett., 2015, 6, 4360 CrossRef CAS PubMed
- Y. S. Park, S. Guo, N. S. Makarov and V. I. Klimov, ACS Nano, 2015, 9, 10386 CrossRef CAS PubMed
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230 CrossRef CAS PubMed
- H. Zhu, K. Miyata, Y. Fu, J. Wang, P. P. Joshi, D. Niesner, K. W. Williams, S. Jin and X. Y. Zhu, Science, 2016, 353, 1409 CrossRef CAS PubMed
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92 CrossRef CAS PubMed
- S. A. Veldhuis, Y. F. Ng, R. Ahmad, A. Bruno, N. F. Jamaludin, B. Damodaran, N. Mathews and S. G. Mhaisalkar, ACS Energy Lett., 2018, 33, 526 CrossRef
- J. C. Blancon, H. Tsai, W. Nie, C. C. Stoumpos, L. Pedesseau, C. Katan, M. Kepenekian, C. M. M. Soe, K. Appavoo, M. Y. Sfeir, S. Tretiak, P. M. Ajayan, M. G. Kanatzidis, J. Even, J. Crochet and A. D. Mohite, Science, 2017, 355, 1288 CrossRef CAS PubMed
- T. A. Berhe, W. N. Su, C. H. Chen, C. J. Pan, J. H. Cheng, H. M. Chen, M. C. Tsai, L. Y. Chen, A. A. Dubaleb and B. J. Hwang, Energy Environ. Sci., 2016, 9, 323 RSC
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286 CrossRef CAS PubMed
- M. De Bastiani, I. Dursun, Y. Zhang, B. A. Alshankiti, X. H. Miao, J. Yin, E. Yengel, E. Alarousu, B. Turedi, J. M. Almutlaq, M. I. Saidaminov, S. Mitra, I. Gereige, A. AlSaggaf, Y. Zhu, Y. Han, I. S. Roqan, J. L. Bredas, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 7108 CrossRef CAS
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924 CrossRef CAS PubMed
- J. Yin, Y. Zhang, A. Bruno, C. Soci, O. M. Bakr, J. L. Brédas and O. F. Mohammed, ACS Energy Lett., 2017, 2, 2805 CrossRef CAS
- J. Yin, P. Maity, M. De Bastiani, I. Dursun, O. M. Bakr, J. L. Brédas and O. F. Mohammed, Sci. Adv., 2017, 3, e1701793 CrossRef PubMed
- S. Kondo, A. Masaki, T. Saito and H. Asada, Solid State Commun., 2002, 124, 211 CrossRef CAS
- M. Nikl, K. Nitsch, J. Chval, F. Somma, A. R. Phani, S. Santucci, C. Giampaolo, P. Fabeni, G. P. Pazzi and X. Q. Feng, J. Phys. Condens. Matter., 2000, 12, 1939 CrossRef CAS
- S. Bhaumik, S. A. Veldhuis, S. K. Muduli, M. Li, R. Begum, T. C. Sum, S. Mhaisalkar and N. Mathews, ChemPlusChem, 2018, 83, 514 CrossRef CAS PubMed
- T. Umebayashi, K. Asai, T. Kondo and A. Nakao, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155405 CrossRef
- S. Wang, Y. Yao, J. Kong, S. Zhao, Z. Sun, Z. Wu, L. Li and J. Luo, Chem. Commun., 2018, 54, 4053 RSC
- X. Lao, Z. Yang, Z. Su, Z. Wang, H. Ye, M. Wang, X. Yao and S. Xu, Nanoscale, 2018, 10, 9949 RSC
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497 RSC
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619 CrossRef CAS PubMed
- D. Han, H. Shi, W. Ming, C. Zhou, B. Ma, B. Saparov, Y. Z. Ma, S. Chen and M. H. Du, J. Mater. Chem. C, 2018, 6, 6398 RSC
- A. Y. Kobitski, K. S. Zhuravlev, H. P. Wagner and D. R. T. Zahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 115423 CrossRef
- J. C. Vial, A. Bsiesy, F. Gaspard, R. Hérino, M. Ligeon, F. Muller, R. Romestain and R. M. Macfarlane, Phys. Rev. B: Solid State, 1992, 45, 14171 CrossRef CAS PubMed
- A. Yangui, D. Garrot, J. S. Lauret, A. Lusson, G. Bouchez, E. Deleporte, S. Pillet, E. E. Bendeif, M. Castro, S. Triki, Y. Abid and K. Boukheddaden, J. Phys. Chem. C, 2015, 119, 23638 CrossRef CAS
- T. Hu, M. D. Smith, E. R. Dohner, M. J. Sher, X. Wu, M. T. Trinh, A. Fisher, J. Corbett, X. Y. Zhu, H. I. Karunadasa and A. M. Lindenberg, J. Phys. Chem. Lett., 2016, 7, 2258 CrossRef CAS PubMed
- B. Minaev, G. Baryshnikovb and H. Agrena, Phys. Chem. Chem. Phys., 2014, 16, 1719 RSC
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603 RSC
- M. Saba, F. Quochi, A. Mura and G. Bongiovanni, Acc. Chem. Res., 2016, 49, 166 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00467g |
| This journal is © The Royal Society of Chemistry 2020 |