
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegeneration of a deactivated surface functionalised polyacrylonitrile supported Fenton catalyst for use in wastewater treatment
Caroline A. Akinremiab,
Sanaa Rashida,
Pushpa D. Upretia,
George T. Chia and
Katherine Huddersman *a
*a
aSchool of Pharmacy, Faculty of Health and Life Sciences, De Montfort University, The Gateway, Leicester LE1 9BH, UK. E-mail: huddzeo1@dmu.ac.uk
bDepartment of Chemistry, Federal University of Agriculture, Abeokuta, Nigeria
First published on 31st March 2020
Abstract
Successful attempts to regenerate a used surface functionalised nanocoated polyacrylonitrile (PAN) catalyst are described here. During use in wastewater treatment, the novel Fenton catalyst (F1) is deactivated due to iron loss caused by acid hydrolysis. In this study the deactivated catalyst (D1) is subjected to reactions with 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), followed by reactions with either hydroxylamine to give sample T1 or hydroxylamine and hydrazine to give sample T2. The samples were then impregnated with iron(III) salt to give either Fe-T1 or Fe-T2. The catalysts were characterized by Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR), Atomic Absorption (AA) and UV/VIS spectroscopies, Scanning Electron Microscopy (SEM) with energy-dispersive X-ray (EDX) spectroscopy and Electron Spin Resonance (ESR) spectroscopy. The iron on the regenerated catalyst was found to be in complexed form but had deposited iron oxide species as well. The catalysts were tested in batch mode and compared with the fresh modified PAN catalyst in the degradation of the dye Reactive Orange 16 (RO-16) with analysis by UV/VIS spectroscopy. The reactivated catalysts prepared with EDC were found to be more active and faster (as measured at 120 min) in decolourising RO-16 than the fresh catalytic mesh but also with a higher degree of Fe leaching (0.85% loss of iron per gram of Fe-T2 catalyst over 6 cycles compared to 0.32% loss of Fe per gram of F1 catalyst over 6 cycles). This leaching was found not to contribute significantly to degradation of the dye and the preliminary results suggest that the regime can be used for catalyst regeneration encouraging industrial uptake.
1 Introduction
Fenton heterogeneous catalysts are becoming more popular as a green method of wastewater treatment in Advanced Oxidation Processes (AOP).1 In heterogeneous Fenton reactions, highly reactive hydroxyl radicals (˙OH) are produced which can degrade stable organic pollutants in water.2 The catalysts can be made of a modified material to which iron is complexed and available for Fenton like reactions in the presence of hydrogen peroxide. One of the materials successfully used is a surface functionalised nanocoated modified polyacrylonitrile (MPAN) fibre in the form of a mesh.3–6 The PAN mesh was modified to introduce surface functionalisation by amidoximation with a mixture of hydrazine and hydroxylamine at alkaline pH to allow surface complexation with iron(III) sulphate salt.3,7–9 This introduces groups such as amidoxime, amidrazone, amide and carboxylate onto the fibres of the PAN mesh.5 An Extended X-ray Absorption Fine Structure (EXAFS) study showed that the iron is surrounded by six atoms which are either O, or N or a mixture of both. When used with hydrogen peroxide at pH 3, the catalyst is seen to give excellent oxidation of organic pollutants in wastewater.3–6The catalyst degrades pollutants via a heterogeneous Fenton-like mechanism (eqn (1) and (2)), which is known to be slightly slower in comparison to a homogenous Fenton's degradation. However, as the iron in this study is chelated to a PAN support, the amount of iron leaching and hence secondary pollution is reduced drastically. The heterogeneous Fenton-like system also allows catalysis to be undertaken at a slightly higher pH range (3–4) which enables catalyst recycling unlike traditional homogenous Fenton catalysis which would precipitate out as an iron hydroxide sludge.
| Fe3+ + H2O2 → Fe2+ + HO2˙ + H+ | (1) |
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (2) |
However, as with most heterogeneous catalysts in a continuous flow wastewater treatment system, the catalytic activity reduces over time. This could be attributed to acid hydrolysis of the ligating groups, which ultimately leads to iron leaching.5 When used for treatment of organic contaminants in wastewater, the final oxidation products are usually formic or acetic acid, which could contribute to the acid hydrolysis of the ligating groups leading to the formation of amides or carboxyl groups on the surface of the mesh.
Carbodiimides are usually used for crosslinking of carboxylic acids to primary amines. It works by activating carboxyl groups for direct reaction with primary amines via amide bond formation. Reactions of carboxylic acids with hydroxylamine usually yield the hydroxamates when activated by carbodiimide 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC); dicyclohexyl carbodiimide (DCC); 1,1′-carbonyldiimidazole (CDI).10–12 The reaction is usually carried out at room temperature under varying pH of 4.5 to 9.0.13 Hydroxamic acids form particularly stable and relatively pH-independent complexes with ferric ions.
Spectroscopic studies have been previously used to determine the functional groups present, iron content and mode of coordination of the metal on MPAN catalyst.3–5 This work is an attempt to regenerate deactivated MPAN catalysts by means of EDC assisted reactions with inorganic amine group donors (hydroxylamine and hydrazine) to convert the carboxyl groups to hydroxamic acids and hydrazides (Schemes 1 and 2). The study also suggests the iron species present on the catalyst from spectral and diffraction studies of both the catalysts and residues from the modification procedures. The regenerated catalysts were also tested for their activity in the degradation of an azo dye, reactive orange 16.
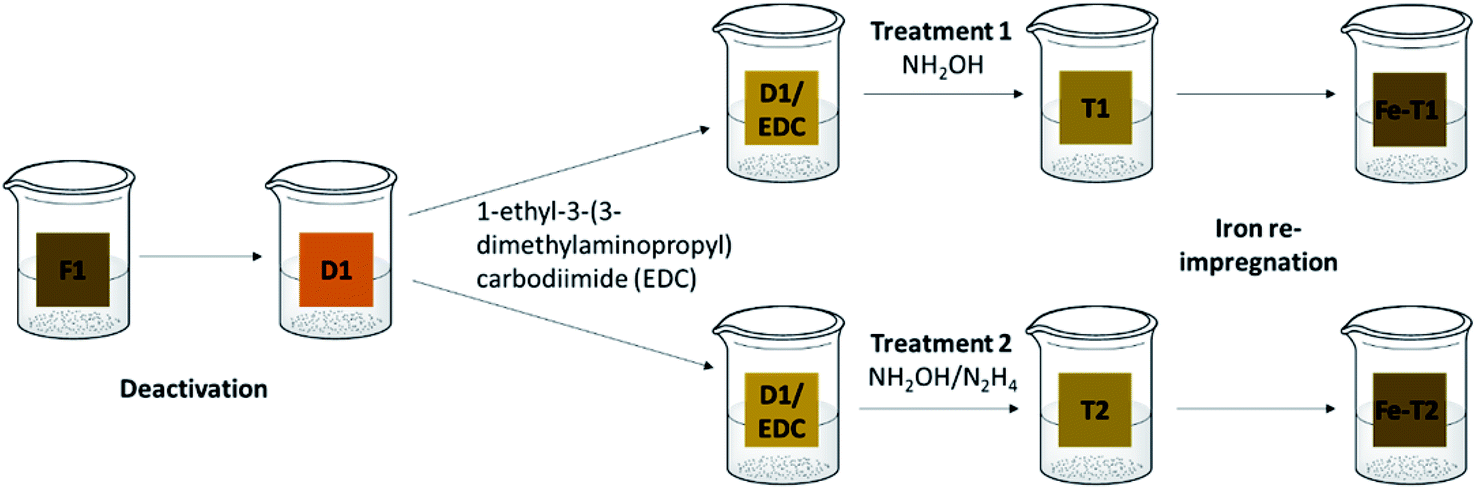 | ||
| Scheme 1 Regeneration procedure. F1: fresh catalyst, D1: deactivated catalyst, T1: treatment 1 (EDC/hydroxylamine), T2: treatment 2: (EDC/hydroxylamine and hydrazine), Fe-T1: iron impregnated T1, Fe-T2: iron impregnated T2. | ||
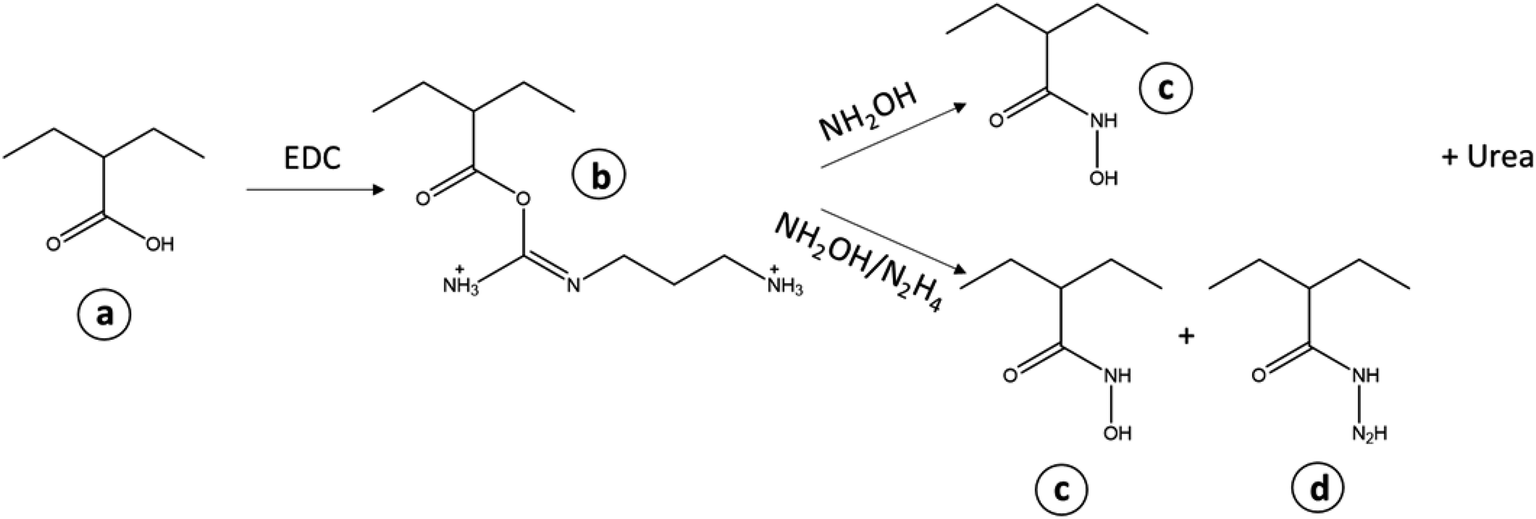 | ||
| Scheme 2 Treatment of the (a) carboxylic acid groups on the deactivated PAN with EDC forming (b) acylisourea active ester and treatment with either hydroxylamine or hydroxylamine and hydrazine. Final products (c) hydroxamic acid and (d) hydrazides. | ||
Many studies into the deactivation and regeneration of supported metal catalysts have been previously carried out but focused primarily on non-ligated metal catalysts on supports. In most cases these supported metal catalysts were found to be deactivated by poisoning.14–17 Utilizing a solvent to remove poisoning agents, such as methanol or aqueous hydrazine to activate a supported palladium catalyst17 has been attempted with good regenerated activity. In literature a Ni/SiO2,14 Rh/CeO2–ZrO215 and Pt/CeO2/Al2O316 to name a few, used air regeneration with heating to high temperatures. This in turn oxidized the poisoning compounds and reactivated the catalysts. In the case of our PAN supported iron catalyst, heating to high temperature is not feasible as it would melt the polymer support. Furthermore, previous work suggests that the deactivation is not due to poisoning but to iron leaching. Iron leaching leads to the proposition that the functional groups have changed and hence the ligated iron is easily lost. In the study by Blanc et al. it was found that leaching from a zinc catalyst was reduced by performing catalysis at a higher pH,18 however this option was not available for the current catalyst in this study. Other studies mention leaching as a cause of activity loss but do not discuss a remedy.19
Studies have also shown how the use of complexing agents such as EDTA20 or S,S-EDDS21 remove interfering metal ions that have deactivated supported metal catalysts.
The novel PAN supported catalyst in this study contains complexed ligated iron. In literature, the authors could find only one study detailing the effect of catalysis on a metal complex catalyst. In the study by Zuleta et al. a cobalt-schiff base catalyst was used to oxidise lignin.22 The catalyst was found to be deactivated by the formation of quinone during lignin oxidation and the quinone oxidized the ligand system in the complex leading to inactive complexes.22 The study did not look into regeneration.
In terms of water remediation, deactivation/regeneration studies have been carried out on TiO2 photocatalysts. The general consensus is that the deactivation of these TiO2 catalysts is due to the buildup of catalytic products on the TiO2 surface.23,24 Studies utilized washing to remove these products such as alkali treatment to remove SiOx build up23 or methanol/hydrogen peroxide to remove methyl orange sorption.25 One study however regenerated the TiO2 catalyst with heating at 420 °C,26 whilst Wang et al. used both NaOH washing and thermal regeneration to return activity.27
Only two studies to date have been found on the regeneration of a Fenton-like catalyst. The study by Chagas et al., used iron/chromium chitosan composite beads alongside hydrogen peroxide to remove methylene blue.28 The study regenerated the beads after six cycles with ethyl alcohol alone.28 Whereas the first cycle delivered 93.8% dye removal, the cycle post regeneration was slightly poorer at only 80.3% removal.28 Another study by Yang et al. used only ethanol washing to remove poisoning from Fe2O3 nanoparticles working with hydrogen peroxide in the oxidation of methylene blue.29 These studies provide insight into Fenton catalyst regeneration in terms of deactivation by poisoning.
This study will detail the regeneration of a supported iron complex Fenton catalyst deactivated by iron leaching.
2 Materials and methods
2.1 Materials and instrumentation
All chemicals and materials were used without further purification. Solutions of the Reactive Orange-16 dye (RO-16) (Sigma-Aldrich) with an assay of 50% were prepared by dissolving the weighed solid in distilled water.The IR spectra was collected on a Bruker Alpha ATR-FTIR spectrometer at a resolution of 4 cm−1. Samples were analysed after normalization of the spectra (allows for differences in sample loading and concentration) at pH = 3. Iron content was determined on PerkinElmer Analyst 200 Atomic Absorption Spectrometer after digestion of the sample with Conc. HCl at 140 °C. UV-VIS measurements were carried out on an Evolution 220 UV-Visible spectrophotometer by Thermo Fisher Scientific at bandwidth of 2 nm. The SEM images were obtained from a Carl Zeiss EVO 15 and the EDX on an Oxford instrument Xmax 80 mm2. The EPR measurements were done on an EMXnano from Bruker at a g factor of 4.00. The XRD measurements were done on a Bruker XRD D2 PHASER 2theta/scan Cu tube with 1.54184 Å with Lynexy (ID mode) detector run from 10 to 80 2theta and resolved on a Diffrac.Suite EVA software (VERSION 4.2.1.10).
2.2 Methods
Pieces of D1 (2.0 g) were placed in water (50 mL) and the pH was adjusted to 4.5 under stirring for few minutes. To D1, 0.2 g of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (equivalent of 1.0 g of catalyst to 0.1 g of EDC) was added and stirring continued for thirty more minutes for activation of the carboxyl group.13
The EDC treated D1 (D1/EDC) was then subjected to either treatment 1 or treatment 2. For treatment 1, 2.1 g of hydroxylamine chloride (HA) was added to the EDC solution3 and the pH adjusted to 6.0 using sodium hydroxide solution with stirring for 2 h, after which the pH was then raised to pH 9.0. The reaction was then left to stir for over 24 h at room temperature. The resultant mesh (T1) was then rinsed in distilled water at pH = 3 until there was no further pH change and air dried. Treatment 2 was carried out on D1 using a combination of hydroxylamine chloride and hydrazine dihydrochloride (HA/HZ) (2.1 g + 1.5 g) to give T2.
2.3 Catalytic degradation of reactive orange-16 (RO-16)
This was carried out on the fresh catalyst and on a number of the regenerated catalysts. A calibration curve for the UV/Vis analysis of the residual dye after catalysis was obtained from standard solutions of the RO-16 at concentrations 0.10, 0.25, 0.50, 1.00, 2.50, 5.00, 10.00, 25.00 and 50.00 mg L−1. To a three necked flask, 100 mL of RO-16 (50 mg L−1) was added with the pH adjusted to 3 followed by the addition of 37.53 μL of hydrogen peroxide (to give a concentration of 125 mg L−1 H2O2). The different catalysts (6 g) were then added to the solution and the temperature maintained to 25 °C. Samples were collected at 0, 5, 10, 20, 30, 40, 60, 80, 100 and 120 minutes. All absorbances were measured on a UV-VIS spectrophotometer (Scan mode) with particular focus on wavelengths at 254 and 493.5 nm.30–33 C0 is the initial concentration of RO-16 (ppm) and Ct corresponds to the concentration of RO-16 at time t (ppm).The catalysts were reused for a number of catalytic cycles under the same conditions. In between the different cycles, the catalysts were rinsed with water. The leached iron content in the treated solution collected after each catalysis was determined using Atomic Absorption Spectroscopy (AAS).
3 Results
3.1 Iron content of iron impregnated samples
The iron content of each of the samples is shown in Fig. 1. The iron content on the deactivated catalyst D1 is highly reduced compared to the fresh catalyst F1 that suggests leaching of iron during usage. The Fe(III) content of the re-impregnated D1 (Fe-D1) and the re-impregnated T1 (Fe-T1) was quite low (2–3 mg g−1) suggesting that these treatments with EDC to reintroduce functionality did not work. However, re-impregnated T2 (Fe-T2) has a much higher Fe content of 13.5 mg g−1, which is quite comparable with F1 at 11 mg g−1.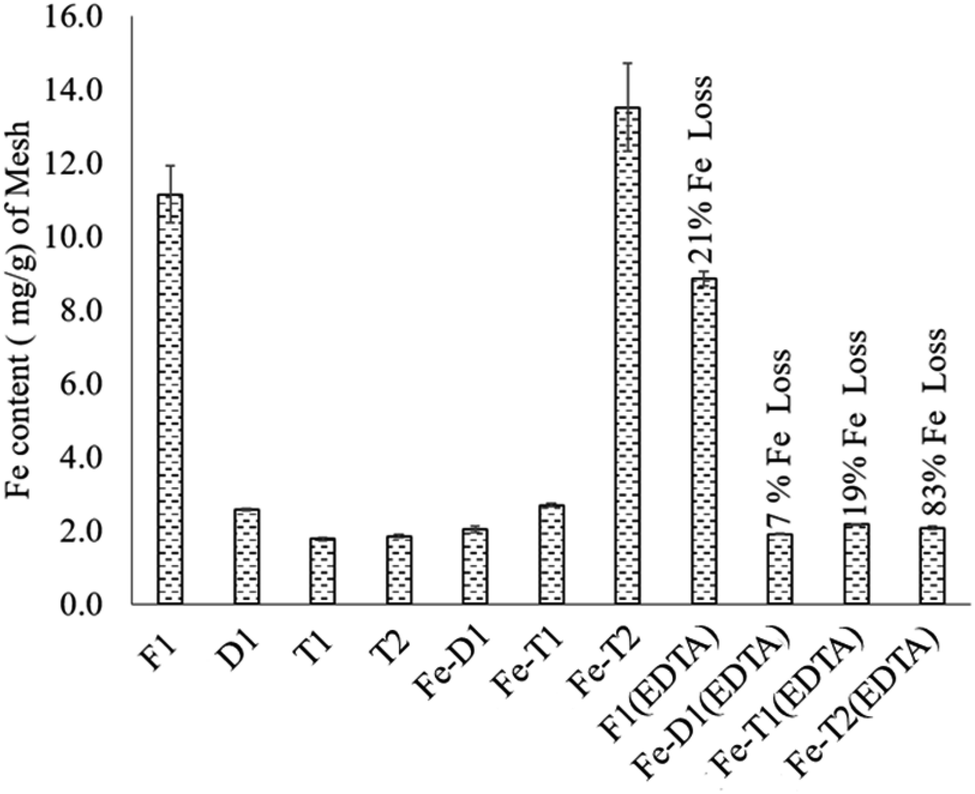 | ||
| Fig. 1 The iron content (mg g−1) of fresh and modified PAN catalysts after iron impregnation and treatment with EDTA. F1: fresh catalyst; D1: deactivated catalyst; T1: treatment 1; T2: treatment 2; Fe-D1: iron impregnated D1; Fe-T1: iron impregnated T1; Fe-T2: iron impregnated T2; F1(EDTA): fresh catalyst treated with EDTA; Fe-D1(EDTA): iron re-impregnated deactivated catalyst treated with EDTA; Fe-T1(EDTA): iron impregnated T1 treated with EDTA; Fe-T2(EDTA): iron impregnated T2 treated with EDTA. | ||
Generally, EDTA treatment of the mesh samples led to significant loss of Fe suggesting that the Fe was loosely held to the polymer support. However, the fresh catalyst F1 had only a 21% loss of Fe (F1(EDTA)) owing to a relatively small amount of loosely held complexed iron or iron oxides. In comparison, the loss of iron for Fe-T2(EDTA) was 83% showing that most of the iron on this catalyst after reimpregnation was loosely held as opposed to that on F1(EDTA). In stark comparison, Fe-T1(EDTA) which had an iron content of 2–3 mg g−1 experienced a 19% loss of iron. Thus D1, Fe-T1, Fe-T1 and Fe-T2(EDTA) ultimately all had similar iron contents.
3.2 EPR analysis
In previous works focusing on the De Montfort University PAN catalyst, it has been theorised that the iron present is chelated to the functional groups on the PAN surface. The EPR spectra of F1, D1 and Fe-T2 were compared with an Fe2(SO4)3 salt and Fe-EDTA complex (Fig. 2). F1, D1 and Fe-T2 all had peaks at around 1600 Gauss attributed to complexed Fe which was also seen in the EPR spectra of [Fe-EDTA] at the same position.31–33 This peak was absent in the image of Fe2(SO4)3 salt. This suggests that the iron present in the catalyst is coordinated (or in complex form) by a mixture of oxygen and nitrogen ligating atoms which has been previously confirmed by EXAFS.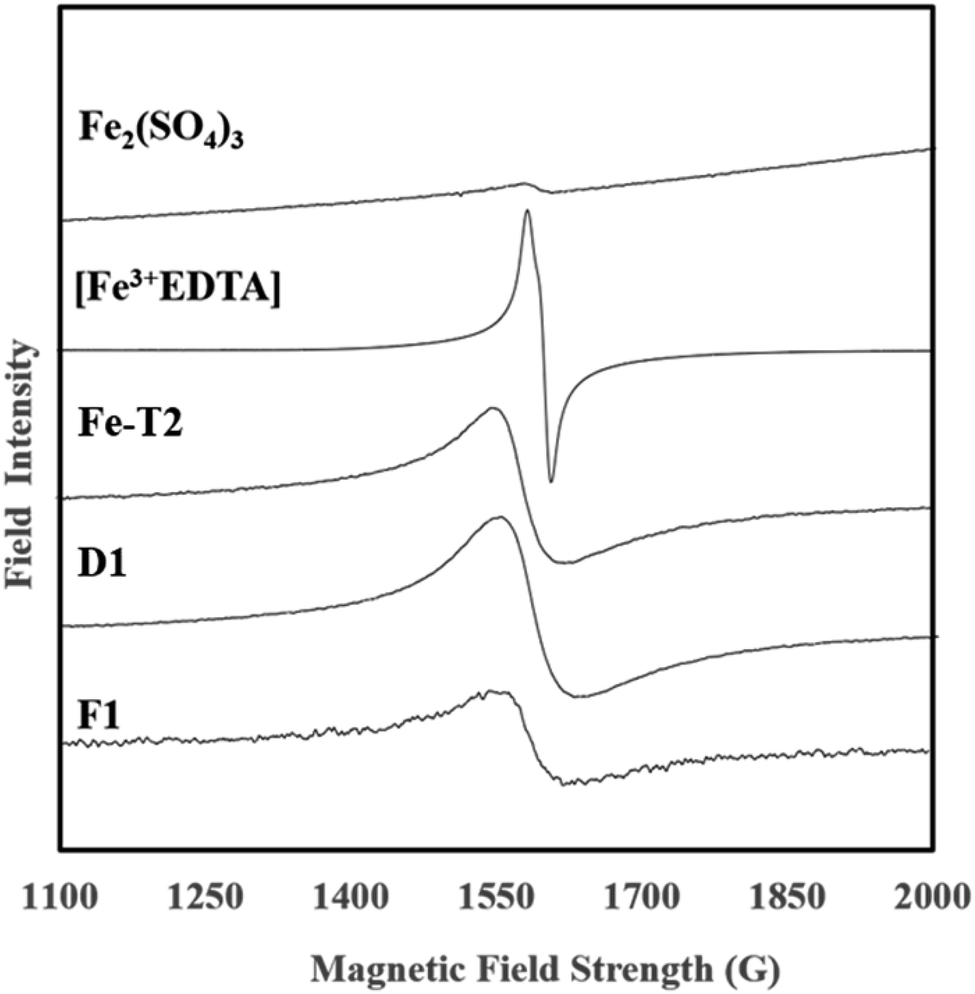 | ||
| Fig. 2 EPR spectra of MPAN catalysts, Fe-EDTA and iron(III) sulphate. | ||
3.3 SEM and EDX
The surface of all catalyst samples were rough, uneven and non-porous.3,34 F1, F1(EDTA) and Fe-T2 all had particulates on their surface (Fig. 3). After treatment with the EDTA, the surface of Fe-T2 becomes much smoother. | ||
| Fig. 3 SEM micrographs of the catalysts after modification, after iron impregnation and after treatment with EDTA. Samples are pH adjusted to 3. | ||
In comparison with the Fe-T2 samples, the particulates on F1 are not removed to the same extent with EDTA as Fe-T2. Hence, this suggests that the iron on the F1 catalyst is in a different form in comparison to that on the regenerated catalysts. In particular it suggests that the iron particulates deposited on F1 are more crystalline and/or of a different speciation in comparison to the particulates on Fe-T2 and therefore less likely to dissolve.
Table 1 shows the content by weight of C, O, N and Fe on the catalyst samples shown in Fig. 3 where the SEM-EDX was targeted on the particulates on the surface of the catalyst fibres. It can be seen that the iron and oxygen content of the Fe-T2 catalyst is higher in comparison to that of the fresh catalyst. This would suggest that the particulates on the surface of the regenerated catalyst, Fe-T2 are likely to have a higher proportion of iron oxides in comparison to the F1 catalyst. Sulphur was also found to be present in the EDX of the particulates but was too low in percentage to be quantified.
| Weight% | Fe | C | N | O |
|---|---|---|---|---|
| F1 | 13.21 | 50.21 | 17.69 | 18.88 |
| Fe-T2 | 30.83 | 26.44 | 10.05 | 32.68 |
3.4 Infrared spectra
Peak 2, between 1650–1530 cm−1 was assigned to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the amide I, the asymmetric stretch of the CO of the carboxylic acid, –CN– of the oxime and –N–H of the amide II group.5,36 It is likely that acid hydrolysis causes deactivation leading to the loss of –CN and the gain of –CO in amide formation (CO(NH2)), as well as a loss of –N–H arising from a loss of the amide group and an increase in the carboxyl groups on the surface of the catalyst which contributes to the loss of Fe.5 As the N–O peak of the oxime group at 910 cm−1 also decreases on going from F1 to D1 which supports the suggestion that the oxime is being converted to amides by acid hydrolysis i.e. CNOH into CO. Thus, acid hydrolysis has resulted in loss of CN and N–H and gain in CO as it is converted to an amide. This results in peak 1 decreasing in intensity whilst peak 2 remains unchanged. On reaction of D1 with EDC followed by treatment with hydroxylamine to form T1, there is a gain in N–H (1650–1530 cm−1) and C–N (1235 cm−1). This could indicate the conversion of the carboxylic group of D1 to hydroxamate. The low energy side of peak 2 increases on going from D1 to T1 due to N–H band at 1650–1530 cm−1, but this is not reflected in peak 1 which does not change.
O of the amide I, the asymmetric stretch of the CO of the carboxylic acid, –CN– of the oxime and –N–H of the amide II group.5,36 It is likely that acid hydrolysis causes deactivation leading to the loss of –CN and the gain of –CO in amide formation (CO(NH2)), as well as a loss of –N–H arising from a loss of the amide group and an increase in the carboxyl groups on the surface of the catalyst which contributes to the loss of Fe.5 As the N–O peak of the oxime group at 910 cm−1 also decreases on going from F1 to D1 which supports the suggestion that the oxime is being converted to amides by acid hydrolysis i.e. CNOH into CO. Thus, acid hydrolysis has resulted in loss of CN and N–H and gain in CO as it is converted to an amide. This results in peak 1 decreasing in intensity whilst peak 2 remains unchanged. On reaction of D1 with EDC followed by treatment with hydroxylamine to form T1, there is a gain in N–H (1650–1530 cm−1) and C–N (1235 cm−1). This could indicate the conversion of the carboxylic group of D1 to hydroxamate. The low energy side of peak 2 increases on going from D1 to T1 due to N–H band at 1650–1530 cm−1, but this is not reflected in peak 1 which does not change.
The sample T2 is the result of the interaction of the carboxylate group of D1 with EDC followed by hydrazine and hydroxylamine. Needless to say, the situation is very complicated but results in the increase of N–H (1650–1530 cm−1) and C–N (1231 cm−1) whilst some OH (∼3000 cm−1) is lost due to reaction with hydrazine (see Scheme 2). This could be attributed to the formation of hydroxamates and hydrazides from the respective reactions with hydroxylamine and hydrazine. As mentioned before, loss in the oxime peak at 910 cm−1 is due to hydrolysis.
On adding Fe to D1, T1 and T2 the ratios of peak 1 to peak 2 remain unchanged but the peak at 1080 cm−1 increases particularly for Fe-T2 which has a significant increase in Fe content as shown in Fig. 1. The FTIR bands of the regenerated samples loaded with iron were compared against those of the fresh catalyst, F1. The spectra are as shown in Fig. 4. There was no significant difference in the spectra of samples D1-T2 except the peak at 1080 cm−1 which is thought to relate to C–O–Fe and the SO42− band which is strong. This peak decreased as the Fe content of the samples reduced as shown in Fig. 4 and markedly with the reduced exposure to sulphate on going from D1 to T1 and T2 (HA and HZ were used as chloride salts to prepare T1 and T2 whereas the sulphate salt was used to prepare D1).
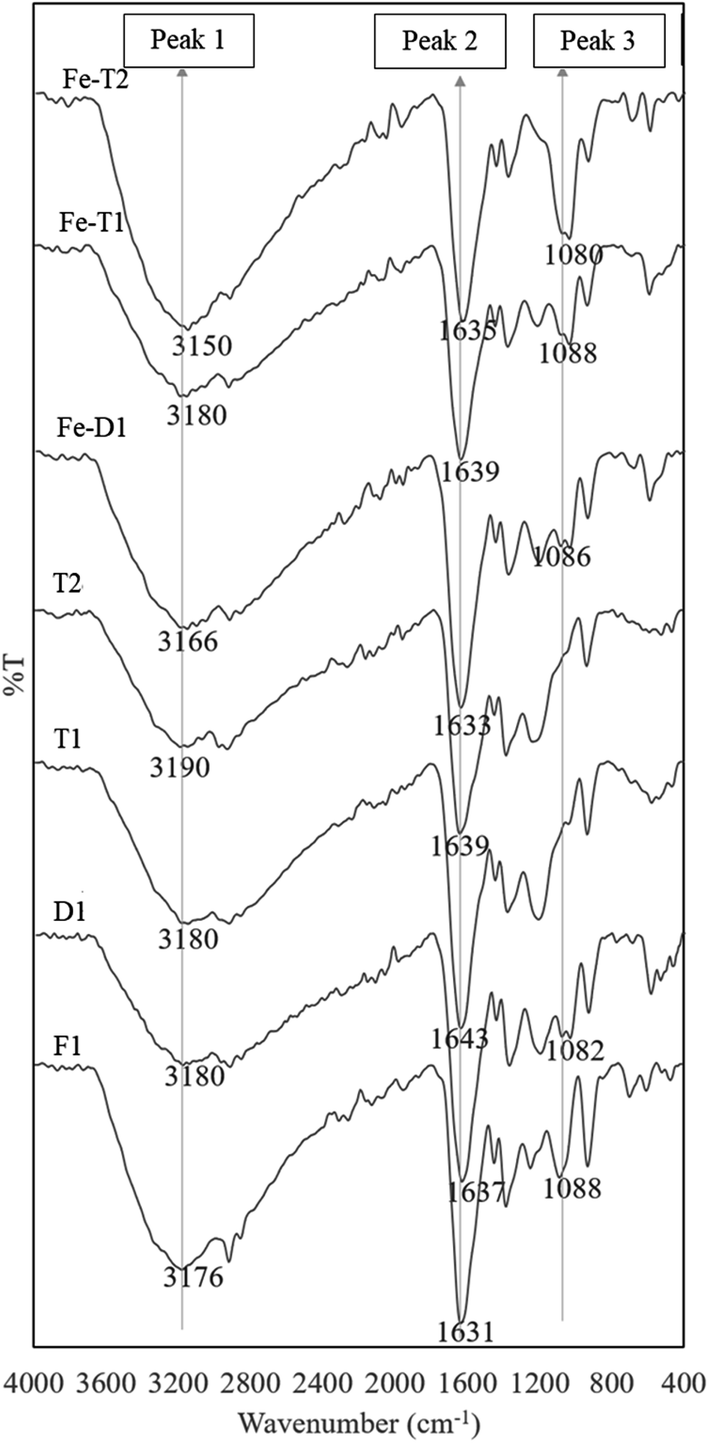 | ||
| Fig. 4 ATR-FTIR spectra of used PAN catalysts after reaction with hydroxylamine/hydrazine activated with EDC and followed by iron impregnation (peak assignments in Table 2). | ||
The peak 3 at 1080 cm−1 assigned to C–O–Fe and SO42− reduced considerably in the EDTA treated meshes due to loss of iron (Fig. 5). Fig. 6 compares the ratio of peak 1 at ∼3000 cm−1 to peak 2 at ∼1650 cm−1 with the Fe content as determined from the AAS analysis of Fig. 1 supporting loss of Fe as the peak ratio decreases (i.e. the greater the intensity of peak 1 is to peak 2 the higher the iron content).
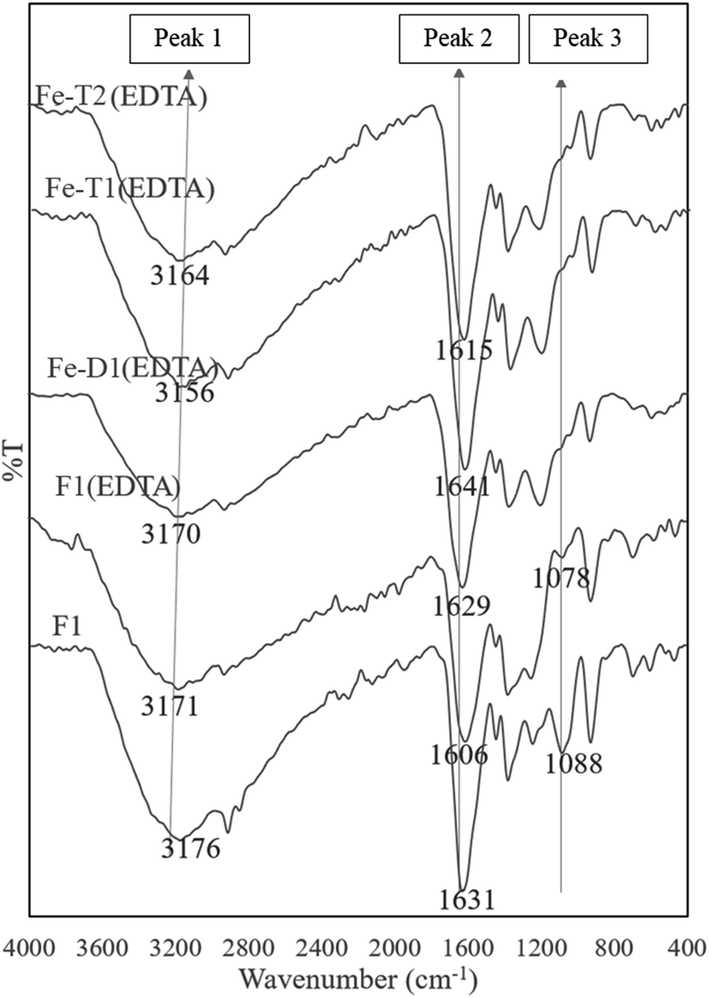 | ||
| Fig. 5 ATR-FTIR spectra of used PAN catalysts after reaction with hydroxylamine/hydrazine activated with EDC, impregnated with iron and thereafter treated with EDTA (peak assignments in Table 2). | ||
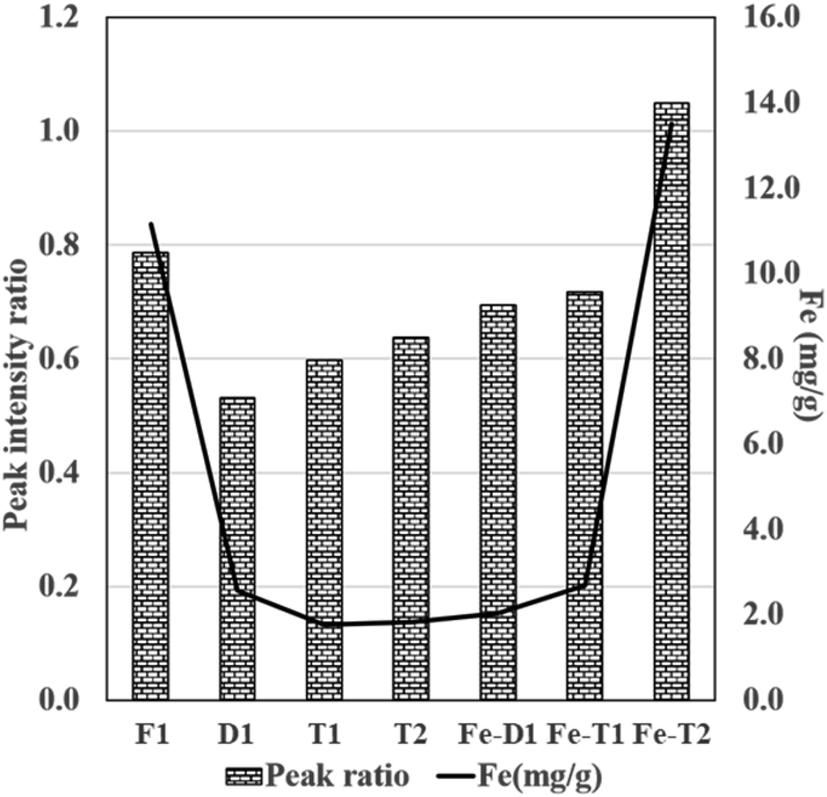 | ||
Fig. 6 Plot of peak ratio of peak 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :peak 2 (LHS) derived from the intensity data presented in ATR-FT-IR spectra of Fig. 4 and 5 and Fe content (RHS) from Fig. 1 (Fe mg g−1 was determined from AAS). :peak 2 (LHS) derived from the intensity data presented in ATR-FT-IR spectra of Fig. 4 and 5 and Fe content (RHS) from Fig. 1 (Fe mg g−1 was determined from AAS). | ||
Table 2 summarises the peak number, wavenumber range and functional group assignment for Fig. 4 and 5.
| Peak number | Assignment | Wavenumber/cm−1 |
|---|---|---|
| 1 | OH/N–H/NH2 | 3300–3100 |
| 2 | CO/CN/N–H |
1645–1600 |
| 3 | C–O–Fe/SO42− | 1090–1080 |
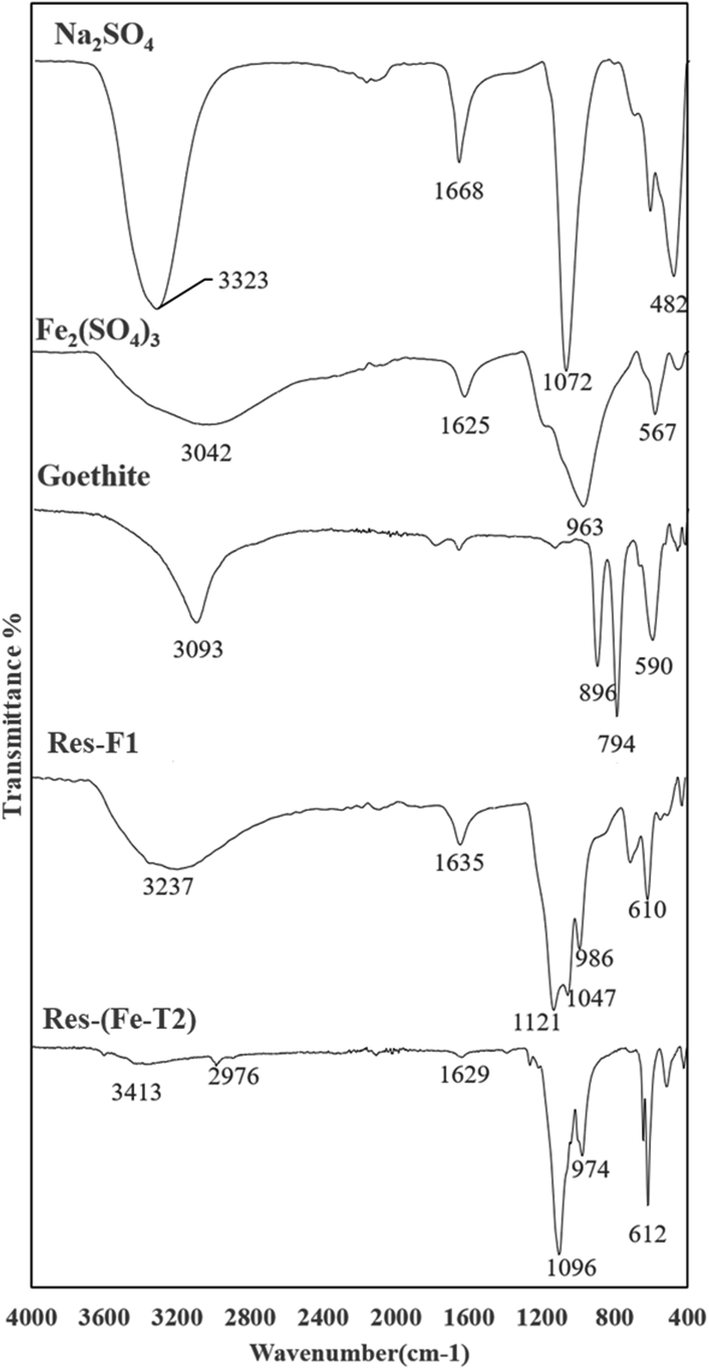 | ||
| Fig. 7 ATR-FTIR spectra of residue (Res-Fe) from iron impregnation compared with some iron oxides, sulphate salts of Fe and Na (peak assignments in Table 3). | ||
As the FTIR results are of the residue produced during impregnation of the catalysts, this can give an indication as to the speciation of the iron in the particulates deposited on the fibres as shown in the SEM results in Section 3.3.
A major indication of schwertmannite, a basic iron sulphate (Fe8O8(OH)8.2x(SO4)X where 1 ≤ x ≤ 1.75) is the presence of the SO42− peaks at 1121 cm−1 for Res-F1 and 1096 cm−1 and 974 cm−1 for Res-(Fe-T2).42 Other than schwertmannite, there is a major presence of goethite. The presence of the SO42− peak in the spectra is due to the residual Fe2(SO4)3 and Na2SO4 from impregnation from which some schwertmannite has been produced.
3.5 XRD images of the residual iron after impregnation on mesh
The XRD powder diffraction patterns of Res-F1, Res-Fe-T2 and a dried iron(III) hydroxy precipitate (prepared by treating Fe2(SO4)3 with a solution of NaOH until precipitation occurred) (Fig. 8) showed very little resemblence to each other. Furthermore, the XRD of the precipitate from F1 and Fe-T2 are clearly different suggesting different species of iron oxides.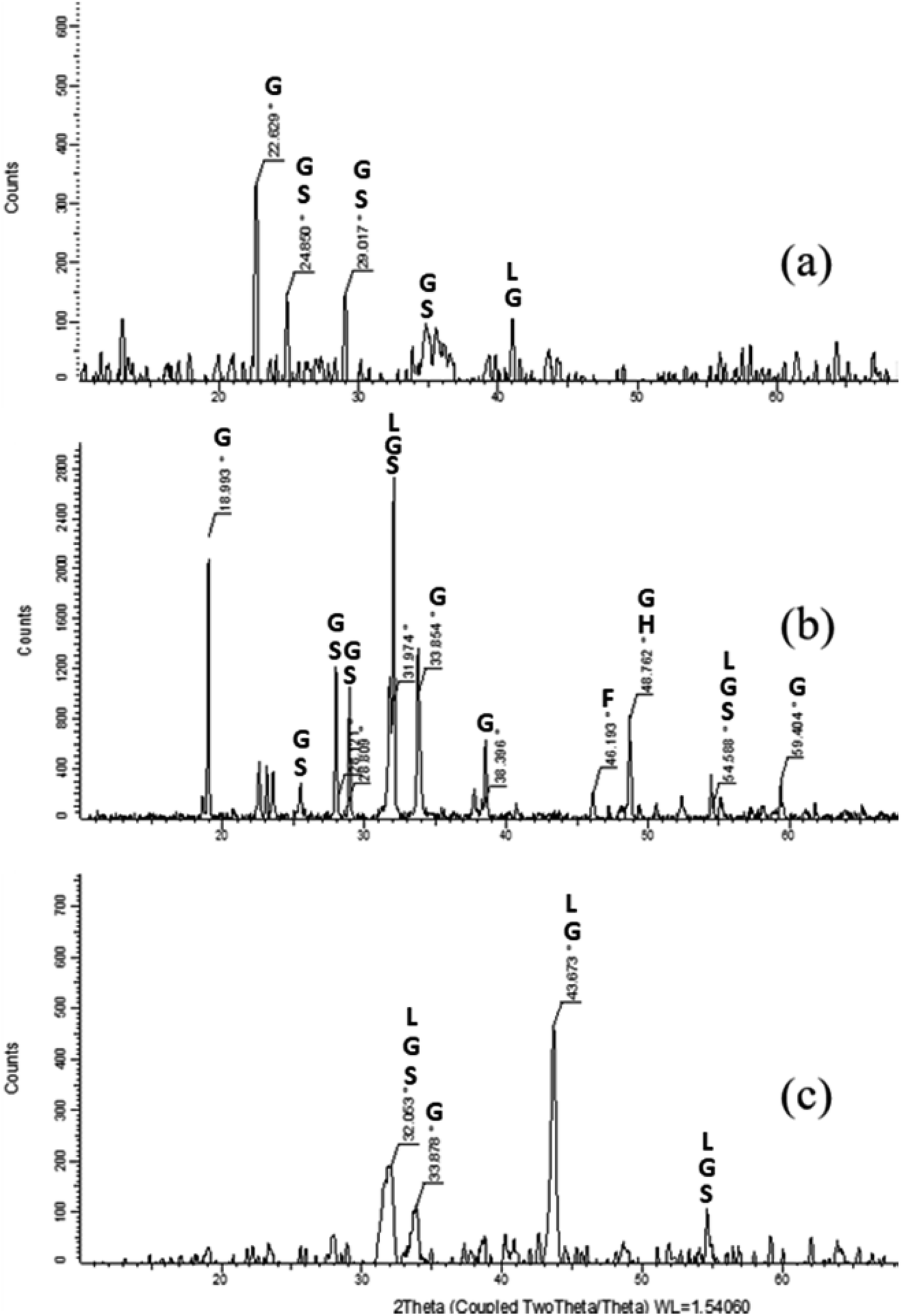 | ||
| Fig. 8 XRD images of residual precipitates from the production of samples (a) Res-F1 (b) Res-Fe-T2 and (c) precipitated iron(III) hydroxide (peak assignments in Table 4). Goethite (G), Schwertmannite (S), Lepidocrocite (L), Ferrihydrite (F) and Hematite (H). | ||
| Res-F1 | Res-Fe-T2 | Precipitate | Assignment |
|---|---|---|---|
| 22.6 | 19 | X | Goethite38,44 |
| 24.9 | 26.1 | X | Schwertmannite39 |
| Goethite45 | |||
| 29 | 28.8 | X | Synthetic schwertmannite46 |
| Synthetic goethite47 | |||
| X | 32 | 32.1 | Synthetic schwertmannite46 |
| Synthetic goethite47 | |||
| Goethite39 | |||
| Lepidocrocite39 | |||
| X | 33.9 | 33.9 | Goethite39,44,45 |
| 35 | X | X | Schwertmannite/goethite40 |
| X | 38.4 | X | Goethite39,44,45 |
| 41 | X | 43.7 | Goethite44,45 |
| Lepidocrocite39 | |||
| X | 46.2 | X | Ferrihydrite39 |
| X | 48.8 | X | Hematite/goethite39 |
| X | 54.6 | 54.5 | Schwertmannite/goethite39 |
| Lepidocrocite39 | |||
| X | 59.4 | X | Goethite39,44 |
As stated previously, the precipitate produced in the impregnating solution is only indicative of the iron oxide speciation deposited on the surface of the PAN. The FTIR indicated that a mixture of schwertmannite, goethite and lepidocrocite and the XRD confirms this mixture.
3.6 Catalysis of reactive orange-16 using fresh (F1) and regenerated (Fe-T2) PAN catalysts
Degradation studies of the catalysts on RO-16 are presented in Fig. 9. The deactivated catalyst, D1, was tested for catalysis, but the resulting activity was very low and ineffective. Fe-T2 showed better decolorisation of the dye (493.5 nm) as well as degradation of the aromatic ring (254 nm) in comparison to the fresh catalyst. With both catalysts, the removal of the aromatic ring (Fig. 9b) is lower in contrast to the decolorisation (Fig. 9a), which happens at a faster rate.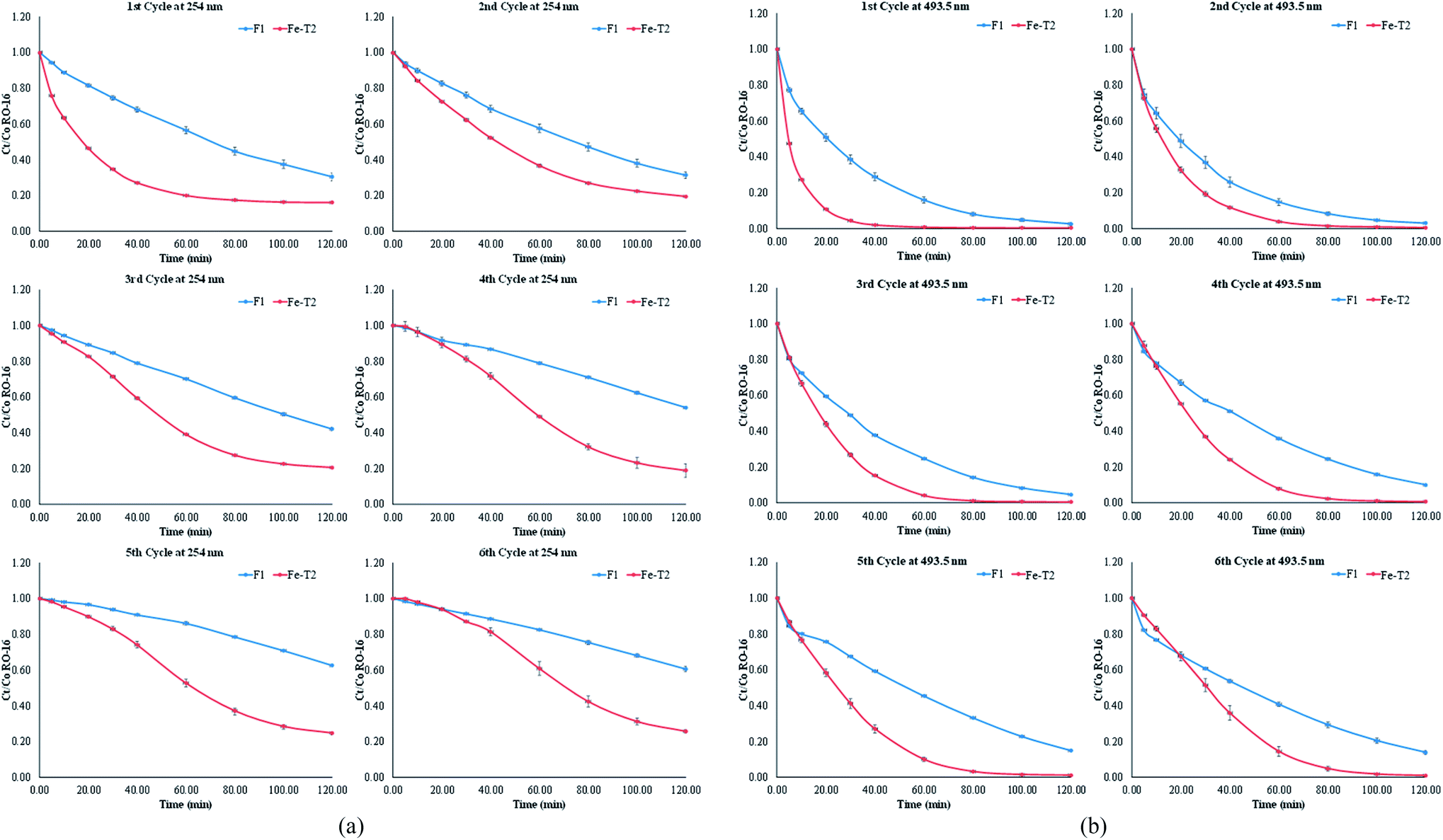 | ||
| Fig. 9 (a) Comparison of F1 and Fe-T2 in the catalytic degradation of RO-16 over six cycles at 254 nm [duration 120 min with starting pH = 3 at 25 °C, stirred at 400 rpm. [H2O2] = 125 ppm; [RO-16] = 50 ppm; volume = 100 mL]. (b) Comparison of F1 and Fe-T2 in the catalytic degradation of RO-16 over six cycles at 493.5 nm [duration 120 min with starting pH = 3 at 25 °C, stirred at 400 rpm. [H2O2] = 125 ppm]; [RO-16] = 50 ppm; volume = 100 mL]. | ||
| Sample | % dye removal | Order | Rate constant | R2 |
|---|---|---|---|---|
| F1 cycle 1 | 97.47 | 1st | 0.0301 min−1 | 0.9989 |
| F1 cycle 6 | 86.16 | 1st | 0.0154 min−1 | 0.9924 |
The catalysis results of Fig. 9b were fitted to kinetic orders as shown in Table 5. The F1 catalyst readily fitted to the 1st order regime with good fit. However, it was more difficult to fit the regenerated catalyst Fe-T2 to any specific kinetic regime. It can be seen from Table 6, that at different points in the reaction, different kinetic regimes are being followed-initially first order and then towards the end of the catalysis, second order.
| Sample | Time points | % dye removal | Order | Rate constant | R2 |
|---|---|---|---|---|---|
| Fe-T2 cycle 1 | 0–40 min | — | 1st | 0.0961 min−1 | 0.9912 |
| 40–100 min | 99.71 | 2nd | 0.1013 mg−1 L min−1 | 0.9915 | |
| Fe-T2 cycle 6 | 0–30 min | — | 1st | 0.0219 min−1 | 0.9931 |
| 30–80 min | — | 1st | 0.0474 min−1 | 0.9948 | |
| 80–120 min | 98.98 | 2nd | 0.0397 mg−1 L min−1 | 0.996 |
On first glance, the higher catalytic activity of the Fe-T2 catalyst may be due to higher leaching (Table 7). Leaching of iron has previously been related to the deactivation of the catalyst. In terms of the percentage loss of the iron from the catalytic system (6 g of catalyst in 100 mL of substrate) over 6 cycles, the Fe-T2 catalyst had a 5.10% loss of iron (0.85% loss of Fe per gram of catalyst over 6 cycles). This was higher in comparison to the 1.96% loss of iron (0.32% loss of Fe per gram of catalyst over 6 cycles) from the fresh catalyst.
| Cycle | Fe leachate (mg L−1) g−1 of catalyst | |
|---|---|---|
| F1 | Fe-T2 | |
| 1 | 0.213 | 1.198 |
| 2 | 0.562 | 1.123 |
| 3 | 0.392 | 1.174 |
| 4 | 0.376 | 1.227 |
| 5 | 0.316 | 1.144 |
| 6 | 0.293 | 1.016 |
It could be argued, that the high leaching from the Fe-T2 catalyst along with its improved catalytic activity, indicates contribution from homogenous Fenton catalysis. However, depending on the type of species leached, the contribution to catalysis may not be significant. An experiment to determine this was carried out below.
It is thought that the species of iron oxides on the catalyst is similar in structure and nature to the residues precipitated on Fe impregnation of the fibres using the iron(III) sulphate/sodium sulphate mixture. To test this theory, dye degradation using iron residues produced from the regenerated PAN catalyst (Fe-T2) were evaluated for catalytic activity. The same amount of iron as Res-Fe-T2 (residue produced during impregnation of the catalyst) was used as leached iron from catalysis with Fe-T2 to degrade the dye. Any leached iron from the surface of the Fe-T2 catalyst is likely to resemble the species on the Res-Fe-T2.
It was found that the Res-Fe-T2 complex (mimicking leachate from Fe-T2) decolorised 91% of the RO-16 dye after 2 hours but only 15% of the aromatic ring whereas Fe-T2 was able to decolorise almost 100% of RO16 and remove 80% of the aromatic ring over two hours.
To see whether the iron that leached from the fresh catalyst (F1) was active in the degradation of the dye RO16, an experiment as described in Section 2.31 was performed. In this experiment the catalyst was exposed to hydrogen peroxide at pH 3 and left for 4 hours. The catalyst was removed and the residual hydrogen peroxide was allowed to decompose over a 24 hour period. At this point fresh dye and fresh hydrogen peroxide solutions were added to see if the leachate from the catalyst was active in the degradation of the dye. It is important to note that in this experiment, the temperature at which catalysis was carried out was 30 °C and hence experienced a faster degradation rate in comparison with the F1 catalysis carried out at room temperature in Fig. 9a and b.
In Fig. 10, it was found that overall, the heterogeneous system (F1) had much better catalytic activity compared to the homogenous system (leachate) of F1. This is distinct at 10 minutes where the decolourisation of the dye (measured at 493.5) with the fresh catalyst was 5 times greater in comparison to that of the leachate. A similar trend is seen for the aromatic ring (254 nm) at 30 minutes.30 Thus again, the heterogenous systems F1 and Fe-T2 showed similar trends to each other in degradation of both the dye chromophore and aromatic ring whilst the leachates were only successful in decolorization but not destruction of the aromatic ring.
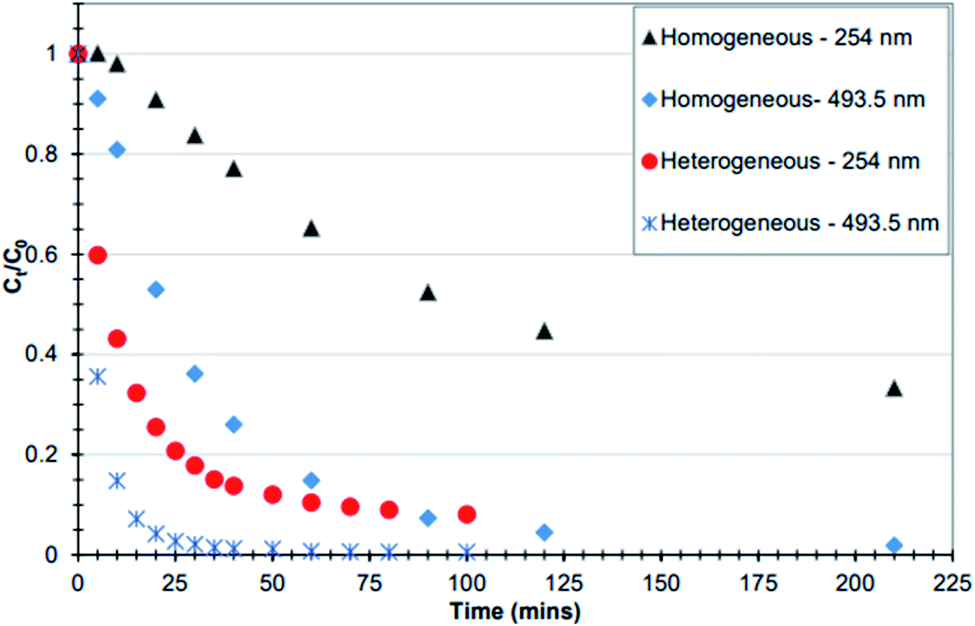 | ||
| Fig. 10 Comparison between homogeneous and heterogeneous catalytic decomposition of RO-16. The heterogeneous system was the F1 catalyst whilst the homogenous system contained leached iron from 6 g of F1 [experimental conditions: the iron in both processes was sourced from 6 g catalyst, pH 3, 125 mg L−1 H2O2 in 100 mL RO-16 (C0 = 50 mg L−1) at temperature = 30 °C] (reproduced from Upreti, 2018 (ref. 30)). | ||
4 Discussion
The deactivated catalyst was treated by EDC followed by either hydroxylamine (T1) or hydroxylamine and hydrazine (T2). The regeneration reactions may have activated the carboxyl groups to form a complex with iron. However, this was difficult to confirm by FT-IR.After reimpregnation, it was seen that there was an increase in the amount of iron which suggested that reintroduction of the functional groups for both T1 and T2 was successful. Fe-T2 has a higher iron content compared to Fe-T1 which suggests that it would be a more efficient catalyst. As Fe-T1 was solely treated with hydroxylamine, it was brittle in nature making it less useful in practical scale-up terms and was therefore not tested for catalytic activity in the degradation of RO-16. The brittleness was due to the crosslinking effect of the hydroxylamine in comparison to hydrazine. Although Fe-T2 had a higher iron content, it also lost 83% of its total content when exposed to EDTA whereas Fe-T1 only lost 19%. From the results of EPR, it was found that some iron species on Fe-T2 were present as a coordinated complex thus such a high proportion of iron to be lost by EDTA treatment is surprising.
The SEM images show particulates on the surface of the Fe-T2 sample and not on the Fe-T1 sample. Whilst the EDX results show that the particulates on the surface of the Fe-T2 catalyst had a higher percentage of iron and oxygen which gives a great deal of support to the theory of the particulates being iron oxide species. Further evidence of the particulates being iron oxide species is their removal on treatment with EDTA as it is known that EDTA can cause dissolution of iron oxides.35,48 This is shown in Fig. 3 where after EDTA treatment the surface of Fe-T2 is smoother. The lack of iron oxide and the low iron content indicates that the iron on the Fe-T1 sample is all coordinated/chelated iron which cannot be easily removed via EDTA dissolution.
As the EPR results confirmed the presence of coordinated iron in the Fe-T2 sample, the following mechanism of iron impregnation is suggested. The authors propose that in the first instance iron is indeed chelated to the functional groups on the surface of the polymer. These groups then provide nucleating sites for iron oxide species deposition and precipitation. It is likely that any leached ammonia or hydrazine from the treated polymer may also leach out and raise the pH to provide an ideal environment for iron oxide species formation.13 These particulates are also seen on the F1 catalyst (Fig. 3) but unlike Fe-T2 are not removed via EDTA treatment. The iron species on the surface of the F1 catalyst may therefore be in a different form than that on Fe-T2 and may be more crystalline in nature and less likely to dissolve. Thus, nucleating sites upon which iron oxides are deposited is likely to be dependent on the nature of iron chelates initially present on the PAN which ultimately effects the species of iron oxide deposited.
In an attempt to determine the species of iron oxides, the precipitate produced in the impregnating solution was analysed. The FTIR data suggested that the precipitate contained mostly a mixture of iron oxides: schwertmannite, goethite and possibly lepidocrocite. The presence of the iron oxide species was confirmed by XRD analysis. Comparing Res-F1 and Res-(Fe-T2) to synthetic iron oxides, it was found that Res-F1 was likely to contain more goethite whilst Res-(Fe-T2) resembled mostly schwertmannite. It is likely that the type of iron oxide deposited is influenced by the pH of the iron(III) sulphate/sodium sulphate solution as well as the pH of the surface of the fibres (dependent on the nature and amount of the different functional groups present). To synthesise the F1 catalyst, the hydrazine and hydroxylamine treatment is carried out at pH > 9 and then placed into the impregnating iron solution. However, the Fe-T2 catalyst was pH adjusted to 3 before impregnation. From literature, it is likely that the form of iron oxide species in acidic conditions is schwertmannite whilst at alkaline conditions it is converted more to goethite.43 The presence of schwertmannite may also be caused by the ammonia leaching out of the catalyst30 into the impregnating solution leading to the iron(III) sulphate precipitating into schwertmannite.42 In regards to the removal of iron from Fe-T2 with EDTA in Section 3.3, as schwertmannite is more soluble compared to goethite due to its poor crystallinity, it is more likely to be dissolved by EDTA treatment.
Catalytic activity of the regenerated catalyst Fe-T2 was compared to that of the fresh F1 catalyst in batch recycling. Compared to the fresh catalyst, the regenerated Fe-T2 sample performed better but had higher leaching (0.85% loss of Fe per gram of Fe-T2 catalyst over 6 cycle compared to 0.32% loss of Fe per gram of F1 catalyst over 6 cycles). However, utilising the Res-Fe-T2 precipitate as a catalyst showed lower catalytic activity compared to the actual Fe-T2 catalyst. This suggests that the Res-Fe-T2 complexes, similar to the species leached from Fe-T2, is catalytically active but not to the same extent as the complexed iron present on the surface of Fe-T2. Therefore, the contribution from the leached iron oxide species on the catalyst does not provide as an extensive oxidative degradation as that of the catalyst itself. This suggests that there is a significant contribution to catalysis from the ligated Fe species on the catalyst and less from the leached iron species.
In regards to the difference in activity of the F1 catalyst with the Fe-T2, iron on the Fe-T2 catalyst is held less strongly in comparison with the speciation of the iron on the fresh catalyst F1, hence the high leaching and greater catalytic activity for Fe-T2. An equivalent amount of Fe as iron(III) sulphate salt was not used for the comparison of leached iron species as the speciation of iron is likely to be very different from that leached off the fresh catalyst F1. This is because, the leached iron is likely to be in the form of complexed iron-possibly ligated to dye and other intermediary catalytic products as well as leached ammonia, hydroxylamine and hydrazine. These complexed iron leachates may therefore have very different catalytic activity to those of the iron sulphate salts. Fig. 10 suggests that any contribution from purely homogenous Fenton will be negligible in the degradation of RO-16 by F1.30
From an industrial scale-up perspective, the F1 catalyst has already shown promise in performing efficient catalysis in continuous flow set-up. However, regeneration is an added cost to any process. In reference to the system set out in this study, the use of EDC as a cross-linking agent is costly and therefore a cheaper compound with similar properties should be used. Furthermore, the process would need to be optimised to minimise downtime which would suggest a reduction in the number of steps or perhaps combining stages. As this study is focused on catalyst regeneration, this would mean future work to determine catalyst lifetime would need to be done in continuous flow. This would include taking fresh catalyst to deactivation, regeneration of deactivated catalyst and finally taking the regenerated catalyst to exhaustion. Only this would truly indicate the scale-up potential of the regeneration procedure.
5 Conclusions
This study has shown that the deactivated modified PAN catalyst can be reactivated using room temperature activation with EDC carried out together with inorganic amines, in this case, hydroxylamine or/and hydrazine. Nevertheless, the regeneration with both hydrazine and hydroxylamine with EDC resulted in a catalyst with a high iron content (13.5 mg g−1). Iron on the catalysts were determined by EPR to be complexed and the results of SEM showed in addition the presence of particulates on the surface of the PAN. FTIR and XRD suggested that these particulates were a mixture of schwertmannite, or other basic iron sulphates and goethite iron oxides. During catalysis with the regenerated catalysts, the degradation efficiency was better in comparison to the fresh catalyst (F1) however this was in part a result of significantly higher leaching from the regenerated catalyst. Nevertheless, the leaching from the regenerated catalyst is likely to be in a similar form to that of the residue formed on iron impregnation of the regenerated catalysts (Res-Fe-T2) which was found to be catalytically active but to a lesser degree compared to the catalyst Fe-T2. It was also determined, that the leached iron from the fresh catalyst F1 which is likely to be in complexed form had negligible contribution to the catalytic system and therefore that the majority of the catalytic process was heterogenous and not homogenous Fenton's.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the research grant provided by Schlumberger Faculty of the Future fellowship for Dr Caroline Akinremi and the SEM/EDX facility at De Montfort University Leicester.References
- Y. Deng and R. Zhao, J. Ind. Eng. Chem., 2015, 1, 167–176 Search PubMed
.
- J. He, X. Yang, B. Men and D. Wang, J. Environ. Sci., 2016, 39, 97–106 CrossRef PubMed
- V. V. Ishtchenko, K. D. Huddersman and R. F. Vitkovskaya, Appl. Catal., A, 2003, 242, 123–137 CrossRef
- V. V. Ishtchenko, R. F. Vitkovskaya and K. D. Huddersman, Appl. Catal., A, 2003, 242, 221–231 CrossRef CAS
- G. T. Chi and K. D. Huddersman, J. Adv. Oxid. Technol., 2011, 14, 235–243 CAS
- G. T. Chi, Z. K. Nagy and K. D. Huddersman, Prog. React. Kinet. Mech., 2011, 36, 189–214 CrossRef CAS
- Y.-C. Dong, F. Du, H.-X. Ma, Z.-B. Han and Y.-N. Cao, Guocheng Gongcheng Xuebao, 2008, 8, 359–365 CAS
- K. Saeed, S. Haider, T. J. Oh and S. Y. Park, J. Membr. Sci., 2008, 322, 400–405 CrossRef CAS
- B. Li, Y. Dong and Z. Ding, J. Environ. Sci., 2013, 25, 1469–1476 CrossRef CAS
- N. Usachova, G. Leitis, A. Jirgensons and I. Kalvinsh, Synth. Commun., 2010, 40(6), 927–935 CrossRef CAS
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef PubMed
- C. R. Cammarata, M. E. Hughes and C. M. Ofner, Mol. Pharm., 2015, 12, 783–793 CrossRef
- K. Aiedeh and M. O. Taha, Eur. J. Pharm. Sci., 2001, 13, 159–168 CrossRef
- R. Aiello, J. E. Fiscus, H. C. Zur Loye and M. D. Amiridis, Appl. Catal., A, 2000, 192, 227–234 CrossRef
- H. S. Roh, A. Platon, Y. Wang and D. L. King, Catal. Lett., 2006, 110, 1–6 CrossRef
- D. J. Fullerton, A. V. K. Westwood, R. Brysdon, M. V. Twigg and J. M. Jones, Catal. Today, 2003, 81, 659–671 CrossRef
- Y. Diao, P. Yang, R. Yan, L. Jiang, L. Wang, H. Zhang, C. Li, Z. Li and S. Zhang, Appl. Catal., B, 2013, 142, 329–336 CrossRef
- B. Blanc, A. Bourrel, P. Gallezot, T. Haas and P. Taylor, Green Chem., 2000, 2, 89–91 RSC
- P. Gallezot, P. J. Cerino, B. Blanc, G. Fleche and P. Fuertes, J. Catal., 1994, 146, 93–102 CrossRef CAS
- Y. Yu, J. Wang, J. Chen, X. He, Y. Wang, K. Song and Z. Xie, J. Environ. Sci., 2016, 47, 100–108 CrossRef
- B. Subramanian, S. Y. Christou, A. M. Efstathiou, V. Namboodiri and D. D. Dionysiou, J. Hazard. Mater., 2011, 186, 999–1006 CrossRef CAS
- E. C. Zuleta, G. A. Goenaga, T. A. Zawodzinski, T. Elder and J. J. Bozell, Catal. Sci. Technol., 2020, 10, 403–413 RSC
- R. D. Sun, A. Nakajima, T. Watanabe and K. Hashimoto, J. Photochem. Photobiol., A, 2003, 154, 203–209 CrossRef CAS
- M.
G. Jeong, E. J. Park, H. O. Seo, K. D. Kim, Y. D. Kim and D. C. Lim, Appl. Surf. Sci., 2013, 271, 164–170 CrossRef CAS
- X. Yan, J. Li, C. Ma, Y. Tang, X. Kong and J. Lu, Water Sci. Technol., 2020 DOI:10.2166/wst.2020.091
- L. Cao, Z. Gao, S. L. Suib, T. N. Obee, S. O. Hay and J. D. Freihaut, J. Catal., 2000, 196, 253–261 CrossRef CAS
- H. Wang, Q. Liu and C. You, Appl. Catal., A, 2019, 572, 15–23 CrossRef CAS
- P. M. B. Chagas, A. A. Caetano, A. A. Tireli, P. H. S. Cesar, A. D. Corrêa and I. do Rosário Guimarães, Sci. Rep., 2019, 9, 1–15 CrossRef
- Z. Yang, J. Qian, A. Yu and B. Pan, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6659–6664 CrossRef CAS
- P. D. Upreti, PhD thesis, De Montfort University, 2018
- F. A. Taiwo, Spectroscopy, 2003, 17, 53–63 CrossRef CAS
- J. T. Weisser, M. J. Nilges, M. J. Sever and J. J. Wilker, Inorg. Chem., 2006, 45, 7736–7747 CrossRef CAS
- C. T. Migita, K. Ogura and T. Yoshino, J. Chem. Soc., Dalton Trans., 1985, 1077–1080 RSC
- Y.-C. Dong, Z.-B. Han, C.-Y. Liu and F. Du, Sci. Total Environ., 2010, 408, 2245–2253 CrossRef CAS
- B. Nowack and L. Sigg, Geochim. Cosmochim. Acta, 1997, 61, 951–963 CrossRef CAS
- N. M. El-Sawy, Polym. Int., 2000, 49, 533–538 CrossRef CAS
- E.-S. H. El-Mossalamy, F. M. Al-Nowaiser, S. A. Al-Thabaiti, A. O. Al-Youbi, S. N. Baschel and A. Y. Obaid, Monatshefte für Chemie - Chemical Monthly, 2007, 138, 853–857 CrossRef CAS
- F. Huang, Y. Xu, S. Liao, D. Yang, Y. Lo Hsieh and Q. Wei, Materials, 2013, 6, 969–980 CrossRef CAS
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory: Preparation and Characterization, John Wiley & Sons, Weinheim, 2000 Search PubMed
- P. S. R. Prasad, K. S. Prasad, V. K. Chaitanya, E. V. S. S. K. Babu, B. Sreedhar and S. Ramana Murthy, Journal of Asian Earth Sciences, 2005, 27, 503–511 CrossRef
- M. Veneranda, J. Aramendia, L. Bellot-Gurlet, P. Columban, K. Castro and J. M. Madariaga, Corros. Sci., 2018, 133, 68–77 CrossRef CAS
- Z. Zhang, X. Bi, X. Li, Q. Zhao and H. Chen, RSC Adv., 2018, 8, 33583–33599 RSC
- J. F. Boily, P. L. Gassman, T. Peretyazhko, J. Szanyi and J. M. Zachara, Environ. Sci. Technol., 2010, 44, 1185–1190 CrossRef CAS
- Y. Wang, Y. Gao, L. Chen and H. Zhang, Catal. Today, 2015, 252, 107–112 CrossRef CAS
- C. Blanco-Andujar, D. Ortega, Q. A. Pankhurst and N. T. K. Thanh, J. Mater. Chem., 2012, 22, 12498–12506 RSC
- C. L. Vithana, L. A. Sullivan, R. T. Bush and E. D. Burton, Geoderma, 2015, 249, 51–60 CrossRef
- P. Pulišová, B. Máša, E. Michalková, E. Večerníková, M. Maříková, P. Bezdička, N. Murafa and J. Šubrt, J. Therm. Anal. Calorim., 2014, 116, 625–632 CrossRef
- K. Norén, J. S. Loring, J. R. Bargar and P. Persson, J. Phys. Chem. C, 2009, 113, 7762–7771 CrossRef
| This journal is © The Royal Society of Chemistry 2020 |