
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple and efficient synthesis of bicyclic enol-carbamates: access to brabantamides and their analogues†
Ondrej Záborský *a,
Ľudmila Petrovičováa,
Jana Doháňošováb,
Ján Moncolc and
Róbert Fischera
*a,
Ľudmila Petrovičováa,
Jana Doháňošováb,
Ján Moncolc and
Róbert Fischera
aInstitute of Organic Chemistry, Catalysis and Petrochemistry, Slovak University of Technology in Bratislava, Radlinského 9, Bratislava, 81237, Slovak Republic. E-mail: ondrej.zaborsky@stuba.sk
bCentral Laboratories, Slovak University of Technology in Bratislava, Radlinského 9, Bratislava, 81237, Slovak Republic
cInstitute of Inorganic Chemistry, Technology and Materials, Slovak University of Technology in Bratislava, Radlinského 9, Bratislava, 81237, Slovak Republic
First published on 13th February 2020
Abstract
A novel synthetic approach towards the formation of the unusual bicyclic enol-carbamates, as found in brabantamides A–C, is reported. The bicyclic oxazolidinone framework was obtained in very good yield and with high E/Z selectivity from a readily available β-ketoester under mild reaction conditions using Tf2O and 2-chloropyridine tandem. The major E isomer was used in the synthesis of the brabantamide A analogue.
Brabantamides A–C (1–3) (Fig. 1) were first isolated in 2000 from the culture extracts of Pseudomonas fluorescens.1 They displayed nanomolar inhibitory activity towards lipoprotein-associated phospholipase A2 (Lp-PLA2) and therefore they could be used in the treatment of the inflammatory diseases such as atherosclerosis.1–3 It was found that the sugar moiety is not necessary for the biological activity against Lp-PLA2. In contrast, deglycosylated brabantamides A and C showed improved inhibitory activity compared to their natural counterparts.3 Moreover, simplified brabantamide analogues with amide functional group and long alkyl side chains showed even higher inhibitory effect.4 Brabantamides A–C also exhibit significant activity against Gram-positive bacteria, fungi, and oomycetes.5,6 A recent study confirmed that the bicyclic scaffold and the long lipophilic side chain are essential for the antibacterial activity.7
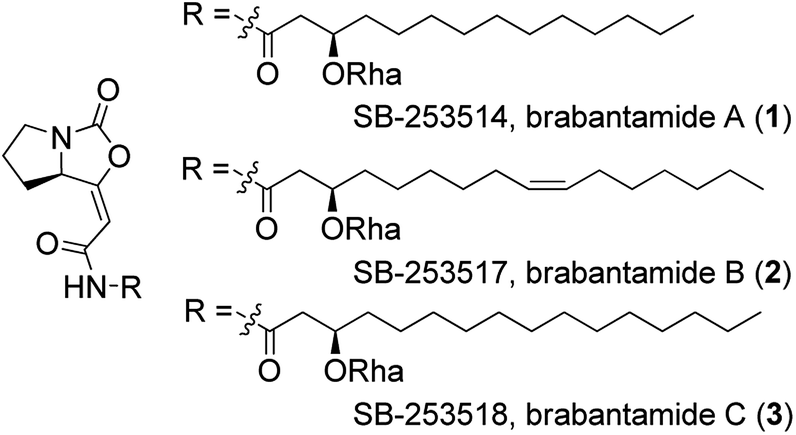 | ||
| Fig. 1 Structures of brabantamides A–C (1–3). Rha = rhamnose. | ||
Surprisingly, only five reports concerning the synthesis of the bicyclic oxazolidinone with an exocyclic double bond have been reported to date.
In 2000, Pinto et al. prepared a series of analogues of brabantamide A where the enol-carbamate 5 was first obtained by direct iodocyclization from acetylene derivative 4 and then converted into key ester 6 by carbonylation of the corresponding vinyl iodide in the presence of 2-(trimethylsilyl)ethanol and PdCl2 (Scheme 1a).4 Another synthesis of the related esters 9 has been reported by Snider et al. in 2006, employing Wittig reaction between stabilized ylides and bicyclic oxazolidindione 8 synthesized from L-proline 7 (Scheme 1b).8 Shortly after, bicyclic enol-carbamate 10 with an exocyclic methylene group was synthesized in one step from acetylene derivative 4 using gold(I) catalysts (Scheme 1c).9 Very recently, Witte et al. reported the synthesis of series of Z-analogues of brabantamides 12 by cyclization of β-ketoamides 11 using CDI (Scheme 1d).7 As a part of our ongoing research program aimed at the utilization of trifluoromethanesulfonic anhydride (Tf2O)/2-halopyridine (2-XPy) tandem in the synthesis of bioactive natural products and their analogues, we envisioned that similar reaction conditions could be effectively used to form the bicyclic oxazolidinone framework as found in brabantamides.
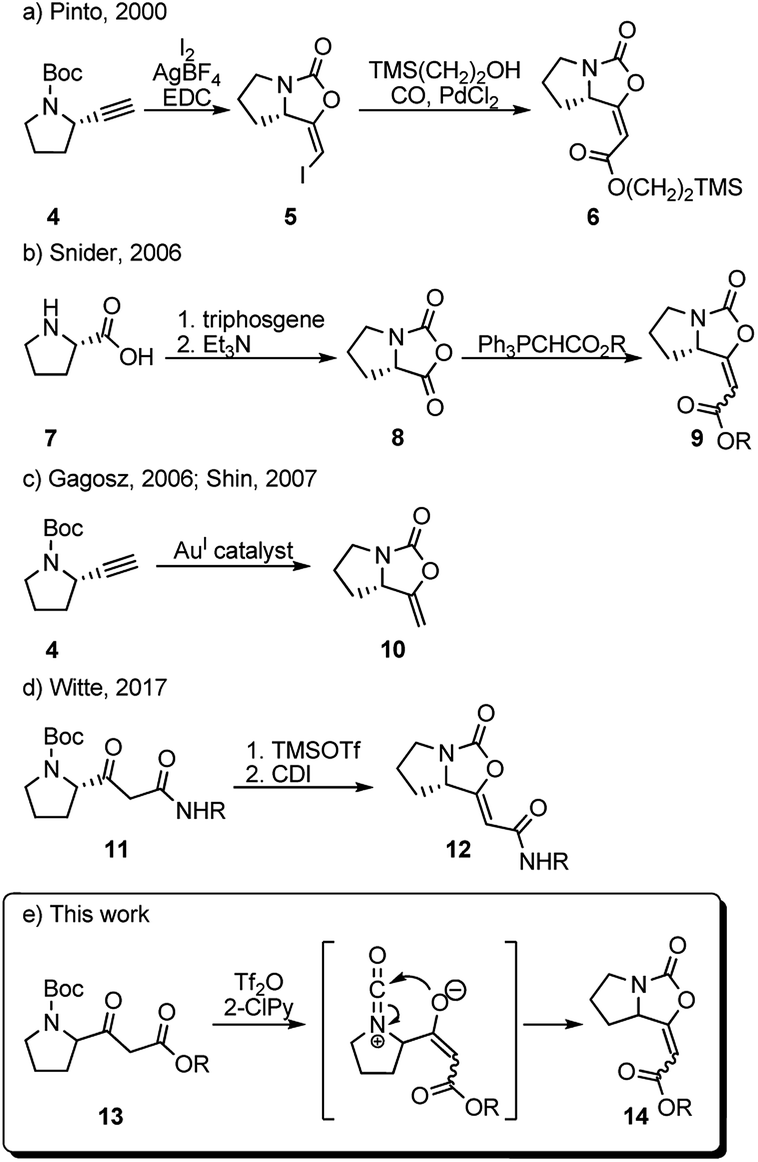 | ||
| Scheme 1 Literature syntheses of bicyclic enol-carbamates and method proposed herein. | ||
The combination of Tf2O/2-XPy was extensively and successfully used in the amide activation10,11 as well as in generating isocyanate species from N-Boc and N-Cbz protected amines.12 We anticipated that the N-Boc-protected β-ketoester 13 could react in its enolate form with in situ generated isocyanate ion by intramolecular 5-endo-dig cyclization to give bicyclic enol-cyclocarbamate 14 (Scheme 1e).
At the start of our investigation, model β-ketoester 15, prepared from N-Boc-L-proline,7 was chosen as the model substrate to identify optimal reaction conditions (Table 1). According to Kokotos' protocol,12c the initial using of 1.5 equivalents of Tf2O and 3 equivalents of 2-ClPy led to a full conversion of the substrate 15 in 15 minutes (monitored by TLC) (Table 1, entry 7). The inspection of 1H NMR spectrum of the crude reaction mixture confirmed the presence of only desired product 16 almost exclusively as E isomer (E/Z ratio 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) which was isolated in 53% combined yield. Gratifyingly, lowering the amount Tf2O (1.1 equiv.) resulted in a significant increase of the yield up to 80% with the slight decrease of E isomer 16a (Table 1, entry 8).13 Any variation of the amount of 2-ClPy did not have any positive impact on the reaction (Table 1, entries 9 and 10). The use of other 2-halopyridines reduced yield of 16 and prolonged reaction times were observed (Table 1, entries 11–13). For comparison, when we applied Witte's reaction conditions, yield dropped remarkably and E/Z selectivity disappeared completely (Table 1, entry 14). At last, we tested other bases commonly used in the combination with Tf2O. Triethylamine, 4-dimethylaminopyridine, pyridine, and 2,6-lutidine resulted only in traces of product 16 (Table 1, entries 2–5), as well as when no base was used (Table 1, entry 1). Using DBU, enol-carbamate 16 was formed in slightly improved E/Z ratio (Table 1, entry 6). Nevertheless, 1H NMR spectrum of the crude reaction mixture showed the formation of a large amount of unidentified by-products and desired product was isolated only in 41% yield.
:7) which was isolated in 53% combined yield. Gratifyingly, lowering the amount Tf2O (1.1 equiv.) resulted in a significant increase of the yield up to 80% with the slight decrease of E isomer 16a (Table 1, entry 8).13 Any variation of the amount of 2-ClPy did not have any positive impact on the reaction (Table 1, entries 9 and 10). The use of other 2-halopyridines reduced yield of 16 and prolonged reaction times were observed (Table 1, entries 11–13). For comparison, when we applied Witte's reaction conditions, yield dropped remarkably and E/Z selectivity disappeared completely (Table 1, entry 14). At last, we tested other bases commonly used in the combination with Tf2O. Triethylamine, 4-dimethylaminopyridine, pyridine, and 2,6-lutidine resulted only in traces of product 16 (Table 1, entries 2–5), as well as when no base was used (Table 1, entry 1). Using DBU, enol-carbamate 16 was formed in slightly improved E/Z ratio (Table 1, entry 6). Nevertheless, 1H NMR spectrum of the crude reaction mixture showed the formation of a large amount of unidentified by-products and desired product was isolated only in 41% yield.
|
|||||
|---|---|---|---|---|---|
| Entry | Tf2O | Base | Time (min) | 16a:16ba |
Yieldb (%) |
| a Ratio determined by 1H NMR of the crude reaction mixture.b Isolated combined yield.c Traces of products.d Reactions performed with 1.1 equiv. of Tf2O did not lead to full conversion of ester 15.e Reactions were performed on 1 mmol of ester 15.f Reaction mixture contained a large amount of unidentified by-products.g Reaction conditions: (1) TMSOTf (2 equiv.), CH2Cl2, 0 °C, 1 h. (2) CDI (1.5 equiv.), CH2Cl2, 0 °C – rt, overnight.7 | |||||
| 1 | 1.5 | — | 60 | — | –c |
| 2 | 1.5 | Et3N | 60 | — | –c |
| 3 | 1.5 | DMAP | 60 | — | –c |
| 4 | 1.5 | Pyridine | 60 | — | –c |
| 5 | 1.5d | 2,6-Lutidine | 60 | — | –c |
| 6 | 1.5d | DBU | 60 | 90:10 |
41e,f |
| 7 | 1.5 | 2-ClPy | 15 | 93:7 |
53e |
| 8 | 1.1 | 2-ClPy | 15 | 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :15 :15 |
80e |
| 9 | 1.1 | 2-ClPy (1.5 equiv.) | 40 | 85:15 |
75e |
| 10 | 1.1 | 2-ClPy (5 equiv.) | 15 | 87:13 |
64e |
| 11 | 1.1 | 2-FPy | 15 | 89:11 |
71e |
| 12 | 1.1 | 2-BrPy | 70 | 86:14 |
73e |
| 13 | 1.1 | 2-IPy | 90 | 86:14 |
68e |
| 14 | Witte's protocolg | Overnight | 50:50 |
36e | |
It is noteworthy that the reaction can be performed on a gram scale without affecting the yield and both isomers are easily separable by FCC (see the ESI†).
The 1H and 13C NMR data of the major E isomer 16a were consistent with those published previously.8 Possible racemization in the course of the reaction was dismissed based on the comparing specific optical rotation with the published data for 16a ([α]22D = −261.1 (c 1.01, MeOH); ref. 8: [α]22D = −207 (c 1.0, MeOH)). Most importantly, X-ray crystallographic analysis of 16a (Fig. 2; see the ESI† for further details)14 confirmed its absolute configuration on the C-7a carbon atom.
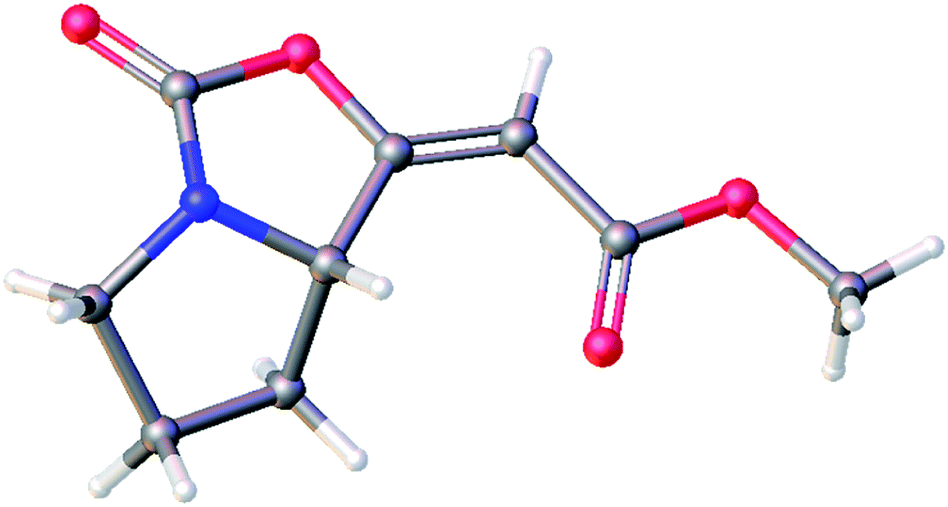 | ||
| Fig. 2 Molecular structure of the enol-carbamate E-16a confirmed by X-ray crystallographic analysis. | ||
The minor Z isomer 16b was isolated for the first time as the pure compound and was fully characterized. Its structure was assigned on the basis of its 1H, 13C, COSY, HSQC, and HMBC NMR spectra.
A plausible mechanism of the cyclization of β-ketoester 15 was based upon previous works12a–c and it is depicted in Scheme 2. Isocyanate cation II, as a key intermediate, can be formed directly from iminium triflate I (path A) or through the formation of carbamoyl triflate III with subsequent elimination of triflate ion spontaneously (path B). Ester enolate moiety IV then reacts as O-nucleophile via 5-endo-dig cyclization and leads predominantly to the formation of the enol-carbamate 16a.
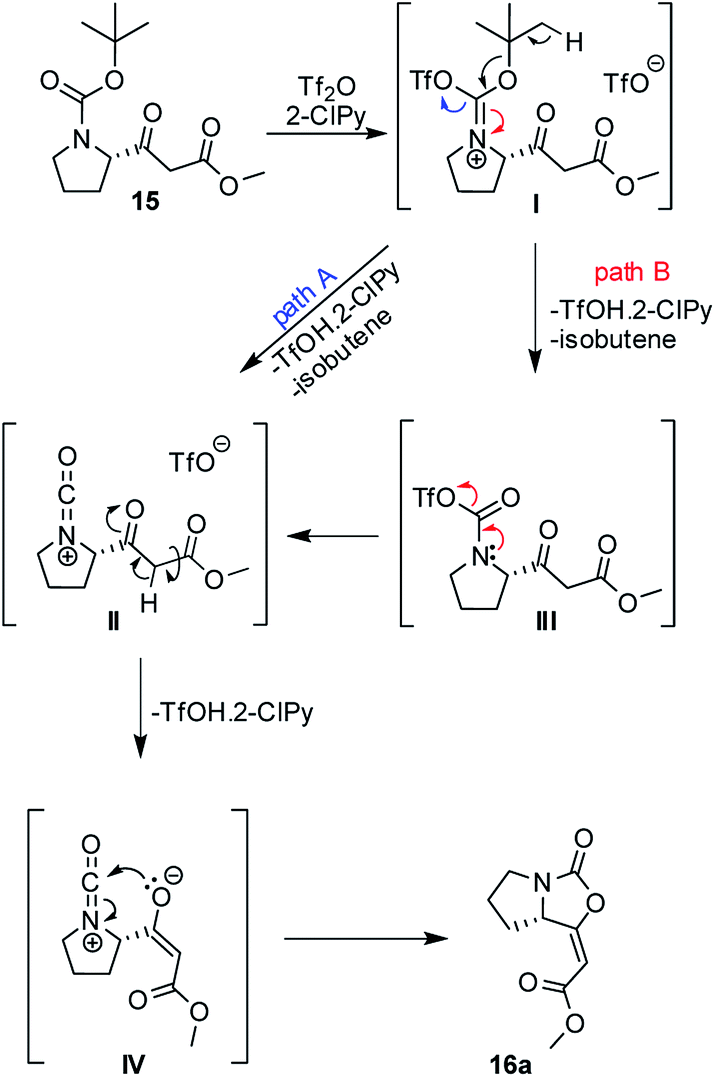 | ||
| Scheme 2 Plausible mechanism of the cyclization β-ketoester 15. | ||
Next, the optimized conditions were briefly applied in the synthesis of the brabantamide A analogue 21 (Scheme 3). Starting β-ketoester 18 was synthetized in two steps in a 70% yield using both commercially available N-Boc-D-proline 17 and 2-(trimethylsilyl)ethanol. It ought to be mentioned that previously examined hydrolysis of the corresponding methyl ester 16a under acidic as well as basic conditions failed due to the instability of the bicyclic enol-carbamate.3,8
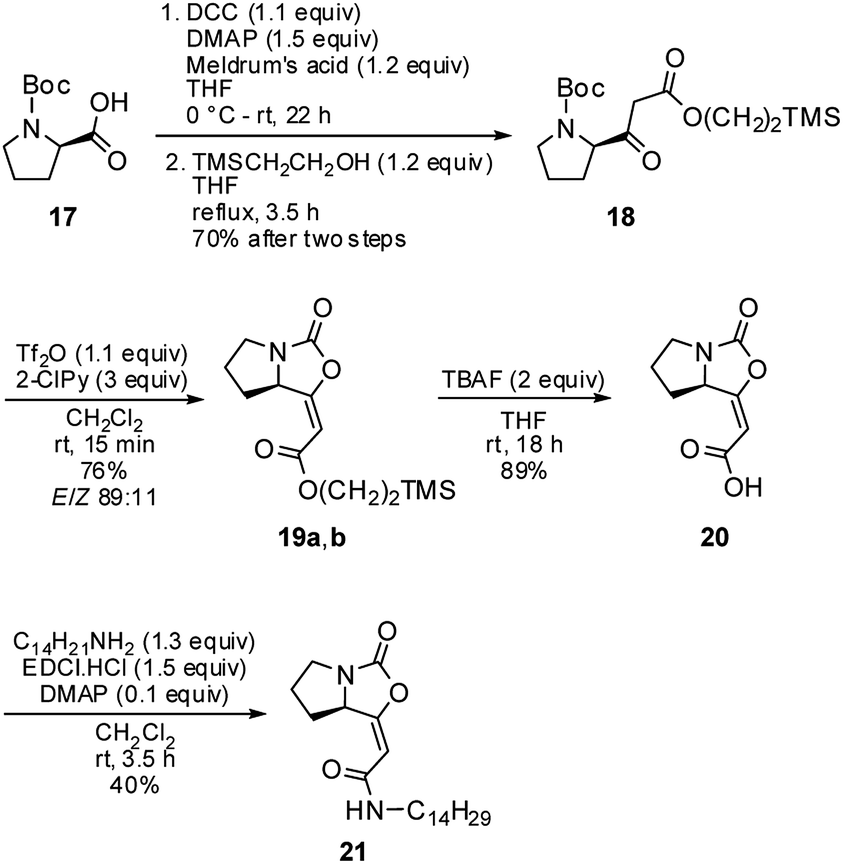 | ||
| Scheme 3 Synthesis of the brabantamide A analogue 21. | ||
Subsequent cyclization of ester 18 using optimized reaction conditions afforded enol-carbamate 19 in 76% yield as a mixture of E and Z isomers in a ratio of 89:11. After isolation of the major isomer E-19a, it was treated with TBAF, providing free acid 20 in 89% yield. Finally, an amidation of 20 with tetradecylamine in the presence of EDCI gave amide 21 in moderate 40% yield. Both free acid 20 and amide 21 were fully characterized for the first time and their structures were assigned on the basis of its 1H, 13C, COSY, HSQC, and HMBC NMR spectra. Moreover, their structures were unambiguously confirmed by X-ray crystallographic analysis (Fig. 3; see ESI† for further details).14
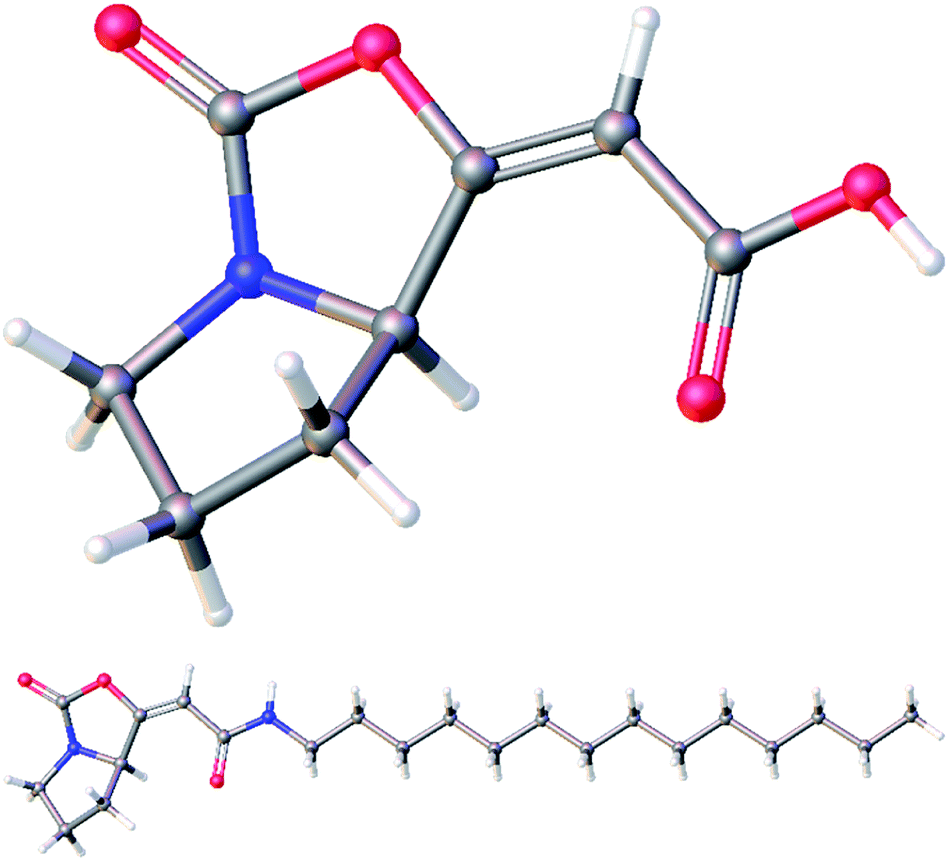 | ||
| Fig. 3 Molecular structures of acid 20 (top) and amide 21 (bottom) confirmed by X-ray crystallographic analysis. | ||
Conclusions
In conclusion, a new method of preparing bicyclic enol-carbamates with exocyclic double bond has been developed. Bicyclic oxazolidinone framework was obtained in one step from readily available β-ketoesters in very good yields and with high E/Z selectivity under mild reaction conditions using Tf2O and 2-chloropyridine tandem. The simplicity of this method was exemplified by a short and effective synthesis of the analogue of brabantamide A from commercially available N-Boc-D-proline in five steps with an overall 17% yield.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Slovak Grant Agency for Science VEGA (project no. 1/0552/18) and the Research and Development Operational Programmes funded by the ERDF (ITMS project no. 26240120001 and 26240120025). This article was also created with the support of the MŠVVaŠ of the Slovak Republic within the Research and Development Operational Programme for the project “University Science Park of STU Bratislava” (ITMS project no. 26240220084) co-funded by the European Regional Development Fund.Notes and references
- J. Thirkettle, E. Alvarez, H. Boyd, M. Brown, E. Diez, J. Hueso, S. Elson, M. Fulston, C. Gershater, M. L. Morata, P. Perez, S. Ready, J. M. Sanchez-Puelles, R. Sheridan, A. Stefanska and S. Warr, J. Antibiot., 2000, 53, 664 CrossRef CAS PubMed.
- D. J. Busby, R. C. B. Copley, J. A. Hueso, S. A. Readshaw and A. Rivera, J. Antibiot., 2000, 53, 670 CrossRef CAS.
- J. Thirkettle, J. Antibiot., 2000, 53, 733 CrossRef CAS PubMed.
- I. L. Pinto, H. F. Boyd and D. M. B. Hickey, Bioorg. Med. Chem. Lett., 2000, 10, 2015 CrossRef CAS.
- K. Reder-Christ, Y. Schmidt, M. Dӧrr, H.-G. Sahl, M. Josten, J. M. Raaijmakers, H. Gross and G. Bendas, Biochim. Biophys. Acta, 2012, 1818, 566 CrossRef CAS PubMed.
- P. F. Andersson, J. Levenfors and A. Broberg, BioControl, 2012, 57, 463 CrossRef CAS.
- P. Dockerty, J. G. Edens, M. B. Tol, D. Morales Angeles, A. Domenech, Y. Liu, A. K. H. Hirsch, J.-W. Veening, D.-J. Scheffers and M. D. Witte, Org. Biomol. Chem., 2017, 15, 894 RSC.
- J. R. Duvall, F. Wu and B. B. Snider, J. Org. Chem., 2006, 71, 8579 CrossRef CAS.
- (a) A. Buzas and F. Gagosz, Synlett, 2006, 17, 2727 Search PubMed; (b) E.-S. Lee, H.-S. Yeom, J.-H. Hwang and S. Shin, Eur. J. Org. Chem., 2007, 3503 CrossRef CAS.
- For reviews, see: (a) D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421 CrossRef CAS PubMed; (b) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899 RSC.
- For selected examples of the amide activation, see: (a) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592 CrossRef CAS PubMed; (b) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254 CrossRef CAS PubMed; (c) M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096 CrossRef CAS PubMed; (d) S.-L. Cui, J. Wang and Y.-G. Wang, J. Am. Chem. Soc., 2008, 130, 13526 CrossRef CAS; (e) O. K. Ahmad, M. D. Hill and M. Movassaghi, J. Org. Chem., 2009, 74, 8460 CrossRef CAS; (f) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228 CrossRef CAS PubMed; (g) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462 CrossRef CAS PubMed; (h) T. Wezeman, S. Zhong, M. Nieger and S. Bräse, Angew. Chem., Int. Ed., 2016, 55, 3823 CrossRef CAS PubMed; (i) S. Shaaban, V. Tona, B. Peng and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 10938 CrossRef CAS PubMed; (j) B. Mátravölgyi, T. Hergert, E. Bálint, P. Bagi and F. Faigl, J. Org. Chem., 2018, 83, 2282 CrossRef PubMed; (k) H. Chen, J.-L. Ye and P.-Q. Huang, Org. Chem. Front., 2018, 5, 943 RSC; (l) Y.-H. Huang, S.-R. Wang, D.-P. Wu and P.-Q. Huang, Org. Lett., 2019, 21, 1681 CrossRef CAS PubMed; (m) T.-T. Chen, A.-E. Wang and P.-Q. Huang, Org. Lett., 2019, 21, 3808 CrossRef CAS PubMed; (n) J. Li, M. Berger, W. Zawodny, M. Simaan and N. Maulide, Chem, 2019, 5, 1883 CrossRef CAS; (o) J. Li, R. Oost, B. Maryasin, L. González and N. Maulide, Nat. Chem., 2019, 10, 2327 Search PubMed.
- (a) S. Hwang, D. Kim and S. Kim, Chem.–Eur. J., 2012, 18, 9977 CrossRef CAS PubMed; (b) J. In, S. Hwang, C. Kim, J. H. Seo and S. Kim, Eur. J. Org. Chem., 2013, 965 CrossRef CAS; (c) C. Spyropoulos and C. G. Kokotos, J. Org. Chem., 2014, 79, 4477 CrossRef CAS; (d) H.-K. Kim and A. Lee, Tetrahedron Lett., 2016, 57, 4890 CrossRef CAS; (e) H.-K. Kim and A. Lee, Org. Biomol. Chem., 2016, 14, 7345 RSC; (f) P. Bana, Á. Lakó, N. Z. Kiss, Z. Béni, Á. Szigetvári, J. Kóti, G. I. Túrós, J. Éles and I. Greiner, Org. Process Res. Dev., 2017, 21, 611 CrossRef CAS; (g) A. Sevšek, L. Šrot, J. Rihter, M. Čelan, L. Q. van Ufford, E. E. Moret, N. I. Martin and R. J. Pieters, ChemMedChem, 2017, 12, 483 CrossRef; (h) H. Valkenier, C. M. Dias, C. P. Butts and A. P. Davis, Tetrahedron, 2017, 73, 4955 CrossRef CAS; (i) G. Pandey, R. Fernandes and D. Dey, Synlett, 2018, 29, 805 CrossRef CAS; (j) G. Pandey, R. Fernandes, D. Dey and B. Majumder, Tetrahedron, 2018, 74, 5752 CrossRef CAS; (k) W. M. Abdelmagid, T. Adak, J. O. Freeman and M. E. Tanner, Biochemistry, 2018, 57, 5591 CrossRef CAS PubMed; (l) S. N. Ramachandra, C. Srinivasulu, B. Hosamani, S. L and V. V. Sureshbabu, ChemistrySelect, 2018, 3, 12089 CrossRef CAS; (m) S. Hirasawa, K. Mukai, S. Sakai, S. Wakamori, T. Hasegawa, K. Souma, N. Kanomata, N. Ogawa, M. Aizawa and M. Emoto, J. Org. Chem., 2018, 83, 14457 CrossRef CAS PubMed.
- Lowering the temperature to 0 °C and −20 °C did not have any impact on the outcome of the reaction.
- CCDC no. 1954308 (16a), 1954309 (20) and 1954310 (21) contain supplementary crystallographic data for this paper. All products were crystallized from the mixture of dichloromethane/n-hexanes as the solvents.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization, copies of 1H, 13C, COSY and HSQC NMR spectra of all compounds (PDF), and crystallographic data for 16a, 20 and 21 (PDF and CIF). CCDC 1954308–1954310. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00796j |
| This journal is © The Royal Society of Chemistry 2020 |