
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResonance Raman view of the active site architecture in bacterial DyP-type peroxidases†
Célia M. Silveiraa,
Elin Moea,
Marco Fraaije b,
Lígia O. Martinsa and
Smilja Todorovic*a
b,
Lígia O. Martinsa and
Smilja Todorovic*a
aInstituto de Tecnologia Química e Biológica António Xavier, Universidade NOVA de Lisboa, Av. da República, 2780-157 Oeiras, Portugal. E-mail: smilja@itqb.unl.pt
bMolecular Enzymology, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands
First published on 17th March 2020
Abstract
Dye decolorizing peroxidases (DyPs) are novel haem-containing peroxidases, which are structurally unrelated to classical peroxidases. They lack the highly conserved distal histidine that acts as an acid-base catalyst in the catalytic reaction of classical peroxidases, which implies distinct mechanistic properties. Despite the remarkable catalytic properties and recognized potential for biotechnology applications, the knowledge of DyP's structural features in solution, which govern the reactivity and catalysis, is lagging behind. Resonance Raman (RR) spectroscopy can reveal fine details of the active site structure in hemoproteins, reporting on the oxidation and spin state and coordination of the haem cofactor. We provide an overview of the haem binding pocket architecture of the enzymes from A, B and C DyP subfamilies, in the light of those established for classical peroxidases and search for subfamily specific features among DyPs. RR demonstrates that multiple spin populations typically co-exist in DyPs, like in the case of classical peroxidases. The haem spin/coordination state is strongly pH dependent and correlates well with the respective catalytic properties of DyPs. Unlike in the case of classical peroxidases, a surprisingly high abundance of catalytically incompetent low spin population is observed in several DyPs, and tentatively related to the alternative physiological function of these enzymes. The molecular details of active sites of DyPs, elucidated by RR spectroscopy, can furthermore guide approaches for biotechnological exploitation of these promising biocatalysts.
Introduction
DyP-type peroxidases (DyPs) are haem b-containing enzymes that like classical peroxidases catalyse the reduction of hydrogen peroxide to water with concomitant oxidation of structurally diverse substrates. DyPs possess a number of unique properties:1–4 (i) an amino acid sequence that shows no homology to classical haem peroxidases; (ii) a broad substrate specificity, which includes anthraquinone-based and azo dyes, complex phenolic and non-phenolic molecules like kraft lignin, as well as carotenoids, phenols, aromatic sulphides and metal ions; (iii) an unusually low value of optimal pH for the catalytic activity (pHopt); (iv) distinct catalytic residues and tertiary structure and (v) still largely unexplored physiological role. While plant and mammalian peroxidases are primarily α-helical proteins, DyPs encompass two domains that contain α-helices and anti-parallel β-sheets, which adopt a ferredoxin-like fold and form a crevice that sandwiches the haem cofactor.5 The proximal axial iron ligand is His, which is hydrogen bonded to an acidic (Arg or Glu) residue, analogous to the members of peroxidase–catalase and peroxidase–cyclooxygenase superfamilies. At the distal haem side DyPs carry a unique GXXDG motif, lacking the highly conserved His, which acts as an acid–base catalyst in the catalytic reaction of classical peroxidases. Instead, the known DyP structures have an Asp (or Glu) and an Arg in the closest proximity of the haem distal face, and more peripherally located, conserved Phe and Asn (Ser or Gly), Fig. 1 and Fig. S1.†1,3 Deprotonated Asp and Arg are weak bases, still capable of H2O2 deprotonation due to lowering of their pKa upon binding to the haem.6 Since the Arg and Phe are, together with the conserved His, present in the distal haem pocket of classical peroxidases, the distal Asp was proposed to be the key residue in the heterolytic cleavage of O–O bond by DyPs.7 Nevertheless, mutagenesis studies of a number of DyPs indicated that either distal Asp or Arg can take up the role of the distal His.8 This however may not be the case in all DyPs, as it was demonstrated that neither Asp nor Arg is individually essential for peroxidase activity of Bacillus subtilis DyP (BsDyP).9 The electrons for H2O2 reduction are provided by the (bulky) substrates that bind close to the haem pocket or at the enzyme surface (for electron delivery via long-range electron transfer).4,10 | ||
| Fig. 1 Detail of the haem binding pocket of Cbo, Tfu, Pp and Vc DyPs indicating the proximal His and the conserved distal residues Asp/Glu, Asn and Arg. | ||
Based on phylogenetic analysis and catalytic and structural characteristics, DyPs have been classified into four subfamilies.3,5 Bacterial enzymes constitute subfamilies A–C while fungal DyPs form subfamily D. Class B and C DyPs are predicted to be cytoplasmic, playing a role in intracellular metabolic pathways, while those belonging to class A contain a Tat-dependent signal sequence, which suggests that they function extracellularly. Crystal structures have been solved for a number of DyPs, often at a relatively high pH.1–3,11–14 However, since the haem spin configuration that governs the H2O2 binding and reactivity is in peroxidases particularly sensitive to pH, temperature, and physical state (solution vs. crystal),17 it is of crucial importance to understand the configuration of DyPs active site in solution. To that end Resonance Raman (RR) spectroscopy has provided a wealth of information about the haem configuration, structure and catalytic mechanism of classical peroxidases.15–17 When excited into the Soret electronic transition band, the high-frequency region (1300–1700 cm−1) of RR spectra of peroxidases reveal core-size marker bands (designated by νi) sensitive to the haem iron configuration and oxidation state.18,19 As previously established for a number of haem proteins and model compounds, the frequencies of these bands can be associated with specific oxidation (e.g. ν4) and spin/coordination states (e.g. ν3 and ν2),18,19 and will be mainly discussed herein. It is noteworthy that the high-frequency region of RR spectra of haem proteins also contains other less well resolved bands (e.g. ν38, ν10 and νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), which are outlined in Table 1. The frequencies of the core-size marker bands are particularly sensitive to the spin and coordination pattern of ferric haem iron, which is in peroxidases catalytically relevant; H2O2 binds to the Fe3+ state and the catalytic reaction proceeds via formation of Compound I (CI), the two equivalent oxidized resting ferric state and Compound II (CII), the one equivalent oxidized ferric state. DyPs can form and stabilize catalytic intermediaries CI and CII in the absence of the 2nd substrate. In some cases, the Fe–ligand stretching modes that indirectly probe the H-bonding and electrostatics of the haem pocket can be observed in the low frequency of RR spectra.18,19
C), which are outlined in Table 1. The frequencies of the core-size marker bands are particularly sensitive to the spin and coordination pattern of ferric haem iron, which is in peroxidases catalytically relevant; H2O2 binds to the Fe3+ state and the catalytic reaction proceeds via formation of Compound I (CI), the two equivalent oxidized resting ferric state and Compound II (CII), the one equivalent oxidized ferric state. DyPs can form and stabilize catalytic intermediaries CI and CII in the absence of the 2nd substrate. In some cases, the Fe–ligand stretching modes that indirectly probe the H-bonding and electrostatics of the haem pocket can be observed in the low frequency of RR spectra.18,19
| Enzyme | BsDyP | CboDyP | TfuDyP | PpDyP | VcDyP | DrDyP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| νi/cm−1 (Δν) | 5cHS | 6cHS | 6cLS | 6cHS | 6cLS | 5cHS | 6cHS | 6cLS | 5cQS | 5cHS | 6cHS | 6cLS | 5cHS | 6cHS | 6cLS |
| ν4 | 1373 (11.7) | 1370 (9.2) | 1377 (11.0) | 1371 (11.5) | 1373 (12.4) | 1375 (15.1) | 1371 (13.9) | 1378 (10.7) | 1376 (11.3) | 1372 (10.5) | 1365 (13.2) | 1378 (11.3) | 1372 | 1372 (12.1) | 1377 (9.3) |
| ν3 | 1488 (14.6) | 1481 (11.6) | 1506 (16.5) | 1481 (12.4) | 1506 (8.5) | 1493 (12.6) | 1481 (13.1) | 1509 (13.6) | 1502 (12.6) | 1493 (12.3) | 1483 (12.6) | 1509 (10.9) | 1491 | 1480 (9.2) | 1505 (12.9) |
| ν38 | — | 1517 (12.3) | — | 1514 (11.0) | — | — | 1517 (14.2) | — | — | — | 1518 (13.1) | — | — | 1514 (8.9) | — |
| ν2 | 1565 (15.9) | 1563 (11.4) | 1582 (15.3) | 1561 (12.8) | 1581 (12.3) | — | 1565 (12.7) | 1585 (14.1) | 1572 (16.3) | 1565 (12.3) | 1559 (12.7) | 1585 | 1565 | 1555 (14.7) | 1587 (13.2) |
| νCC |
1622 (6.1) | 1624 (5.8) | 1623 (6.4) | 1619 (8.1) | 1618 (8.0) | — | 1619 (11.2) | 1619 (7.3) | 1625 (7.6) | 1622 (9.0) | — | 1622 | 1622 | 1623 (10.6) | 1622 (12.6) |
| 1632 (11.2) | 1629 (12.9) | 1631 (9.6) | |||||||||||||
| ν10 | — | — | 1635 (12.7) | — | 1640 (12.5) | — | — | 1640 (9.3) | 1636 (12.7) | 1631 (7.3) | — | — | — | — | 1641 (8.9) |
Here we use RR spectroscopy to describe the active site architecture of a number of DyPs from A, B and C subfamilies in solution and correlate the respective haem configurations with catalytic features. We provide an overview of the haem binding pocket of DyPs in the light of those established for classical peroxidases and furthermore search for subfamily specific features among the studied DyPs.
A-type DyPs
BsDyP
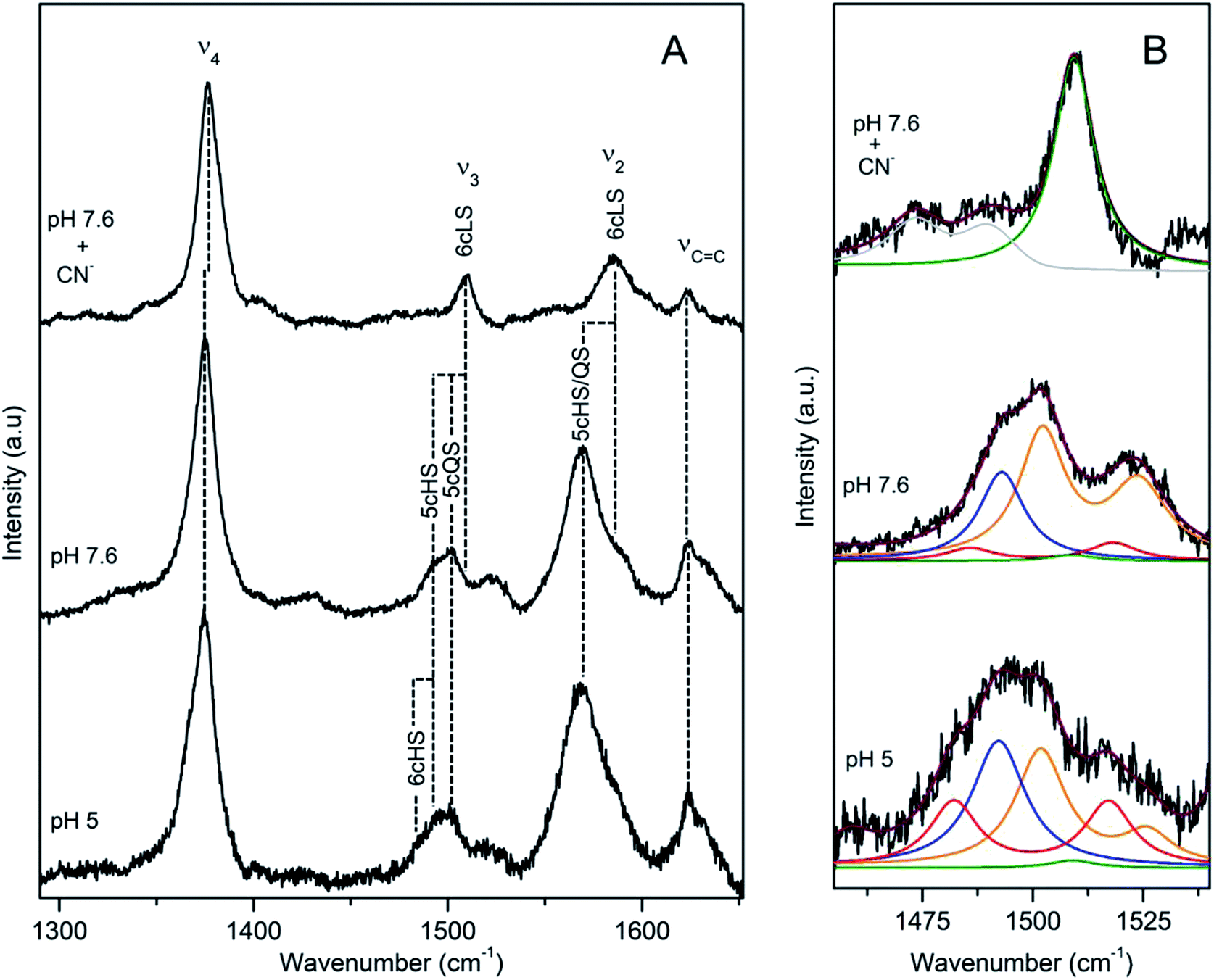
DyP from Bacillus subtilis (BsDyP), a bacterium found in soil and the gastro-intestinal tract, is capable of efficient oxidation of anthraquinonic (AQ) and azo dyes, e.g. reactive blue 5 (RB5), acid blue 62 (AB62), mordant black 9 (MB9), and moderate degradation of the phenolic guaiacol and Mn2+.20 The pHopt is 4 (using ABTS as a substrate).20 The RR frequencies of core size marker bands of BsDyP measured at pH 7.6 indicate a heterogeneous haem coordination, Fig. 2.21 This becomes particularly evident upon band fitting analysis that reveals a presence of three co-existing spin populations, Fig. 2b. The most abundant is the six coordinated low spin (6cLS) state, represented by 1377 (ν4), 1506 (ν3), 1582 (ν2) cm−1; the 6c high spin (6cHS) population, with 1370 (ν4), 1481 (ν3) and 1565 cm−1 (ν2) modes is less populated, while the five coordinated HS (5cHS) one, with 1373 (ν4), 1488 (ν3) and 1565 cm−1 (ν2) modes, shows minor contribution, Table 1.21 The 6cLS species is catalytically incompetent, as the haem carries a strong ligand in the 6th axial position that impedes H2O2 from binding. The HS populations either carry a weak ligand (e.g. H2O molecule) or have a vacant 6th axial position, giving origin to 6c and 5cHS species, respectively that both readily bind H2O2. Spin population distribution is highly pH dependent in peroxidases.17 However, unlike plant peroxidases, which show a complete transition to the 6cLS state at high pH, RR spectra of BsDyP at pH 10 indicate only a moderate increase of the LS configuration at the expense of the 6cHS population, suggesting alkaline transition at pH > 10.21 In the presence of a strong ligand (imidazole) all spin populations collapse into the 6cLS configuration. At pH 5, close to pHopt the catalytically competent 6cHS population becomes predominant, Fig. 2. | ||
| Fig. 2 RR spectra of ferric BsDyP. (A) High frequency region for the resting state enzyme at pH 7.6 in the presence/absence of imidazole and at pH 5. (B) Component analysis of ν3/ν38 region; the component spectra represent the 6cLS (green) ν3 1506 cm−1, 6cHS (red) ν3 1481 cm−1 and ν38 1516 cm−1, 5cHS (blue) ν3 1488 cm−1 populations, overall fit (purple) and non-assigned bands (gray). Spectra were measured with 413 nm excitation.21 | ||
As in the case of classical peroxidases, the coordination pattern of the haem iron in BsDyP is also strongly influenced by temperature,17 which has been attributed to the effect of packing forces. Both the frequencies of the RR bands and equilibria of spin and coordination states gradually change as the temperature decreases from RT to −190 °C, Fig. S2;† the most sensitive band to the spin/coordination state(s) of the haem, ν3, upshifts for 4–13 cm−1. The change of relative intensities of ν3 vs. ν2 bands is indicative of a conversion of the 5cHS population into the 6cLS population as the temperature is lowered.21
RR furthermore helped evaluate the impact of site-directed mutagenesis of the conserved residues (i.e. distal Asp240, Arg339 and Asn244 and proximal Asp383) on the haem cavity architecture of BsDyP.9 Single D240N, D383N, N244L and R339L mutants and D240N-R339L double mutant lacking both Asp and Arg, were constructed in order to address the role of each individual amino acid in catalysis. The study actually provided the first evidence that none of the distal residues is indispensable for promoting H2O2 (de)protonation and O–O bond cleavage in a DyP. RR spectra of N244L, R339L, D383N and even D240N-R339L, measured at neutral pH, show remarkable similarities. They are nevertheless distinct from the RR fingerprint of the wild type BsDyP, in particular with respect to the relative intensities of ν2 (HS) vs. ν2 (LS). Component analysis of the spectra reveals a higher abundance of the 6cHS species in the variants, which is in direct correlation with a significant improvement of their catalytic efficiencies.9
CboDyP
DyP from the alkaliphilic cellulomonad, Cellulomonas bogoriensis (CboDyP) is capable of oxidation of anthraquinone, azo and indigoid dyes, its pHopt is 5 (for Reactive Blue 19, RB19, as oxidizing substrate).11 The elucidated X-ray structure of CboDyP shows Glu, Arg and Phe in the distal haem cavity; the presence of a Glu instead of conserved Asp, indicates a unique GXXEG distal motif.11The high frequency region of RR spectra of CboDyP shows narrow, well resolved core size bands at 1371 (ν4), 1481 (ν3) and 1561 cm−1 (ν2) at neutral and pHopt, Fig. 3, which are indicative of a uniform and homogeneous 6cHS population,18,19 Table 1. Spectral features indicative of 5cHS species18,19 become apparent at pH 3.5 (ν3 at 1493 cm−1), although the enzyme is catalytically inactive at this pH (Fig. S3†).11 No alterations are observed in the RR spectra upon lowering the T down to −80 °C (Fig. S3†). The 6cLS state, with ν4, ν3, and ν2 modes at 1373, 1506, and 1581 cm−1, respectively, becomes partially populated only at very low T, co-existing with the 6cHS species at −190 °C. It is also readily formed in the presence of strong ligand (CN−) at RT, but not at pH 10 (Fig. S3†), indicating an alkaline transition at pH > 10.
 | ||
| Fig. 3 RR spectra of ferric CboDyP. High frequency region for the resting state enzyme at pH 7.5 in the presence/absence of CN− and at pHopt (pH 5). Spectra were measured with 413 nm excitation; experimental details can be found in ESI.† | ||
It is noteworthy that the structure of CboDyP, resolved for crystals obtained at pH 8.5, reveals that the distal side of the haem cavity has a somewhat ‘reduced volume’.11 At the distal haem face CboDyP houses Glu201 and Arg307, the former is actually bulkier than the Asp found in other known DyPs. In comparison with the structures of other DyPs, the extra methyl group of Glu201 positions the oxygens of the carboxylate group closer to the haem Fe, which is also the case of the carboxylate group and the guanidine group of the distal Arg. The unique residues found in the second sphere of the CboDyP haem pocket, such as Thr330 (the other DyPs carry Cys, Ser or Ala) induce further adjustment of the nearby residues and contribute to more tightly packed distal cavity.11 These structural elements most likely constrain the flexibility of the CboDyP haem pocket, which is reflected in the homogeneous spin population in RR spectra.
TfuDyP
A cellulose degrading thermophilic actinomycete Thermobifida fusca expresses DyP (TfuDyP), which is capable of oxidation of veratryl alcohol, o-phenylenediamine, and 3,3-diaminobenzidine, AQ dyes, RB19 and Reactive Blue 4 (RB4), and shows modest activity towards typical plant peroxidase substrates, e.g. guaiacol and 2,6-dimethoxyphenol. The pHopt of TfuDyP (for RB19 as oxidizing substrate) is 3.5.13RR spectra of the resting TfuDyP at pH 7.5 indicate a single spin population with core size marker bands at 1378 (ν4), 1509 (ν3) and 1585 cm−1 (ν2), which are characteristic of 6cLS configuration,18,19 Fig. 4 and Table 1. The same frequencies are observed upon binding of a strong ligand, such as CN−, to the haem. A presence of a diatomic 6th axial ligand at the distal face of the haem was actually observed in the X-ray structure of TfuDyP obtained at 1.8 Å resolution using crystals grown at pH 8.5.22 The presence of the ligand was initially rationalized in terms of possible formation of dioxygen bound to ferrous species generated by photo-reduction during the data collection or to iron(III)-superoxide species.22 However, RR spectra clearly demonstrate that the 6cLS species is the major species in TfuDyP in solution at neutral physiological like conditions.
 | ||
| Fig. 4 RR spectra of ferric TfuDyP. High frequency region of TfuDyP in the resting state at pH 7.5 in the presence/absence of CN− and at pHopt (pH 3.5). Inset: component analysis of ν3/ν38 region; the component spectra represent the 6cHS (red) ν3 1481 cm−1 and ν38 1516 cm−1, 5cHS (blue) ν3 1493 cm−1 populations and the overall fit (purple). Spectra were measured with 413 nm excitation; experimental details can be found in ESI.† | ||
At pHopt TfuDyP undergoes a complete transition to HS state, which is particularly evident from the presence of two ν3 modes, found at 1481 cm−1 and 1493 cm−1 that are characteristic for 6cHS and 5cHS states,18,19 respectively, and a 5 cm−1 downshift of ν4, Fig. 4 and Table 1. This is most likely due to an equilibrium between H2O bound 6cHS and unbound 5cHS states. No traces of 6cLS species can be detected in the spectra at pH 3.5, which corroborates the high activity of the enzyme. RR spectra of TfuDyP are sensitive to T; as it decreases, RR modes slightly up-shift (ca. 3 cm−1) and the amount of the 6cLS population increases at pH 3.5. This is particularly clear at −190 °C, from the higher intensities of ν3 and ν2 6cLS modes at 1512 and 1591 cm−1, respectively, Fig. S4.†
B-type DyPs
PpDyP
DyP from the soil bacterium Pseudomonas putida (PpDyP) exhibits exceptionally high activities and a broad range of substrates, which include AQ dyes (RB5, AB62), azo dyes (MB9), phenols (guaiacol, syringaldehyde and acetosyringone) and metal ions (e.g. Mn2+).23 The pHopt of PpDyP is 5 (for ABTS as a substrate).20RR spectra of resting state PpDyP measured at pH 7.6, show broad and asymmetric ν3 band indicative of multiple species, Fig. 5. Component analysis of the spectra reveals two major ν3 bands, at 1493 cm−1 and at 1502 cm−1, which are assigned to 5cHS and five-coordinated quantum mechanically mixed-spin (5cQS) ferric haem populations, respectively.24 The 1502 cm−1 frequency falls into the range attributable to 6cLS species and can be easily confused with it. However, the frequency of ν3 mode of pure 6cLS species formed in e.g. PpDyP–imidazole or CN− complex is 1509 cm−1. Further confirmation of the assignment of 1502 cm−1 band to ν3 QS originates from UV-Vis spectra that lack features characteristic for LS haem population.24
 | ||
| Fig. 5 RR spectra of ferric PpDyP. (A) High frequency region for the resting state enzyme at pH 7.6 in the presence/absence of CN− and at pHopt. (B) Component analysis of ν3/ν38 region; the component spectra represent the 6cLS (green) ν3 1509 cm−1, 6cHS (red) ν3 1483 cm−1 and ν38 1516 cm−1, 5cHS (blue) ν3 1493 cm−1, 5cQS (orange) ν3 1502 cm−1 and ν38 1525 cm−1 populations, overall fit (purple) and non-assigned bands (gray). Spectra were measured with 413 nm excitation.21 | ||
This unusual spin state results from an admixture of in biological molecules very rarely populated intermediate spin (S = 3/2) and HS haem states. It has been also observed in class III plant peroxidases and catalase peroxidases (e.g. KatG).16,21 Actually the major contribution in the RR spectra of the resting PpDyP at pH 7.6 originates from the QS species, with the core size bands at 1376 (ν4), 1502 (ν3) and 1572 cm−1 (ν2), less abundant is the 5cHS species with 1372 (ν4), 1493 (ν3) and 1565 cm−1 (ν2) modes, Table 1. In addition, a very minor contribution of the 6cHS population (ν4, ν3, and ν2 modes at 1365, 1483, and 1559 cm−1, respectively) and 6cLS (ν4, ν3, and ν2 modes at 1378, 1509, and 1585 cm−1, respectively) are also detected in the spectra. At pH 10, the amount of LS population moderately increases. At pHopt, a partial conversion of QS to 6cHS state occurs, Fig. 5.21 It is noteworthy that Raman spectroscopy provided the first unambiguous evidence that the unusual QS species is actually catalytically competent.25 Surface enhanced RR experiments (SERR), employing PpDyP attached to biocompatible plasmonic metal, revealed that the QS species is capable of H2O2 binding and formation of the catalytic intermediate Compound I, which has a distinct (SERR) fingerprint.25
Apart from RR band shifts, a temperature decrease induces a spin transition from the 5cHS to the 6cLS state at a fixed temperature (−45 °C) in PpDyP. The bands characteristic for 5cHS (e.g. ν2 at 1565 cm−1 and ν3 at 1492 cm−1) disappear from spectra, while those indicative of 6cLS species (ν2 at 1585 cm−1 and ν3 at 1509 cm−1) gain intensity. The ν3 (QS) undergoes an upshift for more than 10 wavenumbers at −190 °C; a similar tendency of the QS population was previously reported for barley peroxidase.16,21
The roles of conserved distal residues, Asp132, Asn136 and Arg214, in the catalytic mechanism of PpDyP were addressed by generation of site-directed variants, and the mutation-induced alterations of the active site were evaluated by RR.23 It was demonstrated that the distal Arg in particular plays an essential role in PpDyP, as in the case of DyPB from Rhodococcus jostii RHA1 but not in DyPs from the A and D subfamilies. The D132N and N136L mutations have a substantial effect on the geometry of the distal side of the active site. The component analysis of the spectra reveals co-existing 5cQS, 6cHS, 5cHS and 6cLS populations, but with different relative abundance in comparison with the wild type PpDyP. In case of D132N, in which the 6cLS is the major species, impeded H2O2 binding to the haem active site was reflected in the measured Km values.23
VcDyP
The B-type DyP from the pathogen Vibrio cholera (VcDyP) shows relatively low efficiency towards RB19 and guaiacol even at pHopt (pHopt 4 for RB19 substrate).26 RR spectra of the ferric VcDyP measured at pH 8 show marker bands at 1372 (ν4), 1491 (ν3) and 1565 cm−1 (ν2), together with 1581 (ν37) and 1624 cm−1 (νCC) modes, Fig. 6 and Table 1. These frequencies are indicative of 5cHS ferric haem, which is consistent with the vacant 6th axial coordination, observed in crystallographic structure of VcDyP. (Note that RR spectra for different pH values and T are not available.) The ferrous VcDyP obtained by chemical reduction reveals RR modes at 1352 (ν4), 1470 (ν3) and 1565 cm−1 (ν2). This is consistent with a presence of a single 5cHS haem population, which allows for indirect probing of the proximal haem environment through the low frequency Fe–ligand stretching modes of the 5cHS Fe2+ species. The Fe–His mode (νFe–His) is sensitive to H bonding interactions.17,27,28 This mode, typically found in 200–250 cm−1 region, is in VcDyP observed at 229 cm−1, which is slightly higher than that of myoglobin (220 cm−1), but not as high as in classical haem peroxidases (∼245 cm−1). The frequency is comparable with the value reported for chlorite dismutase (Cld) in which the hydrogen bonding between the distal axial and proximal residues is weak.29 The ferrous 5cHS haem also readily binds CO. The frequency of νFe–CO stretching mode of the haem–CO adduct is sensitive to distal electrostatic interactions.17,27,28 The νFe–CO band of VcDyP is comprised of two bands at 505 and 518 cm−1 (Fig. 6). The pH dependence of the band intensities could be correlated with pH dependence of the enzymatic activity and the protonation of the haem distal ligand at low pH.26
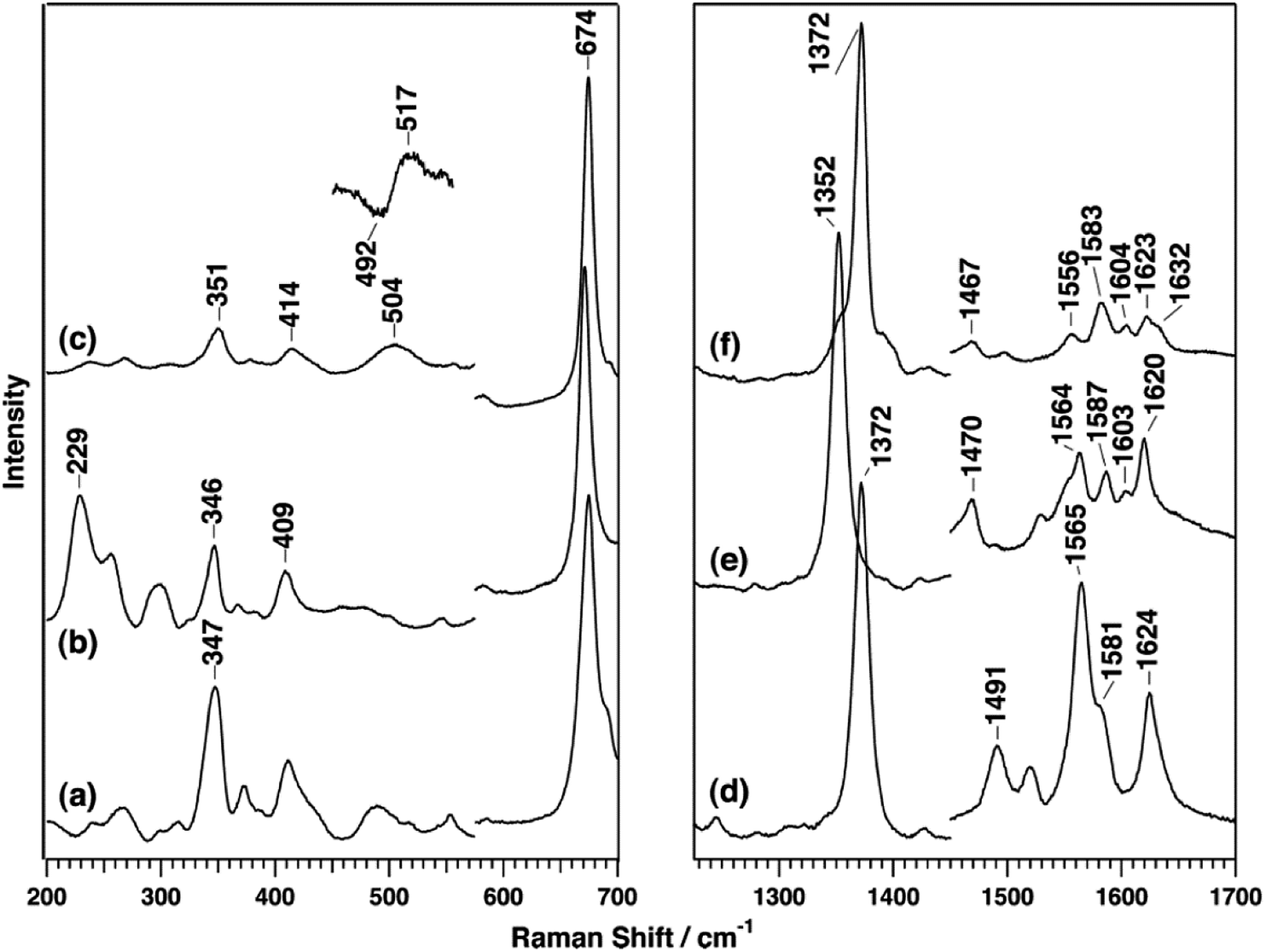 | ||
| Fig. 6 RR spectra of VcDyP in the low-frequency (left) and high-frequency (right) regions. Spectra of ferric (a and d), ferrous (b and e), and ferrous CO-bound (c and f) forms are measured with 413.1 nm excitation wavelength for ferric and CO-bound forms and 441.6 nm for the ferrous form at pH 8. Reprinted with permission from (T. Uchida, et al., Biochemistry, 2015, 54, 6610–6621). Copyright (2020) American Chemical Society.26 | ||
C-type DyPs
DrDyP
DyP from the extremely radiation resistant bacterium Deinococcus radiodurans, DrDyP, shows no catalytic activity at physiological pH and moderate activity at low pH (unpublished results from our group). It has a unique DyP fingerprint motif, GXXDM, carrying a Met (M190) instead of a conserved Gly.30 RR spectra of DrDyP measured at pH 7.5, reveal haem marker bands centred at 1377 (ν4), 1505 (ν3), 1555 (ν11), 1579 (ν2) and 1641 (ν10) cm−1, Fig. 7 and Table 1, characteristic of a single ferric 6cLS spin population.18,19 The assignment of the marker bands to 6cLS state is confirmed by the RR fingerprint obtained in the presence of a strong ligand (CN−) and UV-Vis data which suggest that DrDyP is in a LS state above pH 4.30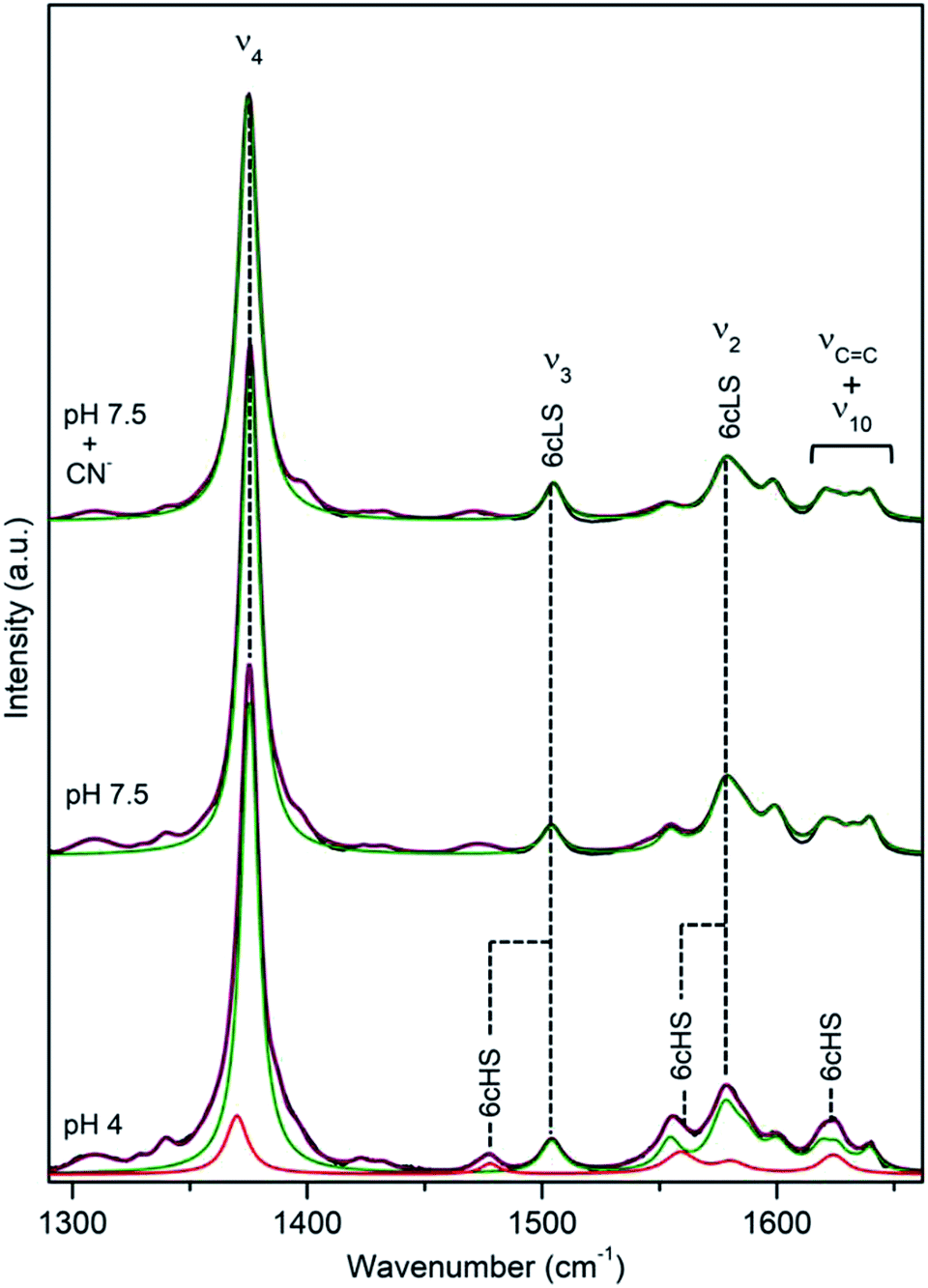 | ||
| Fig. 7 RR spectra of ferric DrDyP. High frequency region for the resting enzyme at pH 7.5 in the presence/absence of CN− and at pH 4. The component spectra represent the 6cLS (green) and 6cHS (red) populations, overall fit (purple) and non-assigned bands (gray). Spectra were measured with 413 nm excitation; experimental details can be found in ESI.† | ||
Upon lowering of pH towards pHopt, additional modes, indicative of a 6cHS ferric haem (ν4 at 1372 cm−1, ν3 at 1480 cm−1 and ν10 at 1624 cm−1) appear in the spectra, Fig. 7. Considering the relative intensities of the ν4 mode only, it can be estimated that 6cHS population represents ∼15% of overall population at pH 4.0 and ∼30% at pH 3.0, Table 2, which is in agreement with slightly increased peroxidase activity observed below pH 4. Noticeably, at low temperatures and low pH the equilibrium between LS and HS populations is inverted, Fig. S5.† A maximum of 70% 6cHS state is observed at −50 °C, which decreases to ca. 35% below −80 °C. At pH 7.5 and low T, the spin population distribution remains the same as at RT, with up to 4 cm−1 upshifts of the vibrational modes.
| Subfamily | DyP | pH 7 | pHopt |
|---|---|---|---|
| A | BsDyP | 60% HS + 40% LS (35% 6cHS + 22% 5cHS) | 80% HS + 20% LS (68% 6cHS + 8% 5cHS) |
| TfuDyP | 100% LS | 100% HS (60% 6cHS + 40% 5cHS) | |
| CboDyP | 100% HS | 100% HS | |
| B | PpDyP | 98% HS (63% 5cQS + 29% 5cHS + 6% 6cHS) | 99% HS (33% 5cQS + 36% 5cHS + 30% 6cHS) |
| VcDyP | (5c)HS | nd | |
| C | DrDyP | 100% LS | 30% (6c)HS + 70% LS |
Discussion
In order to provide a place for hydrogen peroxide binding, the active site of classical peroxidases either has a vacant sixth axial position, giving origin to 5cHS ferric haem iron, or carries a loosely bound water molecule, yielding 6cHS configuration.15–17,31 X-ray structural analysis of HRP,32 CcP33 and a number of other peroxidases reveals a 5cHS configuration of haem iron, with a proximal histidine ligand and a water molecule in the distal side that in the majority of cases is not coordinated to the iron; e.g. the oxygen of the water molecule is 3.20 Å from the iron atom in HRP and at 2.40 Å in CcP.32,33 RR spectroscopy has a unique capacity to sensitively probe the haem configurations that peroxidases adopt in solution, providing a physiologically relevant picture, which is not necessarily identical to the crystal state.21,34 It reveals that the distal cavity of peroxidases in solution is actually highly flexible, allowing for a co-existence of multiple spin states and their interconversion.17 RR characterization of ferric HRP isozymes, such as HRPC and HRPA2, shows a mixture of 6cHS and 5cHS forms in the former, while in the latter these two forms coexist together with 5cQS configuration.15 The 5cHS species is dominant in HRPC, in agreement with the X-ray structure.15 The three populations observed in HRPA2 were also identified in soy bean peroxidase, but with different abundance, while in barley peroxidase 5cHS and 5cQS forms coexist. The LS configuration is in native classical peroxidases typically observed only upon alkaline transition, which results in reversible enzyme inactivation due to formation of 6cLS Fe(III)–OH species; in HRPC it occurs at pH 11.15Here we demonstrate that RR spectra of a number of representative members of A, B and C DyP subfamilies share several common features with classical peroxidases. Like in the latter, DyPs are capable of adopting several haem configurations in the resting state, including 5/6cHS and QS. The spin population distribution is sensitive to pH, temperature, and solution vs. crystal state,17,21 which has to be taken into consideration in X-ray structural analysis of these enzymes. Surprisingly, a significant number of DyPs has a substantial amount of catalytically incompetent 6cLS population in the resting state (Table 2), which is highly uncommon for classical peroxidases. We observe DyPs that are actually trapped in LS configuration in the resting state at neutral pH and undergo a complete (TfuDyP) or moderate (DrDyP) transition to HS state as the pH lowers and approaches pHopt. A systematic study of predominantly 5cHS CcP (MI) and its variants reveals a formation of the 6cLS species at neutral pH in some mutants.35 This was associated with the structural changes that accompany disruption of H-bonding network that tightly connects proximal and distal sides, and as a consequence, results in formation of OH− and/or aquo–Fe3+ LS complexes. In variants in which the H-bonding between the distal His and a haem propionate is compromised (loss of ‘anchoring H-bond’), the Fe3+ ion is free to move into the haem plane which facilitates its coordination by a distal ligands.4 We speculate that enzyme dependent fine tuning of the already quite particular H-bonding network observed in DyPs,36 also plays a role in populating the 6cLS state in some of these enzymes. The H network that involves distal amino acid residues, haem propionates and H2O molecule(s) in DyPs imposes constraints to propionate chains that result in unusual high energy conformations,37 which are therefore, unlike in the case of other peroxidases and Clds, forced into atypical dispositions in order to accommodate the H-bonding. The T dependences of RR features associated with particular spin states are clearly distinct in the described DyPs, which implies different packing forces upon T lowering due to characteristic H bonding network in each enzyme. Several available DyP structures actually indicate a presence of a solvent molecule at the distal haem face, including in Shewanella oneidensis TyrA, Rhodococcus jostii DyPB, Amycolatopsis sp. DyP2 and Auricularia auricula-judae DyP.12,14,37,38 The water molecule can be held in place by hydrogen bonding network37,39 at Fe–O distances as short as 2.1 Å in DyPDec1 and DyPB.3,12 Even shorter distal Fe–OH2 bond length of 1.98 Å was observed in DyP from Streptomyces lividans, DtpA, giving origin to 6c configuration of the haem active site.36 Molecular oxygen has also been found to coordinate the DyP haem iron, which was nevertheless associated with photoreduction of the haem by the X-ray beam during data collection or the crystallization conditions.12,22,40 We demonstrate that this finding is not an artefact, as the presence of the 6cLS population is also observed in solution. Although the nature of the 6th axial ligand that gives origin to 6cLS state cannot be determined from RR spectra at this point, we propose a water molecule or OH− ion as the likely candidates. Note that although most commonly imposing a weak ligand field, H2O can also act as a strong haem ligand.41 At low pH the distal charged residues of DyPs are protonated, which facilitates the removal of the 6th axial ligand and results in species capable of substrate binding. It is tempting to speculate that in exclusively 6cLS DyPs, this configuration could be associated with a gating role, in which the peroxidase activity would be triggered only in certain conditions. To that end, it was reported that in Halobacterium salinarum NRC-1 a putative DyP-type peroxidase (VNG0798H) becomes transcriptionally induced in response to oxidative stress, providing additional protection when H2O2 content exceeds the detoxification capability of the main peroxidase PerA.2 Similarly, DtpA has been suggested to contribute to protection of S. lividans from oxidative damage imposed by metabolically generated H2O2, simultaneously playing a role in a copper trafficking pathway.42 One cannot completely rule out a possibility that (6cLS) DyPs have an alternative role. Namely, since the DyP physiological substrates are largely unknown, it has been suggested that the apparent peroxidase activity might not be related to their physiological function.2,3 YcdB has, for example, been proposed to function as an electron acceptor by virtue of its peroxidase activity.2 The hypothesis that 6cLS state has a distinct role, lays emphasis on the redox potential of Fe2+/3+ couple that differs in species with different spin states. For instance, the 5cHS cyt c has a significantly downshifted redox potential than the native 6c His/Met coordinated one, and shows microperoxidase activity.43,44 One can envisage a similar scenario in the case of LS DyPs, however, we clearly need to investigate more of these enzymes in order to test this hypothesis.
In conclusion, the comparative RR study of several bacterial DyPs from 3 subfamilies reveals that (i) the haem active site architecture of DyPs in solution, as portrayed by RR, does not appear to be subfamily dependent, as the members of the same subfamily populate distinct spin states; (ii) the nature of amino acid residues that line up the distal side also cannot be directly correlated with spin configurations, since the DyPs that e.g. house Asp, Arg, Asn and Phe in the distal site populate distinct spin states that follow very different pH dependences; (iii) there is a surprisingly high abundance of 6cLS populations in several DyPs, which we attribute to the specific H-bonding network in the vicinity of the haem active site. The pronounced pH dependence of the spin state population distribution emphasizes the importance of the second sphere arrangements in fine-tuning the pKa of the distal residues, which ultimately governs the capacity of DyPs to bind and reduce H2O2. It appears that very subtle structural changes in the distal haem pocket have a remarkable effect on spin state configurations, fine tuning hydrogen peroxide reactivity.36
Overall, we summarize here the intricacies of the active site architecture that DyPs adopt in solution, which is crucial for full comprehension of the physiological role of these enzymes. Importantly, this knowledge will also guide further advances in biotechnological applications of DyPs. Continuous efforts are being made to explore novel biocatalysts and in that respect DyPs represent a particularly promising alternative to classical peroxidases, due to improved stability and catalytic performance, broad range of substrates, easy overexpression and manipulation to produce specific variants.45 Understanding molecular details of the respective active site structures will facilitate selection of DyPs that represent the most favourable candidates for the construction of biosensors of hydrogen peroxide and other applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Project LISBOA-01-0145-FEDER-007660 (Microbiologia Molecular, Estrutural e Celular) funded by FEDER funds through COMPETE 2020-Programa Operacional Competitividade e Internacionalização (POCI); by national funds through FCT – Fundação para a Ciência e a Tecnologia (PTDC/BBBEBB/0122/2014, IF/00710/2014, PTDC/BII-BBF/29564/2017 and PTDC/BIA-BFS/31026/2017) We acknowledge TIMB3 and B-LigZymes projects support, which have received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreements No 810856 and 824017, respectively.References
- Y. Sugano, Cell. Mol. Life Sci., 2009, 66, 1387–1403 CrossRef CAS PubMed.
- T. Yoshida and Y. Sugano, Arch. Biochem. Biophys., 2015, 574, 49–55 CrossRef CAS PubMed.
- R. Singh and L. D. Eltis, Arch. Biochem. Biophys., 2015, 574, 56–65 CrossRef CAS PubMed.
- D. Linde, R. Pogni, M. Cañellas, F. Lucas, V. Guallar, M. C. Baratto, A. Sinicropi, V. Sáez-Jiménez, C. Coscolín, A. Romero, F. J. Medrano, F. J. Ruiz-Dueñas and A. T. Martínez, Biochem. J., 2015, 466, 253–262 CrossRef CAS PubMed.
- D. I. Colpa, M. W. Fraaije and E. van Bloois, J. Ind. Microbiol. Biotechnol., 2014, 41, 1–7 CrossRef CAS PubMed.
- P. Jones and H. B. Dunford, J. Inorg. Biochem., 2005, 99, 2292–2298 CrossRef CAS PubMed.
- Y. Sugano, R. Muramatsu, A. Ichiyanagi, T. Sato and M. Shoda, J. Biol. Chem., 2007, 282, 36652–36658 CrossRef CAS PubMed.
- R. Singh, J. C. Grigg, Z. Armstrong, M. E. Murphy and L. D. Eltis, J. Biol. Chem., 2012, 287, 10623–10630 CrossRef CAS PubMed.
- S. Mendes, T. Catarino, C. Silveira, S. Todorovic and L. O. Martins, Catal.: Sci. Technol., 2015, 5, 5196–5207 RSC.
- M. Pérez-Boada, F. J. Ruiz-Dueñas, R. Pogni, R. Basosi, T. Choinowski, M. J. Martínez, K. Piontek and A. T. Martínez, J. Mol. Biol., 2005, 354, 385–402 CrossRef PubMed.
- M. H. Habib, H. J. Rozeboom and M. W. Fraaije, Molecules, 2019, 24, 1208 CrossRef PubMed.
- J. N. Roberts, R. Singh, J. C. Grigg, M. E. Murphy, T. D. Bugg and L. D. Eltis, Biochemistry, 2011, 50, 5108–5119 CrossRef CAS PubMed.
- E. van Bloois, D. E. Torres Pazmino, R. T. Winter and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2010, 86, 1419–1430 CrossRef CAS PubMed.
- C. Zubieta, S. S. Krishna, M. Kapoor, P. Kozbial, D. McMullan, H. L. Axelrod, M. D. Miller, P. Abdubek, E. Ambing, T. Astakhova, D. Carlton, H. J. Chiu, T. Clayton, M. C. Deller, L. Duan, M. A. Elsliger, J. Feuerhelm, S. K. Grzechnik, J. Hale, E. Hampton, G. W. Han, L. Jaroszewski, K. K. Jin, H. E. Klock, M. W. Knuth, A. Kumar, D. Marciano, A. T. Morse, E. Nigoghossian, L. Okach, S. Oommachen, R. Reyes, C. L. Rife, P. Schimmel, H. van den Bedem, D. Weekes, A. White, Q. Xu, K. O. Hodgson, J. Wooley, A. M. Deacon, A. Godzik, S. A. Lesley and I. A. Wilson, Proteins, 2007, 69, 223–233 CrossRef CAS PubMed.
- A. Feis, B. D. Howes, C. Indiani and G. Smulevich, J. Raman Spectrosc., 1998, 29, 933–938 CrossRef CAS.
- B. D. Howes, C. B. Schiodt, K. G. Welinder, M. P. Marzocchi, J. G. Ma, J. Zhang, J. A. Shelnutt and G. Smulevich, Biophys. J., 1999, 77, 478–492 CrossRef CAS PubMed.
- G. Smulevich, A. Feis and B. D. Howes, Acc. Chem. Res., 2005, 38, 433–440 CrossRef CAS PubMed.
- F. Siebert and P. Hildebrandt, in Vibrational Spectroscopy in Life Science, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 227–282, DOI:10.1002/9783527621347.ch7.
- S. Todorovic, in Radiation in Bioanalysis: Spectroscopic Techniques and Theoretical Methods, ed. A. S. Pereira, P. Tavares and P. Limão-Vieira, Springer International Publishing, Cham, 2019, pp. 111–145, DOI:10.1007/978-3-030-28247-9_4.
- A. Santos, S. Mendes, V. Brissos and L. O. Martins, Appl. Microbiol. Biotechnol., 2014, 98, 2053–2065 CrossRef CAS PubMed.
- M. Sezer, A. Santos, P. Kielb, T. Pinto, L. O. Martins and S. Todorovic, Biochemistry, 2013, 52, 3074–3084 CrossRef CAS PubMed.
- R. Rahmanpour, D. Rea, S. Jamshidi, V. Fulop and T. D. Bugg, Arch. Biochem. Biophys., 2016, 594, 54–60 CrossRef CAS PubMed.
- S. Mendes, V. Brissos, A. Gabriel, T. Catarino, D. L. Turner, S. Todorovic and L. O. Martins, Arch. Biochem. Biophys., 2015, 574, 99–107 CrossRef CAS PubMed.
- M. Sezer, T. Genebra, S. Mendes, L. O. Martins and S. Todorovic, Soft Matter, 2012, 8, 10314–10321 RSC.
- S. Todorovic, P. Hildebrandt and L. O. Martins, Phys. Chem. Chem. Phys., 2015, 17, 11954–11957 RSC.
- T. Uchida, M. Sasaki, Y. Tanaka and K. Ishimori, Biochemistry, 2015, 54, 6610–6621 CrossRef CAS PubMed.
- T. G. Spiro and A. A. Jarzecki, Curr. Opin. Chem. Biol., 2001, 5, 715–723 CrossRef CAS PubMed.
- T. G. Spiro and I. H. Wasbotten, J. Inorg. Biochem., 2005, 99, 34–44 CrossRef CAS PubMed.
- B. R. Streit, B. Blanc, G. S. Lukat-Rodgers, K. R. Rodgers and J. L. DuBois, J. Am. Chem. Soc., 2010, 132, 5711–5724 CrossRef CAS PubMed.
- K. S. T. Frade, A. C. P. Fernandes, C. M. Silveira, C. Frazao and E. Moe, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2018, 74, 419–424 CrossRef CAS PubMed.
- G. Smulevich, J. M. Mauro, L. A. Fishel, A. M. English, J. Kraut and T. G. Spiro, Biochemistry, 1988, 27, 5486–5492 CrossRef CAS PubMed.
- M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith and T. L. Poulos, Nat. Struct. Biol., 1997, 4, 1032–1038 CrossRef CAS PubMed.
- B. C. Finzel, T. L. Poulos and J. Kraut, J. Biol. Chem., 1984, 259, 13027–13036 CAS.
- G. Smulevich, J. Porphyrins Phthalocyanines, 2019, 23, 691–700 CrossRef CAS.
- G. Smulevich, M. A. Miller, J. Kraut and T. G. Spiro, Biochemistry, 1991, 30, 9546–9558 CrossRef CAS PubMed.
- M. Lucic, A. K. Chaplin, T. Moreno-Chicano, F. S. N. Dworkowski, M. T. Wilson, D. A. Svistunenko, M. A. Hough and J. A. R. Worrall, Dalton Trans., 2020, 49, 1620–1636 RSC.
- E. Strittmatter, C. Liers, R. Ullrich, S. Wachter, M. Hofrichter, D. A. Plattner and K. Piontek, J. Biol. Chem., 2013, 288, 4095–4102 CrossRef CAS PubMed.
- M. E. Brown, T. Barros and M. C. Chang, ACS Chem. Biol., 2012, 7, 2074–2081 CrossRef CAS PubMed.
- C. Zubieta, R. Joseph, S. Sri Krishna, D. McMullan, M. Kapoor, H. L. Axelrod, M. D. Miller, P. Abdubek, C. Acosta, T. Astakhova, D. Carlton, H.-J. Chiu, T. Clayton, M. C. Deller, L. Duan, Y. Elias, M.-A. Elsliger, J. Feuerhelm, S. K. Grzechnik, J. Hale, G. W. Han, L. Jaroszewski, K. K. Jin, H. E. Klock, M. W. Knuth, P. Kozbial, A. Kumar, D. Marciano, A. T. Morse, K. D. Murphy, E. Nigoghossian, L. Okach, S. Oommachen, R. Reyes, C. L. Rife, P. Schimmel, C. V. Trout, H. van den Bedem, D. Weekes, A. White, Q. Xu, K. O. Hodgson, J. Wooley, A. M. Deacon, A. Godzik, S. A. Lesley and I. A. Wilson, Proteins: Struct., Funct., Bioinf., 2007, 69, 234–243 CrossRef CAS PubMed.
- X. Liu, Q. Du, Z. Wang, D. Zhu, Y. Huang, N. Li, T. Wei, S. Xu and L. Gu, J. Biol. Chem., 2011, 286, 14922–14931 CrossRef CAS PubMed.
- S. Todorovic, C. Jung, P. Hildebrandt and D. H. Murgida, J. Biol. Inorg Chem., 2006, 11, 119–127 CrossRef CAS PubMed.
- M. L. C. Petrus, E. Vijgenboom, A. K. Chaplin, J. A. R. Worrall, G. P. van Wezel and D. Claessen, Open Biol., 2016, 6, 150149 CrossRef PubMed.
- D. A. Capdevila, W. A. Marmisollé, F. Tomasina, V. Demicheli, M. Portela, R. Radi and D. H. Murgida, Chem. Sci., 2015, 6, 705–713 RSC.
- C. M. Silveira, M. A. Castro, J. M. Dantas, C. Salgueiro, D. H. Murgida and S. Todorovic, Phys. Chem. Chem. Phys., 2017, 19, 8908–8918 RSC.
- C. Barbosa, C. M. Silveira, D. Silva, V. Brissos, P. Hildebrandt, L. O. Martins and S. Todorovic, Biosens. Bioelectron., 2020, 112055, DOI:10.1016/j.bios.2020.112055.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00950d |
| This journal is © The Royal Society of Chemistry 2020 |