
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of α-indolylacrylates as potential anticancer agents using a Brønsted acid ionic liquid catalyst and the butyl acetate solvent†
Ahmed El-Harairy *ab,
Mennatallah Shaheenc,
Jun Lib,
Yuzhou Wub,
Minghao Lib and
Yanlong Gubd
*ab,
Mennatallah Shaheenc,
Jun Lib,
Yuzhou Wub,
Minghao Lib and
Yanlong Gubd
aEnvironmental, Energy and Green Chemistry Laboratory, Faculty of Agriculture, Damietta University, 34511, Damietta, Egypt. E-mail: ahmed.elharairy@hotmail.com
bSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, 430074, Wuhan, China
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Horus University, 34511, New Damietta, Damietta, Egypt
dState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Lanzhou, 730000, China
First published on 2nd April 2020
Abstract
In this study, new α-indolylacrylate derivatives were synthesized by the reaction of 2-substituted indoles with various pyruvates using a Brønsted acid ionic liquid catalyst in butyl acetate solvent. This is the first report on the application of pyruvate compounds for the synthesis of indolylacrylates. The acrylate derivatives could be obtained in good to excellent yields. A preliminary biological evaluation revealed their promising anticancer activity (IC50 = 9.73 μM for the compound 4l) and indicated that both the indole core and the acrylate moieties are promising for the development of novel anticancer drugs. The Lipinski's rule and Veber's parameters were assessed for the newly synthesized derivatives.
Introduction
The chemistry of indoles is an extensive research topic in organic synthesis because indole derivatives have unique biological activities.1 In the diverse chemical libraries of reported potent antitumor agents, several indole-based derivatives have been described as effective antitumor agents.2 Moreover, acrylate-containing derivatives have been proven to display prominent antitumor potency.3 Caffeic acid phenethyl ester (CAPE) (Fig. 1) is the main constituent of propolis, a resinous substance used in folk medicine for treating various ailments. CAPE has been widely reported to possess antitumor activities.2a | ||
| Fig. 1 Chemical structure of caffeic acid phenethyl ester (CAPE). | ||
Herein, our rational design is based on molecular hybridization. Molecular hybridization is a useful approach to design new biologically active agents. It involves the combination of two or more pharmacophoric entities with relevant biological activities to attain new hybrids with improved activity, selectivity, and safety.4 In addition, two structural motifs were considered in the design of the proposed scaffold: the indole moiety and the acrylate structure. Although the acrylic linkage has significant potency, a literature survey revealed that only few studies have been reported on the synthesis of α-indolylacrylate analogs5 and their biological activity as promising anticancer agents was evaluated in 2020 for the first time (Fig. 2).6
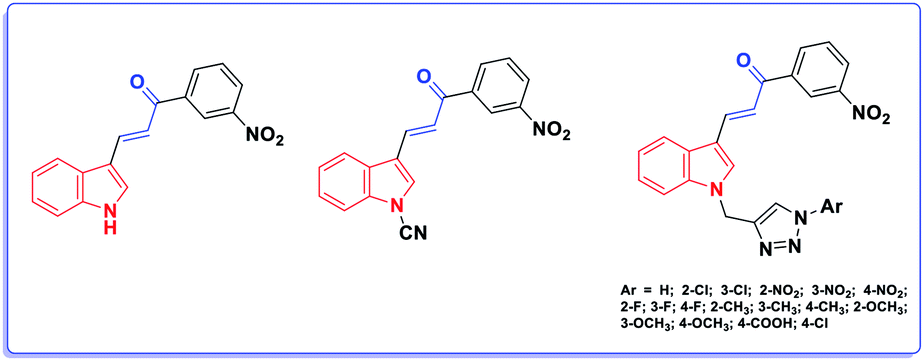 | ||
| Fig. 2 The reported indole acrylate-based anticancer agents. | ||
Some methods, such as a straightforward strategy involving the use of a Michael acceptor for the synthesis of α,β-unsubstituted indoles, have been developed to transform C–H bonds into C–C bonds by Friedel–Crafts alkylation and acylation reactions,7 also called cross-dehydrogenative coupling (CDC) reactions.8
Most of the widely used methods involve the conjugate addition of heterocycles to α,β-unsaturated carbonyl compounds; however, most of these methods require different catalysts such as chiral transition-metal catalysts and organocatalysts.9 Moreover, in another reported study, directing groups or auxiliary amine catalysts were used to convert the β-position to the α-position through dehydrogenation at the β-position; however, studies based on this strategy are rare.10 Recently, some studies have reported the use of acrylate derivatives for the synthesis of indolylacrylates under harsh conditions such as the use of expensive catalysts, toxic solvents, and high temperatures (Scheme 1).11 Recently, we proposed the use of a sulfone-containing imidazolium-based Brønsted acid ionic liquid as a catalyst and butyl acetate as a solvent to replace dipolar and aprotic solvents.12 In this study, we investigated the possibility of using reactive carbonyl compounds, i.e. pyruvates, for the synthesis of α-indolylacrylate derivatives under green conditions, and to our delight, highest reactivity of pyruvate with indole was observed in a biphasic system with an ionic liquid as a catalyst and butyl acetate as a solvent.
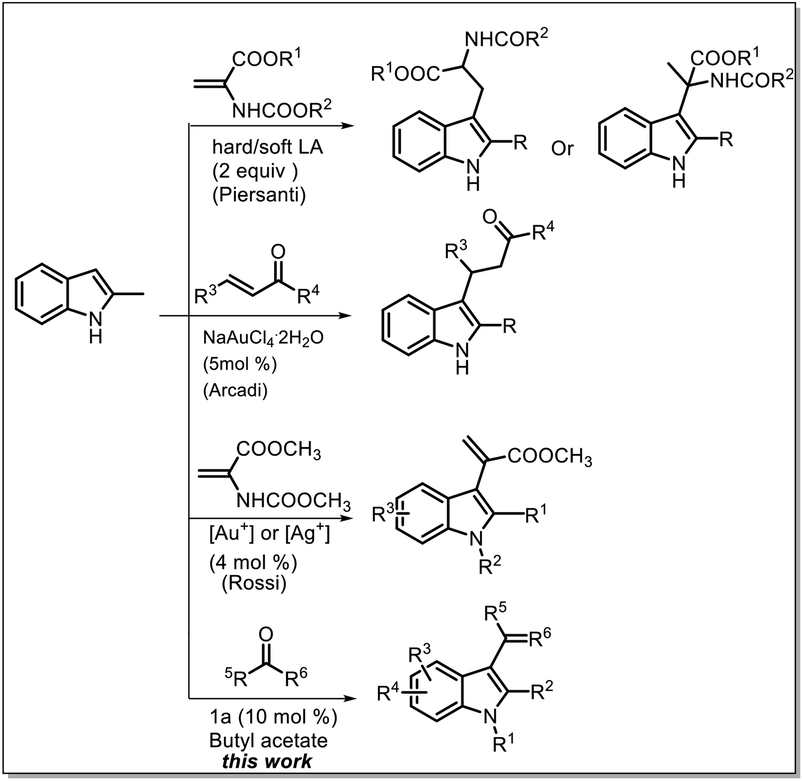 | ||
| Scheme 1 Routes for the synthesis of C3-functionalized indoles. | ||
Results and discussion
We used our system 1a/butyl acetate12 in a simple protocol involving the reaction of 2-methyl indole with ethyl pyruvate as a model reaction (Table 1); this is the first report on the application of pyruvate compounds for the synthesis of α-indolylacrylate derivatives and replacement of dipolar, aprotic, and hazardous solvents by butyl acetate.
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield (%) |
| a Reaction conditions: 2a, 0.3 mmol; 3a, 0.3 mmol; 4a, 0.3 mmol; catalyst, 0.03 mmol; medium, 0.5 ml; temperature, 80 °C; and time, 2 h.b Temperature, 60 °C.c Time, 1 h. | |||
| 1a | 1a | Butyl acetate | 91 |
| 2 | 1b | Butyl acetate | 25 |
| 3 | 1c | Butyl acetate | 65 |
| 4 | 1a | Ethyl acetate | 70 |
| 5 | TfOH | Butyl acetate | 50 |
| 6 | PTSA | Butyl acetate | 47 |
| 7 | TfOH | EtOH | 35 |
| 8 | TfOH | Anisole | 40 |
| 9 | TfOH | Nitromethane | 10 |
| 10 | TfOH | 1,2-Dichloroethane | 30 |
| 11 | TfOH | 1,4-Dioxane | 7 |
| 12 | TfOH | Toluene | 70 |
| 13 | TfOH | Benzene | 65 |
| 14b | 1a (15%) | Butyl acetate | 91 |
| 15c | 1a (5%) | Butyl acetate | 72 |
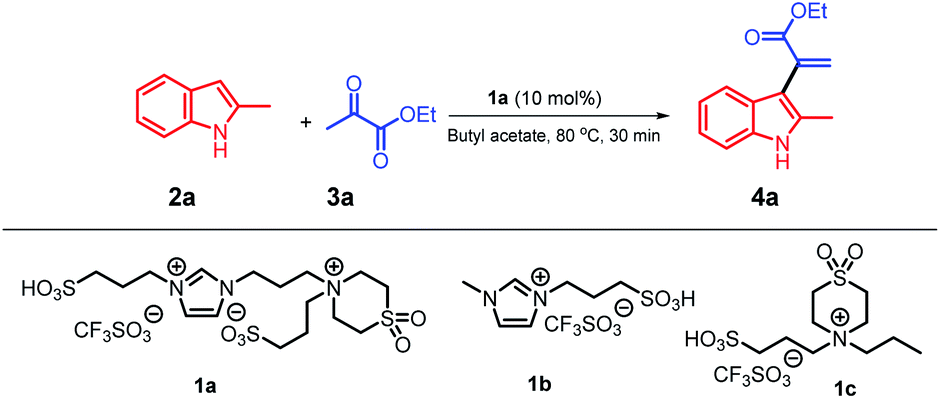
The reaction of 2-methylindole 2a with ethyl pyruvate 3a was conducted in a biphasic system at 80 °C for 30 minutes in the presence of 10 mol% of ionic liquid 1a as a catalyst and butyl acetate as a solvent; the ratio of 2a/3a was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the product 4a was obtained in an excellent yield (91%) (Table 1, entry 1). With the Forbes's ionic liquid 1b,12 4a was obtained in a 25% yield (entry 2), whereas with the ionic liquid 1c,12 the yield of 4a was increased to 65% (entry 3). Note that when we replaced the butyl acetate solvent with the ethyl acetate solvent, the yield of 4a reached 70% (entry 4). Strong acids, such as triflic acid and PTSA, were used with the butyl acetate solvent, and 4a was obtained in 50% and 47% yields (entries 5 and 6), respectively. We examined other green solvents such as ethanol and anisole, and 4a could be isolated in 35% and 40% yields, respectively. Nitromethane was tested as a polar solvent, and 4a was obtained in a 10% yield; moreover, when other dipolar and aprotic solvents, such as 1,2-dichloroethane (DCE) and 1,4-dioxane, were used, the yields of 4a were 30% and 7% (entries 10 and 11), respectively. These results indicate that the abovementioned reaction does not require dipolar and aprotic solvents, and the product 4a can be obtained in an excellent yield using weakly-to-moderately polar butyl acetate. Moreover, we investigated the reactivity of the substrate using non-polar solvents such as toluene and benzene, and the yields of 4a were significantly increased to 70 and 65% (entry 12 and 13), respectively. In addition, we increased the amount of the ionic liquid catalyst 1a; however, no further increase in the yield was observed (entry 14). On the other hand, when we decreased the amount of the ionic liquid catalyst 1a to 5 mol%, 4a was obtained in a 72% yield (entry 15). Moreover, the results of this reaction completely confirmed that a balance between butyl acetate as a solvent and ionic liquid 1a as a catalyst was required and the substrates were more active in this biphasic system. Moreover, the recyclability of the ionic liquid 1a was studied for conducting this model reaction on a large scale using 10 mmol of the substrate to obtain the target product 4a in a 91% yield; the results indicated that 1a could be recycled 8 times without any significant loss in its activity, and the recovery of the butyl acetate solvent reached about 96%.
:1, and the product 4a was obtained in an excellent yield (91%) (Table 1, entry 1). With the Forbes's ionic liquid 1b,12 4a was obtained in a 25% yield (entry 2), whereas with the ionic liquid 1c,12 the yield of 4a was increased to 65% (entry 3). Note that when we replaced the butyl acetate solvent with the ethyl acetate solvent, the yield of 4a reached 70% (entry 4). Strong acids, such as triflic acid and PTSA, were used with the butyl acetate solvent, and 4a was obtained in 50% and 47% yields (entries 5 and 6), respectively. We examined other green solvents such as ethanol and anisole, and 4a could be isolated in 35% and 40% yields, respectively. Nitromethane was tested as a polar solvent, and 4a was obtained in a 10% yield; moreover, when other dipolar and aprotic solvents, such as 1,2-dichloroethane (DCE) and 1,4-dioxane, were used, the yields of 4a were 30% and 7% (entries 10 and 11), respectively. These results indicate that the abovementioned reaction does not require dipolar and aprotic solvents, and the product 4a can be obtained in an excellent yield using weakly-to-moderately polar butyl acetate. Moreover, we investigated the reactivity of the substrate using non-polar solvents such as toluene and benzene, and the yields of 4a were significantly increased to 70 and 65% (entry 12 and 13), respectively. In addition, we increased the amount of the ionic liquid catalyst 1a; however, no further increase in the yield was observed (entry 14). On the other hand, when we decreased the amount of the ionic liquid catalyst 1a to 5 mol%, 4a was obtained in a 72% yield (entry 15). Moreover, the results of this reaction completely confirmed that a balance between butyl acetate as a solvent and ionic liquid 1a as a catalyst was required and the substrates were more active in this biphasic system. Moreover, the recyclability of the ionic liquid 1a was studied for conducting this model reaction on a large scale using 10 mmol of the substrate to obtain the target product 4a in a 91% yield; the results indicated that 1a could be recycled 8 times without any significant loss in its activity, and the recovery of the butyl acetate solvent reached about 96%.
Based on these results, we selected the condition stated in entry 1 as an identical condition to extend the scope of the substrate by smoothly reacting different varieties of indole substituents with ethyl pyruvate 3a (Scheme 2); moreover, different positions of indole 2a–k were investigated for this reaction, and the corresponding α-indolylacrylates 4a–k with generally excellent to good yields were obtained. Both electron-rich (2a, 2c, and 2e–g) and moderately electron-poor (2b, 2d, and 2h) indoles readily participated in the reaction; however, when different positions of electron-poor indoles were used, the yield of 4k was affected and reached 70%. Moreover, when we changed the group at the C5 position to hydrogen and methoxy, 4i and 4j were obtained with increased yields, respectively. When electron-rich indoles were used, 4c and 4i were obtained in excellent yields (96% and 93%), respectively; using indoles with a phenyl group at the C2 position and different groups at the C1 position, the corresponding products 4e, 4f, and 4g could be obtained in 88%, 91%, and 90% yields, respectively. When the moderately electron-poor indoles 2b, 2d, and 2h with different groups were used, the corresponding products 4b, 4d, and 4h were obtained in very satisfactory yields (90%, 93%, and 92%), respectively, and no detectable formation of decomposition products occurred.
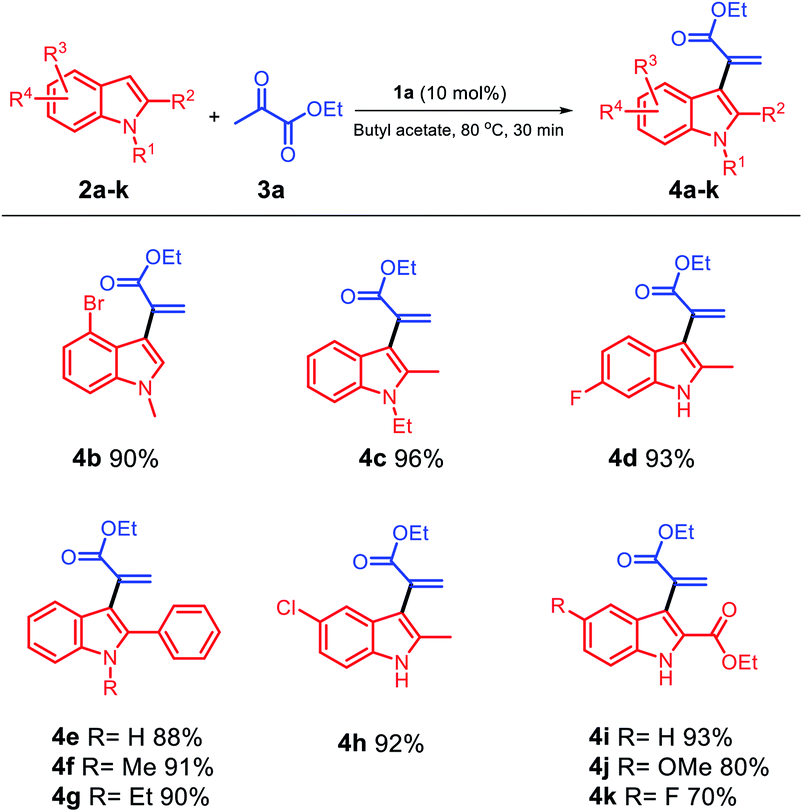 | ||
| Scheme 2 Scope of the substrates with respect to the indole component. | ||
Then, we probed the scope of the reaction with respect to the ketone component. The reaction of methyl pyruvate with methoxy pyruvate was also investigated, and the products 4l and 4m were obtained in excellent yields (Scheme 3). Moreover, we used a pyruvate substrate with a phenyl group at the C5 position, and the obtained product 4n was isolated with a 92% yield. In addition, trans-3-hexenyl pyruvate (3e) smoothly reacted with 2-methylindole using butyl acetate as a solvent and 1a as a catalyst, and the trans-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond was effectively retained in the product 4o isolated with an excellent yield of 94%. Ethyl 2-oxohexanoate was examined in the biphasic system of 1a/butyl acetate, and 4p was obtained in a 95% yield. We used trifluoroacetone to see if it would react in the 1a/butyl acetate biphasic system, and to our delight, the target product 4q was obtained in an 88% yield after 8 h.
C bond was effectively retained in the product 4o isolated with an excellent yield of 94%. Ethyl 2-oxohexanoate was examined in the biphasic system of 1a/butyl acetate, and 4p was obtained in a 95% yield. We used trifluoroacetone to see if it would react in the 1a/butyl acetate biphasic system, and to our delight, the target product 4q was obtained in an 88% yield after 8 h.
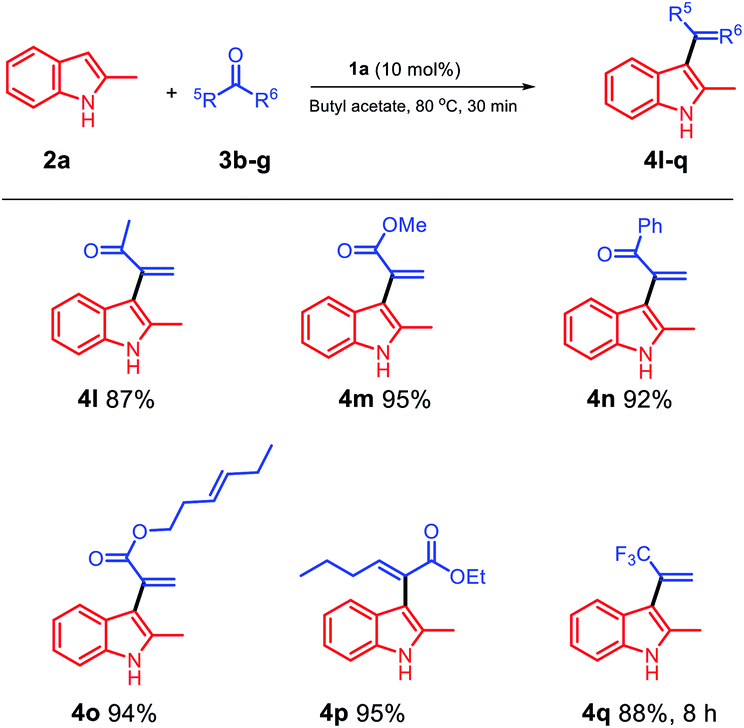 | ||
| Scheme 3 Scope of the substrates with respect to the ketone component. | ||
Based on the abovementioned results, a plausible mechanism was proposed (Scheme 4). The C3 position of 2-methyl indole is a reactive nucleophilic site and can attack the ketone carbonyl group of ethyl pyruvate. The initial episode of the reaction involved the formation of an intermediate hydroxyl group (I) from 2a and 3a in the presence of the acid catalyst 1a. The intermediate (I) can be possibly converted to 4a by eliminating one molecule of water.
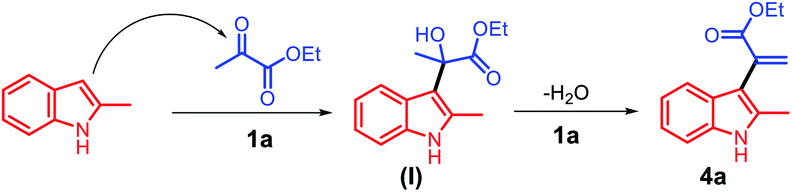 | ||
| Scheme 4 The proposed reaction mechanism for the synthesis of α-indolylacrylates. | ||
When an electro-rich group, such as methyl and methoxy group, was used at the C3 position, different structures were obtained; moreover, two molecules of ethyl pyruvate were reacted with one molecule of a 2-methylindole derivative (Scheme 5) to optimize the conditions for the solvents and acid catalysis, and the final products 4r and 4s were obtained in excellent yields. However, when a dipolar solvent was used (Scheme 5), 4r and 4s were obtained in poor yields; moreover, using the non-polar solvent toluene, the yield of 4s was increased to 70%. As observed from the table shown in Scheme 5, the 1a/butyl acetate system is a unique system because it not only can replace the dipolar aprotic solvent but can also replace a non-polar toxic solvent such as toluene.
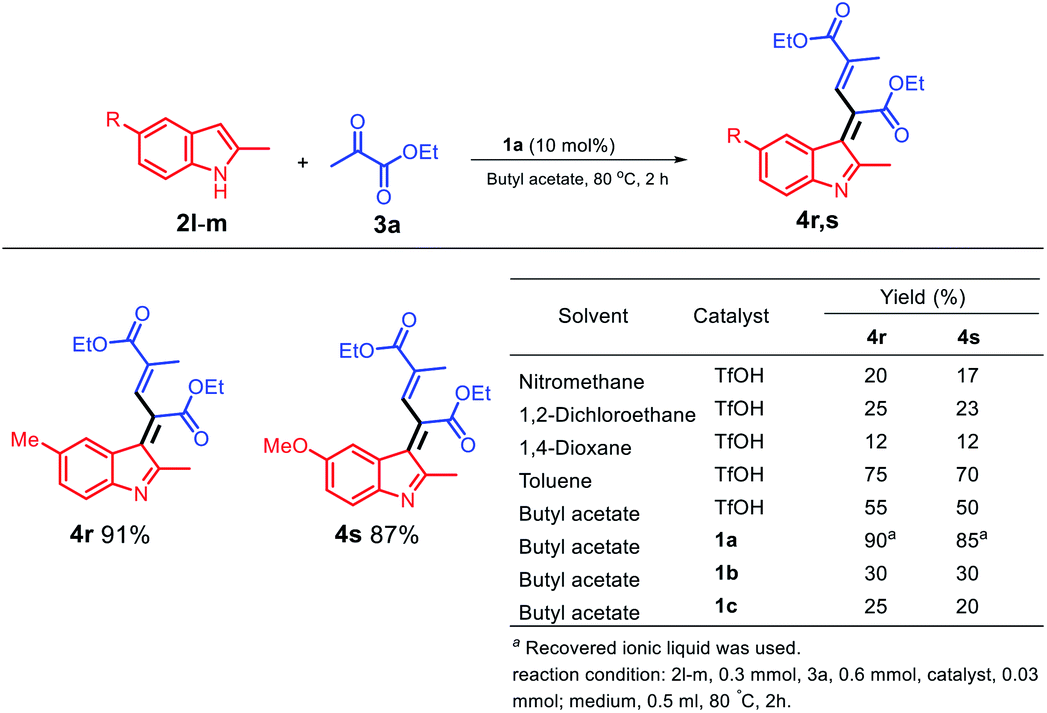 | ||
| Scheme 5 Schematic for the synthesis of electro-rich indolylacrylates. | ||
Considering the formation of the two products 4r and 4s, the nucleophilicities of 2l and 2m in the presence of acid can be considered as the key for rendering the reaction possible. The C3 positions of 2l and 2m are reactive nucleophilic sites that can attack the electrophilic carbonyl group of 3a; using the acid ionic liquid catalyst 1a, the intermediate (I) can be converted to intermediate (II), which has a nucleophilic carbon and can attack the carbonyl group of another molecule of 3a and form the intermediate (III). The abstraction of the NH proton by the negatively charged oxygen using the Brønsted acid ionic liquid catalyst 1a leads to the formation of (IV), which undergoes water elimination to finally afford the products 4r and 4s (Scheme 6).
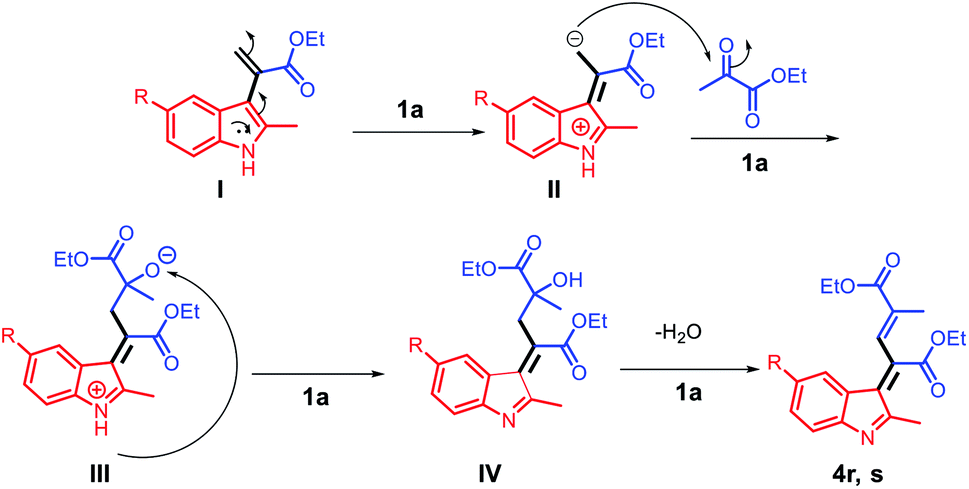 | ||
| Scheme 6 The proposed reaction mechanism for the synthesis of the compounds 4r and 4s. | ||
We studied the reaction of 2-unsubstituted indole with ethyl pyruvate, and the C3 position of the indole nucleus reacted with the α-carbon of ethyl pyruvate to form a new carbon–carbon bond. We optimized the conditions of this reaction using different solvents and catalysts; the ionic liquid was immiscible with the organic phase, thus forming a biphasic reaction system. The designed structure of the ionic liquid 1a involved a cyclic sulfone fragment, which enabled the ionic phase to provide a suitable reaction microenvironment for the stabilization of the reaction intermediate. Other ionic liquids, such as 1b and 1c, do not have this property due to the lack of a strongly polar part. Moreover, the biphasic system of 1a/butyl acetate is the best condition for this reaction (Table 2, entry 8).
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield |
| a 2a, 0.3 mmol; 3a, 0.3 mmol, TfOH, 0.03 mmol; 1a, 0.03 mmol; solvent, 10 ml, 80 °C, 1 h. | |||
| 1 | TfOH | 1,4-Dioxane | 10 |
| 2 | TfOH | 1,2-Dichloroethane | 42 |
| 3 | TfOH | MeNO2 | 20 |
| 4 | TfOH | EtOH | 38 |
| 5 | TfOH | Anisole | 15 |
| 6 | TfOH | Ethyl acetate | 35 |
| 7 | TfOH | Butyl acetate | 42 |
| 8 | 1a | Butyl acetate | 92 |
| 9 | 1b | Butyl acetate | 33 |
| 10 | 1c | Butyl acetate | 63 |
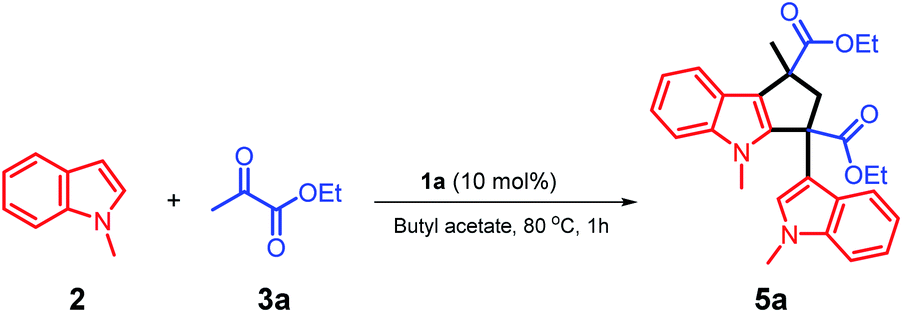
The unique acidic environment offered by the ionic liquid catalyst 1a should be the key to initiate the dimerization of two molecules of intermediate (I) to form the diester indole intermediate (II); the C2 position of the indole ring will act as a nucleophilic site to attack the positively charged carbon of (II) to afford the product 5a (Scheme 7). The crystal and molecular structures of 5a were further confirmed using single-crystal X-ray diffraction (ESI†).
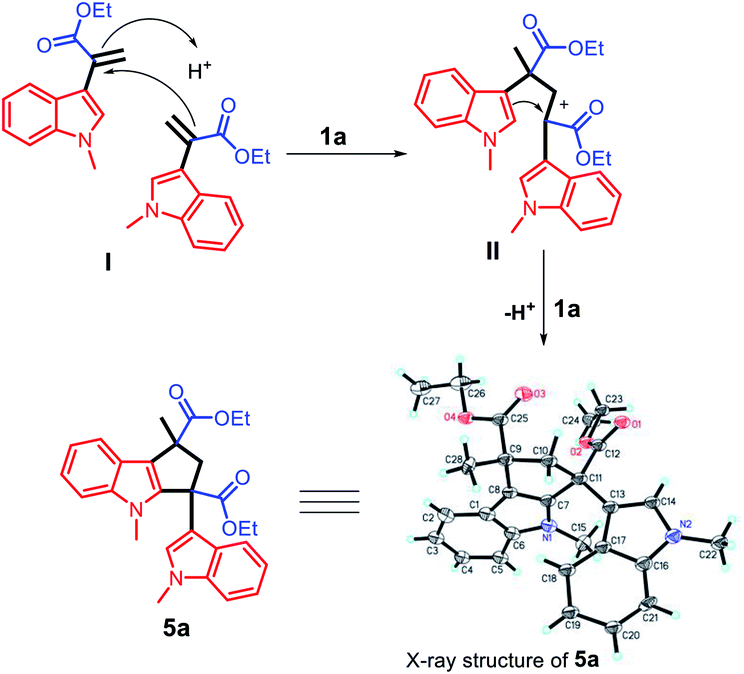 | ||
| Scheme 7 The proposed reaction mechanism for the synthesis of compound 5a. | ||
Since many organic acrylates and indole-containing compounds have anticancer properties, the newly synthesized α-indolylacrylates 4a–s and 5a were screened for their in vitro growth inhibitory potential against the human cervical adenocarcinoma (HeLa) cell line using the 3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The MTT cell proliferation assay has been widely accepted as a reliable way to measure the cell proliferation rate when metabolic events lead to apoptosis or necrosis. After incubation with the abovementioned compounds at different concentrations for 24 h, the cells were treated with MTT to measure their growth/viability using a spectrophotometer.13 The results showed that the synthesized compounds 4f, 4h, 4l, 4o, and 4p have maximum percentage cytotoxicity at 100 μg ml−1 (Fig. 3). These five compounds were subjected to further cytotoxicity studies against the HeLa cell line to determine the IC50 values. Among these derivatives, the compound 4I exhibited a potent anticancer activity with the IC50 value of 9.73 μg (Fig. 4). In future, several derivatives of 4l analogs will be subjected to further tests for cytotoxic activity against multiple human cancer cell lines and inhibitory activity against several enzymes to study a comprehensive SAR.
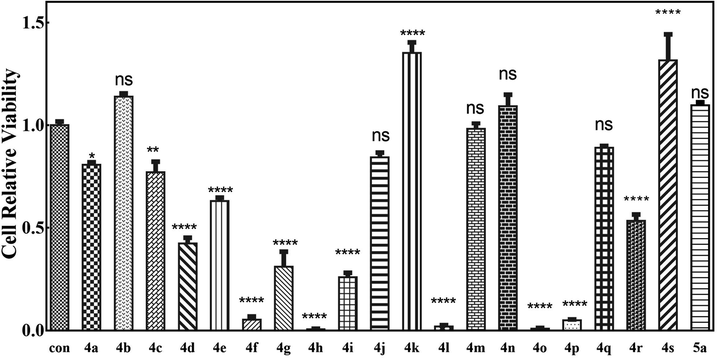 | ||
| Fig. 3 MTT assays of cell relative viability. (a) Herein, 100 μM 4a–s and 5a were incubated with the HeLa cells for 48 h. ns represents P > 0.05, * represents p value < 0.05, ** represents p value < 0.01, and **** represents p value < 0.001. The significant difference was compared with the control group. | ||
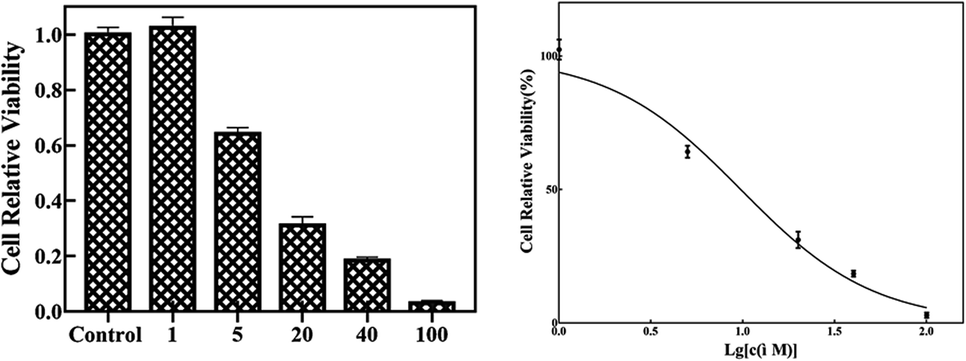 | ||
| Fig. 4 The compound 4l at different concentrations was incubated with the HeLa cells for 48 h. The IC50 is 9.73 μM. | ||
In silico studies
| Comp. no. | Molecular properties | ||||||
|---|---|---|---|---|---|---|---|
| TPSAa | Nrotbb | milogPc |
nOH–NHd | nO–Ne | M. wtf | nVsg | |
| a Topological polar surface area.b Number of rotatable bonds.c The parameter of lipophilicity.d Number of hydrogen bond donor sites.e Number of hydrogen bond acceptor sites.f Molecular weight.g Number of violations from the Lipinski's rule of five. | |||||||
| 4a | 42.10 | 4 | 2.90 | 1 | 3 | 229.28 | 0 |
| 4b | 31.24 | 4 | 3.51 | 0 | 3 | 308.18 | 0 |
| 4c | 31.24 | 5 | 3.35 | 0 | 3 | 257.33 | 0 |
| 4d | 42.10 | 4 | 3.04 | 1 | 3 | 247.27 | 0 |
| 4e | 42.10 | 5 | 4.36 | 1 | 3 | 291.35 | 0 |
| 4f | 31.24 | 5 | 4.42 | 0 | 3 | 305.38 | 0 |
| 4g | 31.24 | 6 | 4.80 | 0 | 3 | 319.40 | 0 |
| 4h | 42.10 | 4 | 3.56 | 1 | 3 | 263.72 | 0 |
| 4i | 68.40 | 7 | 3.13 | 1 | 5 | 287.31 | 0 |
| 4j | 77.64 | 8 | 3.16 | 1 | 6 | 317.34 | 0 |
| 4k | 68.40 | 7 | 2.30 | 1 | 5 | 305.31 | 0 |
| 4l | 32.86 | 2 | 2.21 | 1 | 2 | 199.25 | 0 |
| 4m | 42.10 | 3 | 2.53 | 1 | 3 | 215.25 | 0 |
| 4n | 32.86 | 3 | 3.81 | 1 | 2 | 261.32 | 0 |
| 4o | 42.10 | 7 | 4.20 | 1 | 3 | 283.37 | 0 |
| 4p | 42.10 | 6 | 4.45 | 1 | 3 | 271.36 | 0 |
| 4q | 15.79 | 2 | 3.59 | 1 | 1 | 225.21 | 0 |
| 4r | 65.50 | 7 | 4.35 | 0 | 5 | 341.41 | 0 |
| 4s | 74.74 | 8 | 3.96 | 0 | 6 | 357.41 | 0 |
| 5a | 62.48 | 7 | 5.45 | 0 | 6 | 458.56 | 1 |
Conclusions
Herein, using an ionic liquid as a catalyst and butyl acetate as a solvent, we successfully synthesized various α-indolylacrylate derivatives via the dehydrative alkenylation of indole derivatives with inexpensive, convenient, and simple ketones. Compared with the established method for the synthesis of α-indolylacrylates, the method proposed herein is a green approach involving a cost-effective reaction, high synthetic efficiency, recyclable catalyst, recyclable solvent, and easy product isolation. The newly synthesized compounds were evaluated for their in vitro anticancer activity against a human cervical adenocarcinoma (HeLa) cell line, and the results revealed that the compound 4l was the most active member with the IC50 value of 9.73 μM. Moreover, based on the results of the Molinspiration calculations, the compound 4l was predicted to have good oral absorption. Based on the abovementioned results, 4l is considered to be a promising lead for the future development of new potent antitumor agents.Experimental
General remarks
1-(3-Aminopropyl)imidazole, divinyl sulfone, 1,3-propane sulfonate, trifluoromethanesulfonic acid, n-butylamine, 1-methylimidazole, 2-methyl-1H-indole, 1-methyl-1H-indole, 2-phenyl-1H-indole, 1-methyl-2-phenyl-1H-indole, 1-ethyl-2-phenyl-1H-indole, 4-bromo-1-methyl-1H-indole, ethyl 1-methyl-1H-indole-2-carboxylate, 1-ethyl-2-methyl-1H-indole, 6-fluoro-2-methyl-1H-indole, 5-methoxy-2-methyl-1H-indole, 2,5-dimethyl-1H-indole, ethyl 5-fluoro-1H-indole-2-carboxylate, ethyl 5-methoxy-1H-indole-2-carboxylate, and 5-chloro-2-methyl-1H-indole were purchased from Energy Chemical Company. Ethyl pyruvate, biacetyl, 1-phenylpropane-1,2-dione, methyl pyruvate, ethyl 2-oxohexanoate, (E)-hex-3-en-1-yl 2-oxopropanoate, and 1,1,1-trifluoropropan-2-one were purchased from Alfa Aesar Chemical Company. Butyl acetate, ethyl acetate, petroleum ether, sodium chloride, methanol, acetonitrile, acetone, 1,2-dichloroethane, anisole, 1,4-dioxane, and ethanol were purchased from Sinopharm Chemical Reagents Limited Company (SCRC). The 1H, 13C, and 19F NMR spectra were obtained using Bruker AV-400 (400 MHz 1H, 100 MHz 13C, and 375 MHz 19F) at room temperature. Fourier transform infrared (FTIR) spectra were acquired via FTIR Bruker (VERTEX 70) using the liquid film technology. A high-resolution mass spectrum (HRMS) was obtained using the Bruker micrOTOF-Q II instrument. Melting points of the products were determined by a microscopic melting point meter (Yu Hua Instrument, X-4).A typical procedure for the reaction of 2-methyl-1H-indole with ethyl pyruvate (the model reaction)
The reaction was carried out in a 10 ml V-type flask equipped with triangle magnetic stirring. In a typical reaction, 1a (19.11 mg, 0.03 mmol) was mixed with 2-methylindole 2a (39.354 mg, 0.3 mmol) and ethyl pyruvate 3a (34.857 mg, 0.3 mmol) in 0.5 ml of butyl acetate. The mixture was stirred for 30 min at 80 °C. After the completion of the reaction, the mixture was cooled down to room temperature. Butyl acetate was removed under reduced pressure and then extracted with EtOAc (3 × 0.5 ml). The product was obtained by isolation with preparative TLC (eluting solution: petroleum ether/ethyl acetate = 10/1 v/v). The desired product 4a was obtained in a 90–91% yield. At the end, the Brønsted acid ionic liquid catalyst was recycled several times; a sodium chloride solution (3 × 0.5 ml) was added to dissolve the ionic liquid; then, EtOAc (3 × 0.5 ml) was added to extract the remaining desired product. Tests for substrate scope were achieved according to the model reaction conditions.A typical procedure for the reaction of 2,5-dimethyl-1H-indole/5-methoxy-2-methyl-1H-indole with ethyl pyruvate
The reaction was carried out in a 10 ml V-type flask equipped with triangle magnetic stirring. In a typical reaction, 1a (19.11 mg, 0.03 mmol) was mixed with 2,5-dimethyl-1H-indole (43.561 mg, 0.3 mmol)/5-methoxy-2-methyl-1H-indole (48.361 mg, 0.3 mmol) and ethyl pyruvate 3a (34.857 mg, 0.3 mmol) in 0.5 ml of butyl acetate. The mixture was stirred for 2 h at 80 °C. After the completion of the reaction, the mixture was cooled down to room temperature. Butyl acetate was removed under reduced pressure and then extracted with EtOAc (3 × 0.5 ml). The product was obtained by isolation with preparative TLC (eluting solution: petroleum ether/ethyl acetate = 10/1 v/v). The desired products 5r and 5s were obtained in a 91% and 87% yield, respectively.A typical procedure for the reaction of 1-methyl-1H-indole with ethyl pyruvate
The reaction was carried out in a 10 ml V-type flask equipped with triangle magnetic stirring. In a typical reaction, 1a (19.11 mg, 0.03 mmol) was mixed with 1-methylindole (39.354 mg, 0.3 mmol) and ethyl pyruvate 3a (34.857 mg, 0.3 mmol) in 1 ml of butyl acetate. The mixture was stirred for 1 h at 80 °C. After the completion of the reaction, the mixture was cooled down to room temperature. Butyl acetate and the product were separated from the ionic liquid phase to reuse the ionic liquid, and then, butyl acetate was distilled for reuse; subsequently, the product remained in the ionic liquid phase, which was dissolved in EtOAc (1 ml). The product was obtained by isolation with preparative TLC (eluting solution: petroleum ether/ethyl acetate = 10/1 v/v). The desired product 5a was obtained in an 89–91% yield. At the end, the Brønsted acid ionic liquid catalyst was recycled several times; after this, a sodium chloride (3 × 0.5 ml) solution was added to dissolve the ionic liquid; then, EtOAc (3 × 0.5 ml) was added to extract the remaining desired product.Cell lines
The HeLa cells and A549 cells were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. The cells were cultured in the Dulbecco's modified Eagle medium (Hyclone, Thermo Scientific) and supplemented with 10% fetal bovine serum (Zhejiang Tianhang Biotechnology Co., Ltd). The cells were cultured under a 5% CO2 atmosphere at 37 °C.In vitro cytotoxicity study (MTT assay)
Cytotoxicity was estimated using an MTT assay. The cells were seeded in 96-well plates and cultured overnight to reach a ∼80% confluency. Fresh media containing the compounds were incubated with cells for 48 h. Then, 100 μl of 0.5 mg ml−1 thiazolyl blue tetrazolium bromide (MTT, in the DMEM medium) solution was added to each well, followed by a 4 h incubation at 37 °C. Then, the medium was removed, and 150 μl dimethyl sulfoxide (DMSO) was added. The optical density of the final solution was measured by a micro plate reader at the wavelength of 490 nm.Spectroscopic data of the obtained products
351, 1708, 1614, 1550, 1459, 1370, 1281, 1177. HRMS-ESI (m/z) calcd for C14H15NO2, [M + Na]+ 229.1103, found 252.0997.Conflicts of interest
The authors declare that they have no competing financial interest.Notes and references
- (a) J. M. Finefield, J. C. Frisvad, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2012, 75, 812–833 CrossRef CAS PubMed; (b) M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed; (c) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed; (d) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (e) P. S. Bhadury and J. Pang, Curr. Org. Chem., 2014, 18, 2108–2124 CrossRef CAS.
- (a) M. Fantacuzzi, B. De Filippis, M. Gallorini, A. Ammazzalorso, L. Giampietro, C. Maccallini, Z. Aturki, E. Donati, R. S. Ibrahim, E. Shawky, A. Cataldi and R. Amoroso, Eur. J. Med. Chem., 2020, 185, 111815 CrossRef PubMed; (b) R. I. Al-Wabli, A. A. Almomen, M. S. Almutairi, A. B. Keeton, G. A. Piazza and M. I. Attia, Drug Des. Dev. Ther, 2020, 14, 483–495 CrossRef PubMed; (c) M. Demurtas, A. Baldisserotto, I. Lampronti, D. Moi, G. Balboni, S. Pacifico, S. Vertuani, S. Manfredini and V. Onnis, Bioorg. Chem., 2019, 85, 568–576 CrossRef CAS PubMed; (d) N. M. Cury, R. M. Capitão, R. do C. B. de Almeida, L. L. Artico, J. R. Corrêa, E. F. Simão dos Santos, J. A. Yunes and C. R. D. Correia, Eur. J. Med. Chem., 2019, 181, 111570 CrossRef PubMed; (e) W. Li, H. Sun, F. Xu, W. Shuai, J. Liu, S. Xu, H. Yao, C. Ma, Z. Zhu and J. Xu, Bioorg. Chem., 2019, 85, 49–59 CrossRef CAS PubMed; (f) G. La Regina, R. Bai, A. Coluccia, V. Naccarato, V. Famiglini, M. Nalli, D. Masci, A. Verrico, P. Rovella, C. Mazzoccoli, E. Da Pozzo, C. Cavallini, C. Martini, S. Vultaggio, G. Dondio, M. Varasi, C. Mercurio, E. Hamel, P. Lavia and R. Silvestri, Eur. J. Med. Chem., 2018, 152, 283–297 CrossRef CAS PubMed.
- (a) M. F. Khan, T. Anwer, A. Bakht, G. Verma, W. Akhtar, M. M. Alam, M. A. Rizvi, M. Akhter and M. Shaquiquzzaman, Bioorg. Chem., 2019, 87, 667–678 CrossRef CAS PubMed; (b) I. D. Vlaicu, R. Olar, C. Maxim, M. C. Chifiriuc, C. Bleotu, N. Stănică, G. Vasile Scăeţeanu, C. Dulea, S. Avram and M. Badea, Appl. Organomet. Chem., 2019, 33, 1–13 CrossRef; (c) G. Verma, G. Chashoo, A. Ali, M. F. Khan, W. Akhtar, I. Ali, M. Akhtar, M. M. Alam and M. Shaquiquzzaman, Bioorg. Chem., 2018, 77, 106–124 CrossRef CAS PubMed; (d) S. Fang, L. Chen, M. Yu, B. Cheng, Y. Lin, S. L. Morris-Natschke, K. H. Lee, Q. Gu and J. Xu, Org. Biomol. Chem., 2015, 13, 4714–4726 RSC; (e) C. Yu-Jen, M.-S. Shiao, M.-L. Hsu, T.-H. Tsai and W. Sheng-Yuan, J. Agric. Food Chem., 2001, 49, 5615–5619 CrossRef PubMed.
- (a) L. K. Gediya and V. C. Njar, Expert Opin. Drug Discovery, 2009, 4, 1099–1111 CrossRef CAS PubMed; (b) C. V. Junior, A. Danuello, V. dS. Bolzani, E. J. Barreiro and C. A. M. Fraga, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef PubMed.
- A. Arcadi, G. Bianchi, M. Chiarini, G. D'Anniballe and F. Marinelli, Synlett, 2004, 6, 944–950 CrossRef.
- S. K. Yadav, R. K. Yadav and U. Yadava, DNA Repair, 2020, 86, 102765 CrossRef PubMed.
- (a) R. J. Sundberg, Indoles, Elsevier, 1996 Search PubMed; (b) E. C. Taylor, A. H. Katz, H. Salgado-Zamora and A. McKillop, Tetrahedron Lett., 1985, 48, 5963–5966 CrossRef.
- (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (b) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436 CrossRef CAS PubMed; (c) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- (a) D. A. Evans, K. R. Fandrick, H. Song, K. A. Scheidt and R. Xu, J. Am. Chem. Soc., 2007, 129, 10029–10041 CrossRef CAS PubMed; (b) B. M. Trost and C. Muller, J. Am. Chem. Soc., 2008, 130, 2438–2439 CrossRef CAS PubMed; (c) T. Arai and N. Yokoyama, Angew. Chem., Int. Ed., 2008, 47, 4989–4992 CrossRef CAS PubMed; (d) R. Rasappan, M. Hager, A. Gissibl and O. Reiser, Org. Lett., 2006, 8, 6099–6102 CrossRef CAS PubMed; (e) J. Zhou and Y. Tang, J. Am. Chem. Soc., 2002, 124, 9030–9031 CrossRef CAS PubMed.
- S. Ueno, R. Shimizu and R. Kuwano, Angew. Chem., Int. Ed., 2009, 48, 4543–4545 Search PubMed.
- (a) H. Hikawa and Y. Yokoyama, RSC Adv., 2013, 3, 1061–1064 Search PubMed; (b) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750–5753 Search PubMed; (c) Y. Lian and H. M. L. Davies, Org. Lett., 2012, 14, 1934–1937 Search PubMed; (d) K. Aikawa, K. Honda, S. Mimura and K. Mikami, Tetrahedron Lett., 2011, 52, 6682–6686 Search PubMed; (e) Y. Cai, S.-F. Zhu, G.-P. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2011, 353, 2939–2949 Search PubMed; (f) T. Goto, Y. Natori, K. Takeda, H. Nambu and S. Hashimoto, Tetrahedron: Asymmetry, 2011, 22, 907–915 Search PubMed; (g) A. N. Campbell, E. B. Meyer and S. S. Stahl, Chem. Commun., 2011, 47, 10257–10259 RSC; (h) Q. Liu, G. Li, H. Yi, P. Wu, J. Liu and A. Lei, Chem.–Eur. J., 2011, 17, 2353–2357 CrossRef CAS PubMed; (i) H. Liu and D.-M. Du, Eur. J. Org. Chem., 2010, 11, 2121–2131 CrossRef; (j) G. Blay, I. Fernandez, M. C. Munoz, J. R. Pedro and C. Vila, Chem.–Eur. J., 2010, 16, 9117–9121 CrossRef CAS PubMed; (k) V. Pirovano, D. Facoetti, M. Dell'Acqua, E. Della Fontana, G. Abbiati and E. Rossi, Org. Lett., 2013, 15, 3812–3815 CrossRef CAS PubMed.
- (a) A. El-Harairy, B. Lai, L. Vaccaro, M. Li and Y. Gu, Adv. Synth. Catal., 2019, 361, 3342–3350 CrossRef CAS; (b) A. El-Harairy, M. Yue, W. Fan, F. Popowycz, Y. Queneau, M. Li and Y. Gu, ChemCatChem, 2019, 11, 4403–4410 CrossRef CAS; (c) A. El-Harairy and Y. Gu, ISGC-International Symposium on Green Chemistry, La Rochelle, France, May 2019, https://isgc-symposium-2019.livescience.io/medias/isgc2019/abstract_submission/_pdf/abstract-892-8B539.pdf Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- https://www.molinspiration.com/cgi-bin/properties.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1980793. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00990c |
| This journal is © The Royal Society of Chemistry 2020 |