
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTreasures old and new: what we can learn regarding the macrocyclic problem from past and present efforts in natural product total synthesis
Maryam Sadat Alehashem *a,
Azhar Bin Ariffinb,
Paul B. Savagea,
Wageeh Abdulhadi Yehya Dabdawbc and
Noel Francis Thomas*d
*a,
Azhar Bin Ariffinb,
Paul B. Savagea,
Wageeh Abdulhadi Yehya Dabdawbc and
Noel Francis Thomas*d
aDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, USA. E-mail: maryam.alehashem@yahoo.com
bDepartment of Chemistry, Faculty of Science, University Malaya, 50603 Kuala Lumpur, Malaysia
cNanotechnology and Catalysis Research Centre (NANOCAT), University of Malaya, 50603 Kuala Lumpur, Malaysia
dAmerican Degree Transfer Program, Methodist College Kuala Lumpur, 50470 Kuala Lumpur, Malaysia. E-mail: noelfthomas@yahoo.com
First published on 17th March 2020
Abstract
In this review the strategies leading to successful macrocyclization, in the context of total synthesis are discussed. These synthetic endeavors will be discussed paying particular attention to the methods employed, and including the type of reactive intermediates that could play a key role in key cyclization steps. In many cases “simple” macrocyclization methods were found to be inadequate, and alternative creative approaches were required. For example, we describe Boger's imaginative development of the intramolecular version of the Larock annulation which yielded the chloropeptin 1 DEF macrocycle. Peptide coupling approaches were unsuccessful. In another example, a key macrocyclic domain within diazonamide was beautifully installed (Nicolaou, et al.) by single electron oxidation/reduction (Witkop reaction), thereby establishing a crucial biaryl functionality. In contrast, oxidative methodologies failed to deliver the distorted biaryl found in haouamine, and Baran, et al. subsequently exploited a spectacular pyrone N-butyne intramolecular Diels–Alder reaction to install this biaryl moiety. Other unexpected and mechanistically intriguing observations will be described throughout the review.
1. Introduction
The construction of macrocycles can present formidable synthetic challenges. The improvement in cyclisation efficiency at the expense of intermolecular reactions is a crucial goal in macrocyclisations that was tackled traditionally by high dilution and/or slow addition of one or more reagents. In the process of macrocyclization, the entropic consequences of ring/chain equilibrium must be carefully considered. Where the transformation involves a macrocyclization catalyst, selecting reaction conditions that facilitate separation of the catalyst from the substrate is an important consideration. Recently James and co-workers1 have employed the parameter Emac (efficiency of macrocyclization) where the relationship between calculated and experimentally determined yields can be exploited to compare different macrocyclization protocols. Another area of study that is becoming more important in the synthesis of macrocyclic peptides is the concept of pre-organization, described by Yudin, et al.,1 in which hydrogen bonding in linear precursors prior to or even during macrocyclization1,2 plays a critical role. This review examines several total syntheses with a view to describing the particular reactions by which the macrocyclic entity was constructed (or the key ring closure realized). Even rates of addition can have profound effects on macrocyclization outcomes. For example, in the course of this synthesis of bielschowsky model systems, Nicolaou found that when specific furan ketoester precursors where exposed to ceric ammonium nitrate, introduced by syringe addition at 0 °C, intramolecular cyclisation occurred, but if the reagent was added in one portion at 0 °C, intermolecular reactions occurred, followed by macrocyclisation resulting in even large macrocycles.Macrocyclic entities have increased in importance due to recorded anticancer, antifungal and immunosuppressive activities and because more than 100 marketed macrocyclic drugs isolated from natural products are known.3–6 The diversity-oriented synthesis of natural product-based libraries as part of drug discovery programmes has increased the focus on macrocycles in part because the conformational restriction inherent in macrocyclic structures seems to enhance binding affinity6–8 and bioavailability when compared to drug like molecules of more “conventional” structure.9
In the 1970's and in more recent developments,10,11 the highly original methodology exploiting, for example, 2,2-dipyridyl disulphide by Corey and Nicolaou, subsequently named after these inventors, solved several macrolactonisation problems in circumstances where other macrolactonisation protocols failed.12–14 On the other hand, conventional methods, e.g. intramolecular amide construction (peptide coupling), may fail rather unexpectedly in certain situations. For example, during Boger's synthesis of the chloropeptin 1 DEF macrocycle,15 he discovered that conventional peptide coupling (and free radical protocols) were unsuccessful. The macrocyclization problem was solved by an imaginative exploitation of Larock annulation (Scheme 1). The intriguing observation that free radical protocols failed to deliver the macrocycles that were the target of Boger's investigations raises the question as to whether radical cation protocols might offer unique advantages (vide infra). The macrocyclic Larock annulation16 described by Boger proceeded with good atropodiastereoselectivity and rivals in efficiency the better known Stille17 and Suzuki18,19 cross-coupling reactions. In this process, oxidative insertion, palladium ligand exchange, carbopalladation etc. are key mechanistic events.20 For selected papers related to the original Larock annulation the following papers should be consulted Larock,21–24 Cacchi,25,26 Gribble27 and Poli.28
 | ||
| Scheme 1 Efficient macrocyclisation achieved by the Larock annulation.15 | ||
In the first part of this review, although we examine an interesting oxidative cleavage process, we wish to highlight mainly electron transfer processes involving oxidative and/or reductive protocols. At specific points we will include carbon centred radical protocols exemplified by the use of tributyltin hydride for comparison. Although such examples will be presented as possible alternatives to intramolecular peptide coupling, lactamisation and lactonization methods, these latter methods will be discussed in situations where the use of these “more conventional” methods has been singularly instructive and efficient. In the second part, we will attempt to examine other strategies for macrocyclic construction more broadly. This will include exploitation of the Tsuji–Trost reaction, Rh(III) catalyzed processes, HATU catalyzed macrolactamisation, Witkop processes and imaginative applications of the intramolecular Diels Alder reaction to list only a few examples. Occasionally, we include a few non – macrocyclic syntheses where these have a bearing on mechanistic questions (for example the chemistry in Scheme 14).
1.1. Macrocycles accessed by means of ring cleavage/expansion protocols
Tan et al.9 has developed an approach in which polycyclic enol ethers and enamines were exposed to RuCl3 and NaIO4. The process resulting in novel macrocycles is illustrated in Scheme 2. | ||
| Scheme 2 Tan's oxidative ring cleavage expansion approach.29 | ||
For certain substrates, oxone based systems proved very effective. The mechanism most likely involves a cyclic ruthenium complex (Scheme 3).30
 | ||
| Scheme 3 The oxidative cleavage-ring expansion mechanism.9 | ||
This method implies a creative alternative to macrocyclic lactonization and lactamisation approaches. For further examples of ring expansion reactions creatively applied to the macrocyclic problem, including non-oxidative variants, the review by Unsworth is highly informative and thought provoking.31 Having examined an oxidation cleavage process exemplified by the Tan chemistry, we now turn to a single electron transfer oxidative cyclisation developed by the Nicolaou group32 during their investigation of the synthesis of complex furanocembranoids.
1.2. Exploitation of CAN (ceric ammonium nitrate) promoted single electron oxidation in macrocyclic synthesis
The furanocembranoids are marine diterpenoids possessing a C14 cembrane skeleton. Examples of such structures are found in the publications of Baran,33 Rodriguez34 and Stonik.35 In the course of synthetic studies related to the bielschowskysin model system, Nicolaou et al., discovered that when furans tethered to β-ketoesters are exposed to ceric ammonium nitrate, oxidation of the active methylene gave rise to radicals that subsequently attacked the furan moiety. For example, substrate (10) produced (11) when exposed to CAN (Scheme 4). | ||
| Scheme 4 Exploitation of tethered β-ketoesters and furans in macrocyclic synthesis.36 | ||
In contrast to methods yielding the monomeric product (11), the addition of the reagent to the substrate at a concentration of 0.05 M produced the dimeric product (13). Addition of the reagent in one portion at 0 °C was crucial to a successful outcome (Scheme 5).
 | ||
| Scheme 5 A 27 membered macrocycle produced by CAN oxidation of a tethered furan/ester.32 | ||
A typical example of this methodological differentiation is shown below:
The Nicolaou group32,33,36,37 described several examples of macrocycles exemplified by (11) and (13) prepared in good to excellent yields. It is interesting to note that other reagents were tried by Nicolaou et al., such as manganese(III), iron(III) and cobalt(II) complexes but the results were disappointing. It may be that the furan ring is not an efficient acceptor of the radical under these conditions. The “hardness” or “softness” of the radical may well depend of the oxidizing agent employed. Nicolaou38 proposed the following mechanism for the transformations in (Schemes 4 and 6).
 | ||
| Scheme 6 Furan trapping of a doubly activated carbon radical.37 | ||
In is interesting to observe that α-methylene radicals generated by means of Mn(OAc)3 are efficiently trapped by indole rings as Kerr et al.39 has demonstrated (Scheme 7), although macrocycles were not the intended targets.
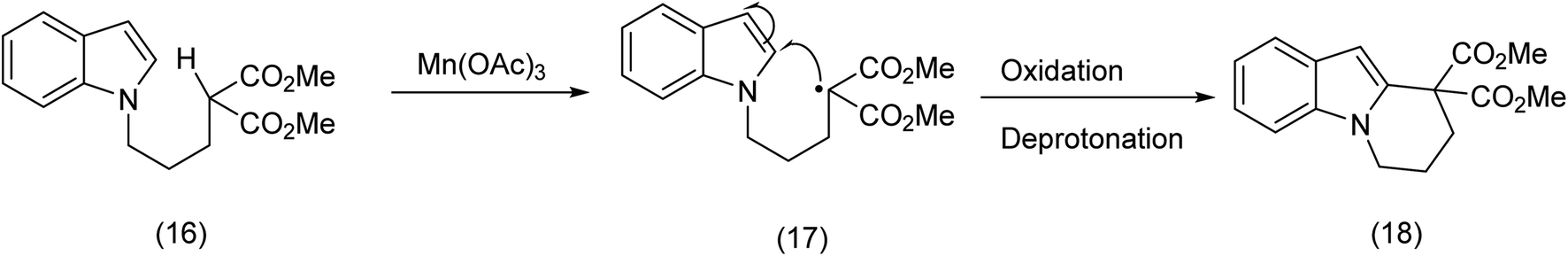 | ||
| Scheme 7 Indole trapping of a beta ketoester (malonyl) – carbon radical.39 | ||
Nishino et al.40 has developed a macrocyclization procedure that exploits tethered bis ketoesters and tethered terminal alkenes which react in the presence of Mn(OAc)3/AcOH at 100 °C in an argon atmosphere (Scheme 8). Remarkably this was an intermolecular process with a spectacular outcome. For further examples and applications of related chemistry from the Nishino group (see ref. 41–51).
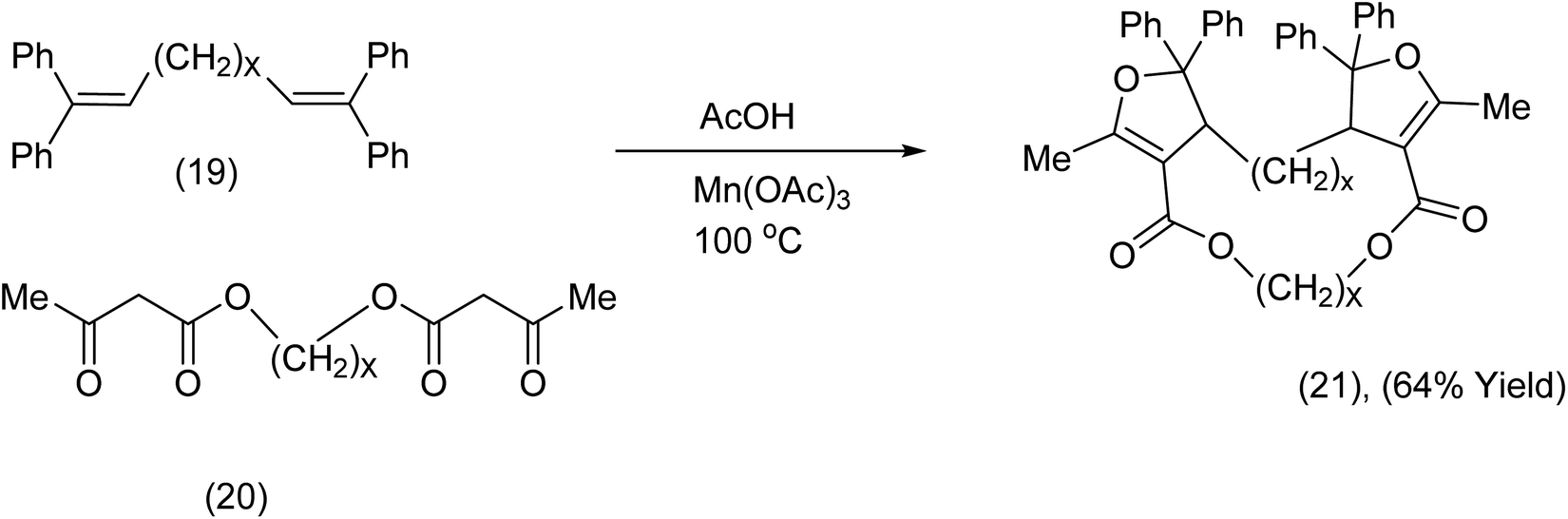 | ||
| Scheme 8 Manganese triacetate promoted of a bisdihydrofuran-diester macrocycle.46 | ||
From a mechanistic standpoint, a ketoenolate MnIII complex ((23) Scheme 9) is formed by reaction of the Mn(OAc)3 with the keto ester moiety. The complex breaks down to yield the methylene radical (24) which then attacks the olefin to yield doubly stabilized radical. We have previously invoked such MnIII keto enolates to explain the regioselective lactonization of stilbenes appropriately substituted.52
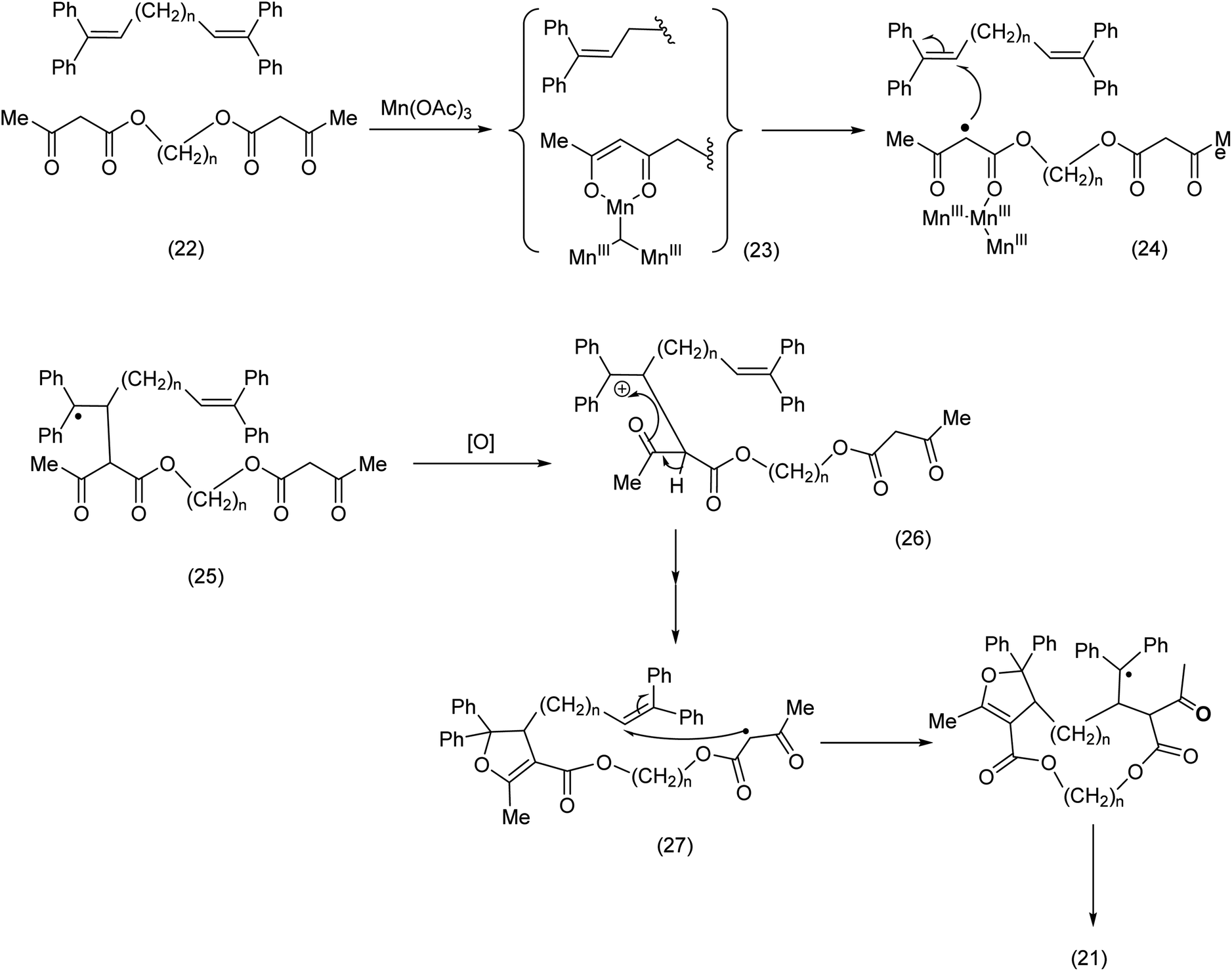 | ||
| Scheme 9 Intermolecular trapping in the course of an active methylene radical-pathway.51 | ||
For articles related to manganese triacetate promoted reactions more generally, the reviews by Snider53–59 should be consulted.
1.3. Reductive radical generation protocols for macrocyclic construction
In contrast to the addition of oxidatively generated radicals to furans as a creative entry to macrocycles (see the Nicolaou chemistry (Scheme 4)), we now turn to reductive generation of the radical and its synthetic utility. The Pattenden group60 had for a number of years studied radical mediated macrocyclisation–transannulation.61–64 Later they reported the addition of vinylic radicals to furans resulting in a cascade culminating in the tetracyclic diketone (33) (Scheme 10). It is instructive to compare this sequence with Parson's furan iodovinyl to indole construction (Scheme 11), in which vinyl radical addition to the furan resulted in a spirodihydrofuran (compare (36) Scheme 11 with (31) Scheme 10) electrocyclic cascade. Although Parson's chemistry was not directed at macrocyclisation, it is nonetheless mechanistically significant. In both sequences the vinylic radical was generated differently, Pattenden exploited a 12 endo diagonal ring closure of a methylene radical60 obtained by exposure of an iodomethyl moiety to tributyltin hydride. Parsons subjected a vinyl iodide to tributyl hydride to generate his vinyl radical.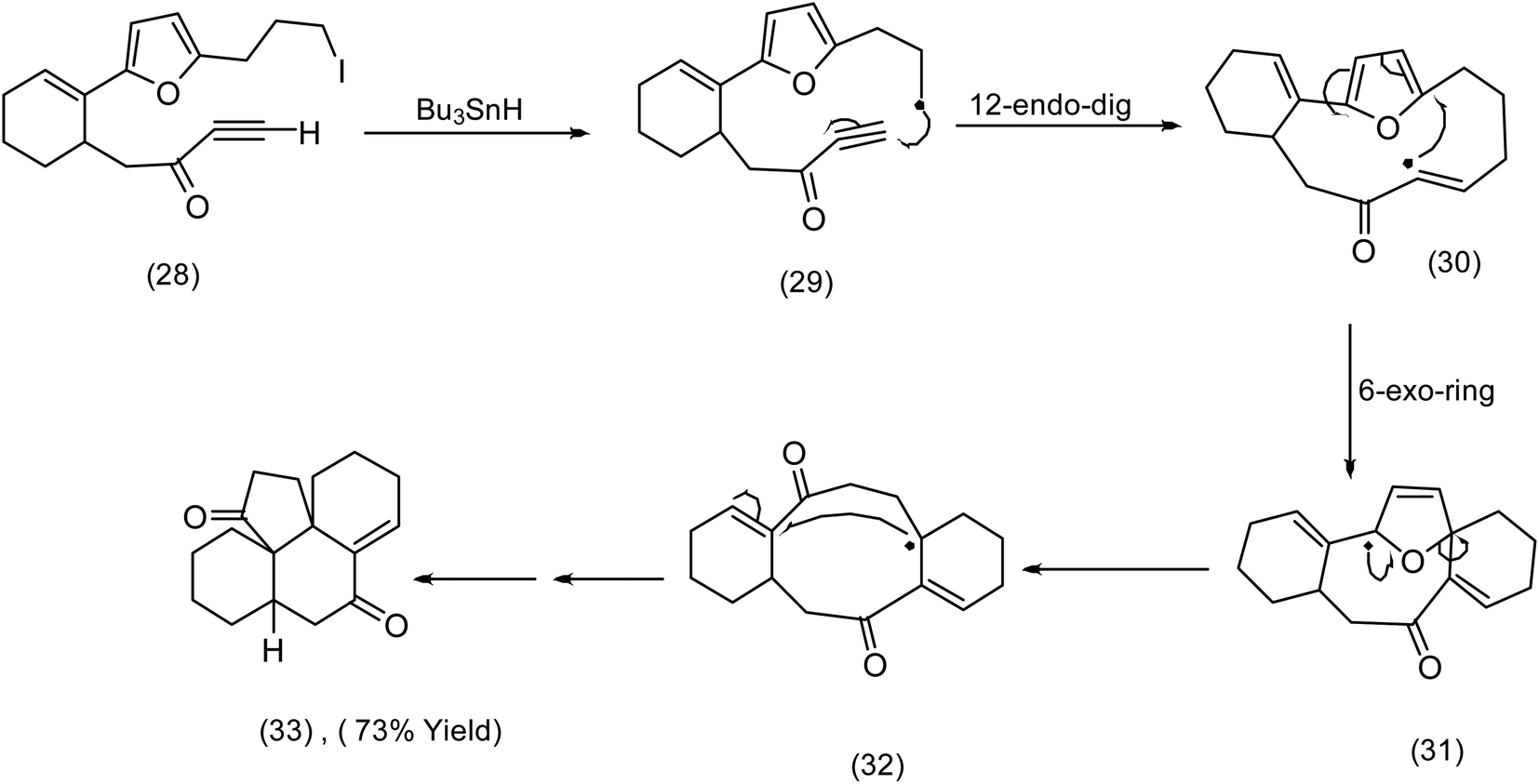 | ||
| Scheme 10 Pattenden's elegant reductive radical generation – cascade cyclisation.64 | ||
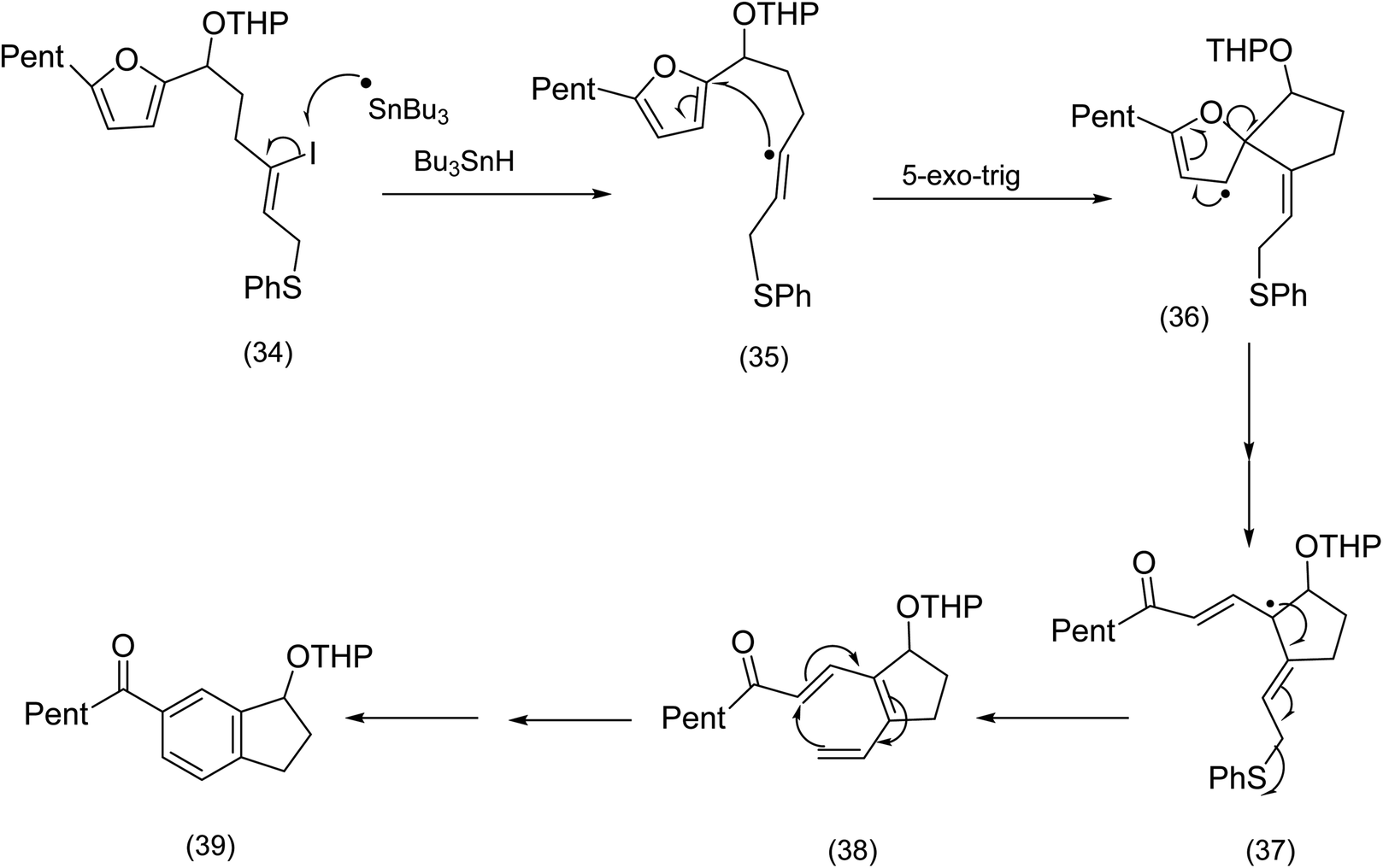 | ||
| Scheme 11 Parson's reductive radical generation/cyclisation (non – macrocyclic)64 | ||
In a fascinating example of the subtle interplay of electronic and conformational factors influencing macrocyclization, Pattenden examined two acyclic trienone systems incorporating seleno ester and vinyl cyclopropane moieties. The close proximity of the acyl radical to the α,β unsaturated ester was clearly critical to the initial 6-endo trigonal ring closure and the ensuing cascade sequence that produced the steroid skeleton (Scheme 12).
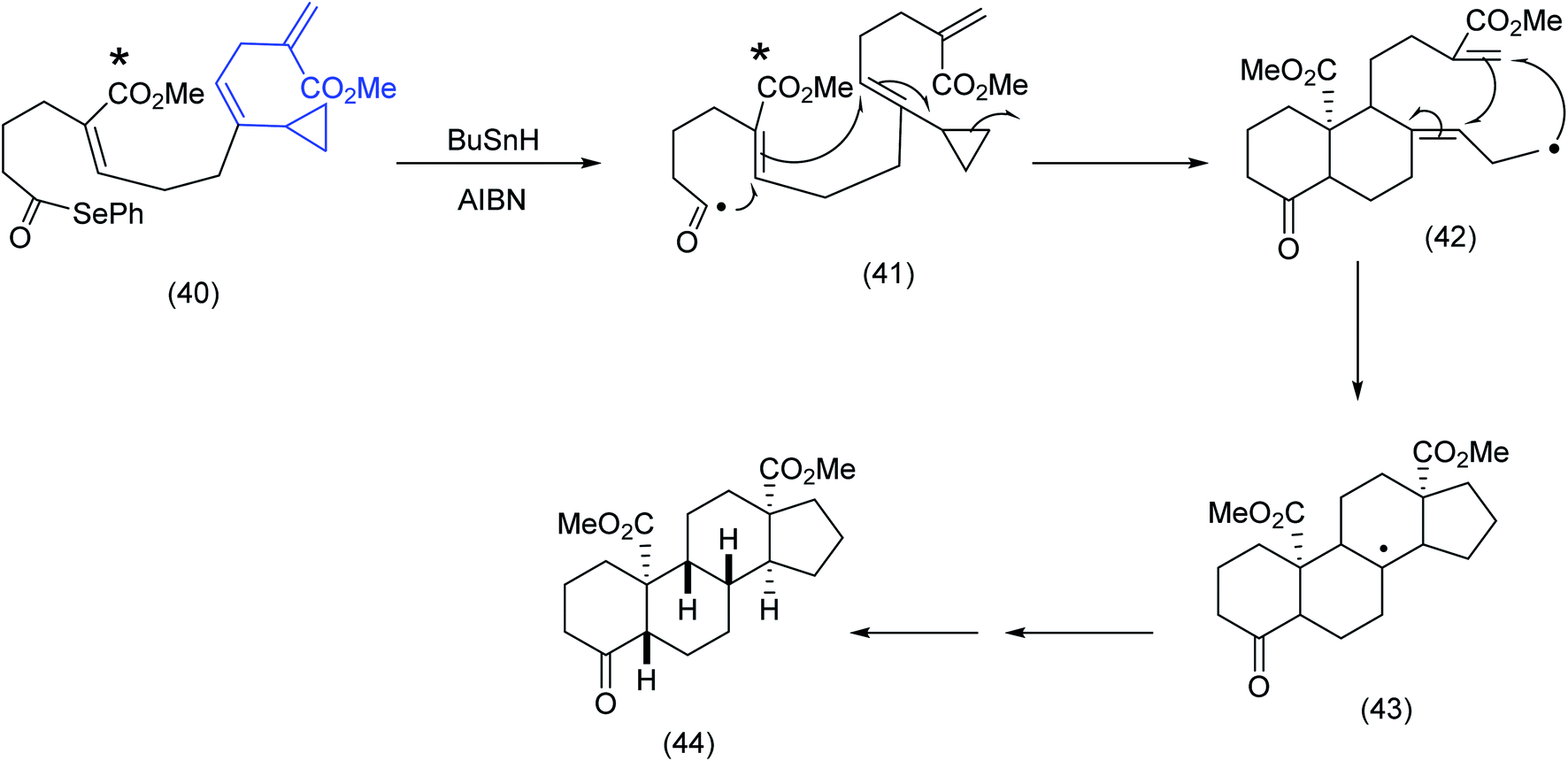 | ||
| Scheme 12 Radical cascade differentiation: acyl radicals to steroids.64 | ||
In comparing (40), Scheme 12 with (45), Scheme 13, the ester group (note asterisk) has now been replaced by a methyl group (see the asterisked carbon). The original 6-endotrigonal cascade (40) to (41) (Scheme 12), has now been suppressed and replaced by the sequence (46) to (47), Scheme 13. The juxtaposition of α,β unsaturated ketone in relation to the cyclopropane in (45) is a likely contributing factor in the very different course the reaction has taken.
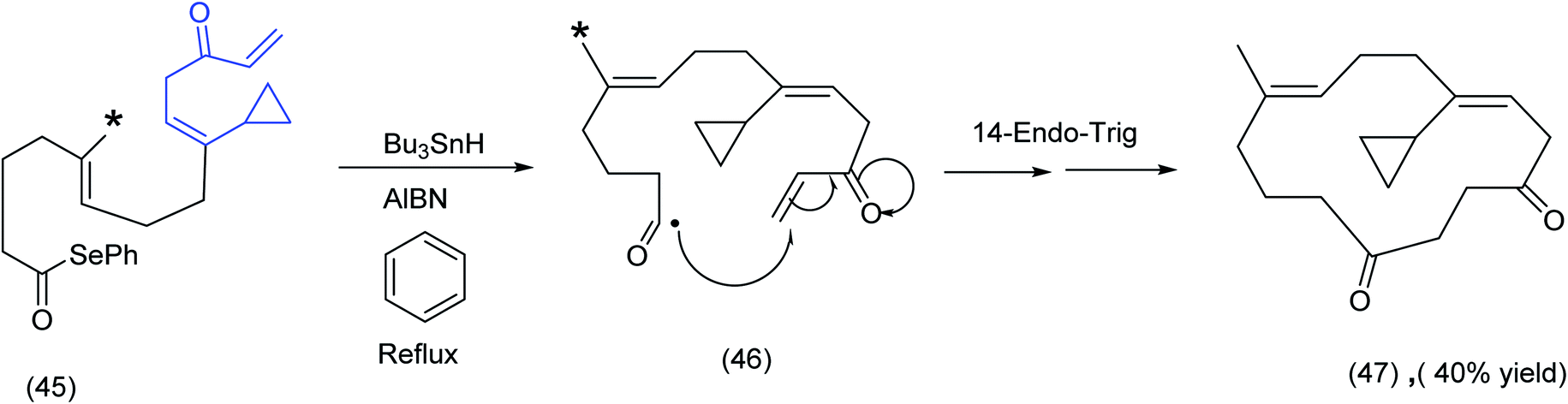 | ||
| Scheme 13 Radical cascade differentiation: acyl radicals to macrocyclic diketones.65 | ||
1.4. Oxidative generation of radical cations in pseudomacrocyclic and macrocyclic synthesis
Our own unexpected synthesis of a pseudomacrocycle resulted66 from exposure of an apparently “innocuous” stilbene to ferric chloride. In contrast to other stilbene derivatives, the 3,5-dimethoxy-10-acetamido stilbene, when exposed to ferric chloride hexahydrate, gave rise to four products, one of which was the pseudomacrocyclic indolo-stilbene atropodiasteroisomer ((56) Scheme 14), for which we proposed the following cascade sequence below: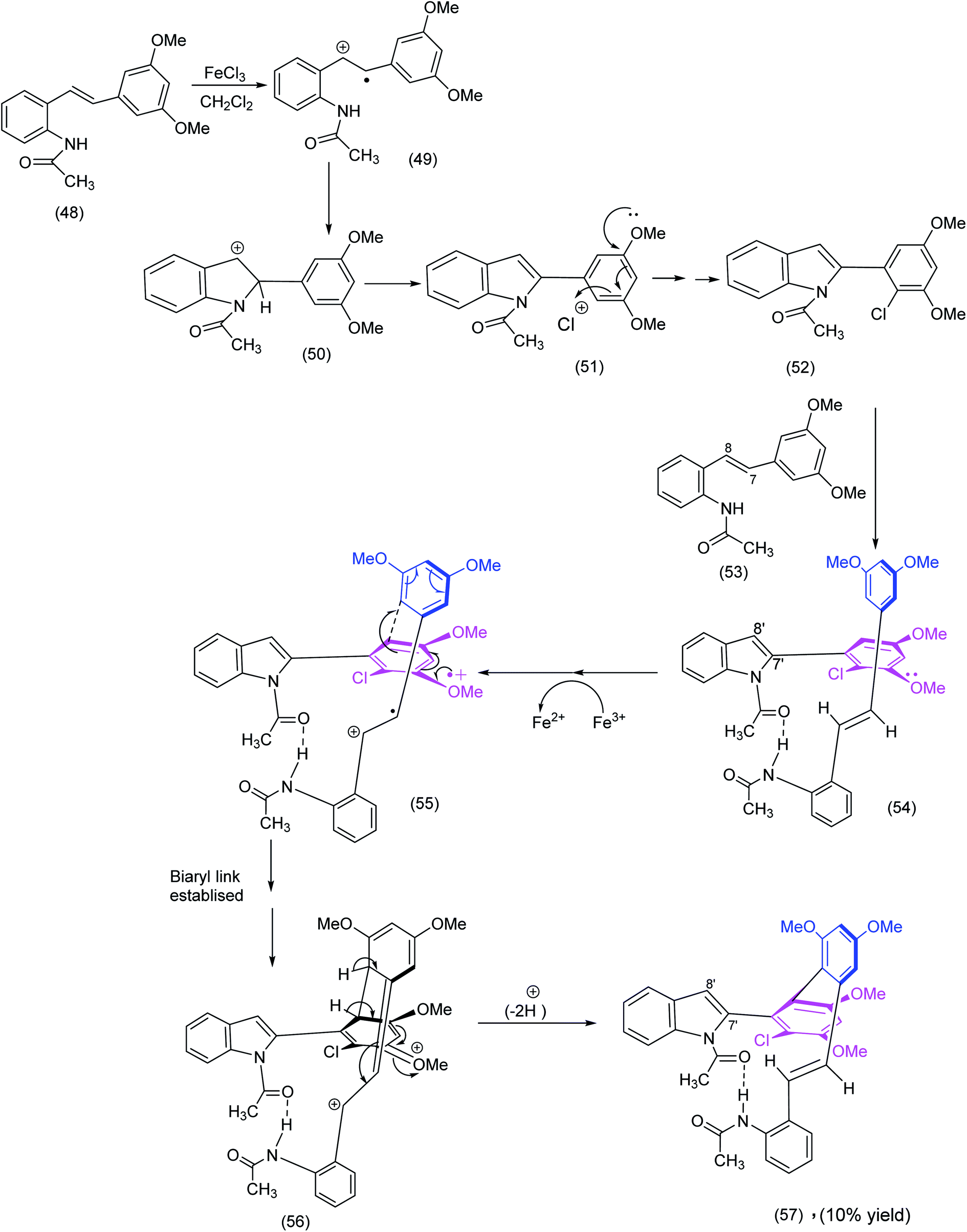 | ||
| Scheme 14 A radical cation mediated construction of a biaryl system in an unusual indolostilbene (57) synthesis.66 | ||
We recognize of course that the sequence depicted in Scheme 14 does not end in a true macrocycle, the function of the scheme is nevertheless threefold.
(1) The intramolecular hydrogen bonding (see intermediates (54) to (57)) have conformational implications and brings to mind the kind of pre organization well known in the synthesis of cyclic peptides (Fig. 1 in the Yudin review1).
(2) The establishment of the biaryl link via oxidative coupling (radical cations as reactive intermediates) – structure (55) – Scheme 14 brings to mind an intriguing biaryl coupling (Witkop cyclisation) exploited with profit by Nicolaou et al. (Scheme 19) in his diazonamide synthesis.
(3) Both of the syntheses described above are in stark contrast to a brilliant non-oxidative solution to a unique biaryl system in Baran's haouamine synthesis (Scheme 31).
Parker et al.67 devised an imaginative radical cation Diels Alder approach that resulted in the macrocyclic intermediate (63) leading ultimately to kingianin A.
For additional examples of the radical cation Diels–Alder reaction see the work of Bauld,68,69 Lin,70,71 Yoon,72 MacMillan,73 and Wiest.74 For major critical reviews on transition metal photocatalysis in relation to radical cation generation and the utility of visible light photocatalysis the publications of Yoon75–77 must be consulted.
1.5. Synthetic utility of π allyl complexes and C–H activation in macrocyclic synthesis: the roles of palladium and rhodium
The next sub section describes macrocyclizations in which π-allyl complexes and C–H activation (metal catalyzed) play a critical role. With regard to the former, the Sorensen chemistry described below is instructive. The key macrocyclization step in the synthesis of the potent microtubule stabilizing agent FR182877 by the Sorensen78 group was a beautifully executed and chemoselective Trost–Tsuji reaction (Scheme 16). Nucleophilic attack by the enolate (66) on an electrophilic π-allyl complex is the key step leading to the advanced intermediate (67). | ||
| Scheme 15 Parker's radical cation mediated macrocyclization exploiting a diester tether.67 | ||
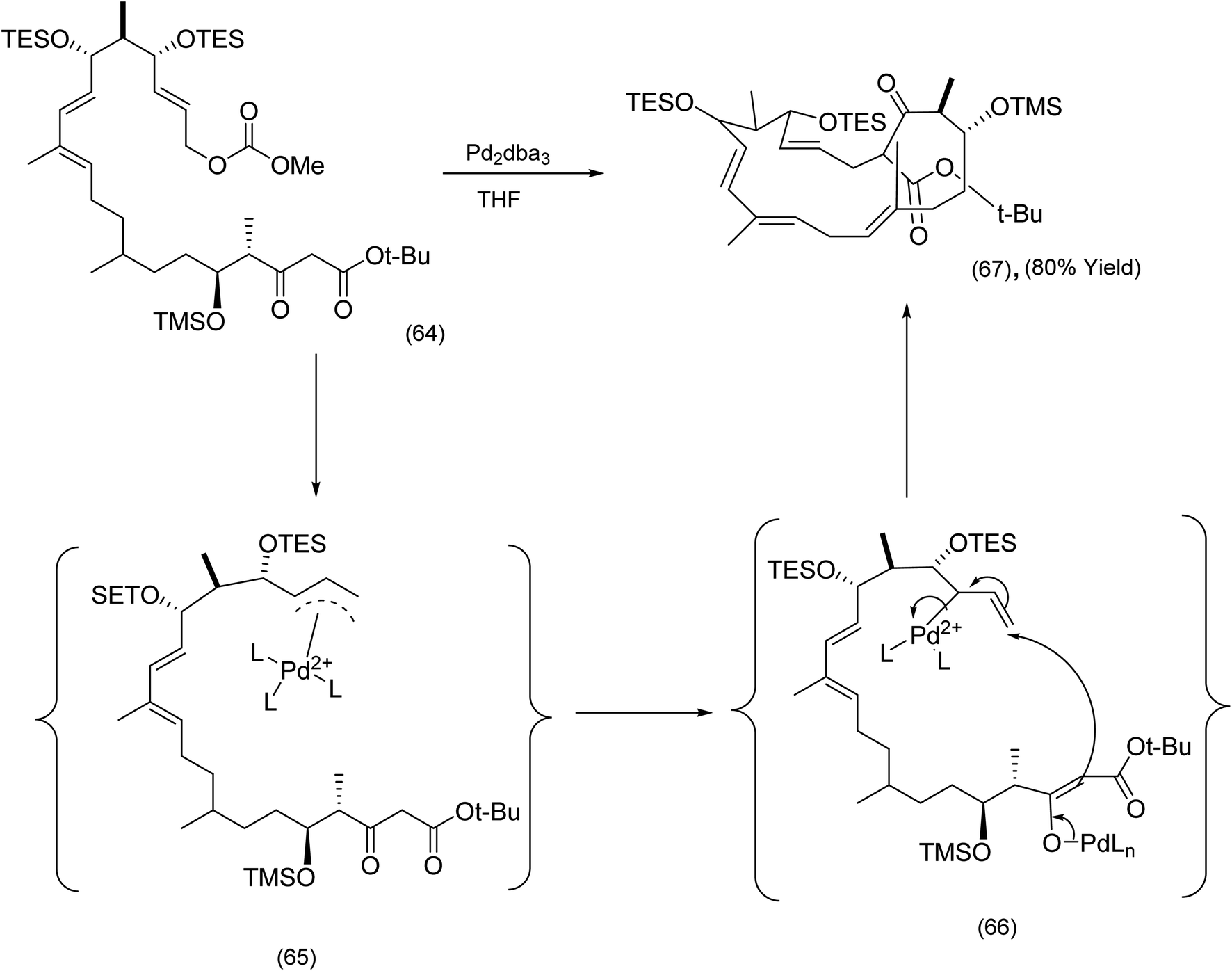 | ||
| Scheme 16 Utilization of the intramolecular Trost–Tsuji reaction enolate to FR182877.78 | ||
Cossy et al.,79 has exploited Rh(III) catalyzed activation for the construction of macrocyclic pyridones. The precursor, incorporating both terminal α,β unsaturated amides and alkynes, was exposed to [Cp*RhCl2]2/CsOAc leading to wide range of macrocyclic pyridones e.g. (71). Mechanistically the reaction proceeds through 5 and 7 membered ring rhodium lactam complexes (69) and (70), (rhodacyclic enamide species) see Scheme 17.
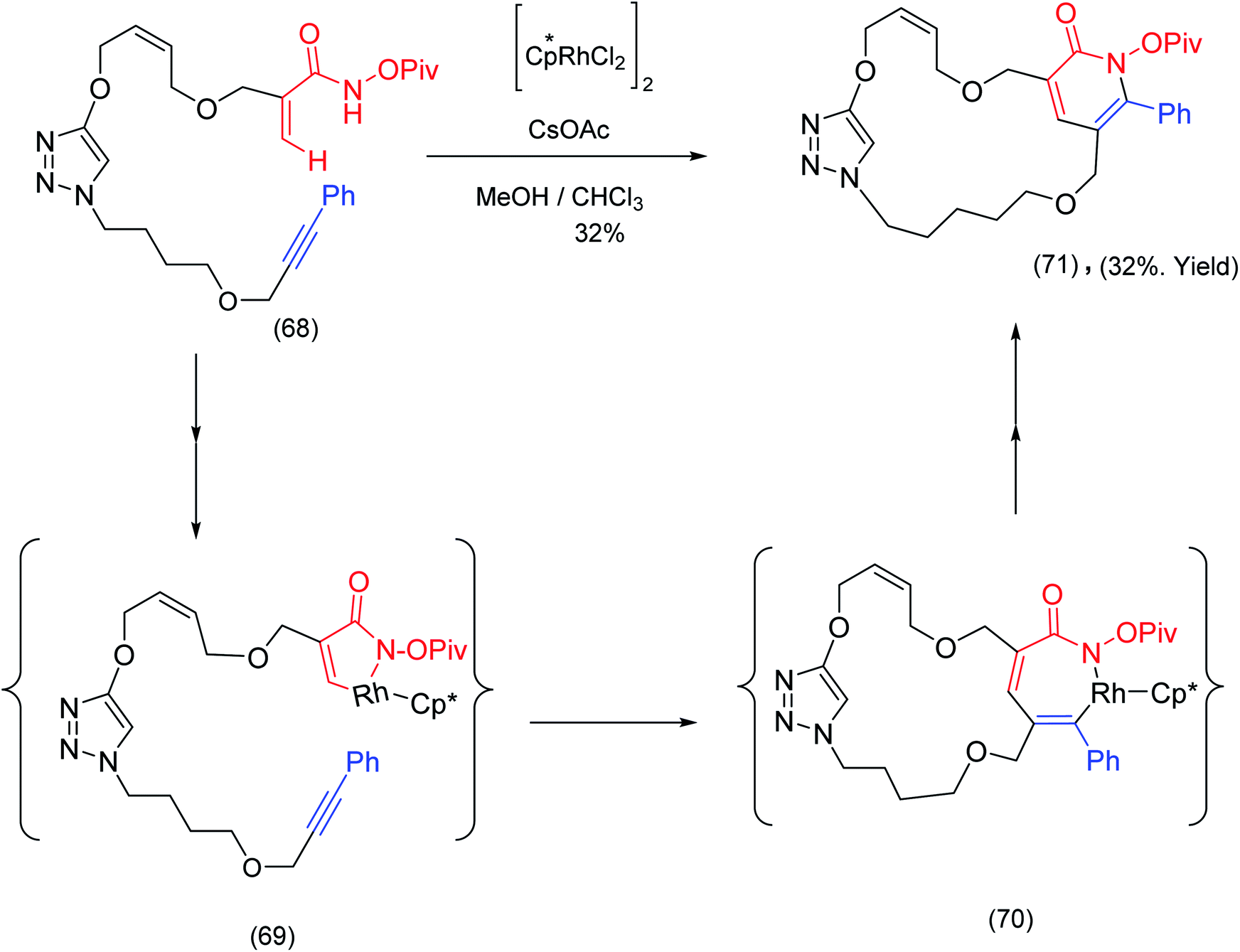 | ||
| Scheme 17 Rh(III) catalyzed intramolecular pyridones formation – a macrocycle forming transformation.79 | ||
Jiang et al.,80 has also reported a rather different Rh catalyzed macrocyclization via oxidative cross coupling of the olefinic moieties. This highly efficient cross coupling is illustrated by the example below (Scheme 18). The mechanism presented below is slightly different from the mechanism proposed by Jiang et al.80
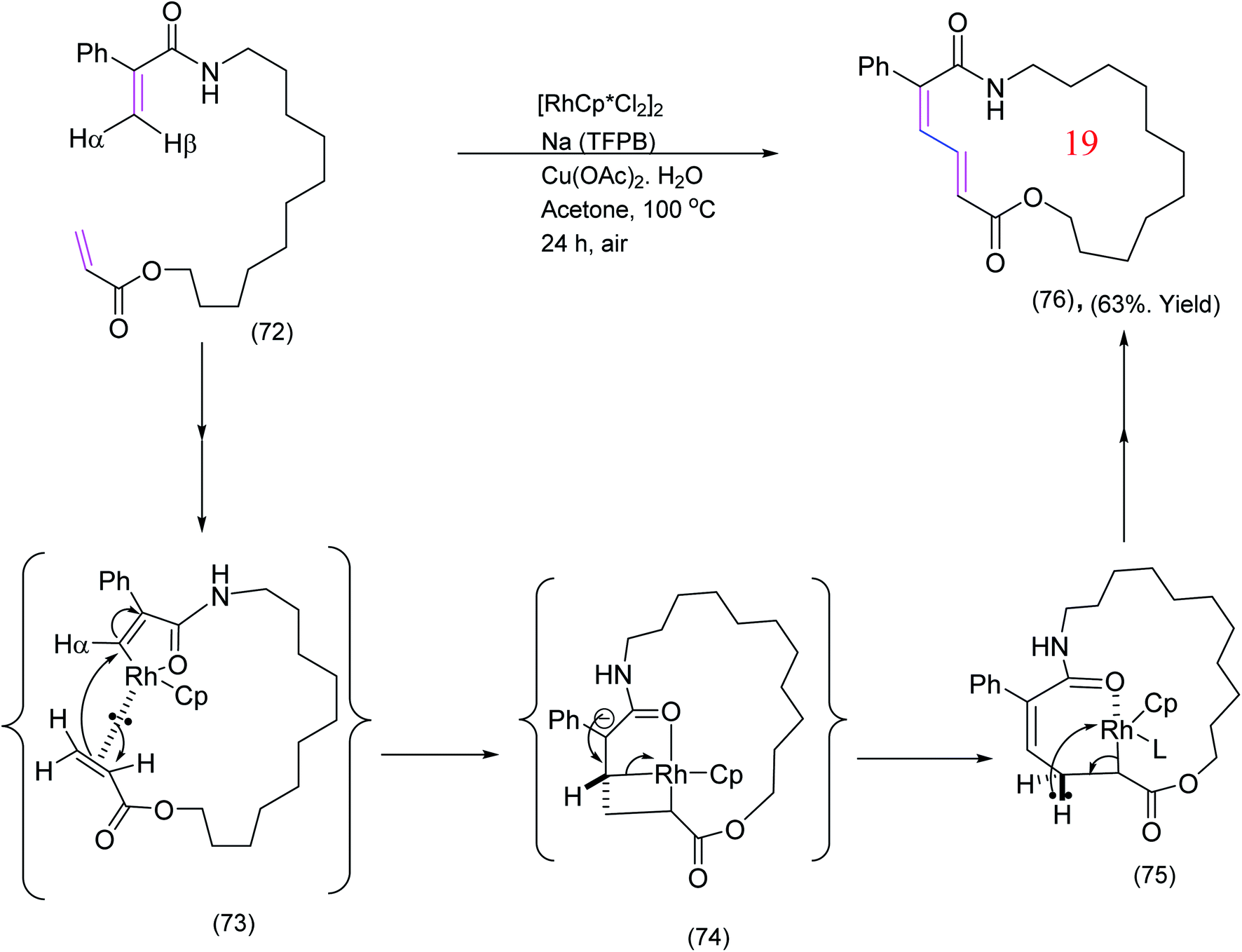 | ||
| Scheme 18 Rhodium catalyzed olefin cross coupling providing access to macrocyclic dienamides.80 | ||
As a result of kinetic isotope effect (KIE) studies,80 it was proposed that the active catalyst [Cp*Rh (OAc)][BARF] coordinates to the enamide moiety by oxidative insertion (removal) of the syn-hydrogen (syn to the amide carbonyl i.e. Hβ) to yield (73). The next step could be viewed as a migratory insertion of the Rh into α-CH bond of the αβ-unsaturated ester. Alternatively, one may envisage the process as a 1,4-type addition to the enamide followed by fragmentation of the rhodium-complex [(73) to (74) to (75)] but this is not certain and other pathways are possible. Decomplexation via β-hydride elimination which does not preclude other possibilities completes the formation of the desired product. Reductive elimination and reoxidation regenerates the active catalyst.
1.6. Distinctive macrocyclic domains and the most appropriate methodology: HATU/amidation and the Witkop coupling
The following example of macrocyclisation from the Nicolaou group is instructive because the target molecule, diazonamide A, possesses two distinctive macrocyclic domains, each constructed by a different method, one of which involved an intriguing biaryl coupling. The total synthesis of diazonamide by the Nicolaou group81 was challenging and provided an excellent opportunity to test various macrocyclization protocols (Scheme 19). In a molecule consisting of two macrocyclic submits, the selective deprotection of terminal BOC amide and methyl ester moieties (TFA and LiOH treatment (77) to (78)) lead to an intermediate which underwent smooth macrolactamisation by means of the HATU/collidine procedure under carefully controlled conditions. Thus one of the two macrocyclic submits was successfully installed. The installation of the second macrocyclic domain within diazonamide A exploited the rarely used Witkop cyclisation. This is a photochemically induced process that involves both a single electron oxidation and a single electron reduction producing a radical cation and radical anion ((89) to (84) (Scheme 19)).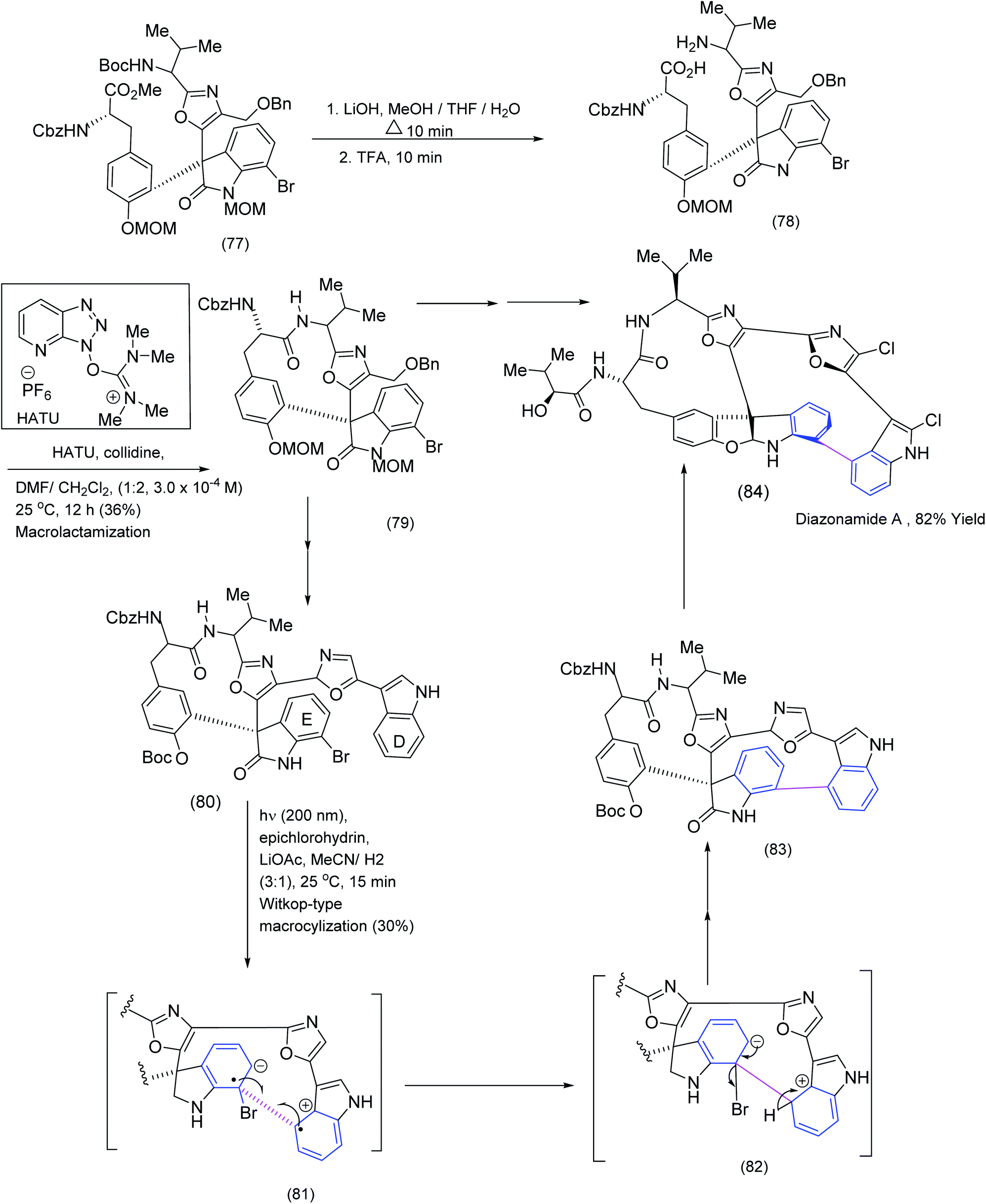 | ||
| Scheme 19 Exploitation of HATU and Witkop cyclisations to install the two distinctive macrocyclic domains in diazonamide.82 | ||
1.7. The contributions of Nicolaou, Schreiber, Baran and Kishi: enediyne macrocycles, bent biaryls and the Diels Alder reaction
The revisiting of the calicheamicin ϒ1/esperamicin (Fig. 1) story allows us to compare two conceptually different but equally fascinating approaches to the elaboration of calicheamicinone (or its analogue) – incorporating the remarkable enediyne moiety. This remarkable class of molecule demonstrates a high potency against tumour cells by a mechanism that involves intramolecular conjugate addition and Bergman cyclization/aromatization leading to the formation of diradicals which induce double strand cuts in DNA83–85 (Schemes 20 and 21).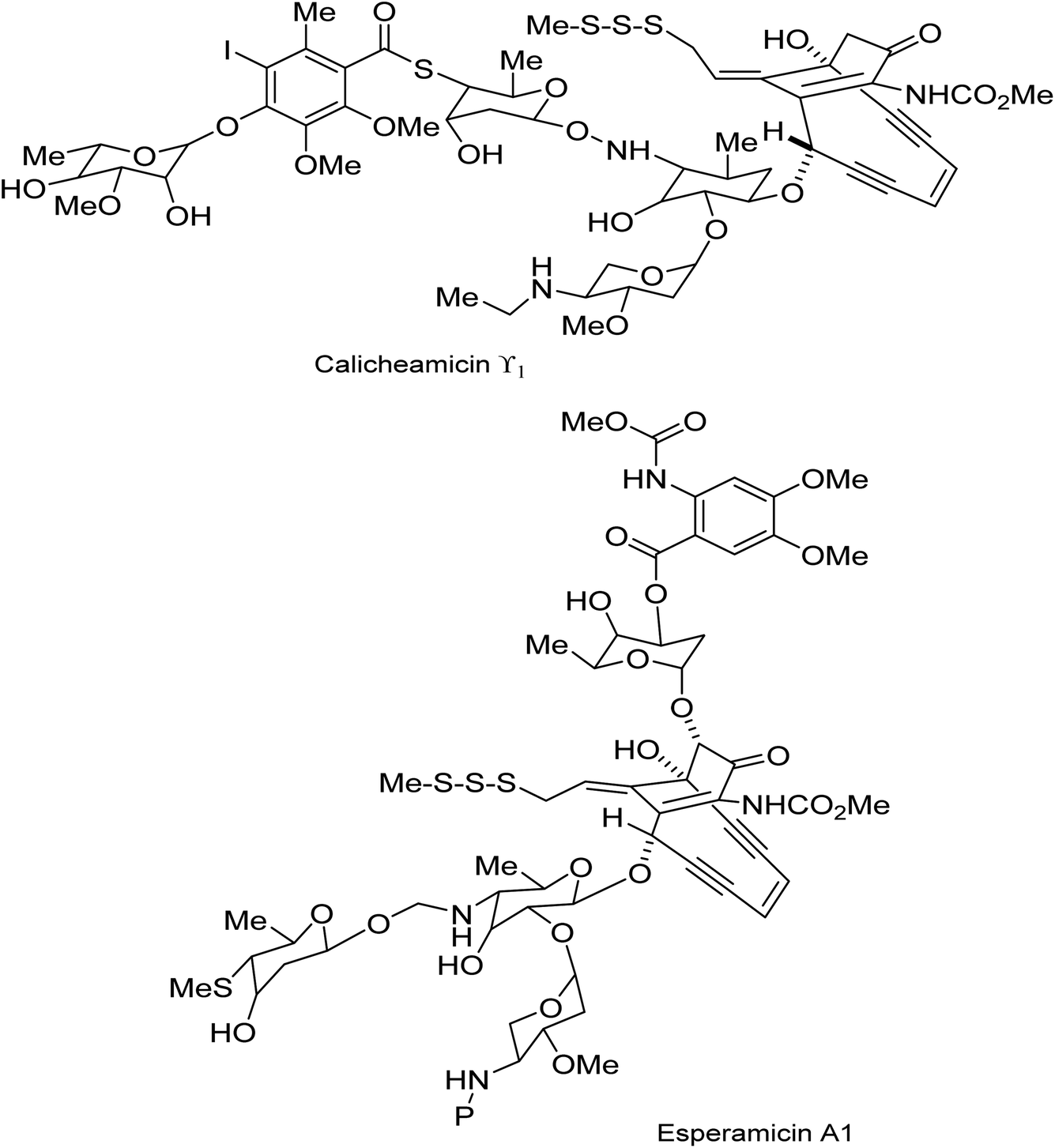 | ||
| Fig. 1 Structure of calicheamicin ϒ1 and esperamicin A1 anticancer agents.85 | ||
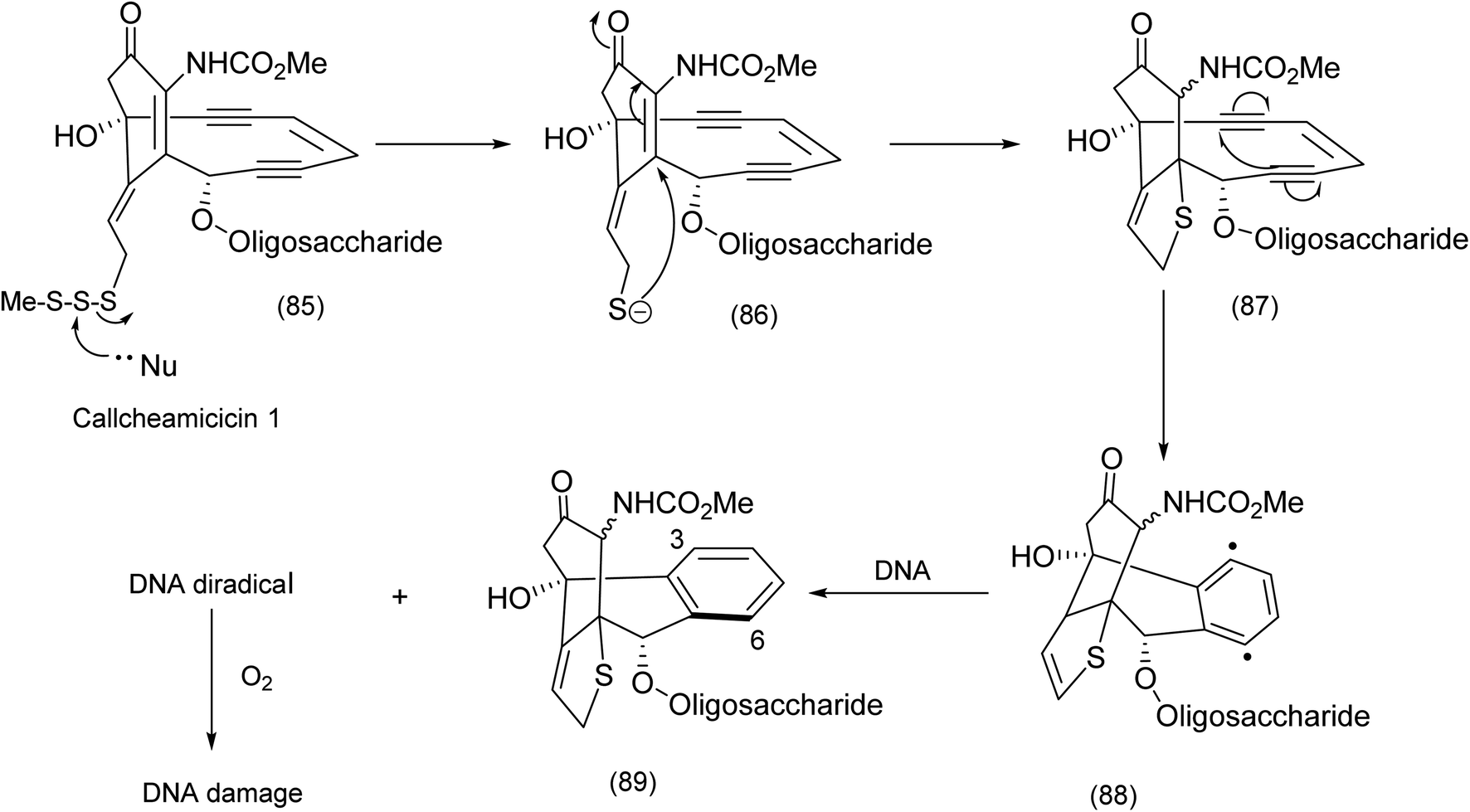 | ||
| Scheme 20 Biochemical intramolecular conjugate addition – Bergman cyclization diradical formation.85 | ||
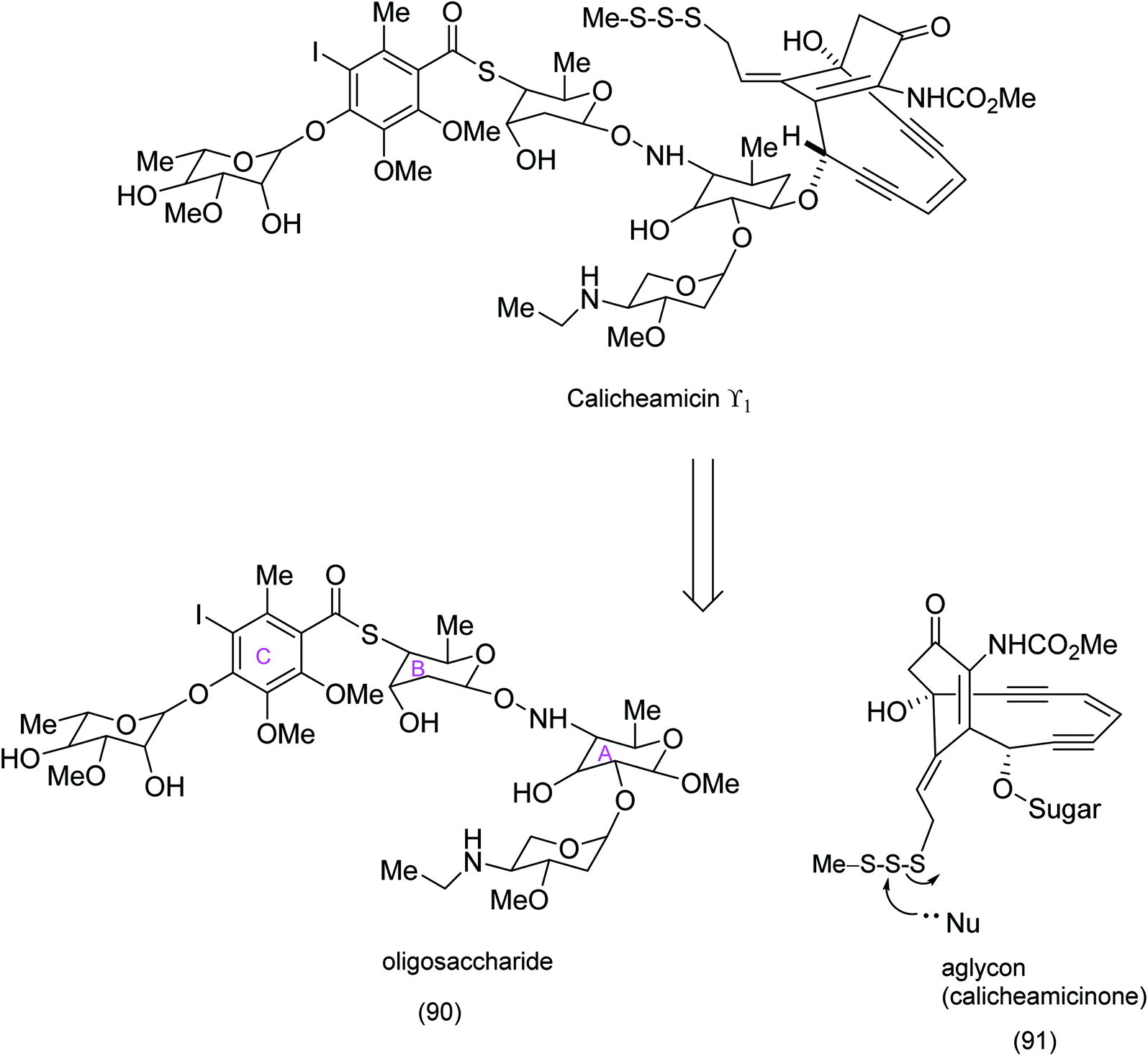 | ||
| Scheme 21 Identification of calicheamicinone as a key intermediate enroute to the synthesis of calicheamicin ϒ1.85 | ||
Both calicheamicin and the related esperamicin incorporate a strained enediyne and a α-carbamato cyclohexenone. Nicolaou's approach to calicheamicinone (Scheme 22), depended critically on the choice of tetronic acid (96) as a starting material, the application of H. C. Brown's (Z)-diisopinocamphenyl allyl borane/aldehyde coupling – a nucleophilic addition that has been shown to produce syndiols of high enantiomeric purity and an intramolecular nitrile oxide cycloaddition (INOC). This chemistry yields a bicyclic isooxazoline (94) which is a masked α-carbamato cyclohexenone characteristic of calicheamicinone (i.e. (103) to be unmasked later).
 | ||
| Scheme 22 Generation of the functionalized bicyclic oxazoline (94) and incorporation of the endiyne moiety.86 | ||
From the product of the intramolecular nitrile oxide cycloaddition (94), the completion of the installation of the enediyne moiety (Nicolaou et al.86) necessitated at least two key steps: addition of lithium acetylide to the functionalized cyclohexanone (95) to produce (96), and palladium/CuI catalyzed coupling of the Z-enyne (98) to the ethynyl t-butylsilyl ether (97) to produce (99) (Scheme 22). Further transformations including isoxazole cleavage produced the intermediate from which the corresponding ethynyl anion (101) was generated (after desilylation of (99)) by exposure to potassium hexamethyl disilazide (KHMDS) prior to intramolecular attack on the aldehyde (101) to set up elegantly, the macrocyclic enediyne core of calicheamicinone (103) (Scheme 23).84
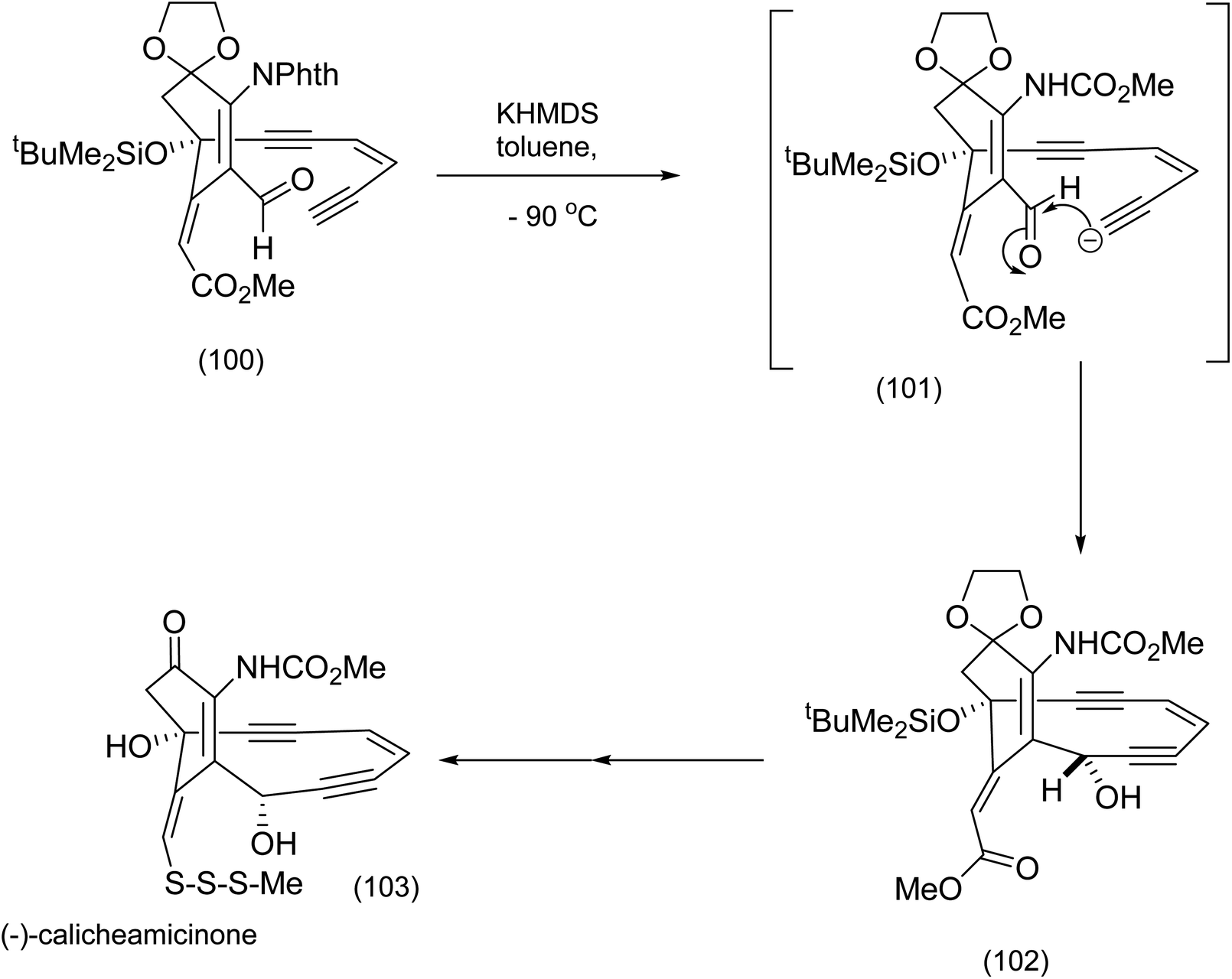 | ||
| Scheme 23 Final stages in (−)-calicheamicinone construction.86 | ||
An alternative approach to a molecule closely related to calicheamicinone was reported by the Schreiber group. This chemistry, beautiful in conception, is described below87 (Scheme 24). Schreiber developed a sequence of reactions leading to an endiyne tethered diene and α,β unsaturated ester moieties. An intramolecular Diels Alder reaction would deliver (it was assumed, not unreasonably) the bridged bicyclic core of a calicheamicinone analogue in a single reaction.87 The Z-dichloroethene (104) was transformed by successive Castro Stevens cross couplings to produce the protected enediyne (105). Acetal deprotecting and metalation of 1-methoxy-3-bromo-1,3-butadiene with n-butyllithium and subsequent addition resulted in intermediate (106). Further Castro Stevens coupling produced the desired diene and dienophile moieties separated by the enediyne tether (106) to (107).87
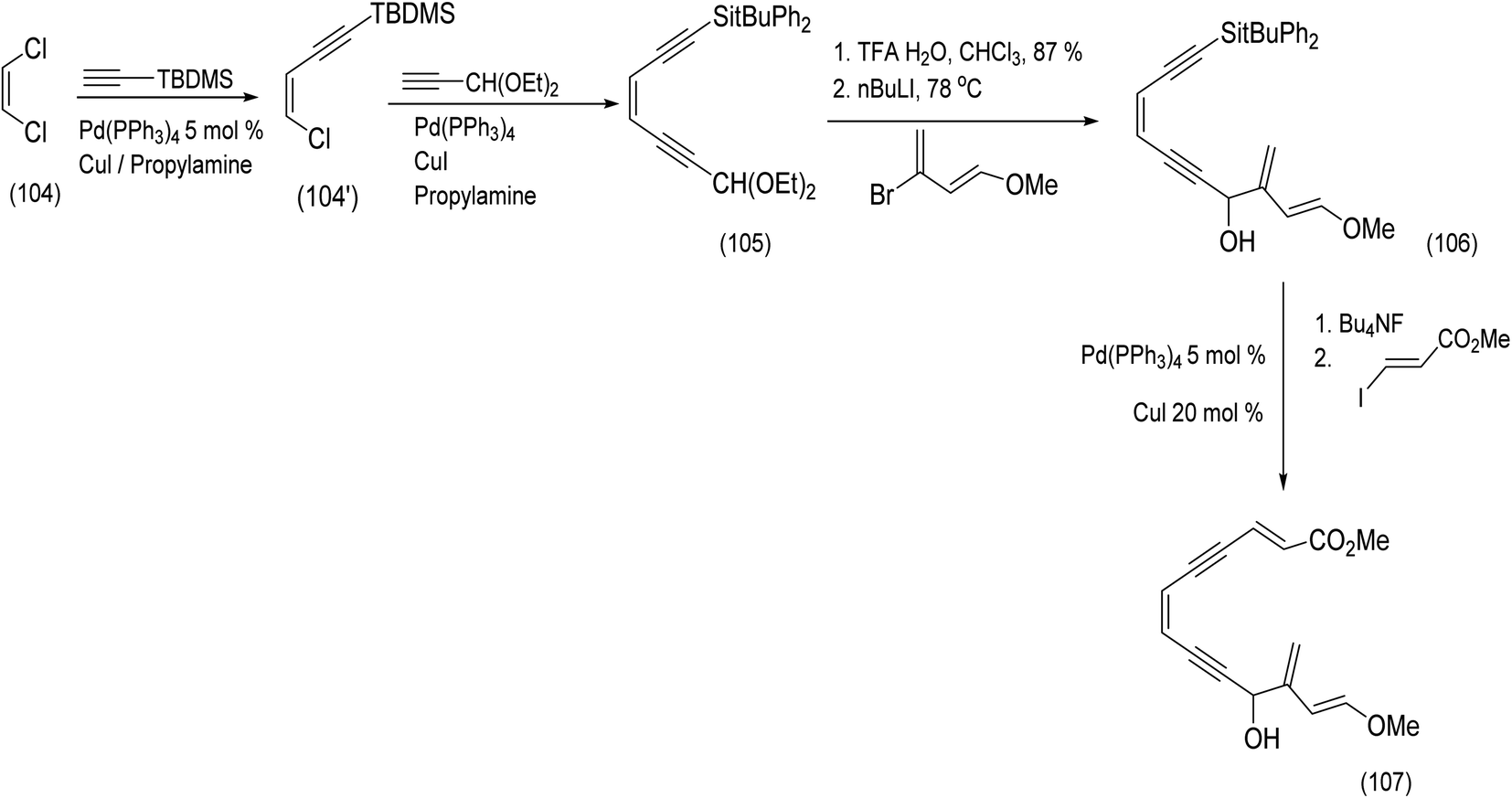 | ||
| Scheme 24 Application of the Castro Steven's coupling to enediyne construction.87 | ||
After protection of the secondary alcohol, exposure of the product to Kish's radical inhibitor (as a precaution) and heating in benzene at 80 °C effected a remarkable Diels Alder reaction believed at the time to have resulted in the desired bridged bicycle (109) (Scheme 25).
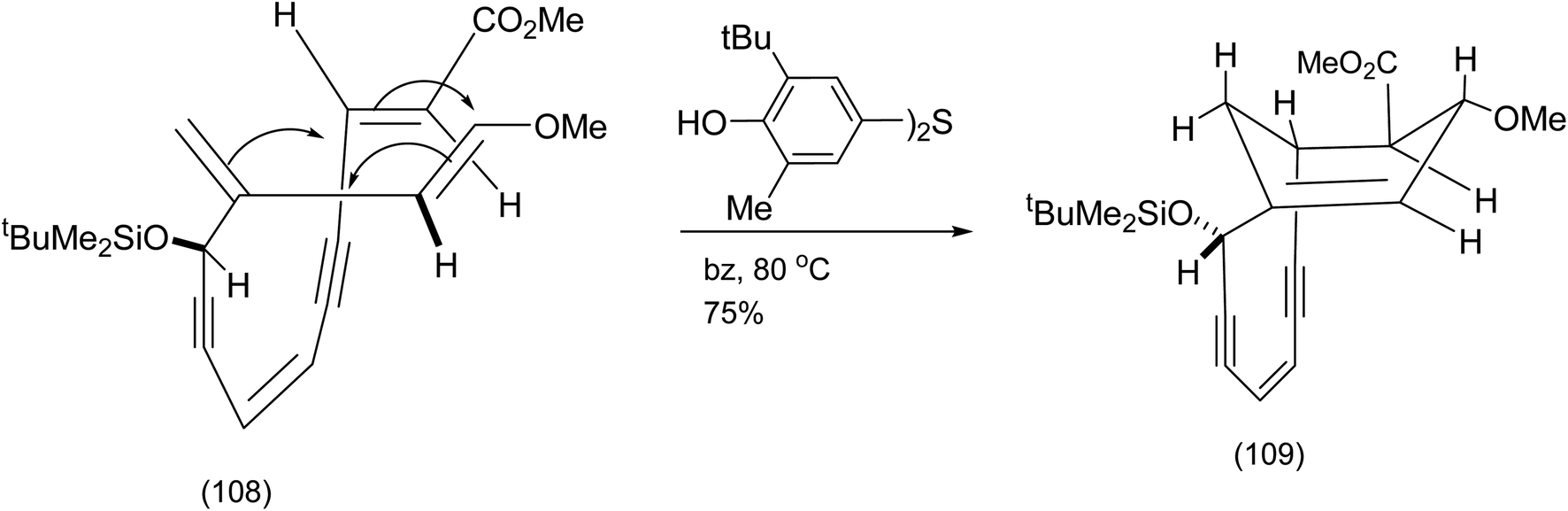 | ||
| Scheme 25 The first attempted 4 + 2 cycloaddition of tethered endiyne.87 | ||
The Schreiber group adopted a similar strategy for the synthesis of (110) which was efficiently transformed into the bridged enone (111),87 (Scheme 26).
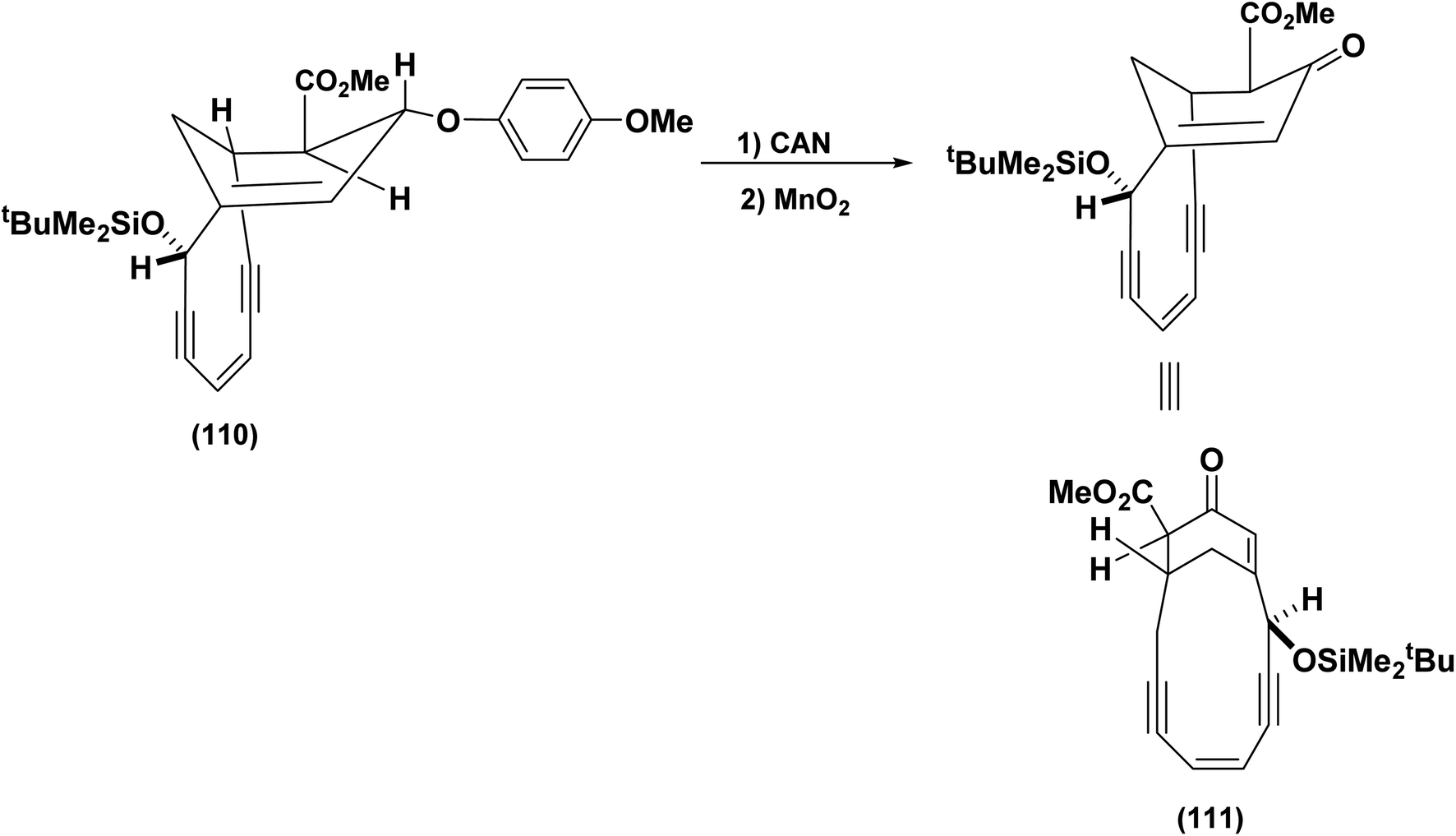 | ||
| Scheme 26 Deprotection/oxidation to generate the bicyclic enone.88 | ||
After the work above was published, further investigations of the presumed structure (111) and the critical observation that it failed to undergo the Bergman cyclisation, led to the conclusion that the Diels–Alder reaction depicted in scheme (25) had followed a different course leading to the alternative regioisomer. In short structure (109) was incorrect. The actual path the Diels–Alder reaction had followed is shown in Scheme 27.
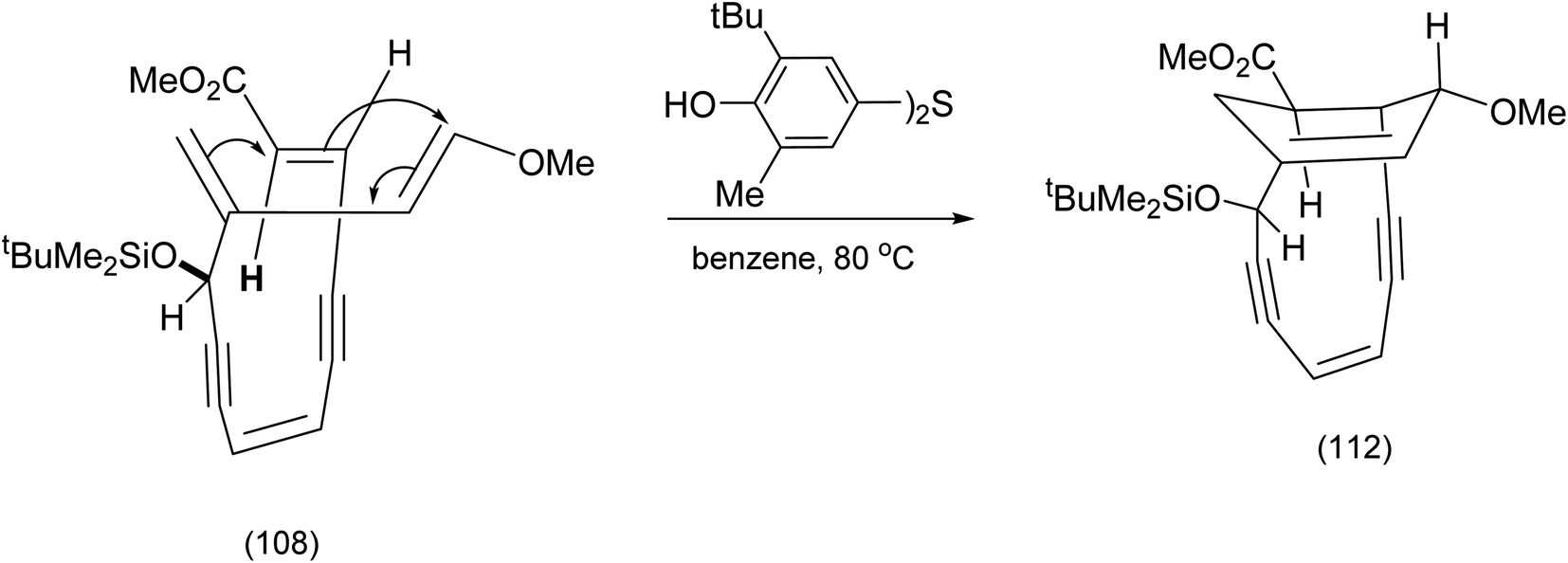 | ||
| Scheme 27 Recognition of the unexpected regiochemical outcome for the intramolecular endiyne tethered Diels Alder reaction.88 | ||
The judicious selection of reactions that revealed the original structural misassignment was insightfully described in a paper by Schreiber et al.88 The same authors pondered the possibility of carefully manipulating the Diels–Alder reaction conditions with a view to generating the correct regiochemistry. As it turned out the breathtakingly ingenious solution to the problem depended on the application of the Tsuchihashi pinacol rearrangement.89 Schreiber generated a modified version of his tethered diene and dienophile by means of the following sequence of reactions. Coupling of the readily available aldehyde (114) to the vinyl lithium species (112), (obtained by treatment of the vinyl stannane with butyl lithium) gave the diene-endiyne (116). A palladium catalysed coupling of the bromovinylene carbonate (118) to (117)89 gave the crucial Diels–Alder precursor (119). Subjection of (119) to the Diels Alder first conditions shown in Scheme 27 produced in good yield the cyclisation product ((120) Scheme 28).
 | ||
| Scheme 28 Installation of the cyclic enecarbonate into the endiyne tethered.88 | ||
The selective base promoted cleavage of the cyclic enecarbonate and selective mesylation of the axial hydroxy group probably proceeds by the following mechanism ((121) to (123) Scheme 29).
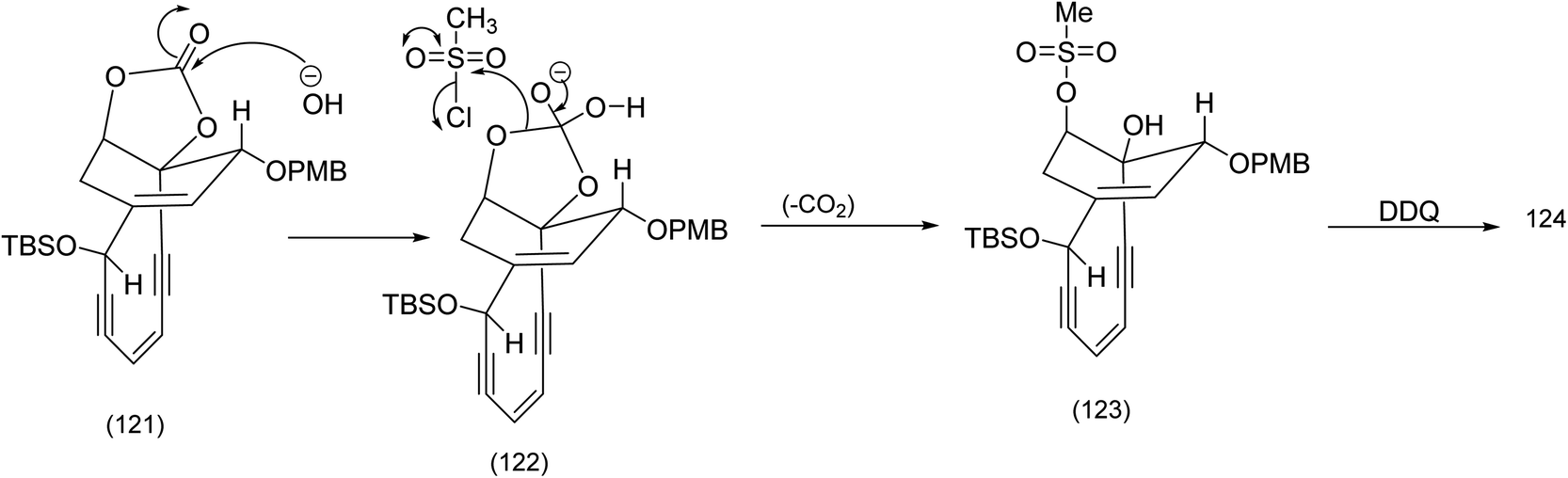 | ||
| Scheme 29 Selective installation of the axial mesylate set up for the Tsuchihashi rearrangement.90 | ||
Treatment of with (123) six equivalents of diethyl aluminum chloride gave rise to the desired bridged enone (128) via the mechanism described below.89 The combination of an imaginative type II Diels Alder reaction and an ingenious pinacol rearrangement have combined to make this a memorable achievement (Scheme 30).
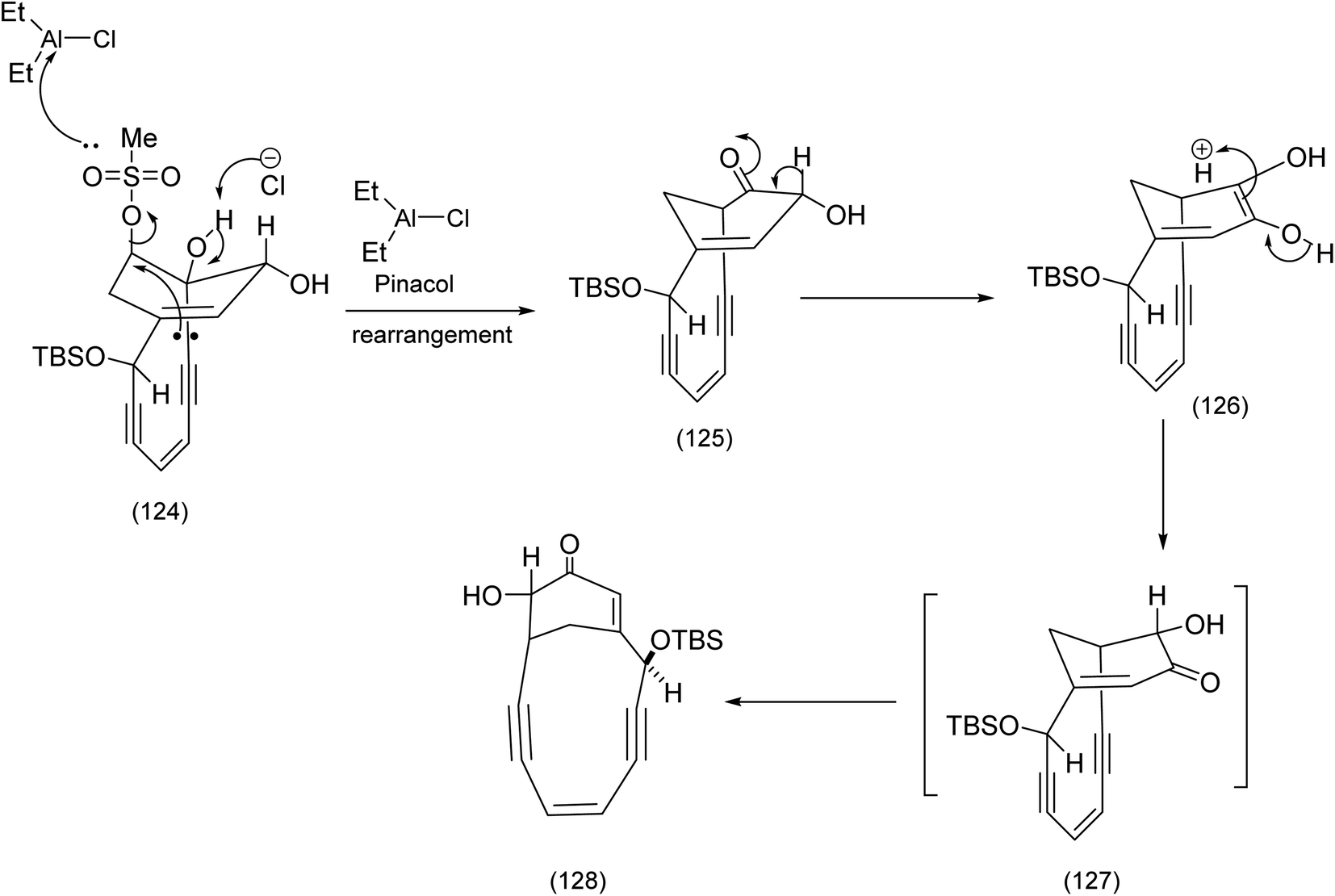 | ||
| Scheme 30 The Tsuchihashi pinacol rearrangement and the effective regioisomeric correction.90 | ||
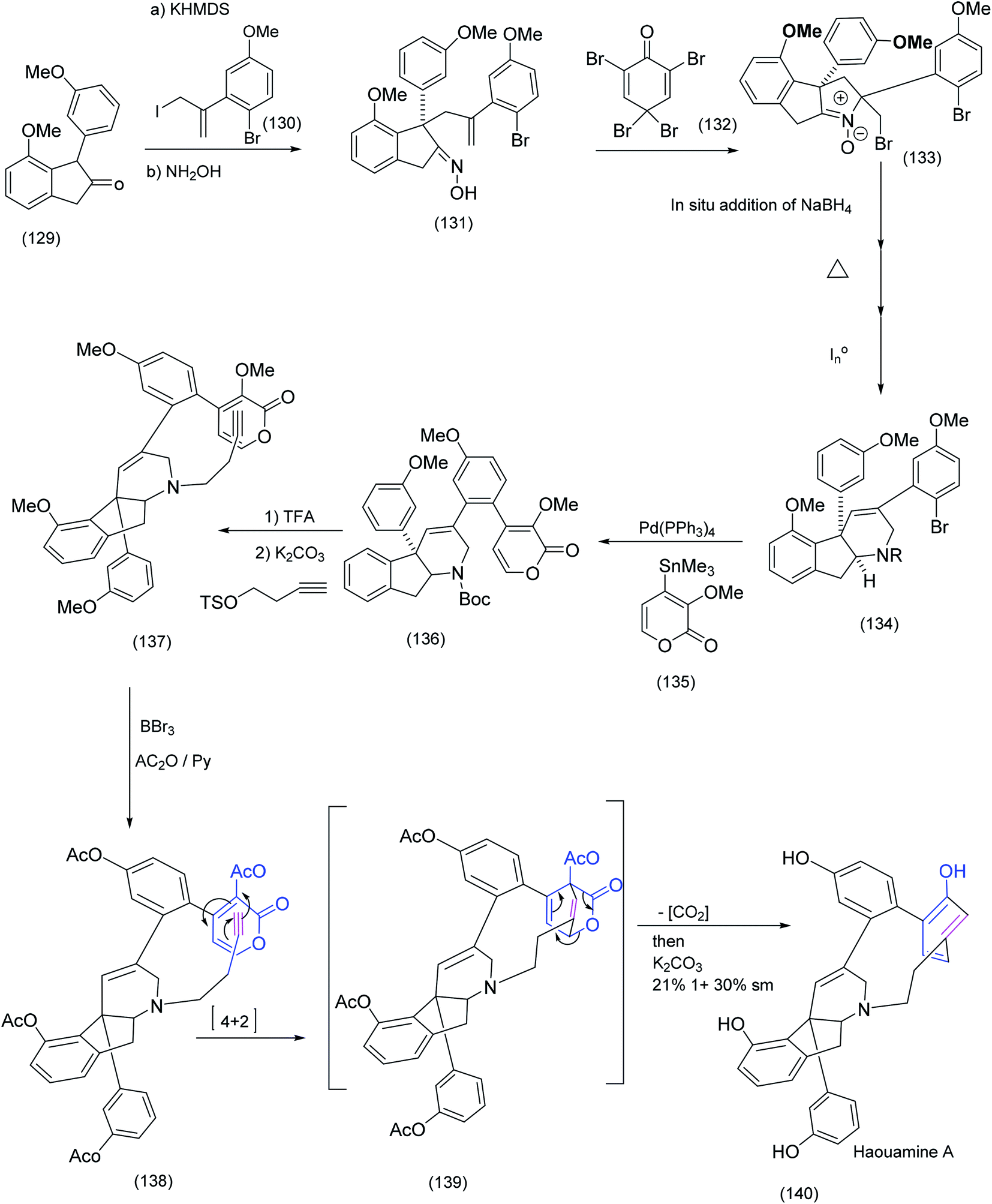 | ||
| Scheme 31 Key steps in Baran's synthesis of the tricyclic azabutyne/pyrone intermediate, reaction and the subsequent macrocyclising Diels Alder.92 | ||
This type of pinacol rearrangement was first reported on different system by Tsuchihashi.90
The next macrocyclic construction, culminating in the molecule Haouamine, gave us pause as we could not quite decide where in this account to insert it. On the one hand, the presence of the biaryl moiety in Haouamine suggested that Baran's synthesis could have been discussed after Nicolaou's Diazonamide synthesis in which the installation of the biaryl was achieved by a single electron oxidation/reduction. On the other hand, Baran's solution to the problem of the strained biaryl moiety within Haouamine involved such an ingenious use of Diels Alder chemistry, that it seemed appropriate to discuss this innovation after Schreiber's91 exhilarating Diels Alder accomplishment.90
Haouamine A is a structurally unique alkaloid. The most outstanding feature of this alkaloid is the 11 membered ring that links an indeno-tetrahydropyridine and a strained biaryl moiety (an aza-paracyclophane) in which one of the benzene rings of the biaryl system is in the boat conformation. The Baran group92 rose to the challenge of this unusual system. Oxidative coupling (single electron oxidation) has proven to be effective in the synthesis of a number of bicyclic systems (see our own endeavours in the area, as depicted in scheme (14), in which a FeCl3 promoted biaryl construction leading to a unique chloindolostilbene hybrid is described). Suzuki and Stille couplings have also been successfully employed in biaryl synthesis and, as we previously described, the Witkop cyclisation was exploited with profit in Nicolaou's Diazonamide construction resulting in the successful installation of the biaryl system. Nevertheless Baran et al.92 reported that more conventional methods failed to achieve the successful construction of the strained biaryl in Haouamine. The creative solution to this problem was found in the pyrone-alkyne Diels–Alder reaction that delivers, at least initially, a cyclohexadiene in the boat conformation. The sequence of steps in this approach is described below.
Coupling of tricyclic ketone (129) with allylic iodide (130) proceeded smoothly to yield the intermediate tetracyclic oxime (131). A series of transformations that included bromination/nucleophilic attack by the oxime, NaBH4 addition, then heat followed by chemoselective N–O bond reduction produced advanced intermediate (134) which underwent Stille coupling to the pyrone (135) to give (136) which, on further treatment with TFA followed by N-alkylation, successfully installed the terminal ethynyl dienophile (137). Demethylation produce the tetraol which was subsequently acetylated to produce (138). At this point, the moment of truth – the pyrone alkyne Diels Alder reaction – had arrived. Heating of (138) proceeded as expected to yield the tetra acetylated Diels Alder adduct (complete with the strained aza-paracyclophane) which on treatment with K2CO3 gave Haouamine. The last two synthesis we have examined from the groups of Schreiber and Baran were beautiful but contrasting examples of the use of the Diels Alder reaction in macrocyclic construction. In this next example Kishi et al.86 successfully effected a remarkably stereoselective late stage intramolecular Diels Alder reaction to yield an advanced intermediate ultimately transformed to the formidable target molecule, pinnatoxin A (Scheme 32).
 | ||
| Scheme 32 A spectacular intramolecular Diels Alder reaction (Diels Alder based self construction) enroute to pinnatoxin.86 | ||
1.8. The vinblastine story according to Fukuyama: macrocycles as synthetic intermediates (a second example see Scheme 15)
In this final example we have chosen, the medium-sized ring was generated in elegant fashion by an intramolecular nucleophilic attack of a nitrobenzenesulphonamide on an epoxide to yield the 11 membered ring. Fukuyama exploited this intermediate in the course of an efficient synthesis of vinblastine. Thus the medium-size ring was not an end itself but a lightly creative means to a significant end. We have previously seen another example of the exploitation of macrocycles as invaluable intermediates in Parker's syntheses that involved a radical cation Diels–Alder transformation (compound 62, Scheme 15). Fukuyama recognised the role played by 11-membered ring indolic macrocycles for the generation of medium-sized ring azabenzfulvenes, which are highly electrophilic species. Prior to Fukuyama's synthesis,93,94 Potier had effected the fragmentation of catharanthine – N-oxide by means of a trifluoroacetic anhydride leading to the macrocyclic azabenzfulvene (147), which was subsequently attacked by vindoline to yield anhydrovinblastine (149) after NaBH4 reduction (Scheme 33).93,95,96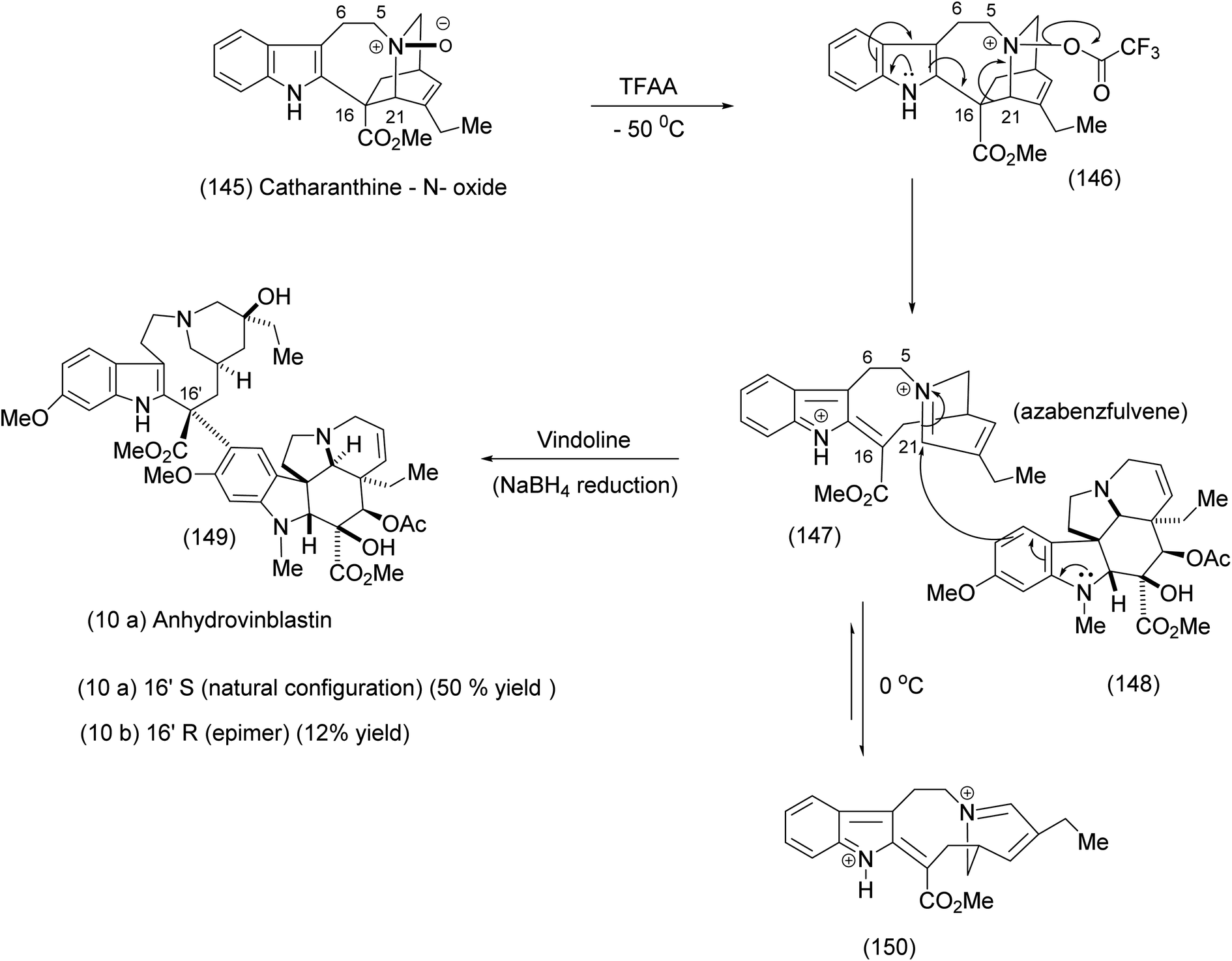 | ||
| Scheme 33 The Potier TFAA fragmentation of catharanthine – N-oxide.93 | ||
Fukuyama's synthesis commenced with the treatment of the functionalised octanoate easter (151) with LDA, THF-78 °C followed by isothiocyanate (152) to generate thioamide (153) which was transformed by means of tributyl tin hydride/triethylborane to yield the corresponding functionalised indole (153) in 67% yield (the Fukuyama indole synthesis). Further functional group manipulations produced the triol (155). Selective C (3′)-tosylation was efficiently performed by means of the five membered tin acetal to yield (154 and 156). The nitrobenzene sulphonamide was then introduced at C (6′) by the Mitsunobu procedure, whose intriguing mechanism is depicted (see (160) to (163)). It is noteworthy that in the presence of NaHCO3/DMF, (169) (Scheme 35) was obtained via the epoxide (164) (see Scheme 35 for full mechanistic details). Alternative pathways via intermediates (157) or (158) were suppressed (Scheme 34), thus enabiliting installation of the nitrobenzene sulphonamide ((162), Scheme 35) by the Mitsunobu protocol. Treatment of epoxide (164) with K2CO3 in DMF at 80 °C proceeded smoothy to produce the desired macrocycle (159). As a demonstration of the value of electrophilic macrocyclic reactive intermediates in complex alkaloid synthesis, (166) was exposed to tertiary butyl hypochlorite. This gave rise to the azabenzfulvene (167) which on further exposure to vindoline to gave rise to vinblastine (169) after judicious functional group manipulations (Scheme 35).
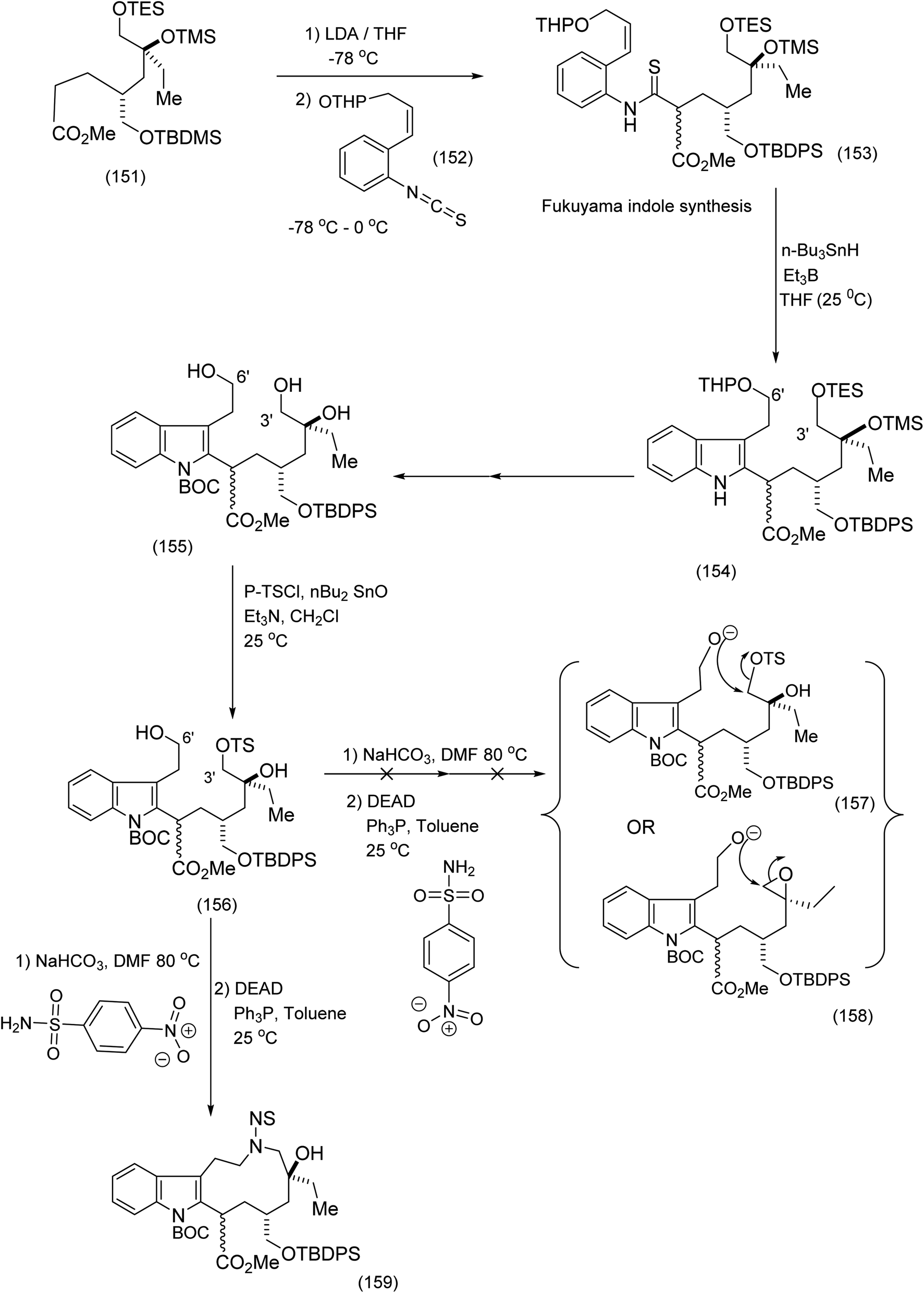 | ||
| Scheme 34 Key steps in the construction of the indolic macrocyclic intermediate93 (159). | ||
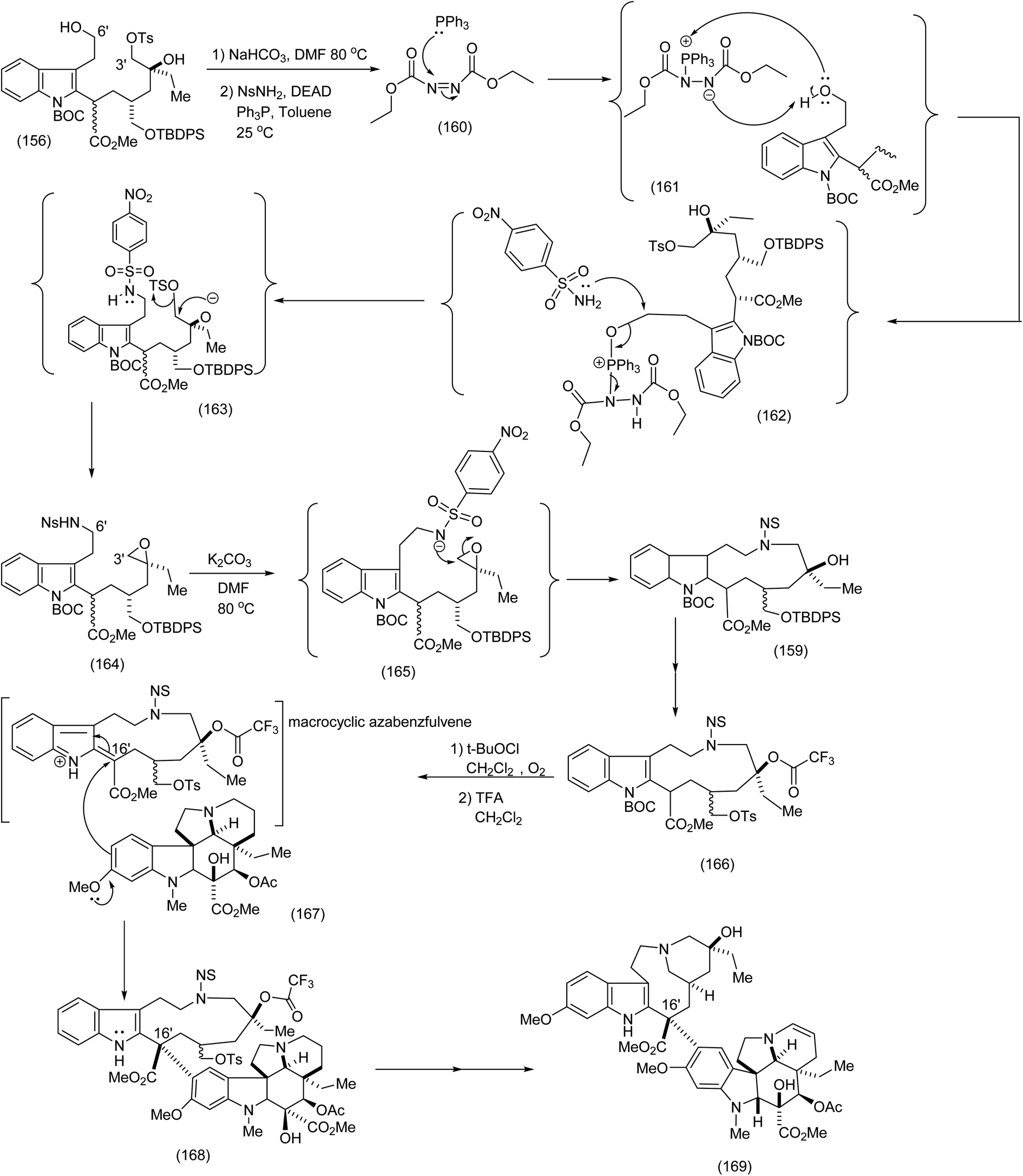 | ||
| Scheme 35 The critical chemoselective installation of the nitrobenzene sulphonamide (Mitsunobu reaction) and final steps leading to vinblastine.93 | ||
2. Conclusion
This review has acknowledged more recent solutions to macrocylic syntheses problems. On the one hand the elegant exploitation of Rh(III) complexes in macrocyclisation from the work of Loh and independently Cossy is detailed. On the other, the older literature has not been ignored as the description of Schreiber's application of pinacol rearrangement to enediyne problem demonstrates. The biaryl coupling in macrocyclic construction highlights the work of Nicolaou and Baran on different targets.Conflicts of interest
There are no conflicts.Acknowledgements
The authors are thankful to Professor, Fei Chen from Wayne State University, MI and Shawn Gubler for help with manuscript preparation.References
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
- J. J. Ciardiello, W. R. Galloway, C. J. O'Connor, H. F. Sore, J. E. Stokes, Y. Wu and D. Spring, Tetrahedron, 2016, 72, 3567–3578 CrossRef CAS.
- D. J. Newman and G. M. Cragg, Drugs, 2014, 12(1), 255–278 Search PubMed.
- J. Mallinson and I. Collins, J. Med. Chem., 2012, 4, 1409–1438 CAS.
- C. M. Madsen and M. H. Clausen, Eur. J. Org. Chem., 2011, 3107–3115 CrossRef CAS.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- K. M. G. O'Connell, H. S. G. Beckmann, L. Laraia, H. T. Horsley, A. Bender, A. R. Venkitaraman and D. R. Spring, Org. Biomol. Chem., 2012, 10, 7545–7551 RSC.
- F. Kopp, C. F. Stratton, L. B. Akella and D. S. Tan, Nat. Chem. Biol., 2012, 8, 358–365 CrossRef CAS PubMed.
- E. J. Corey and D. J. Brunelle, Tetrahedron Lett., 1976, 17, 3409–3412 CrossRef.
- E. J. Corey, D. J. Brunelle and P. J. Stork, Tetrahedron Lett., 1976, 17, 3405–3408 CrossRef.
- K. C. Nicolaou, M. E. Bunnage, D. G. McGarry, S. Shi, P. K. Somers, P. A. Wallace, X. J. Chu, K. A. Agrios, J. L. Gunzner and Z. Yang, Chem.–Eur. J., 1999, 5, 599–617 CrossRef CAS.
- T. Sasaki, M. Inoue and M. Hirama, Tetrahedron Lett., 2001, 42, 5299–5303 CrossRef CAS.
- T. V. Hansen and Y. Stenstrøm, Tetrahedron Lett., 2001, 12, 1407–1409 CrossRef CAS.
- S. P. Breazzano, Y. Poudel and D. L. Boger, J. Am. Chem. Soc., 2013, 135, 1600 CrossRef CAS PubMed.
- R. Larock, E. Yum and M. D. Refvik, J. Org. Chem., 1998, 63, 7652–7662 CrossRef CAS.
- J. K. Stille and M. Tanaka, J. Am. Chem. Soc., 1987, 109, 3785–3786 CrossRef CAS.
- N. Miyaura, T. Ishiyama, H. Sasaki, M. Ishikawa, M. Sato and A. Suzuki, J. Am. Chem. Soc., 1989, 111, 314–321 CrossRef CAS.
- T. Ohe, N. Miyaura and A. Suzuki, J. Org. Chem., 1993, 58, 2201–2208 CrossRef CAS.
- L. Kurti and B. Czakó, Strategic applications of named reactions in organic synthesis, Elsevier, San Diego, 2005 Search PubMed.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689–6690 CrossRef CAS.
- R. C. Larock, Pure Appl. Chem., 1999, 71, 1435–1442 CAS.
- R. C. Larock, M. J. Doty and X. Han, J. Org. Chem., 1999, 64, 8770–8779 CrossRef CAS PubMed.
- R. C. Larock and J. Zenner, J. Org. Chem., 1995, 60, 482–483 CrossRef CAS.
- S. Cacchi, J. Org. Chem., 1999, 576, 42–64 CrossRef CAS.
- S. Cacchi, G. Fabrizi and L. M. Parisi, Heterocycles, 2002, 58, 667–682 CrossRef CAS.
- G. W. Gribble, Org. Synth., 2000, 1045–1075 CAS.
- G. Poli, G. Giambastiani and A. Heumann, Tetrahedron Lett., 2000, 56(33), 5959–5989 CrossRef CAS.
- F. Kopp, C. F. Stratton, L. B. Akella and D. Tan, Nat. Chem. Biol., 2012, 8, 358 CrossRef CAS PubMed.
- D. Yang and C. Zhang, J. Org. Chem., 2001, 66, 4814–4818 CrossRef CAS PubMed.
- J. R. Donald and W. P. Unsworth, Chem.–Eur. J., 2017, 23, 8780–8799 CrossRef CAS PubMed.
- K. C. Nicolaou, C. R. Hale, C. Ebner, C. Nilewski, C. F. Ahles and D. Rhoades, Angew. Chem., Int. Ed., 2012, 51, 4726–4730 CrossRef CAS PubMed.
- J. Marrero, A. D. Rodriguez, P. Baran, R. G. Raptis, J. A. Sánchez, E. Ortega-Barria and T. Capson, Org. Lett., 2004, 6, 1661–1664 CrossRef CAS PubMed.
- A. D. Rodriguez and Y. Shi, J. Org. Chem., 2000, 65, 5839–5842 CrossRef CAS PubMed.
- V. A. Stonik, I. I. Kapustina, A. I. Kalinovsky, P. S. Dmitrenok and B. Grebnev, Tetrahedron Lett., 2002, 43, 315–317 CrossRef CAS.
- Y. Li, G. Pattenden and J. Rogers, Tetrahedron Lett., 2010, 51, 1280–1283 CrossRef CAS.
- K. C. Nicolaou, V. A. Adsool and C. R. H. Hale, Angew. Chem., 2011, 50, 5149–5152 CrossRef CAS PubMed.
- Y. Ito, T. Yoshinaga and H. Nishino, Tetrahedron, 2010, 66, 2683–2694 CrossRef CAS.
- J. Magolan and M. Kerr, Org. Lett., 2006, 8, 4561–4564 CrossRef CAS PubMed.
- Y. Ito and H. Nishino, Heterocycl. Commun., 2009, 15, 467–471 CAS.
- H. Nishino, H. Kamachi, H. Baba and K. Kurosawa, J. Org. Chem., 1992, 57, 3551–3557 CrossRef CAS.
- H. Nishino, K. Ishida, H. Hashimoto and K. Kurosawa, Synthesis, 1996,(7), 888–896 CrossRef CAS.
- J. Ouyang, H. Nishino and K. Kurosawa, J. Heterocycl. Chem., 1997, 34, 81–86 CrossRef CAS.
- V.-H. Nguyen, H. Nishino and K. Kurosawa, Synthesis, 1997, 1997, 899–908 CrossRef.
- V.-H. Nguyen, H. Nishino and K. Kurosawa, Heterocycles, 1998, 3, 465–480 Search PubMed.
- F. A. Chowdhury, H. Nishino and K. Kurosawa, Tetrahedron Lett., 1998, 39, 7931–7934 CrossRef CAS.
- F. A. Chowdhury, H. Nishino and K. Kurosawa, Heterocyclic, 1999, 30, 111–112 Search PubMed.
- R. Kumabe, H. Nishino, M. Yasutake, V.-H. Nguyen and K. Kurosawa, Tetrahedron Lett., 2001, 42, 69–72 CrossRef CAS.
- M. T. Rahman, H. Nishino and C. Y. Qian, Tetrahedron Lett., 2003, 44, 5225–5228 CrossRef CAS.
- R. Kumabe and H. Nishino, Tetrahedron Lett., 2004, 45, 703–706 CrossRef CAS.
- T. Tsubusaki and H. Nishino, Tetrahedron, 2009, 65, 3745–3752 CrossRef CAS.
- N. F. Thomas, K. C. Lee, T. Paraidathathu, J. F. F. Weber and K. Awang, Tetrahedron Lett., 2002, 43, 3151–3155 CrossRef CAS.
- B. B. Snider, Chem. Rev., 1996, 96, 339–364 CrossRef CAS PubMed.
- B. B. Snider, Tetrahedron, 2009, 65, 10738–10744 CrossRef CAS PubMed.
- B. B. Snider and S. O'Neil, Tetrahedron, 1995, 51, 12983–12994 CrossRef CAS.
- B. B. Snider, J. E. Merritt, M. A. Dombroski and B. O. Buckman, J. Org. Chem., 1991, 56, 5544–5553 CrossRef CAS.
- B. B. Snider and R. B. Smith, Tetrahedron, 2002, 58, 25–34 CrossRef CAS.
- B. B. Snider, L. Han and C. J. Xie, J. Org. Chem., 1997, 62, 6978–6984 CrossRef CAS.
- B. B. Snider and B. M. Cole, J. Org. Chem., 1995, 60, 5376–5377 CrossRef CAS.
- G. Pattenden, D. A. Stoker and N. M. Thomson, Org. Biomol. Chem., 2007, 5, 1776–1788 RSC.
- B. D. Boeck, N. M. Harrington-Frost and G. Pattenden, Org. Biomol. Chem., 2005, 3, 340–347 RSC.
- H. M. Boehm, S. Handa, G. Pattenden, L. Roberts, A. Blake and W. Li, J. Chem. Soc., Perkin Trans. 1, 2000, 3522–3538 RSC.
- S. Hitchcock, S. Houldsworth, D. Pryde, N. Thomson and A. J. Blake, J. Chem. Soc., Perkin Trans. 1, 1998, 3181–3206 RSC.
- G. Pattenden, M. A. Gonzalez, S. McCulloch, A. Walter and S. J. Woodhead, Proceedings of the National Academy of Sciences, 2004, 101, 12024–12029 CrossRef CAS PubMed.
- S. Handa and G. Pattenden, Chem. Commun., 1998, 311–312 RSC.
- K. Ahmad, N. F. Thomas, M. R. Mukhtar, I. Noorbatcha, J.-F. F. Weber, M. A. Nafiah, S. S. Velu, K. Takeya, H. Morita and C.-G. Lim, Tetrahedron, 2009, 65, 1504–1516 CrossRef CAS.
- H. N. Lim and K. A. Parker, Org. Lett., 2012, 15, 398–401 CrossRef PubMed.
- D. J. Bellville, D. W. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718–720 CrossRef CAS.
- N. L. Bauld, D. J. Bellville, R. Pabon, R. Chelsky and G. J. Green, J. Am. Chem. Soc., 1983, 105, 2378–2382 CrossRef CAS.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- S. Lin, C. E. Padilla, M. A. Ischay and T. P. Yoon, J. Am. Chem. Soc., 2012, 53, 3073–3076 CAS.
- H. Yoon and W. J. Chae, Tetrahedron Lett., 1997, 38, 5169–5172 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- O. Wiest and E. J. Steckhan, Tetrahedron Lett., 1993, 34, 6391–6394 CrossRef CAS.
- T. P. Yoon, ACS Catal., 2013, 3, 895–902 CrossRef CAS PubMed.
- Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- C. D. Vanderwal, D. A. Vosburg, S. Weiler and E. J. Sorensen, J. Am. Chem. Soc., 2003, 125, 5393–5407 CrossRef CAS PubMed.
- J.-P. Krieger, D. Lesuisse, G. Ricci, M.-A. Perrin, C. Meyer and J. Cossy, Org. Lett., 2017, 19, 2706–2709 CrossRef CAS PubMed.
- B. Jiang, M. Zhao, S. S. Xu and T. P. Loh, Angew. Chem., 2018, 130, 564–568 CrossRef.
- K. Nicolau and S. J. Snyder, J. Am. Chem. Soc., 2003, 443–463 Search PubMed.
- K. Nicolaou, M. Bella, D. Y. Chen, X. Huang, T. Ling and S. A. Snyder, Angew. Chem., Int. Ed., 2002, 41, 3495–3499 CrossRef CAS PubMed.
- K. Nicolaou, C. Hummel, M. Nakada, K. Shibayama, E. Pitsinos, H. Saimoto, Y. Mizuno, K. Baldenius and A. J. Smith, J. Am. Chem. Soc., 1993, 115, 7625–7635 CrossRef CAS.
- A. Smith, E. Pitsinos, C. Hwang, Y. Mizuno, H. Saimoto, G. Scarlato, T. Suzuki and K. J. Nicolaou, J. Am. Chem. Soc., 1993, 115, 7612–7624 CrossRef CAS.
- R. Groneberg, T. Miyazaki, N. Stylianides, T. Schulze, W. Stahl, E. Schreiner, T. Suzuki, Y. Iwabuchi, A. Smith and K. J. Nicolaou, J. Am. Chem. Soc., 1993, 115, 7593–7611 CrossRef CAS.
- K. C. Nicolaou and J. S. Chen, Classics in total synthesis III: further targets, strategies, methods, John Wiley & Sons, 2011 Search PubMed.
- S. L. Schreiber and L. L. Kiessling, J. Am. Chem. Soc., 1988, 110, 631–633 CrossRef CAS.
- S. L. Schreiber and L. L. Kiessling, Tetrahedron Lett., 1989, 30, 433–436 CrossRef CAS.
- F. J. Schoenen, J. A. Porco Jr, S. L. Schreiber, G. D. VanDuyne and J. J. Clardy, Tetrahedron Lett., 1989, 30, 3765–3768 CrossRef CAS.
- K. Suzuki, E. Katayama and G.-i. Tsuchihashi, Tetrahedron Lett., 1983, 24, 4997–5000 CrossRef CAS.
- B. M. Benjamin, H. J. Schaeffer and C. J. Collins, J. Am. Chem. Soc., 1957, 79, 6160–6164 CrossRef CAS.
- P. S. Baran and N. Z. Burns, J. Am. Chem. Soc., 2006, 128, 3908–3909 CrossRef CAS PubMed.
- M. S. Alehashem, C.-G. Lim and N. F. Thomas, RSC Adv., 2016, 6, 18002–18025 RSC.
- N. Langlois, F. Gueritte, Y. Langlois and P. J. Potier, J. Am. Chem. Soc., 1976, 98, 7017–7024 CrossRef CAS PubMed.
- M. S. Alehashem, A. Ariffin, A. A. Q. Al-Khdhairawi and N. F. Thomas, RSC Adv., 2018, 8, 2506–2520 RSC.
- M. S. Alehashem, A. Ariffin and N. F. Thomas, AIP Conf. Proc., 2018, 020017 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |