DOI:
10.1039/D0RA01176B
(Paper)
RSC Adv., 2020,
10, 10461-10470
New triterpenoid saponin glycosides from the fruit fibers of Trichosanthes cucumerina L.†
Received
6th February 2020
, Accepted 3rd March 2020
First published on 11th March 2020
Abstract
Five new triterpenoid saponin glycosides, trichocucumerisides A–E (1–5), together with eleven known compounds (6–16) were isolated from Trichosanthes cucumerina fruit fibers. The structures of the new compounds were elucidated by detailed analysis of NMR and mass spectroscopic data as well as chemical reactions. The anti-inflammatory study against nitric oxide (NO) production in lipopolysaccharide (LPS)-stimulated RAW264.7 cells shows that compounds 7 and 9 exhibited stronger NO inhibitory activity, with IC50 values of 3.0 and 2.7 μM, respectively, with comparison to positive references Celecoxib and aminoguanidine (IC50 values 75.7 and 75.0 μM, respectively). Compounds 7 and 9 also possessed a greater selectivity index (SI) of approximately 3–4-fold activity than that of the positive references.
Introduction
Trichosanthes cucumerina L. is a Thai medicinal plant in the Cucurbitaceae family and the fruits are known for their strong bitter taste. Trichosanthes is mainly distributed in the tropical zone of Southeast Asia and Australia, and it is the largest genus of the family with over 100 species, 17 species of which are found in Thailand.1 Previous reports revealed its pharmacological effects including cytotoxic,2 anti-inflammatory,3 antidiabetic,4,5 hypoglycemic,6 hepatoprotective,7 gastroprotective,8 antifertility9 and larvicidal10 activities. The main chemical constituents present in T. cucumerina are triterpenoids, especially cucurbitacin groups,2 which make the plant pharmacologically and therapeutically important. Studies on the cytotoxic activities of cucurbitacins from T. cucumerina have been reported.11–13 However, the phytochemical investigation of the constituents of this plant has not much been undertaken. In the present study, we wish to report five new triterpenoid saponin glycosides 1–5 together with eleven known compounds 6–16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14–24 from the fruit fibers of T. cucumerina. These compounds were elucidated by analysis of varying spectroscopic evidence, as well as comparison with the literature data of known compounds. All these compounds were evaluated against nitric oxide (NO) production in lipopolysaccharide (LPS)-stimulated RAW264.7 cells.
14–24 from the fruit fibers of T. cucumerina. These compounds were elucidated by analysis of varying spectroscopic evidence, as well as comparison with the literature data of known compounds. All these compounds were evaluated against nitric oxide (NO) production in lipopolysaccharide (LPS)-stimulated RAW264.7 cells.
Results and discussion
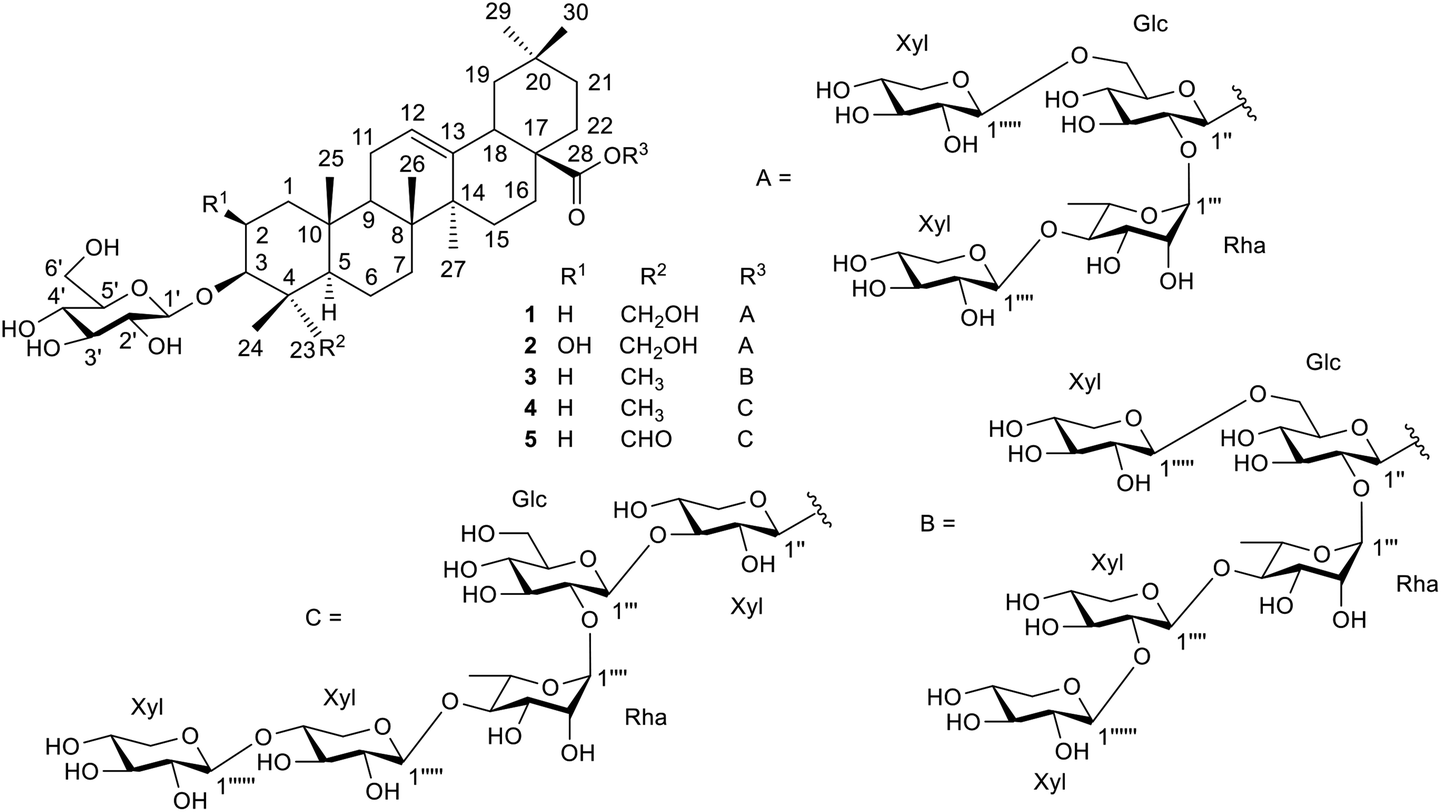
The crushed fruit fiber of T. cucumerina was extracted successively with n-hexane, EtOAc and MeOH. The extracts were repeatedly subjected to silica gel, Sephadex LH-20 and reversed phase RP C-18 column chromatography to afford five new triterpenoid saponin glycosides (1–5), two known steroids (6 and 13) and nine known cucurbitacins (7–12 and 14–16). The structures of the new compounds were elucidated by spectroscopic techniques and chemical means to be hederagenin-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→6)-[β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside (1), bayogenin-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→6)-[β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside (2), oleanolic acid-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→6)-[β-D-xylopyranosyl-(1→2)-β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside (3), oleanolic acid-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→4)-β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)-β-D-glucopyranosyl-(1→3)-β-D-xylopyranoside (4), and gypsogenin-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→4)-β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)-β-D-glucopyranosyl-(1→3)-β-D-xylopyranoside (5) (Fig. 1). The NMR and mass spectra of the new compounds are included in the ESI.† The known compounds were identified as α-spinasterol (6),14 cucurbitacin B (7),15–17 cucurbitacin E (8),17–19 isocucurbitacin B (9),20 isocucurbitacin D (10),21 cucurbitacin D (11),17,18,22 25-acetyl cucurbitacin F (12),23 α-spinasterol-3-O-β-D-glucopyranoside (13),14 2-O-β-D-glucopyranosyl-cucurbitacin B (14),24 2-O-β-D-glucopyranosyl-cucurbitacin D (15),24 and 2-epi-O-β-D-glucopyranosyl-cucurbitacin B (16)24 by spectroscopic data comparisons with those of the literature values (Fig. S1†).
 |
| | Fig. 1 Structures of compounds 1–5. | |
Compound 1 was isolated as a white powder, [α]26D −17.2° (c 0.39 in MeOH). Its ESI-TOF-MS data showed a sodium adduct ion [M + Na]+ at m/z 1229.5924 (calcd for C58H94NaO26, 1229.5926), corresponding to the molecular formula of C58H94O26. The IR spectrum indicated the presence of hydroxyl (3337 cm−1), ester (1734 cm−1) and olefinic (1639 cm−1) groups. The 1H NMR data (Table 1, the NMR spectra are presented in Fig. S19–S42†) showed six tertiary methyl groups at δ 0.91 (3H, s, H-24), 0.94 (3H, s, H-25), 1.11 (3H, s, H-26), 1.18 (3H, s, H-27), 0.80 (3H, s, H-29) and 0.90 (3H, s, H-30) and characteristic signals for olefinic group at δ 5.40 (1H, brt, J = 3.2 Hz, H-12). The primary alcoholic function placement at C-23 was consistent with the down-field shift of CH2-23 at δ 3.74 and 4.29 (each 1H, d, J = 10.9 Hz). The HMBC correlations between these two protons with C-3 at δC 82.8 confirmed the location of the alcoholic function at C-23. The secondary alcoholic function in the aglycone unit was at C-3 (δ 4.26, 1H, m, H-3). HMBC correlation between CH3-24 (δH 0.91) and C-3 (δC 82.8), and between CH2-23 and C-3 allowed the location of this alcoholic function at C-3. The presence of the ester group (νmax CO at 1727 cm−1 and δC CO at 177.1) was proven to be at C-28 by HMBC correlation between H-16 (δH 2.07) and C-28 (Fig. S34–S38†). The aglycone moiety of 1 was therefore identified to be hederagenin (1a, Fig. S2†).25
Table 1 1H NMR data for aglycone moieties of compounds 1–5 in pyridine-d5 at 400 MHz (δ in ppm, J in Hz)
| Position |
1 |
2 |
3 |
4 |
5 |
| Signals overlapped. |
| 1 |
0.94a |
1.30 m |
0.91a |
0.85 m |
0.88 m |
| 1.45 m |
2.32 dd (12.7, 4.1) |
1.44 m |
1.41a |
1.40a |
| 2 |
1.89a |
4.17 m |
1.84 m |
1.83a |
1.84 m |
| 2.23 m |
|
2.24 m |
2.43 dd (11.8, 3.2) |
2.17 m |
| 3 |
4.26 m |
4.25 m |
3.39 dd (11.3, 4.6) |
3.31 dd (11.8, 3.9) |
4.04 m |
| 5 |
1.66 m |
1.84a |
0.84 m |
0.74 brd (11.4) |
1.66 dd (9.2, 8.1) |
| 6 |
1.45 m |
1.49a |
1.44 m |
1.31a |
1.06a |
| 1.89a |
1.92a |
1.68 m |
1.51 m |
1.40a |
| 7 |
1.76 m |
1.77a |
1.82a |
1.41 m |
1.25a |
| 2.02 m |
2.06 m |
2.08 m |
1.47 m |
1.43a |
| 9 |
1.75a |
1.84a |
1.67 m |
1.60 t (8.1) |
1.36 m |
| 11 |
1.89a |
1.94a |
1.97 m |
1.87 m |
1.84 m |
| 1.94 m |
2.08 m |
|
|
|
| 12 |
5.40 brt (3.2) |
5.40 brt (3.2) |
5.46 brs |
5.46 brt (3.2) |
5.44 brs |
| 15 |
1.46 m |
1.49 m |
1.57 m |
1.27a |
1.22a |
| 2.05 m |
2.08a |
2.12 m |
2.11 m |
2.07 m |
| 16 |
1.92a |
1.96a |
2.00 m |
2.03 m |
2.02 m |
| 2.05 m |
2.04a |
2.16 m |
2.11 m |
2.10a |
| 18 |
3.13 dd (14.0, 4.3) |
3.14 dd (13.9, 3.6) |
3.18 brd (12.1) |
3.31 dd (13.7, 3.9) |
3.28 brd (11.6) |
| 19 |
1.14 m |
1.19a |
1.25 m |
1.27a |
1.22a |
| 1.68 m |
1.68 brd (13.6) |
1.80a |
1.81a |
1.78 m |
| 21 |
1.11a |
1.14a |
1.19 m |
1.18 brd (12.7) |
1.75a |
| 1.26 brd (12.0) |
1.30 m |
1.35a |
1.41a |
2.01 m |
| 22 |
1.75a |
1.49 m |
1.44a |
1.75 m |
1.17 m |
| |
1.77a |
1.75 m |
2.11 m |
1.40a |
| 23 |
3.74 d (10.9) |
3.76 d (10.6) |
0.99 s |
1.12 s |
9.95 s |
| 4.29 d (10.9) |
4.51 d (10.6) |
|
|
|
| 24 |
0.91 s |
0.98 s |
1.32 s |
1.34 s |
1.43 s |
| 25 |
0.94 s |
1.06 s |
0.91 s |
0.82 s |
0.79 s |
| 26 |
1.11 s |
1.14 s |
1.20 s |
1.07 s |
1.02 s |
| 27 |
1.18 s |
1.16 s |
1.32 s |
1.29 s |
1.25 s |
| 29 |
0.80 s |
0.83 s |
0.89 s |
0.94 s |
0.94 s |
| 30 |
0.90 s |
0.93 s |
0.96 s |
1.01 s |
1.00 s |
The anomeric protons at δ 5.10 (1H, d, J = 7.2 Hz, H-1′), 6.14 (1H, d, J = 8.0 Hz, H-1′′), 6.46 (1H, brs, H-1′′′), 5.05 (1H, d, J = 7.5 Hz, H-1′′′′) and 4.87 (1H, d, J = 7.4 Hz, H-1′′′′′) suggested that 1 had five sugar units. The five anomeric proton signals at δ 5.10, 6.14, 6.46, 5.05 and 4.87 showed correlations with five anomeric carbon signals at δ 106.3 (C-1′), 95.1 (C-1′′), 101.9 (C-1′′′), 108.2 (C-1′′′′) and 106.1 (C-1′′′′′), respectively, in the HMQC spectra (Fig. S30–S33†). The sugar moieties were initially deduced from the coupling constants of anomeric protons, followed by acid hydrolysis of 1 with TFA and comparison with standard sugars (see Experimental section) and D-glucose, L-rhamnose and D-xylose, together with the aglycone hederagenin (1a, Fig. S2†).25 The magnitude of the coupling constants of the anomeric protons of the sugar units mentioned above were therefore concluded to exist as two β-D-glucose, one α-L-rhamnose and two β-D-xylose moieties.26–28 In the HMBC spectra (Fig. S34–S38†) of 1, the correlations between signal at δH 5.10 (Glc-1′) and δC 82.8 (C-3) was observed, which indicated that the glucose unit was attached at the 3-position of the aglycone. The NMR data did not show any connection of this glucose unit with other sugar moiety, suggesting that the sugar residue at the 3-position was a monoglucoside. The correlations of signals between δH 6.16 (Glc-1′′) and δC 177.1 (C-28), δH 6.46 (Rha-1′′′) and δC 76.7 (Glc-2′′), δH 5.05 (Xyl-1′′′′) and δC 86.0 (Rha-4′′′), and δH 4.87 (Xyl-1′′′′′) and δC 69.4 (Glc-6′′) characterized that the sequence of the sugar chain was β-D-Xyl-(1→6)-[β-D-Xyl-(1→4)-α-L-Rha-(1→2)]-β-D-Glc and that the glycosidic site was at C-28. The sugar moieties at C-3 and C-28 were further confirmed by alkaline hydrolysis of 1 with NaOH (see Experimental section) to give hederagenin-3-O-β-D-glucopyranoside (1b, Fig. S2†),29 the structure of which was confirmed by NMR data (Tables S1 and S2†). The β-orientation of the C-3 hydroxyl group in 1b was confirmed by the large coupling constant (11.9 Hz) of H-3 at δ 4.32 with H-2ax (Table S1†). The presence of a mono glucose unit at the 3-position of 1 was therefore confirmed. In addition, 1H–1H COSY correlations H-1′/H-2′, H-2′/H-3′, H-5′/H-4′, H-5′/H-6′, H-1′′/H-2′′, H-2′′/H-3′′, H-5′′/H-4′′, H-5′′/H-6′′, H-1′′′/H-2′′′, H-2′′′/H-3′′′, H-3′′′/H-4′′′, H-4′′′/H-5′′′, H-5′′′/H-6′′′, H-1′′′′/H-2′′′′, H-2′′′′/H-3′′′′, H-3′′′′/H-4′′′′, H-4′′′′/H-5′′′′, H-1′′′′′/H-2′′′′′, H-2′′′′′/H-3′′′′′, H-4′′′′′/H-5′′′′′ were observed (Fig. 2). Moreover, the NOESY correlations of H-1′/H-3, H-1′′′/H-2′′, H-1′′′′/H-4′′′, and H-1′′′′′/H-6′′ (Fig. 2) were also key interactions to support this gross structure. Accordingly, compound 1 was identified as hederagenin-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→6)-[β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside and was named trichocucumeriside A.
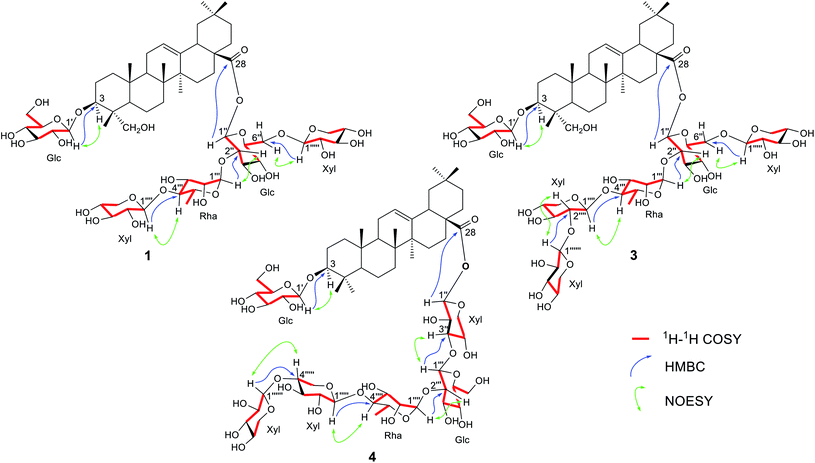 |
| | Fig. 2 Key correlations of compounds 1, 3 and 4. | |
Compound 2 was isolated as a white powder, [α]26D −22.7° (c 0.24 in MeOH). Its ESI-TOF-MS data showed a sodium adduct ion [M + Na]+ at m/z 1245.5869 (calcd for C58H94NaO27, 1245.5875), corresponding to the molecular formula of C58H94O27. The IR spectrum indicated the presence of hydroxyl (3442 cm−1), ester (1727 cm−1) and olefinic (1643 cm−1) groups. The 1H NMR data (Table 1 and Fig. S44–S66†) showed very similar spectral feature to that of compound 1. Comparison of the molecular formula of compound 2 with that of compound 1 indicated that 2 constituted one oxygen atom more than 1, suggesting the presence of one additional hydroxyl group in the aglycone unit. The large down-field shifts at C-2 signals of 2 (δH 4.17 and δC 67.6) comparing with those of 1 (δH 1.89, 2.23 and δC 26.4) were observed. Considerable down-field shifts of proton and carbon resonances at C-1 and C-3 of 2 (Tables 1 and 3) were also noted. These observations indicated that the hydroxyl group should be located at the 2-position. This was confirmed by the 1H–1H COSY correlation of H-2 and H-3, and HMBC correlation between H-2 and C-3.
Table 2 1H NMR data for sugar moieties of compounds 1–5 in pyridine-d5 at 400 MHz (δ in ppm, J in Hz)
| Position |
1 |
2 |
3 |
4 |
5 |
| Glc′ |
Glc′ |
Glc′ |
Glc′ |
Glc′ |
| Signals overlapped. |
| 1′ |
5.10 d (7.2) |
5.20a |
4.94 d (7.8) |
4.93 d (7.6) |
4.83 d (7.6) |
| 2′ |
4.04 t (8.3) |
4.11 m |
4.06 m |
4.20 d (8.2) |
4.12 m |
| 3′ |
4.18 d (8.8) |
4.13 t (8.4) |
4.31 m |
4.34 t (8.8) |
4.27 t (8.8) |
| 4′ |
4.26 m |
4.27 t (10.1) |
4.27 m |
4.07 m |
4.06 m |
| 5′ |
3.90 ddd (9.0, 4.6, 2.5) |
3.93 ddd (9.4, 5.0, 2.2) |
4.02 t (7.7) |
3.95 ddd (9.5, 5.0, 1.9) |
3.87 m |
| 6′ |
4.40 brd (10.2) |
4.32 dd (10.2, 4.9) |
4.43 dd (11.8, 5.0) |
4.37 dd (8.8, 4.5) |
4.34 dd (8.8, 3.8) |
| 4.51 m |
4.52 dd (9.5, 6.5) |
4.62 brd (11.0) |
4.57 m |
4.55 m |
| Position |
1 |
2 |
3 |
4 |
5 |
| Glc′′ |
Glc′′ |
Glc′′ |
Xyl′′ |
Xyl′′ |
| 1′′ |
6.14 d (8.0) |
6.16 d (7.8) |
6.16 d (8.0) |
6.54 d (7.7) |
5.24 d (7.1) |
| 2′′ |
4.29 d (8.3) |
4.32 m |
4.32 m |
4.60 t (9.6) |
4.59 m |
| 3′′ |
4.21 m |
4.24 m |
4.26 t (8.9) |
4.23 m |
4.15 m |
| 4′′ |
4.29 m |
4.11 m |
4.36 t (8.7) |
4.08 m |
4.07 t (8.1) |
| 5′′ |
4.04 t (8.3) |
4.05 m |
4.07 m |
4.00 dd (10.4, 4.0) |
3.99 dd (7.0, 3.5) |
| |
|
|
4.39 m |
4.45 m |
| 6′′ |
4.28 m |
4.30 m |
4.31 m |
|
|
| 4.64 dd (9.6, 1.6) |
4.67 dd (11.3, 1.6) |
4.66 brd (10.9) |
|
|
| Position |
1 |
2 |
3 |
4 |
5 |
| Rha′′′ |
Rha′′′ |
Rha′′′ |
Glc′′′ |
Glc′′′ |
| 1′′′ |
6.46 brs |
6.47 brs |
6.47 brs |
5.24 d (7.6) |
5.18 d (7.2) |
| 2′′′ |
4.82 brs |
4.84 brs |
4.84 m |
4.64 t (9.2) |
4.59 m |
| 3′′′ |
4.70 brd (8.8) |
4.72 brd (9.4) |
4.72 m |
4.74 brs |
4.61 m |
| 4′′′ |
4.37 t (9.4) |
4.39 t (9.3) |
4.49 dd (9.2, 3.4) |
4.19 d (8.6) |
4.17 m |
| 5′′′ |
4.50 t (6.4) |
4.52 t (9.5) |
4.53 m |
4.07 t (6.6) |
4.06 m |
| 6′′′ |
1.82 d (6.1) |
1.84 d (6.1) |
1.81 d (5.9) |
4.64 m |
4.17 m |
| |
|
|
4.42 m |
4.56 m |
| Position |
1 |
2 |
3 |
4 |
5 |
| Xyl′′′′ |
Xyl′′′′ |
Xyl′′′′ |
Rha′′′′ |
Rha′′′′ |
| 1′′′′ |
5.05 d (7.5) |
5.08 d (7.2) |
5.08 d (7.0) |
5.78 brs |
5.82 brs |
| 2′′′′ |
4.09 m |
4.08 m |
4.09 m |
4.60 m |
4.61 m |
| 3′′′′ |
4.09 m |
4.24 m |
4.09 m |
4.45 m |
4.45 m |
| 4′′′′ |
4.46 m |
4.32 m |
4.36 t (8.7) |
4.41 m |
4.41 m |
| 5′′′′ |
3.52 t (10.3) |
3.55 t (10.2) |
3.57 t (10.8) |
4.41 m |
4.40 m |
| 4.26 m |
4.27 t (10.1) |
4.31 t (9.7) |
|
|
| 6′′′′ |
|
|
|
1.79 d (5.0) |
1.76 d (4.7) |
| Position |
1 |
2 |
3 |
4 |
5 |
| Xyl′′′′′ |
Xyl′′′′′ |
Xyl′′′′′ |
Xyl′′′′′ |
Xyl′′′′′ |
| 1′′′′′ |
4.87 d (7.4) |
4.90 d (7.4) |
4.90 d (7.4) |
5.19a |
5.15 d (7.5) |
| 2′′′′′ |
3.97 t (7.9) |
3.99 t (7.8) |
4.00 t (8.1) |
4.07 m |
4.07 m |
| 3′′′′′ |
4.10 m |
4.11 m |
4.17 t (8.7) |
4.59 m |
4.59 m |
| 4′′′′′ |
4.17 t (8.8) |
4.19 m |
4.21 t (8.2) |
4.08 m |
4.07 m |
| 5′′′′′ |
3.63 t (10.6) |
3.67 t (10.8) |
3.66 t (10.6) |
3.48 t (9.8) |
3.51 t (10.1) |
| 4.29 t (10.8) |
4.32 t (8.2) |
4.31 m |
4.23 t (9.4) |
4.24 t (9.4) |
| Position |
1 |
2 |
3 |
4 |
5 |
| Xyl′′′′′′ |
Xyl′′′′′′ |
Xyl′′′′′′ |
| 1′′′′′′ |
|
|
5.23 d (7.5) |
5.28 d (7.6) |
5.23a |
| 2′′′′′′ |
|
|
4.13 t (8.4) |
4.07 m |
4.06 m |
| 3′′′′′′ |
|
|
4.22 m |
4.19 m |
4.17 m |
| 4′′′′′′ |
|
|
4.09 m |
4.20 m |
4.19 m |
| 5′′′′′′ |
|
|
3.53 t (10.0) |
3.70 t (10.0) |
3.70 t (10.0) |
| |
|
|
4.31 t (9.7) |
4.31 m |
4.29 t (10.4) |
Table 3 13C NMR data for aglycone moieties of compounds 1–5 in pyridine-d5 at 100 MHz (δ in ppm)
| Position |
1 |
2 |
3 |
4 |
5 |
| 1 |
39.3 |
47.9 |
39.4 |
39.2 |
38.6 |
| 2 |
26.4 |
67.6 |
27.2 |
27.1 |
25.4 |
| 3 |
82.8 |
89.1 |
89.6 |
89.4 |
83.7 |
| 4 |
43.9 |
45.2 |
40.1 |
40.0 |
55.6 |
| 5 |
48.3 |
47.9 |
56.5 |
56.3 |
48.7 |
| 6 |
18.9 |
18.9 |
19.2 |
18.9 |
21.0 |
| 7 |
32.9 |
32.9 |
33.0 |
33.7 |
33.0 |
| 8 |
40.5 |
40.5 |
40.5 |
40.3 |
40.6 |
| 9 |
48.7 |
48.6 |
48.6 |
48.4 |
48.2 |
| 10 |
37.5 |
38.6 |
37.6 |
37.4 |
36.7 |
| 11 |
24.4 |
23.8 |
24.4 |
24.2 |
24.2 |
| 12 |
123.3 |
123.2 |
123.5 |
123.5 |
123.1 |
| 13 |
144.5 |
144.5 |
144.6 |
144.7 |
144.8 |
| 14 |
42.7 |
42.8 |
42.9 |
42.6 |
42.6 |
| 15 |
29.1 |
29.1 |
29.0 |
28.7 |
28.7 |
| 16 |
23.8 |
24.4 |
24.0 |
23.6 |
23.6 |
| 17 |
47.7 |
47.7 |
47.8 |
47.8 |
47.8 |
| 18 |
42.4 |
42.3 |
42.5 |
42.2 |
42.2 |
| 19 |
46.8 |
46.8 |
47.0 |
46.7 |
46.7 |
| 20 |
31.2 |
31.2 |
31.3 |
31.4 |
31.4 |
| 21 |
34.4 |
34.4 |
34.5 |
34.6 |
33.2 |
| 22 |
33.6 |
33.4 |
33.7 |
33.2 |
34.6 |
| 23 |
65.2 |
64.4 |
17.6 |
17.3 |
210.2 |
| 24 |
14.2 |
15.3 |
28.8 |
28.7 |
11.4 |
| 25 |
16.8 |
18.0 |
16.2 |
16.0 |
16.2 |
| 26 |
18.1 |
18.1 |
18.1 |
18.0 |
17.9 |
| 27 |
26.5 |
26.4 |
26.6 |
26.5 |
26.5 |
| 28 |
177.1 |
177.1 |
177.3 |
176.8 |
176.8 |
| 29 |
33.6 |
33.6 |
33.7 |
33.7 |
33.7 |
| 30 |
24.4 |
24.4 |
24.4 |
24.2 |
24.2 |
The orientation of H-2 was difficult to determine owing to the obscured nature of its 1H signal. However, bayogenin-3-O-β-D-glucopyranoside (2b, Fig. S2†),30 the alkaline hydrolysis product of compound 2 (see Experimental section), exhibited well-resolved 1H-NMR spectrum (Table S1 and Fig. S13†) and H-2 signal (δ 4.24, J = 11.6, 3.9 Hz) showed relatively large coupling constant with the neighbouring H-1ax. It was therefore concluded that H-2 was in the β-orientation. From the existing NMR spectra (Fig. S5 and S6†) and comparison with those of glycosides with bayogenin as the aglycone,31,32 it could therefore be concluded that the aglycone moiety was 2β,3β,23-trihydroxyolean-12-en-28-oic acid (bayogenin) (2a).
For the sugar moiety of compound 2, the 1H NMR data were closely resembled those of compound 1 (Table 2) and the 13C NMR data of the sugar moiety of 2 (Table 4) were almost identical to those of compound 1 (Table 4). Acid hydrolysis of 2 also produced the same sugars, D-glucose, L-rhamnose and D-xylose, together with the aglycone which was identified as bayogenin (2a, Fig. S2†).31,32 HMBC correlations of the proton of one sugar unit to the carbon of the next sugar unit were in the same manner to those occurred in the sugar unit of compound 1. The sugar linkages of compounds 1 and 2 were therefore identical. Alkaline hydrolysis of 2 (see Experimental section) gave bayogenin-3-O-β-D-glucopyranoside (2b, Fig. S2†),30 the structure of which was confirmed by NMR data (Tables S1 and S2†). In addition, the 1H–1H COSY correlation of the sugar units (Fig. 2 and S51–S53†) and the NOESY correlations of the sugar and the aglycone units (Fig. 2 and S64–S66†) confirmed the structure of 2. Compound 2 was therefore identified as bayogenin-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→6)-[β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside and was named trichocucumeriside B.
Table 4 13C NMR data for sugar moieties of compounds 1–5 in pyridine-d5 at 100 MHz (δ in ppm)
| Position |
1 |
2 |
3 |
4 |
5 |
| |
Glc′ |
Glc′ |
Glc′ |
Glc′ |
Glc′ |
| 1′ |
106.3 |
106.3 |
107.4 |
105.5 |
103.4 |
| 2′ |
76.4 |
76.0 |
76.4 |
72.0 |
71.7 |
| 3′ |
79.3 |
79.1 |
79.3 |
78.9 |
78.8 |
| 4′ |
72.1 |
71.8 |
72.3 |
75.8 |
75.5 |
| 5′ |
78.6 |
78.9 |
78.9 |
78.5 |
78.8 |
| 6′ |
63.2 |
62.8 |
63.5 |
63.3 |
63.1 |
| Position |
1 |
2 |
3 |
4 |
5 |
| Glc′′ |
Glc′′ |
Glc′′ |
Xyl′′ |
Xyl′′ |
| 1′′ |
95.1 |
95.1 |
95.2 |
93.8 |
93.9 |
| 2′′ |
76.7 |
76.7 |
76.9 |
70.0 |
76.6 |
| 3′′ |
79.9 |
79.9 |
79.9 |
84.9 |
83.3 |
| 4′′ |
71.4 |
76.7 |
71.2 |
69.5 |
69.5 |
| 5′′ |
78.1 |
78.1 |
78.1 |
63.3 |
63.4 |
| 6′′ |
69.4 |
69.5 |
69.5 |
|
|
| Position |
1 |
2 |
3 |
4 |
5 |
| Rha′′′ |
Rha′′′ |
Rha′′′ |
Glc′′′ |
Glc′′′ |
| 1′′′ |
101.9 |
101.9 |
101.9 |
107.7 |
106.8 |
| 2′′′ |
72.3 |
72.3 |
72.1 |
75.3 |
75.4 |
| 3′′′ |
73.1 |
73.1 |
73.1 |
70.0 |
75.0 |
| 4′′′ |
86.0 |
86.0 |
85.9 |
75.5 |
75.4 |
| 5′′′ |
68.8 |
68.8 |
68.8 |
75.5 |
75.6 |
| 6′′′ |
19.2 |
19.2 |
19.2 |
61.7 |
62.6 |
| Position |
1 |
2 |
3 |
4 |
5 |
| Xyl′′′′ |
Xyl′′′′ |
Xyl′′′′ |
Rha′′′′ |
Rha′′′′ |
| 1′′′′ |
108.2 |
108.2 |
108.2 |
101.4 |
101.5 |
| 2′′′′ |
79.2 |
79.3 |
87.8 |
73.2 |
73.2 |
| 3′′′′ |
76.7 |
76.7 |
75.8 |
66.4 |
66.7 |
| 4′′′′ |
71.6 |
71.4 |
71.4 |
84.3 |
84.4 |
| 5′′′′ |
68.0 |
68.0 |
67.9 |
69.0 |
69.0 |
| 6′′′′ |
|
|
|
18.9 |
18.9 |
| Position |
1 |
2 |
3 |
4 |
5 |
| Xyl′′′′′ |
Xyl′′′′′ |
Xyl′′′′′ |
Xyl′′′′′ |
Xyl′′′′′ |
| 1′′′′′ |
106.1 |
106.1 |
106.1 |
106.9 |
107.6 |
| 2′′′′′ |
75.3 |
75.3 |
75.4 |
77.4 |
76.6 |
| 3′′′′′ |
78.9 |
78.6 |
78.6 |
72.3 |
72.3 |
| 4′′′′′ |
71.4 |
71.6 |
71.6 |
87.5 |
87.6 |
| 5′′′′′ |
67.6 |
67.6 |
67.6 |
67.4 |
67.4 |
| Position |
1 |
2 |
3 |
4 |
5 |
| Xyl′′′′′′ |
Xyl′′′′′′ |
Xyl′′′′′′ |
| 1′′′′′′ |
|
|
106.4 |
106.5 |
106.5 |
| 2′′′′′′ |
|
|
76.6 |
75.6 |
75.8 |
| 3′′′′′′ |
|
|
79.7 |
78.7 |
77.6 |
| 4′′′′′′ |
|
|
78.6 |
71.4 |
71.4 |
| 5′′′′′′ |
|
|
67.5 |
67.8 |
67.8 |
Compound 3 was isolated as a white powder with [α]26D −39.6° (c 0.20 in MeOH). The ESI-TOF-MS gave a sodiated molecular ion at m/z 1345.6331 [M + Na]+ compatible with the molecular formula C63H102O29. The 1H and 13C NMR data (Tables 1–4 and Fig. S68–S90†) revealed that the aglycone moiety of 3 was similar to that of 1. The significant difference was the absence of an oxymethylene group at C-23 and this was replaced by a methyl group at δH 0.99 and δC 17.6. The aglycone moiety of 3 was suggested to be 3β-hydroxyolean-12-en-28-oic acid (oleanolic acid, 3a) from its NMR spectra (Fig. S7 and S8†) and by comparison with the NMR data of the glycosides of oleanolic acid.33 Acid hydrolysis of 3 (see Experimental section) gave D-glucose, L-rhamnose and D-xylose, together with the aglycone oleanolic acid (3a). The NMR data (Tables 2 and 4) of 3 suggested that the sugar chain at C-28 consisted of three β-D-xylose, one β-D-glucose and one α-L-rhamnose and the sugar unit at C-3 was determined as one β-D-glucose in the molecule. The assignments of the sugar moieties at both positions were further confirmed by alkaline hydrolysis (see Experimental section) which yielded oleanolic acid-3-O-β-D-glucopyranoside (3b, Fig. S2†),34 the NMR spectral data of which are presented in Fig. S15, S16, Tables S1 and S2.† The anomeric protons appeared at δH 4.94 (1H, d, J = 7.8 Hz, H-1′), 6.16 (1H, d, J = 8.0 Hz, H-1′′), 6.47 (1H, brs, H-1′′′), 5.08 (1H, d, J = 7.0 Hz, H-1′′′′), 4.90 (1H, d, J = 7.4 Hz, H-1′′′′′) and 5.23 (1H, d, J = 7.5 Hz, H-1′′′′′′). These six anomeric proton signals showed correlations with six anomeric carbon signals at δC 107.4 (C-1′), 95.2 (C-1′′), 101.9 (C-1′′′), 108.2 (C-1′′′′), 106.1 (C-1′′′′′) and 106.4 (C-1′′′′′′), respectively, in the HMQC spectrum (Fig. S78–S81†). These observations suggested that 3 contained six sugar units, with one more sugar unit than that of compound 1. Comparison of the molecular formula of 3 with that of 1 also suggested that the extra sugar moiety was compatible with a xylose unit deduced from the NMR data. The HMBC experiments (Fig. 2) showed correlations between δH 4.94 (Glc-1′) and δC 89.6 (C-3), δH 6.16 (Glc-1′′) and δC 177.3 (C-28), δH 6.47 (Rha-1′′′) and δC 76.9 (Glc-2′′), δH 5.08 (Xyl-1′′′′) and δC 85.9 (Rha-4′′′), and between δH 4.90 (Xyl-1′′′′′) and δC 69.5 (Glc-6′′). The sequence of connection of the sugar units was therefore the same as those of compounds 1 and 2. The additional sixth sugar moiety, the xylose unit, was determined to reside at the lower xylose unit from the HMBC correlation between δH 5.23 (Xyl-1′′′′′′) and δC 87.8 (Xyl-2′′′′). These data have been used to establish the positions of the sugar units in the chain. In addition, 1H–1H COSY correlations of the sugar protons were observed (Fig. 2). Furthermore, key NOESY correlations of the sugar moieties (Fig. 2) were also observed. These correlations were key interactions to support the gross structure of 3. Compound 3 was therefore identified as oleanolic acid-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→6)-[β-D-xylopyranosyl-(1→2)-β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside and was named trichocucumeriside C.
Compound 4 was isolated as a white powder with [α]26D −31.1° (c 0.38 in MeOH). The ESI-TOF-MS gave a sodiated molecular ion at m/z 1345.6340 [M + Na]+ which was compatible with the molecular formula C63H102O29. The 1H and 13C NMR data (Tables 1–4 and Fig. S92–S115†) showed that the aglycone of 4 was almost identical to 3. Acid hydrolysis of 4 (see Experimental section) yielded D-xylose, D-glucose, L-rhamnose and the triterpenoid oleanolic acid (3a).33 Alkaline hydrolysis of 4 (see Experimental section) gave oleanolic acid-3-O-β-D-glucopyranoside (3b, Fig. S2†)34 which was identical to that obtained from alkaline hydrolysis of compound 3. The sugar unit at C-3 was therefore determined as one β-D-glucose and the sugar chain was located at C-28. Six anomeric proton signals at δ 4.93 (1H, d, J = 7.6 Hz, H-1′), 6.54 (1H, d, J = 7.7 Hz, H-1′′), 5.24 (1H, d, J = 7.6 Hz, H-1′′′), 5.78 (1H, brs, H-1′′′′), 5.19 (1H, overlapping signals, H-1′′′′′) and 5.28 (1H, d, J = 7.6 Hz, H-1′′′′′′) showed correlations with six anomeric carbon signals at δ 105.5 (C-1′), 93.8 (C-1′′), 107.7 (C-1′′′), 101.4 (C-1′′′′), 106.9 (C-1′′′′′) and 106.5 (C-1′′′′′′), respectively, in the HMQC spectrum (Fig. S102–S105†). In the HMBC spectrum (Fig. 2 and S106–S112†), the correlation between δH 4.93 (Glc-1′) and δC 89.4 (C-3), which was the mono sugar unit of β-D-glucose, was observed. The correlations between δH 6.54 (Xyl-1′′) and δC 176.8 (C-28), δH 5.24 (Glc-1′′′) and δC 84.9 (Xyl-3′′), δH 5.78 (Rha-1′′′′) and δC 75.3 (Glc-2′′′), δH 5.19 (Xyl-1′′′′′) and δC 84.3 (Rha-4′′′′), δH 5.28 (Xyl-1′′′′′′) and δC 87.5 (Xly-4′′′′′) characterized that the sequence of the sugar chain was β-D-Xyl-(1→4)-β-D-Xyl-(1→4)-α-L-Rha-(1→2)-β-D-Glc-(1→3)-β-D-Xyl and that the glycosidic site was at C-28. In addition, 1H–1H COSY correlations of the sugar protons were observed (Fig. 2) and the NOESY correlations (Fig. 2) were also key interactions to support this gross structure. Compound 3 was therefore assigned as oleanolic acid-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→4)-β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)-β-D-glucopyranosyl-(1→3)-β-D-xylopyranoside and was named trichocucumeriside D.
Compound 5 was isolated as a white powder, [α]26D −11.6° (c 0.30 in MeOH), with the molecular formula of C63H100O30 obtained by the ESI-TOF-MS from the [M + Na]+ ion at m/z 1359.6145 (calcd for C63H100NaO30, 1359.6192). The 13C and 1H-NMR spectra (Tables 1–4) of the aglycone moiety of 5 was similar to that of 4. The major difference was that the chemical shifts of 23-CH3 at δH 1.12 and δC 17.3 of 4 were replaced by the CHO group at δH 9.95 (1H, s, H-23) and δC 210.2 in the 1H and 13C-NMR spectrum, respectively, indicated an aldehydic group in the aglycone moiety of 5 at C-23. The aglycone moiety of 5 was therefore suggested to be gypsogenin (4a, Fig. S2†)35 by comparison of its spectroscopic data with those reported in the literature. Acid hydrolysis of 5 yielded D-xylose, D-glucose, L-rhamnose and the triterpenoid 4a35 (Fig. S2†). Alkaline hydrolysis gave the glycoside gypsogenin-3-O-β-D-glucopyranoside (4b)36 (Fig. S2, Tables S1 and S2†). The 1H–1H COSY (Fig. 2 and S126–S129†), HMBC (Fig. 2 and S134–S141†) and HMQC data (Fig. S130–S133†) confirmed the same sugar moiety of β-D-Glc at C-3. Analysis of the HMBC correlations (Fig. 2 and S134–S141†) supported by the COSY (Fig. 2 and S126–S129†) and NOESY (Fig. 2 and S142–S144†) correlations revealed the sugar linkage at C-28 as β-D-Xyl-(1→4)-β-D-Xyl-(1→4)-α-L-Rha-(1→2)-β-D-Glc-(1→3)-β-D-Xyl, which is the same as that of compound 4. All of the above data led to the formulation of 5 as gypsogenin-3-O-β-D-glucopyranosyl-28-O-β-D-xylopyranosyl-(1→4)-β-D-xylopyranosyl-(1→4)-α-L-rhamnopyranosyl-(1→2)-β-D-glucopyranosyl-(1→3)-β-D-xylopyranoside and was named trichocucumeriside E.
The anti-inflammatory activity of all the isolated compounds was evaluated against nitric oxide (NO) production in LPS-stimulated RAW264.7 cells using the Griess assay37 (Table 5). Compared to the positive references, Celecoxib and aminoguanidine (IC50 75.7 and 75.0 μM, respectively), cucurbitacin B (7) and isocucurbitacin B (9) showed more potent inhibitory activity against NO production in LPS-stimulated RAW264.7 cells with IC50 values of 3.0 and 2.7 μM, respectively. More importantly, both natural compounds showed approximately 3–4-fold higher selectivity index (SI) than that of positive references. Compounds 4, 5 and 10–12 exhibited moderate inhibitory activity towards NO production with IC50 range from 11.3 to 29.2 μM, while compounds 1–3 and 13–16 exhibited weaker activity with the IC50 values of 71.7–153.0 μM.
Table 5 Anti-inflammatory activity of compounds 1–16
| Compound |
(IC50, μM) |
SIc |
| NOa |
MTTb |
| Nitric oxide (NO) production in LPS-stimulated RAW264.7 cells. Cell viability in LPS-stimulated RAW264.7 cells. Selectivity index. Positive references. Mean ± SD, n = 3. |
| 1 |
97.7 ± 12.4 |
>160 |
>1.6 |
| 2 |
99.0 ± 15.6 |
>160 |
>1.6 |
| 3 |
115.7 ± 16.9 |
>160 |
>1.4 |
| 4 |
29.2 ± 5.3 |
49.0 ± 6.4 |
1.7 |
| 5 |
22.5 ± 3.8 |
52.0 ± 3.8 |
2.3 |
| 7 |
3.0 ± 1.1 |
19.4 ± 1.6 |
6.5 |
| 9 |
2.7 ± 0.9 |
17.2 ± 4.5 |
6.4 |
| 10 |
13.3 ± 2.3 |
41.7 ± 9.4 |
3.1 |
| 11 |
21.7 ± 8.1 |
67.0 ± 16.4 |
3.1 |
| 12 |
11.3 ± 3.3 |
19.0 ± 2.3 |
1.7 |
| 13 |
71.7 ± 3.0 |
>160 |
>2.2 |
| 14 |
148.3 ± 6.4 |
141.7 ± 9.3 |
1.0 |
| 15 |
153.0 ± 6.5 |
>160 |
>1.1 |
| 16 |
110.0 ± 9.0 |
>160 |
>1.5 |
| Celecoxibd |
75.7 ± 12.8 |
102.7 ± 8.8 |
1.4 |
| Aminoguanidined |
75.0 ± 15.5 |
>160 |
>2.1 |
Conclusions
In summary, five new triterpenoid saponin glycosides (1–5) have been isolated from T. cucumerina fruit fibers, together with eleven known compounds (6–16) in this work. The anti-inflammatory assays showed that the natural compounds 7 and 9 exhibited more potent NO inhibitory activities than those of positive references Celecoxib and aminoguanidine. In addition, these two compounds possessed approximately 3–4-fold higher selectivity index (SI) than the positive references.
Experimental
General
Melting points were determined with an Electrothermal melting point apparatus and are uncorrected. Optical rotations were acquired using a JASCO P-1020 digital polarimeter. IR spectra were recorded on a PerkinElmer FT-IR 400 spectrophotometer. Spectra of solid sample were recorded in the ATR mode. NMR spectra were recorded on a Bruker AVANCE 400 FT-NMR spectrometer, operating at 400 (1H) and 100 (13C) MHz. For the spectra taken in CDCl3 (δH 7.24 and δC 77.0) and pyridine-d5 (δH 8.74 and δC 150.3), the residual nondeuterated solvent were used as references for 1H and 13C NMR spectra. The electrospray ionization mass spectra (ESIMS) were measured with a Finnigan LC-Q mass spectrometer. High resolution electrospray ionization time-of-flight mass spectra (ESI-TOF-MS) were measured with a Bruker micrOTOF-II mass spectrometer. Column chromatography was carried out using Merck silica gel 60 (<0.063 mm) and reversed phase RP C-18 (40–63 μm, E. Merck). For TLC, Merck precoated silica gel 60 F254 plates and RP-TLC, RP-18 F254 precoated on aluminium plate (E. Merck) were used. Spots on TLC were detected under UV light and by spraying with anisaldehyde–H2SO4 reagent followed by heating. The standard sugars, D(+)-glucose, L(+)-rhamnose and D(+)-xylose, were purchased from Sigma, St. Louis, USA.
Plant material
The dried fruit fibers of T. cucumerina used in this study were collected from Takli district, Nakhon Sawan province, Thailand in 2009 and were identified by Assoc. Prof. Nopporn Damrongsiri, Department of Biology, Faculty of Science, Ramkhamhaeng University. A voucher specimen (Apichart Suksamrarn, No. 058) was deposited at the Faculty of Science, Ramkhamhaeng University, Bangkok, Thailand.
Extraction and isolation
The dried fruit fibers of T. cucumerina (18.0 kg), whose fruit peels and seeds were separated, were chilled in liquid N2 and crushed into small pieces and extracted successively with n-hexane (3 × 50 L), EtOAc (3 × 50 L) and MeOH (3 × 50 L). The extracts were evaporated to dryness under reduced pressure at temperature 40–45 °C to afford the hexane (168.0 g), EtOAc (350.0 g) and MeOH (830.0 g) extracts, respectively. The hexane extract (25.0 g) was subjected to column chromatography (CC) (Merck silica gel 60, 0.063–0.200 mm, 300 g) using gradient elution of n-hexane:EtOAc (100:0 to 0:100, v/v) and EtOAc:MeOH (100:0 to 0:100, v/v) to give 3 fractions (H1 to H3). H1 (8.5 g) was repeatedly CC over silica gel eluted with n-hexane:EtOAc (100:10, v/v) to afford 4 subfractions (H1-1 to H1-4), then H1-3 (1.2 g) was purified by CC eluted with CH2Cl2:MeOH (100:0.4, v/v) to give compound 6 (148.9 mg). H2 (10.8 g) was applied to silica gel using isocratic elution of n-hexane:EtOAc (50:50, v/v) to obtain 4 subfractions (H2-1 to H2-4). H2-3 was identified to be compound 7 (6.5 g). Subfraction H2-1 (0.3 g) was subjected to silica gel CC eluted with n-hexane:EtOAc (50:50, v/v) to yield compound 8 (20.0 mg). H2-2 (3.5 g) was purified by CC eluted with n-hexane:EtOAc (50:50, v/v) to afford compound 9 (2.8 g). The EtOAc extract (140.0 g) was fractionated by CC on silica gel (400 g), eluting with n-hexane:EtOAc (100:0 to 0:100, v/v) and EtOAc:MeOH (100:0 to 0:100, v/v) to give 4 fractions (E1 to E4). E3 (21.4 g) was applied to silica gel using isocratic elution of CH2Cl2:MeOH (98:2, v/v) to yield 4 subfractions (E3-1 to E3-4). E3-2 (1.8 g) was repeated CC with EtOAc:n-hexane (50:35, v/v) to give compound 10 (137.7 mg). E3-3 (3.2 g) was subjected to silica gel CC eluted with EtOAc:n-hexane (50:35, v/v) to yield 3 subfractions (E3-3-1 to E3-3-3). E3-3-3 (0.8 g) was repeatedly CC over silica gel eluting with CH2Cl2:MeOH (98:2, v/v) to afford compounds 11 (30.4 mg) and 12 (6.4 mg). E4 (67.0 g) was subjected to silica gel CC eluted with CH2Cl2:MeOH (95:5, v/v) to yield 4 subfractions (E4-1 to E4-4). E4-2 (5.4 g) was chromatographed on silica column using gradient elution of CH2Cl2:MeOH (98:2 to 95:5, v/v) to give 4 subfractions (E4-2-1 to E4-2-4). E4-2-3 (1.5 g) yielded compound 13 (49.0 mg) by recrystallization with MeOH. The filtrate upon evaporation gave a residue (849.0 mg) which was CC on silica gel eluting under gradient condition of CH2Cl2:MeOH (98:2 to 96:4, v/v) to give compound 14 (217.8 mg). E4-3 (6.9 g) was subjected to silica gel CC eluted with EtOAc:MeOH (97:3, v/v) to yield 4 subfractions (E4-3-1 to E4-3-4). E4-3-3 (3.5 g) was purified by CC eluted with CH2Cl2:MeOH (95:5, v/v) to afford 2 subfractions (E4-3-3-1 and E4-3-3-2). E4-3-3-2 was identified to be compound 15 (34.9 mg). E4-3-3-1 (0.4 g) was purified by CC eluted with CH2Cl2:MeOH (98:2, v/v) to give compound 16 (35.4 mg). The MeOH part (60.0 g) was fractionated by CC on silica gel (300 g), and it was eluted with CH2Cl2:MeOH (100:0 to 0:100, v/v) to give 6 fractions (M1 to M6). M5 (22.9 g) was subjected to reversed phase RP-18 CC eluted with MeOH:H2O (50:25, v/v) to afford 6 subfractions (M5-1 to M5-6). M5-2 (3.2 g) was repeatedly chromatographed on Sephadex LH-20 eluting with 100% MeOH, followed by reversed phase RP-18 CC using MeOH:H2O (50:25, v/v) to afford compound 2 (105.0 mg). M5-3 (1.6 g) was purified by reversed phase RP-18 CC eluted with MeOH:H2O (50:25, v/v) to give compound 1 (46.4 mg). Similarly, M5-4 (5.6 g) was subjected to reversed phase RP-18 gel CC to afford 5 subfractions (M5-4-1 to M5-4-5). M5-4-5 (2.3 g) was separated by reversed phase RP-18 CC eluted with MeOH:H2O (50:20, v/v) to give 3 subfractions (M5-4-5-1 to M5-4-5-3). Then, M5-4-5-3 (0.5 g) was purified by reversed phase RP-18 CC using H2O:ACN (50:25, v/v) to afford compound 4 (26.4 mg). M5-4-3 (2.5 g) was subjected to reversed phase RP-18 CC eluted with MeOH:H2O (50:20, v/v) to obtain 3 subfractions (M5-4-3-1 to M5-4-3-3). Then, M5-4-3-2 (0.4 g) was repeatedly chromatographed on RP-18 column using H2O:ACN (50:25, v/v) to afford compound 5 (42.8 mg) and M5-4-3-3 (0.9 g) was CC using RP-18 with MeOH:H2O (50:25, v/v) as eluting solvent to yield compound 3 (158.8 mg).
Compound 1. White powder; [α]26D −17.2° (c 0.39 in MeOH); Mp 219–222 °C; IR (ATR) νmax: 3337, 2932, 1734, 1639, 1455, 1387, 1255, 1073, 894, 815 cm−1; 1H-NMR (pyridine-d5, 400 MHz) data and 13C-NMR (pyridine-d5, 100 MHz) data: see Tables 1–4; ESI-TOF-MS m/z 1229.5924 [M + Na]+ (calcd for C58H94O26Na, 1229.5926).
Compound 2. White powder; [α]26D −22.7° (c 0.24 in MeOH); Mp 215–217 °C; IR (ATR) νmax: 3442, 2928, 1727, 1643, 1455, 1374, 1258, 1073, 894, 837 cm−1; 1H-NMR (pyridine-d5, 400 MHz) data and 13C-NMR (pyridine-d5, 100 MHz) data: see Tables 1–4; ESI-TOF-MS m/z 1245.5869 [M + Na]+ (calcd for C58H94O27Na, 1245.5875).
Compound 3. White powder; [α]26D −39.6° (c 0.20 in MeOH); Mp 204–206 °C; IR (ATR) νmax: 3332, 2933, 1751, 1639, 1455, 1361, 1258, 1030, 894, 820, 809 cm−1; 1H-NMR (pyridine-d5, 400 MHz) data and 13C-NMR (pyridine-d5, 100 MHz) data: see Tables 1–4; ESI-TOF-MS m/z 1345.6331 [M + Na]+ (calcd for C63H102O29Na, 1345.6399).
Compound 4. White powder; [α]26D −31.1° (c 0.38 in MeOH); Mp 213–215 °C; IR (ATR) νmax: 3338, 2934, 1734, 1639, 1458, 1390, 1258, 1034, 892, 818, 779 cm−1; 1H-NMR (pyridine-d5, 400 MHz) data and 13C-NMR (pyridine-d5, 100 MHz) data: see Tables 1–4; ESI-TOF-MS m/z 1345.6340 [M + Na]+ (calcd for C63H102O29Na, 1345.6399).
Compound 5. White powder; [α]26D −11.6° (c 0.30 in MeOH); Mp 180–182 °C; IR (ATR) νmax: 3344, 2933, 1719, 1640, 1455, 1387, 1261, 1042, 894, 818, 782 cm−1; 1H-NMR (pyridine-d5, 400 MHz) data and 13C-NMR (pyridine-d5, 100 MHz) data: see Tables 1–4; ESI-TOF-MS m/z 1359.6145 [M + Na]+ (calcd for C63H100O30Na, 1359.6192).
Acid hydrolysis
The new compounds 1–5 (5.0 mg each) were individually dissolved in MeOH (0.2 mL) and TFA (2 N, 1 mL each) was added. The mixture was heated at 95 °C for 24 h and the progress of the reaction was monitored by TLC. The residues were evaporated to dryness by repeatedly adding MeOH to remove the acid. The residue was analyzed by TLC over silica gel together with authentic sugar standards using CHCl3:MeOH:H2O (3:1.5:0.2) and the sugars were then separated by silica gel using the same eluting solvent system as the mobile phase.38,39 The monosaccharides of compounds 1–5 were identified as D-glucose, L-rhamnose and D-xylose by TLC and NMR comparisons with those of standard sugars. It was detected at 589 nm on a polarimeter that the optical rotation of 1.02% (w/v) of D-glucose ([α]26D +18.7°),40–43 1.12% (w/v) of L-rhamnose ([α]26D +5.0°)44 and 0.35% (w/v) of D-xylose ([α]26D +11.6°)45 consisting with a homologous standard distilled water solution. Moreover, by comparison of the spectroscopic data (Fig. S3–S10†) with those reported in literature, the aglycones obtained from acid hydrolysis were identified as hederagenin25 (1a, Fig. S2†) from compound 1, bayogenin31,32 (2a, Fig. S2†) from compound 2, oleanolic acid33 (3a, Fig. S2†) from compounds 3 and 4, and gypsogenin35 (4a, Fig. S2†) from compound 5.
Alkaline hydrolysis
The new compounds 1–5 (5 mg each) were dissolved in MeOH (0.2 mL each), then saturated solution of NaOH (1 mL each) were added. The mixture was heated at 60 °C for 24 h, acidified with 1 N HCl and the mixture was then evaporated to dryness in vacuo. The triterpenoid glycosides were separated by silica column chromatography using CHCl3:MeOH:H2O (3:1.5:0.2) as the mobile phase.46 Comparison of these compounds with the corresponding aglycones, the glycosides hederagenin-3-O-β-D-glucopyranoside29 (1b, Fig. S2, Tables S1 and S2†), bayogenin-3-O-β-D-glucopyranoside30 (2b, Fig. S2, Tables S1 and S2†), oleanolic acid-3-O-β-D-glucopyranoside34 (3b, Fig. S2, Tables S1 and S2†) and gypsogenin-3-O-β-D-glucopyranoside36 (4b, Fig. S2, Tables S1 and S2†), which was derived from compounds 1–5, respectively.
Anti-inflammatory assay
Cell culture. RAW264.7 murine macrophages (ATCC, Rockville, MD, USA) were cultured in RPMI-1640 medium (Corning, NY, USA) supplemented with 10% fetal bovine serum, and 1% penicillin/streptomycin (Gibco, MA, USA). Cells were incubated at 37 °C with 5% CO2 in a humidified incubator.
Cell viability test. RAW264.7 cells were grown in a 96-well plate at a density of 1.0 × 105 cells per well for 24 h. Cells were treated with the compounds at various concentrations ranged from 5–160 μM in combination with 10 ng mL−1 of LPS. After 24 h, cells were then incubated with 0.5 mg mL−1 of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution for another 3 h. After removal of MTT solution, the formazan crystal products were dissolved in DMSO (200 μL per well). The absorbance was measured using a microplate reader at a wavelength of 560 nm. Three independent experiments were performed in triplicates. IC50 of cell viability by MTT was calculated from a dose–response curve using GraphPad Prism 6 (GraphPad Software Inc., CA, USA).
Nitric oxide assay. RAW264.7 cells of 1.0 × 105 cells per well were seeded into 96-well plates and grown for 24 h. Cells were pre-treated with various concentrations of compounds (5–160 μM) followed by 10 ng mL−1 LPS stimulation for 24 h. Then, culture supernatants were collected and Griess reagent (1% sulfanilamide and 0.1% naphthylethylene in 2.5% phosphoric acid solution) was added in a 96-well plate according to the described method.37 The absorbance was measured at 540 nm using a microplate reader. Nitrite levels were determined using a NaNO2 standard curve with r2 > 0.999. Data were obtained from three independent experiments in triplicates. IC50 by nitric oxide assay was determined from a dose–response curve using GraphPad Prism 6 (GraphPad Software Inc., CA, USA). The selectivity index (SI) was calculated as the ratio of IC50 (MTT) to IC50 (NO).
Conflicts of interest
Authors declare no conflict of interest.
Acknowledgements
This work was supported by The Thailand Research Fund (TRF, No. DBG 6180030, to AS), National Natural Science Foundation of China (No. 21761142001, to ZJY), Walailak University (No. WU61201, to WC) and the Royal Golden Jubilee (RGJ) Ph.D. Program of TRF (No. PHD 57K0084, to PS). Supports from the Center of Excellence for Innovation in Chemistry, Ministry of Higher Education, Science, Research and Innovation and Ramkhamhaeng University are gratefully acknowledged.
Notes and references
- B. E. E. Duyfjes and K. Pruesapan, Thai Forest Bulletin (Botany), 2004, 32, 76–109 Search PubMed.
- S. Kongtun, W. Jiratchariyakul, T. Kummalue, P. Tan-ariya, S. Kunnachak and A. W. Frahm, Planta Med., 2009, 75, 839–842 CrossRef CAS PubMed.
- R. M. Kolte, V. V. Bisan, C. R. Jangde and A. A. Bhalerao, Indian J. Indig. Med., 1997, 8, 117–121 Search PubMed.
- M. Arawwawala, I. Thabrew and L. Arambewela, Int. J. Biol. Chem. Sci., 2009, 3, 287–296 Search PubMed.
- H. Kirana and B. Srinivasan, Indian J. Pharmacol., 2008, 40, 103–106 CrossRef CAS PubMed.
- A. Kar, B. K. Choudhury and N. G. Bandyopadhyay, J. Ethnopharmacol., 2003, 84, 105–108 CrossRef PubMed.
- S. S. Kumar, B. R. Kumar and G. K. Mohan, J. Ethnopharmacol., 2009, 123, 347–350 CrossRef PubMed.
- L. D. Arawwawala, M. I. Thabrew and L. S. Arambewela, J. Ethnopharmacol., 2010, 127, 750–754 CrossRef CAS PubMed.
- N. D. Kage, V. B. Malashetty, Y. N. Seetharam, P. Suresh and S. B. Patil, Int. J. Morphol., 2009, 27, 173–182 Search PubMed.
- A. A. Rahuman and P. Venkatesan, Parasitol. Res., 2008, 103, 133–139 CrossRef PubMed.
- S. Duangmano, P. Sae-lim, A. Suksamrarn, F. E. Domann and P. Patmasiriwat, BMC Complementary Altern. Med., 2012, 12(185), 1–12 Search PubMed.
- K. Dittharot, S. Dakeng, P. Suebsakwong, A. Suksamrarn, P. Patmasiriwat and M. Promkan, Planta Med., 2019, 85, 370–378 CrossRef CAS PubMed.
- P. Suebsakwong, J. Wang, P. Khetkam, N. Weerapreeyakul, J. Wu, Y. Du, Z.-J. Yao, J.-X. Li and A. Suksamrarn, ACS Med. Chem. Lett., 2019, 10, 1400–1406 CrossRef CAS PubMed.
- H. Kojima, N. Sato, A. Hatano and H. Ogura, Phytochemistry, 1990, 29, 2351–2355 CrossRef CAS.
- H. Jacobs and T. Singh, J. Nat. Prod., 1990, 53, 1600–1605 CrossRef CAS.
- S. Y. Ryu, S. H. Lee, S. U. Choi, C. O. Lee, Z. No and J. W. Ahn, Arch. Pharmacal Res., 1994, 17, 348–353 CrossRef CAS.
- Y. Yamada, K. Hagiwara, K. Iguchi and Y. Takahashi, Chem. Lett., 1978, 319–322 CrossRef CAS.
- V. V. Velde and D. Lavie, Tetrahedron, 1983, 39, 317–321 CrossRef.
- C. Seger, S. Sturm, M. Mair, E. Ellmerer and H. Stuppner, Magn. Reson. Chem., 2005, 43, 489–491 CrossRef CAS PubMed.
- M. Arisawa, J. M. Pezzuto, A. D. Kinghorn, G. A. Cordell and N. R. Farnsworth, J. Pharmaceut. Sci., 1984, 73, 411–413 CrossRef CAS PubMed.
- S. M. Kupchan, H. Meshulam and A. T. Sneden, Phytochemistry, 1978, 17, 767–769 CrossRef CAS.
- C. Seger, S. Sturm, E. Haslinger and H. Stuppner, Monatsh. Chem., 2005, 136, 1645–1649 CrossRef CAS.
- H. Abd El-Fattah, Phytochemistry, 1994, 36, 159–161 CrossRef CAS.
- N. Kawahara, A. Kurata, T. Hakamatsuka, S. Sekita and M. Satake, Chem. Pharm. Bull., 2004, 52, 1018–1020 CrossRef CAS PubMed.
- X. C. Li, C. R. Yang, Y. Q. Liu, R. Kasai, K. Ohtani, K. Yamasaki, K. Miyahara and K. Shingu, Phytochemistry, 1995, 39, 1175–1179 CrossRef CAS.
- Y. Shao, B.-N. Zhou, L.-Z. Lin and G. A. Cordell, Phytochemistry, 1995, 38, 927–933 CrossRef CAS PubMed.
- E. Bedir, N. J. Toyang, I. A. Khan, L. A. Walker and A. M. Clark, J. Nat. Prod., 2001, 64, 95–97 CrossRef CAS PubMed.
- B. Yeskaliyeva, M. A. Mesaik, A. Abbaskhan, A. Kulsoom, G. S. Burasheva, Z. A. Abilov, M. I. Choudhary and Atta-ur-Rahman, Phytochemistry, 2006, 67, 2392–2397 CrossRef CAS PubMed.
- J. M. Augustin, S. Drok, T. Shinoda, K. Sanmiya, J. K. Nielsen, B. Khakimov, C. E. Olsen, E. H. Hansen, V. Kuzina, C. T. Ekstrom, T. Hauser and S. Bak, Plant Physiol., 2012, 160, 1881–1895 CrossRef CAS PubMed.
- Y. Shao, B. N. Zhou, K. Ma and H. M. Wu, Planta Med., 1995, 61, 246–249 CrossRef CAS PubMed.
- R. Kasai, M. Miyakoshi, R. L. Nie, J. Zhou, K. Matsumoto, T. Morita, M. Nishi, K. Miyahara and O. Tanaka, Phytochemistry, 1988, 27, 1439–1446 CrossRef CAS.
- T. Schopke, V. Wray, A. Kunath and K. Hiller, Phytochemistry, 1992, 31, 2555–2557 CrossRef CAS PubMed.
- H. Zhang, A. K. Samadi, K. V. Rao, M. S. Cohen and B. N. Timmermann, J. Nat. Prod., 2011, 74, 477–482 CrossRef CAS PubMed.
- M. Miyakoshi, K. Shirasuna, Y. Hirai, K. Shingu, S. Isoda, J. Shoji, Y. Ida and T. Shimizu, J. Nat. Prod., 1999, 62, 445–448 CrossRef CAS PubMed.
- T. Kuljanabhagavad, P. Thongphasuk, W. Chamulitrat and M. Wink, Phytochemistry, 2008, 69, 1919–1926 CrossRef CAS PubMed.
- B. Yeskaliyeva, M. A. Mesaik, A. Abbaskhan, A. Kulsoom, G. S. Burasheva, Z. A. Abilov, M. I. Choudhary and Atta-ur-Rahman, Phytochemistry, 2006, 67, 2392–2397 CrossRef CAS PubMed.
- J. Sun, X. Zhang, M. Broderick and H. Fein, Sensors, 2003, 3, 276–284 CrossRef CAS.
- A. Rezgui, A. C. Mitaine-Offer, T. Miyamoto, C. Tanaka, S. Delemasure, P. Dutartre and M. A. Lacaille-Dubois, Phytochemistry, 2016, 123, 40–47 CrossRef CAS PubMed.
- Y. Ding, X. R. Tian, H. F. Tang, J. T. Feng, W. Zhang, W. L. Hai, X. Y. Wang and Y. Wang, Phytochem. Lett., 2012, 5, 668–672 CrossRef CAS.
- A. Bagno, F. Rastrelli and G. Saielli, J. Org. Chem., 2007, 72, 7373–7381 CrossRef CAS PubMed.
- A. Penzkofer, J. Anal. Sci., 2013, 3, 234–239 CAS.
- S. Y. Wu, Y. H. Fu, Q. Zhou, M. Bai, G.-Y. Chen, S. Y. Zhao, C. R. Han and X. P. Song, Molecules, 2018, 23, 1–9 Search PubMed.
- H. S. Isbell, Bur. Stand. J. Res., 1929, 3, 1041–1052 CrossRef CAS.
- J. D. Britto, V. S. Manickam, S. Gopalakrishnan, T. Ushioda and N. Tanaka, Chem. Pharm. Bull., 1995, 43, 338–339 CrossRef.
- P. A. Levene and R. S. Tipson, J. Biol. Chem., 1936, 115, 731–747 CAS.
- S. Kirmizigul, H. Anil and M. E. Rose, Phytochemistry, 1995, 39, 1171–1174 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01176b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a