
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-GBq production of the radiotracer [18F]fallypride in a droplet microreactor†
Jia Wang‡
 abd,
Philip H. Chao‡abd,
Roger Slavikc and
R. Michael van Dam*abd
abd,
Philip H. Chao‡abd,
Roger Slavikc and
R. Michael van Dam*abd
aDepartment of Bioengineering, Henry Samueli School of Engineering, UCLA, Los Angeles, CA 90095, USA. E-mail: mvandam@mednet.ucla.edu
bCrump Institute for Molecular Imaging, David Geffen School of Medicine, UCLA, Los Angeles, CA 90095, USA
cAhmanson Translational Imaging Division, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA
dDepartment of Molecular and Medical Pharmacology, David Geffen School of Medicine, UCLA, Los Angeles, CA 90095, USA
First published on 24th February 2020
Abstract
Microfluidics offers numerous advantages for the synthesis of short-lived radiolabeled imaging tracers: performing 18F-radiosyntheses in microliter-scale droplets has exhibited high efficiency, speed, and molar activity as well as low reagent consumption. However, most reports have been at the preclinical scale. In this study we integrate a [18F]fluoride concentrator and a microdroplet synthesizer to explore the possibility of synthesizing patient doses and multi-patient batches of clinically-acceptable tracers. In the integrated system, [18F]fluoride (up to 41 GBq [1.1 Ci]) in [18O]H2O (1 mL) was first concentrated ∼80-fold and then efficiently transferred to the 8 μL reaction chip as a series of small (∼0.5 μL) droplets. Each droplet rapidly dried at the reaction site of the pre-heated chip, resulting in localized accumulation of large amounts of radioactivity in the form of dried [18F]TBAF complex. The PET tracer [18F]fallypride was synthesized from this concentrated activity in an overall synthesis time of ∼50 min (including radioisotope concentration and transfer, droplet radiosynthesis, purification, and formulation), in amounts up to 7.2 GBq [0.19 Ci], sufficient for multiple clinical PET scans. The resulting batches of [18F]fallypride passed all QC tests needed to ensure safety for clinical injection. This integrated technology enabled for the first time the impact of a wide range of activity levels on droplet radiosynthesis to be studied. Furthermore, this substantial increase in scale expands the applications of droplet radiosynthesis to the production of clinically-relevant amounts of radiopharmaceuticals, and potentially even centralized production of clinical tracers in radiopharmacies. The overall system could be applied to fundamental studies of droplet-based radiochemical reactions, or to the production of radiopharmaceuticals labeled with a variety of isotopes used for imaging and/or targeted radiotherapeutics.
1 Introduction
For more than a decade, there has been interest in microreactors for the preparation of radiolabeled probes for positron emission tomography (PET). By performing radiochemical reactions on the microliter scale, the substantial cost of tracer production can be greatly reduced through lower consumption of expensive reagents (e.g. precursor) and a small system footprint that can reduce the amount of shielding needed or can enable many synthesizers to be installed in a single hot cell. Furthermore, yields can be increased due to the higher concentration of radionuclide, rapid mixing of reagents, fast heat transfer and short evaporation times.1Several prototypes of microreactors leveraging those advantages have been reported for preclinical tracer production2–6 but only a few systems were shown to produce doses of both sufficient quantity and quality for clinical use. For example, Lebedev et al. reported a batch-type microfluidic reactor (volume 50 μL) and showed rapid reaction times and high yields for several 18F-labeled tracers.7,8 A concentrator subsystem was incorporated to increase the amount of activity that could be loaded into the 50 μL reactor, and production of [18F]fallypride for clinical imaging was demonstrated. Volume reduction not only reduced the amount of expensive reagents used but also reduced times needed for heating, evaporations, etc. Another microfluidic system (NanoTek, Advion, Inc.) is based on reagents flowing through a pre-heated capillary. Zheng et al. and Liang et al. demonstrated the successful syntheses of [18F]FMISO and [18F]T807, respectively, for clinical use.9,10 This “flow-through” reactor design allows scaling of reaction volumes to adjust the batch size (e.g. changing the volume of radionuclide solution scales the amount of activity), enabling production of small batches for optimization or large batches for clinical doses. However, the large scale reactions (required for producing clinical doses) use similar volumes as conventional systems, generally minimizing reagent savings.9
Recently, several droplet-based platforms have been developed for the synthesis of PET tracers, based on electro-wetting on dielectric (EWOD),2,11–13 passive droplet transport,14 magnetic bead actuation,15 or surface-tension traps.16 These technologies enable reaction volumes to be scaled down to the microliter range (and reagent masses to the 10s of microgram level). The small reaction volume is also advantageous for increasing molar activity, enabling high molar activity even from relatively small batches, something that is not possible with conventional synthesizers,17 and enabling high molar activity in isotopic exchange reactions.18 Furthermore, by removing bulky reagent delivery systems, system size is drastically reduced, enabling safe operation with only small amounts of lead shielding13 or potentially enabling multiple systems to be operated within a single hot-cell.
Radiochemistry performed in these droplet platforms was initially limited to 100 s of MBq [10 s of mCi] due to the small reaction volume compared to the volume of the radioisotope solution (typically ∼1 mL), but several approaches were later described that could modestly increase the production scale. In one approach, a 200 μL droplet of [18F]fluoride solution was loaded at the side of an EWOD chip and heated until its volume shrunk to ∼5 μL, after which it was transferred by EWOD electrodes into the covered portion of the chip and dried at the reaction site.2 Additional aliquots of the radioisotope could, in principle, be loaded and dried to further increase the activity scale, but processing 1 mL would require 1 h (5 × 12 min), leading to significant radioactive decay. In another method, demonstrated on multiple droplet platforms,13,14 a series of small (0.5–5 μL) droplets of [18F]fluoride solution were transported to the reaction site and dried. Though the small droplets can be dried very rapidly, it would take multiple hours to process 1 mL of radioisotope solution. A different strategy was used in the magnetic droplet platform.15 Functionalized magnetic beads were incubated with an initial droplet of radioisotope solution (unspecific volume) to trap the [18F]fluoride on the beads, and the droplet was magnetically transported to a capillary where the liquid was removed via vacuum. The authors suggest that the process can be repeated multiple times and can process 1 mL of liquid in only 5 min; however, the trapping capacity of the beads was limited and could only efficiently sequester ∼100 MBq [∼3 mCi] of activity. Furthermore, the high water content needed to release the trapped [18F]fluoride (50 μL of K2CO3 solution) will not be compatible with many 18F-radiosyntheses. Recently, our lab developed a fully-automated standalone [18F]fluoride concentrator based on a miniaturized strong anion exchange (SAX) cartridge that can trap [18F]fluoride from 1 mL starting volume and efficiently release it into an output volume of 12.4 μL in 10 min.19
Leveraging the latter approach due to its speed and versatility, we study here, for the first time, the performance of droplet-based radiosynthesis starting with 10 s of GBq [100 s of mCi] activity and explore the potential of compact droplet-based radiochemistry platforms to be used in clinically-relevant applications. To accomplish this, we developed a method of rapidly transferring the concentrated radionuclide to the passive-transport-based droplet synthesizer with near quantitative efficiency, and furthermore optimized the synthesis conditions based on the elution conditions needed to efficiently concentrate the [18F]fluoride. In this work, we describe the design and development of the integrated system, characterize the radionuclide concentration process, optimize the radionuclide transfer into the microreactor, and demonstrate the successful droplet-based synthesis of [18F]fallypride using concentrated [18F]fluoride. The synthesis is demonstrated with starting activities up to 41 GBq [1.1 Ci] and we explore the synthesis performance as a function of activity level. Finally, we demonstrate the successful production of clinical-grade [18F]fallypride that passes all quality control (QC) tests in quantities up to 7.2 GBq [0.19 Ci], sufficient for multiple patient scans. The results demonstrate that droplet radiochemistry is not limited only to production of small, preclinical quantities of radiotracers, but can potentially find application in production of clinical tracers on demand at imaging sites or within radiopharmacy distribution networks. Studies are ongoing to assess the performance of droplet synthesis of other radiopharmaceuticals at increased activity levels.
2 Methods
2.1 Materials
Materials can be found in ESI Section 1.†2.2 Apparatus
An integrated system (Fig. 1) was developed comprising an automated radionuclide concentrator (Fig. 2C) coupled to an automated micro-droplet synthesis platform (Fig. 2A).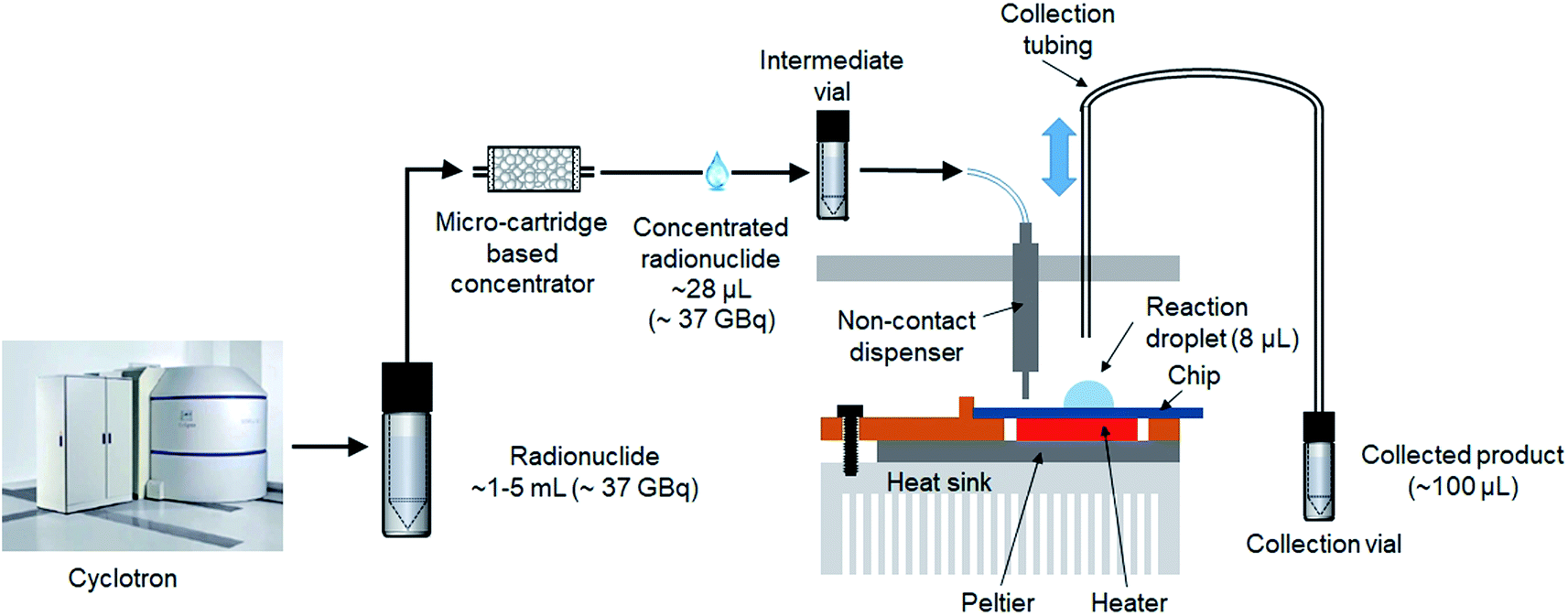 | ||
| Fig. 1 Tracer production scheme using the integrated radionuclide concentrator and microfluidic radiosynthesizer. Radionuclide (e.g. [18F]fluoride) is produced in a cyclotron (1–5 mL) and is concentrated down to 25 μL and then transferred to the droplet-based microfluidic system to perform the radiosynthesis. | ||
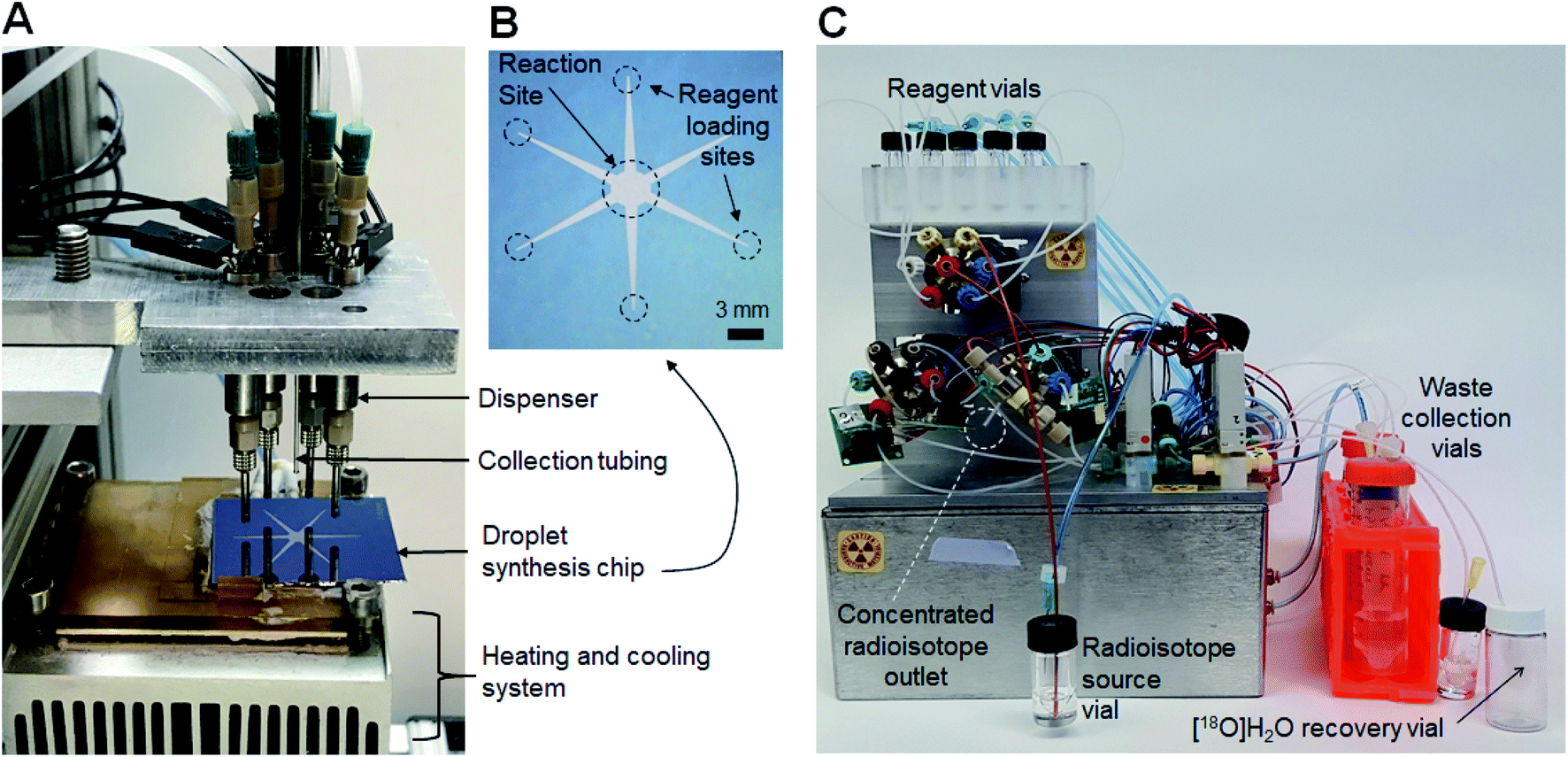 | ||
| Fig. 2 (A) Photograph of the microfluidic radiosynthesis platform. (B) Photograph of the microfluidic chip. (C) Photograph of the radionuclide concentrator. | ||
A detailed report of the design and operation of the [18F]fluoride concentrator, operated here in the “direct loading” configuration, was recently published.19 Briefly, the system relies on a miniature SAX cartridge. The [18F]fluoride in [18O]H2O from the cyclotron is passed through this cartridge to trap the [18F]fluoride, while the [18O]H2O is collected in a waste vial. The trapped [18F]fluoride is then released into a small volume of eluent solution.
Upstream of the concentrator, we incorporated a simple module for strong cation exchange (SCX) cartridge filtration of the initial [18F]fluoride solution to eliminate contaminants and small particles that could potentially interfere with trapping on the micro-cartridge (ESI Section 11†).
The design and operation of the microfluidic chips and microdroplet reaction system were previously published.14 Each microfluidic chip (25.0 × 27.5 mm2) comprises a hydrophobic Teflon-coated silicon surface with a circular hydrophilic (silicon) reaction zone in the center (3 mm diameter), and six tapered hydrophilic pathways for reagent transport from reagent loading sites to reaction zone (Fig. 2B). The chip is affixed atop a heater for temperature control. Reagents are delivered by non-contact liquid dispensers to the reagent loading sites. The crude product is collected from the reaction zone into an evacuated V-vial via a metal collection tubing inserted into the droplet.
The design and evaluation of the interface between the concentrator and droplet synthesis platform is described in detail below.
To prepare for each high activity run, [18F]fluoride solution is loaded in the source vial, reagents needed for elution and reaction are loaded in the corresponding V-vials in the concentrator and droplet synthesizer, and a clean V-vial is installed in the droplet synthesizer for product collection.
2.3 Micro-cartridge fabrication
The micro-cartridges for the radionuclide concentrator were fabricated by a different method than our previous report.19 Due to the high cost and limited re-usability of commercially-packed cartridges (with ∼4 μL bed volume and ∼2 mg of resin), we opted to pack the resin ourselves into short segments of tubing (ESI Section 2†). Cartridges were packed with one of several different resins: Bio-Rad AG-MP1 (200–400 mesh size; Bio Rad, Hercules, CA, USA), resin from Sep-Pak Plus QMA Light Cartridges (37–55 μm particle size; Waters Corporation, Milford, MA, USA), or resin from Oasis MAX Plus short cartridges (30 μm particle size; Waters Corporation). Different resin masses ranging from 2–7 mg were explored.Flow rate of water through cartridges was evaluated (ESI Section 3†) to compare different cartridge geometries (i.e. was the flow rate sufficiently fast to trap the radionuclide in a reasonable time), and to monitor the cartridge-to-cartridge variation.
2.4 Optimization and evaluation of concentrator performance
Prior to use, cartridges were preconditioned with 0.5 mL of K2CO3 followed by 10 mL of deionized (DI) water. Insufficient rinsing was found to adversely impact trapping efficiency (ESI Section 4†). Resin remained hydrated after the preconditioning step and was not air dried before use.The input source vial was loaded with [18F]fluoride in [18O]H2O from the cyclotron (0.011–41 GBq [0.3 mCi to 1.1 Ci]), diluted with DI water if needed to ensure the volume was in the range of 0.5–1.0 mL. For most experiments, [18F]fluoride was first pushed through an SCX filtration module prior to trapping on the SAX cartridge (described in detail in ESI Section 11†). Trapping was performed by flowing this solution at 20 psi through the pre-conditioned micro-cartridge. Water was collected in the [18O]H2O recovery vial. After initial trapping, 0.5 mL of DI water was passed through the system and cartridge to the [18O]H2O recovery vial to recover any residual [18F]fluoride.
Elution efficiency was tested as a function of eluent composition and eluent volume. The performance for different eluent concentrations (TBAHCO3) was explored in order to find the best tradeoff between the amount of TBAHCO3 needed for the elution versus the amount needed for the downstream droplet synthesis. In these experiments, a total of 6 elutions were performed (6.2 μL of eluent per elution), with elution efficiency measured after each pair of elutions (i.e., elutions 1 and 2 together, elutions 3 and 4 together, etc.).
To characterize trapping and elution efficiency, various radioactivity measurements were made with a calibrated dose calibrator (CRC-25 PET, Capintec, Inc., Ramsey, NJ). For the purposes of calculations, all radioactivity measurements were decay-corrected to a common timepoint. Measurements were made of the activity in the [18F]fluoride source vial before trapping (A0 source), activity in the source vial after trapping (Asource), activity in the [18O]H2O recovery vial after trapping (Awaste), and the collected activity after elution (Acollect). The activity on the cartridge after trapping (Acartridge) was determined indirectly (i.e. calculated as A0 source − (Awaste + Asource)) to minimize radiation exposure. This method also proved to be significantly more accurate than directly measuring the cartridge in the dose calibrator, presumably due the differing geometry of the cartridge compared to the vials. Trapping efficiency (%) was computed as Acartridge/(A0 source − Asource). Elution efficiency (%) was calculated as Acollect/Acartridge. Recovery efficiency (%), defined as the amount of activity recovered following elution relative to starting activity, was calculated as trapping efficiency x elution efficiency. Starting activity was defined as A0 source − Asource which can be approximated as A0 source since we found Asource < ∼0.1% of A0 source.
2.5 Interface between concentrator and droplet synthesizer
In our previous work, we showed that multiple 2 μL droplets of the initial [18F]fluoride solution could be sequentially loaded onto the chip, each one spontaneously moving to the reaction site.14 Though synthesis scale of [18F]fallypride was modestly increased in this manner (up to 4× more activity, i.e. 8 μL loaded), we observed a reduction in reaction efficiency as activity increased. We suspect that after drying of the larger radionuclide volumes, the residue was spread over a larger surface area of the chip, making it difficult to efficiently redissolve into the precursor solution for the subsequent reaction. Because the concentrator output volume in this paper (∼25 μL after optimization) was significantly greater than 8 μL, we suspected even larger impact on reaction efficiency.We thus compared several methods of loading and drying larger volumes of [18F]fluoride onto the reaction chip, and used Cerenkov luminescence imaging (CLI)20 to visualize the distribution of activity on the chip after drying.
In the “direct” method (Fig. 3A), the concentrator output tubing (ETFE, 0.01′′ ID, 1/16′′ OD; 1529L; IDEX) was mounted such that the outlet terminated just above the radioisotope loading site of the chip. As each ∼6 μL eluent plug reached the chip it was spontaneously transported to the reaction zone. (All eluent plugs were delivered to the same inlet of the chip.) The chip was heated (to dry the activity) after each pair of eluent plugs.
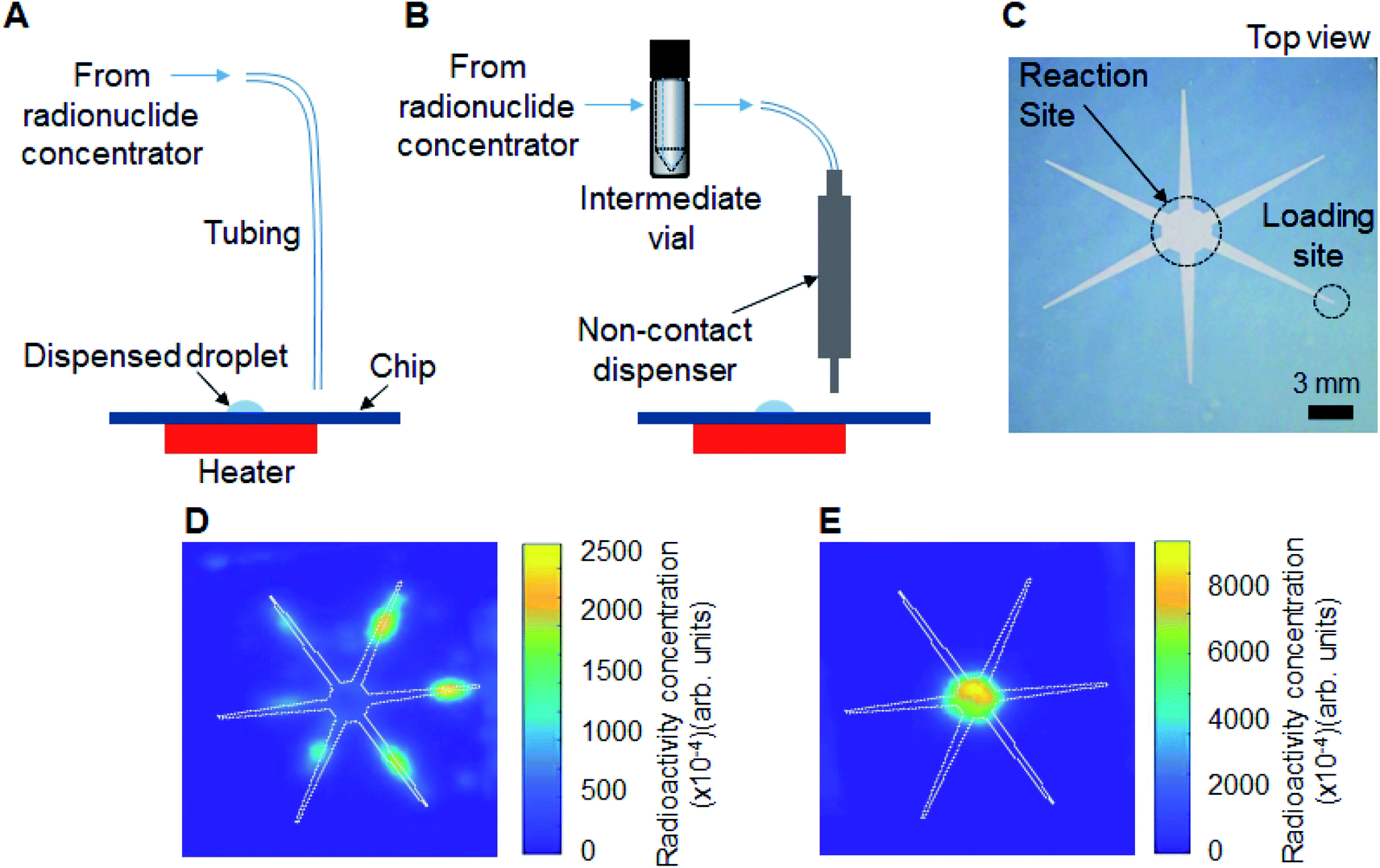 | ||
| Fig. 3 Two different designs of the interface between the radionuclide concentrator and the radioisotope inlet of the droplet-based radiochemistry chip and the resulting distribution of radioactivity on the chip after evaporation following initial [18F]fluoride loading and drying, visualized using Cerenkov imaging. (A) Successive 6.2 μL plugs of solution from the output of the concentrator are deposited directly onto the inlet of the reaction chip. (B) Concentrated activity is first transferred to an intermediate vial (to merge the liquid from individual elution steps into a single liquid plug), and then the vial contents are transferred to the inlet of the chip as a series of small droplets using a piezoelectric dispenser. (C) Top view of the reaction chip showing both the reaction site and the reagent loading site (highlighted by dotted lines). (D) Cerenkov luminescence image after loading and drying the concentrated [18F]TBAF according to the interface method of (A). (E) Cerenkov luminescence image after loading and drying the concentrated [18F]TBAF according to the interface method of (B). The white line indicates the boundary of the hydrophilic pattern on the chip. | ||
In the “dispenser” method (Fig. 3B), the eluent plugs from the concentrator were first transferred via ETFE tubing (0.02′′ ID, 1/16′′ OD; 1516L; IDEX) to an intermediate vial, and then the combined volume of concentrated [18F]fluoride solution was connected to the input of a piezoelectric reagent dispenser (INKX0514300A, Lee Company, Westbrook, CT, USA) in the microdroplet synthesizer via ETFE tubing (0.01′′ ID, 1/16′′ OD; 1529L; IDEX). The intermediate vial was then pressurized (7.5 psi) and concentrated [18F]fluoride was delivered to the radioisotope loading site as a series of smaller droplets. We also explored the development of a specialized droplet merging chip as an alternative to the intermediate vial (see ESI Section 5†). However, the intermediate vial method was ultimately used due to simplicity of operation and higher reliability. Detailed fluidic connections for each droplet merging technique to the dispenser can be seen in ESI Fig. S1.†
2.6 [18F]fallypride synthesis on chip
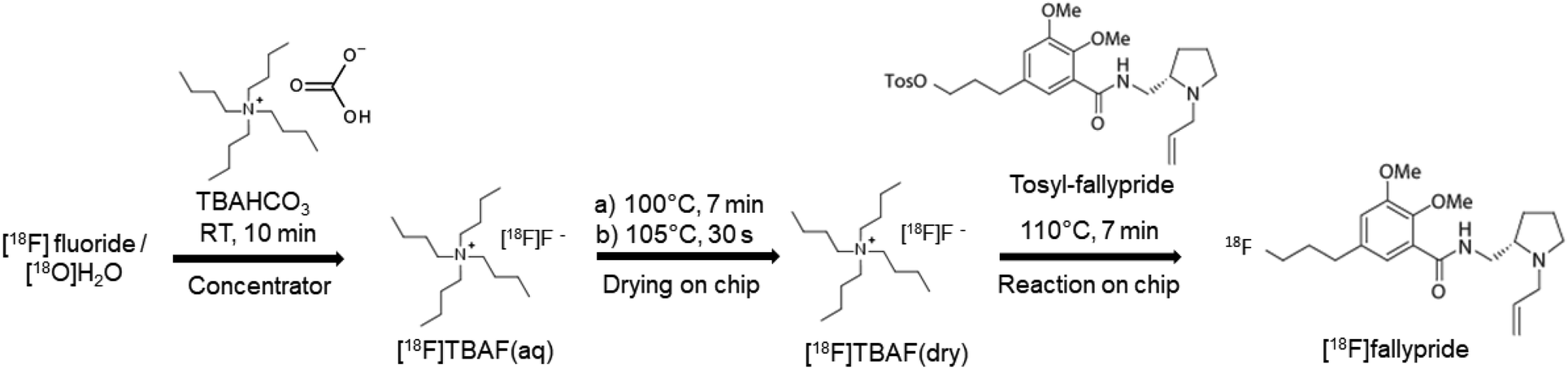 | ||
| Fig. 4 Microdroplet synthesis of [18F]fallypride. | ||
To facilitate the integration with the upstream [18F]fluoride concentrator, further optimization of the synthesis protocol was performed due to the higher salt amount (TBAHCO3) required for efficiently eluting [18F]fluoride from the micro-cartridge. Different [18F]TBAF(aq) stock solutions were prepared by mixing [18F]fluoride/[18O]H2O (11–22 MBq [0.3–0.6 mCi]) with different amounts of 75 mM TBAHCO3 solution to produce final concentrations in the range 0.51–71 mM. For each, a 12.4 μL droplet of [18F]TBAF(aq) solution was manually loaded on the reaction site of the chip and dried at 105 °C for 1 min. Then, a droplet of precursor solution (77 mM, tosyl fallypride dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of MeCN and thexyl alcohol) was loaded and moved to the reaction site automatically, and the chip was heated at 110 °C for 7 min to perform the fluorination step. Different volumes (2–8 μL) of precursor solution were tested. Afterwards, twenty 1 μL droplets of collection solution (9:1 (v/v) mixture of MeOH and DI water) were sequentially deposited at a different reagent loading site and spontaneously moved to the reaction site to dilute the resulting crude reaction mixture. After automatically lowering the collection tubing into the droplet, the diluted droplet was then transferred into the collection vial via negative pressure. The collection process was repeated 4× to maximize recovery of the crude product.
:1 (v/v) mixture of MeCN and thexyl alcohol) was loaded and moved to the reaction site automatically, and the chip was heated at 110 °C for 7 min to perform the fluorination step. Different volumes (2–8 μL) of precursor solution were tested. Afterwards, twenty 1 μL droplets of collection solution (9:1 (v/v) mixture of MeOH and DI water) were sequentially deposited at a different reagent loading site and spontaneously moved to the reaction site to dilute the resulting crude reaction mixture. After automatically lowering the collection tubing into the droplet, the diluted droplet was then transferred into the collection vial via negative pressure. The collection process was repeated 4× to maximize recovery of the crude product.
For [18F]fallypride synthesis of GBq level, we waited several hours for the activity to first decay before taking detailed measurements. To prevent radiolysis during this time, the crude [18F]fallypride was collected into a vial pre-loaded with 2 mL of EtOH.
2.7 Evaluation of synthesis performance
Performance was assessed via measurements of radioactivity, fluorination efficiency (conversion of [18F]fluoride to product), and radioactivity distribution (CLI), using previously described methods14,20 (ESI Section 6†).2.8 Purification, formulation, and quality control testing
For some batches of [18F]fallypride produced at the 15 MBq [0.41 mCi] and 1 GBq [27 mCi] scale, we also performed radio-HPLC purification of the crude product, and analysis of the pure product, both using a previously described analytical HPLC method14 (ESI Section 7†).For purification, the crude [18F]fallypride mixture collected from the chip (80 μL) was diluted with 90 μL mobile phase and manually injected into the HPLC system, and the pure [18F]fallypride fraction (∼2 mL) was collected through a selector valve (Cheminert, Valco Instrument Co. Inc.) based on the gamma detector signal. The product fraction was dried by evaporation of solvent in an oil bath at 110 °C for 8 min with nitrogen flow, and then dissolved in saline. The product dissolved in saline was sterile filtered through a syringe filter (13 mm diameter, 0.22 μm pore size, PVDF; Fisherbrand™, Waltham, MA, USA) into a sterile vial (2 mL; ALK, Denmark).
Finally, a full set of clinical quality control tests were performed on three batches of synthesized and formulated [18F]fallypride. For these experiments, starting activity ranged from 799 MBq [21.6 mCi] to 992 MBq [26.8 mCi]. Details on these quality control tests can be found in Table S5 of the ESI.†
3 Results and discussion
3.1 [18F]fluoride concentrator cartridge optimization
First, due to the change in cartridge fabrication compared to our previous work, we performed optimization of the cartridge design and [18F]fluoride concentration process.Initially, we compared flow rates (of DI water) through the different cartridge designs (resin type, resin mass, tubing inner diameter). The results are tabulated in Table 1. We consider flow rates ≥ 0.5 mL min−1 to be acceptable, which ensures trapping of [18F]fluoride can be completed in a short time. We also found that flow rates in this range gave reliable, repeatable elution compared to cartridges with slow flow rates. For the Bio-Rad AG-MP1 resin, both 2 mg and 3 mg cartridges had suitable flow rates. For the Sep-Pak QMA resin, cartridges with sufficient flow included 3 mg resin in 0.03′′ ID tubing and 5 mg resin in 0.04′′ ID tubing. Lastly, flow rates were adequate for all tested Oasis MAX cartridges (3 mg or 5 mg in 0.03′′ ID tubing and 7 mg in 0.04′′ ID tubing).
| Resin type | Bio-Rad AG-MP1 | Sep-Pak QMA | Oasis MAX | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Resin mass (mg) | 2 | 3 | 4 | 3 | 5 | 5 | 7 | 3 | 5 | 7 |
| Tubing inner diameter (inch) | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.04 | 0.04 | 0.03 | 0.03 | 0.04 |
| Flow rate (mL min−1) | 0.92 ± 0.11 (n = 3) | 0.84 ± 0.06 (n = 4) | 0.47 ± 0.04 (n = 3) | 0.68 ± 0.11 (n = 4) | 0.28 ± 0.11 (n = 2) | 0.56 ± 0.08 (n = 2) | 0.23 ± 0.07 (n = 2) | 0.70 ± 0.03 (n = 2) | 0.50 ± 0.04 (n = 2) | 0.92 ± 0.08 (n = 2) |
It should be noted that these resin masses are all equal to or higher than literature reports where efficient trapping of up to 110 GBq [3 Ci] [18F]fluoride was achieved using 2 mg of various SAX resins.4,8
3.2 Optimization of [18F]fluoride concentration process
Trapping and elution performance was first compared between the 3 resin types, using cartridges containing 3 mg of resin (Table 2). Eluent composition was arbitrarily chosen to be 25 mM TBAHCO3 as a starting point.| Resin type | |||
|---|---|---|---|
| Sep-Pak QMA | AG-MP1 | Oasis MAX | |
| Number of repeats (n) | 2 | 2 | 1 |
| Trapping efficiency (%) | 99.4 ± 0.8 | 96 ± 4 | 99 |
| Partial elution efficiency (E1 + E2) (%) | 92 ± 5 | 21 ± 3 | 65 |
| Partial elution efficiency (E3 + E4) (%) | 6 ± 4 | 68 ± 6 | 34 |
| Partial elution efficiency (E5 + E6) (%) | 0.9 ± 0.4 | 12 ± 8 | 3 |
| Cumulative elution efficiency (E1 to E4) (%) | 98 ± 1 | 89 ± 9 | 100 |
| Cumulative elution efficiency (E1 to E6) (%) | 98.9 ± 0.2 | 101 ± 1 | 103 |
Trapping of fluoride was high for all resins: 99.4 ± 0.8% (n = 2) for the Sep-Pak QMA resin, 96 ± 4% (n = 2) for the Bio-Rad AG-MP1 resin, and 99% for the Oasis MAX resin. However, differences were observed among elution efficiencies. Sep-Pak QMA cartridges released 92 ± 5% (n = 2) of the activity in the first two elutions, while the Bio-Rad AG-MP1 and Oasis MAX cartridges released only 21 ± 3% (n = 2) and 65% (n = 1), respectively. After four elution steps, all cartridges had high cumulative elution efficiencies, i.e. 98 ± 1% (n = 2) and 100% (n = 1) for the Sep-Pak QMA and Oasis MCX resins, respectively, and 89 ± 9% (n = 2) for the Bio-Rad AG-MP1 resin. Due to the high elution efficiency while using minimal eluent volume (only 2 elution steps) of the Sep-Pak QMA resin, further experiments focused on this resin.
In order to explore if eluent concentration could be decreased to reduce the amount of TBAHCO3 that enters the downstream reaction, we explored the effect of eluent concentration (Table 3) using the 3 mg Sep-Pak QMA cartridges. Consistent with the previous experiment, trapping of [18F]fluoride was nearly quantitative for all trials (≥93%). Increasing concentration of TBAHCO3 was found to increase the amount of activity eluted, especially in the first two elution steps. For 3.8 mM TBAHCO3, the efficiency was only 5 ± 1% (n = 3) in the first 2 elutions and only reached 64 ± 4 (n = 3) after 6 elution steps. In the case of 10 mM TBAHCO3, elution efficiency after 2 steps was also low (17 ± 9, n = 3), but increased to ∼89% (n = 3) after 4 elution steps. For additional repeats of 25 mM TBAHCO3, we again observed reliable and high recovery (95 ± 2%, n = 4) within the first two elution steps (12.4 μL). We hypothesized that the missing ∼5% of activity had likely been released from the cartridge but was lost as residual liquid left behind in the system. We explored eluting with two plugs of 25 mM TBAHCO3 followed by two plugs of DI water (12.4 μL; to rinse this residual activity to the concentrator outlet) and found that all of the activity (100 ± 1%, n = 3) was recovered. Because this approach resulted in a total recovery higher than 4 elutions of 10 mM eluent while only increasing total salt content marginally, we focused on this approach for further experiments.
| TBAHCO3 concentration (mM) | ||||
|---|---|---|---|---|
| 3.8 | 10 | 25 | 25a | |
| a In the final column, eluent plugs 1 and 2 were 25 mM TBAHCO3, eluent plugs 3 and 4 were DI water. No further elution steps were performed. | ||||
| Number of repeats (n) | 3 | 3 | 4 | 3 |
| Trapping efficiency (%) | 99.8 ± 0.4 | 99.8 ± 0.2 | 93 ± 5 | 99.6 ± 0.3 |
| Partial elution efficiency (E1 + E2) (%) | 5 ± 1 | 17 ± 9 | 95 ± 2 | 94 ± 3 |
| Partial elution efficiency (E3 + E4) (%) | 18 ± 1 | 72 ± 3 | 2.9 ± 0.4 | 5 ± 1 |
| Partial elution efficiency (E5 + E6) (%) | 41 ± 3 | 12 ± 7 | 0.8 ± 0.3 | N/A |
| Cumulative elution efficiency (E1 to E6) (%) | 64 ± 4 | 101 ± 1 | 99 ± 2 | 100 ± 1 |
3.3 Optimization of [18F]fallypride synthesis conditions
First, the effect of TBAHCO3 amount on fluorination efficiency of [18F]fallypride was investigated (Table 4). Across all conditions, the radioactivity recovery was relatively constant (85–93%), but the fluorination efficiency varied significantly, with a maximum value (99% conversion; 90% crude RCY) for a concentration of 10 mM. With higher concentration, we observed the formation of a radioactive side-product, perhaps due to the base-sensitivity of the precursor. With lower concentration, we did not observe the side product, but the conversion decreased.:1, v/v)). All reported efficiencies and yields are decay-corrected
| Concentration of TBAHCO3 (mM) | ||||||
|---|---|---|---|---|---|---|
| 71 | 25 | 10 | 3.6 | 1.2 | 0.51 | |
| Radioactivity recovery (%) | 88 | 86 | 91 | 93 | 85 | 86 |
| Fluorination efficiency (%) | 12 | 65 | 99 | 47 | 32 | 39 |
| Crude radiochemical yield (%) | 11 | 56 | 90 | 44 | 27 | 33 |
| Residual on chip (%) | 5 | 10 | 5 | 4 | 4 | 3 |
However, as described above, the elution efficiency of the micro-cartridge with two elutions (12.4 μL) of 10 mM TBAHCO3 was very low (17 ± 9%, n = 2). Thus, even with an optimal synthesis, the overall performance (concentrator efficiency and synthesis efficiency) would be expected to be very low (∼17% × 90% = 15%). Comparing instead the elution procedure with 12.4 μL of 25 mM TBAHCO3 followed by 12.4 μL of DI water, the concentrator efficiency was much higher (100 ± 1%, n = 3). Even with the reduced synthesis efficiency with 25 mM TBAHCO3 (65% conversion; 56% crude RCY), the overall performance would be expected to be good (∼100% × 56% = 56%).
Using the condition of 25 mM TBAHCO3 mixed with the [18F]fluoride solution (12.4 μL), we then studied the effect of the amount of precursor (Table 5). Increasing the volume of precursor solution (77 mM) was found to increase the fluorination efficiency and crude RCY. Increasing from 2 μL to 4 μL resulted in a significant improvement (from 57 ± 1%, n = 2 to 91%, n = 1) in crude RCY. Little difference was observed upon further increasing the precursor solution volume from 4 μL to 8 μL, but we elected to use the higher amount to provide a safety factor.
:1, v/v) for all reactionsa
| Precursor volume (μL) | |||
|---|---|---|---|
| 2 (n = 2) | 4 | 8 | |
| a All reported efficiencies and yields are decay-corrected. | |||
| Radioactivity recovery (%) | 88 ± 2 | 93 | 92 |
| Fluorination efficiency (%) | 65 ± 0 | 98 | 99 |
| Crude radiochemical yield (%) | 57 ± 1 | 91 | 92 |
| Residual on chip (%) | 10 ± 1 | 3 | 6 |
One factor to consider is whether the DI water rinse during the radionuclide concentration phase was important, as this resulted in only a modest increase in activity recovered from the concentrator (94 ± 3%, n = 3 to 100 ± 1%, n = 3), but took additional time for elution (∼1 min) and drying (∼1.5 min). Radioactive decay during this added time is less than the gains from the rinsing step and thus the rinsing step has an overall benefit to process efficiency.
3.4 Performance of transfer method between systems
First the “direct” loading method was tested. The output volume of two elutions (12.4 μL) from the concentrator was loaded on the microfluidic chip as two sequential 6.2 μL droplets and dried. The micro-cartridge was then rinsed with two 6.2 μL DI water plugs and this rinse volume was also loaded onto the chip and dried. In this direct loading method, however, the droplet did not remain confined to the reaction zone and spread out along all of the reagent delivery paths. Indeed, CLI imaging of the chip after drying confirmed that radioactivity was distributed across all hydrophilic areas of the chip after the [18F]fluoride drying process (Fig. 3D). This is undesirable as much of the dried [18F]TBAF complex would not be dissolved into the precursor droplet loaded for the subsequent fluorination step. The problem was likely due to the mismatch between the volume (12.4 μL) output from the concentrator and the capacity of the chip (8 μL).Next, the “dispenser” loading method was evaluated. The concentrator output was connected to the dispenser through an intermediate vial, which first collected the full volume of concentrated activity from the 2 eluent plugs and 2 DI water plugs (∼25 μL), and then delivered this volume to the reagent loading site via the dispenser as a series of ∼0.5 μL droplets while the chip was heated at 100 °C. We observed that only 48% of the eluted activity was found to be loaded on the chip – an additional 26% and 20% of the eluted radioactivity were found as residual activity in the dispenser and intermediate vial, respectively. To recover the radioactivity left in the dispenser and the intermediate vial, another 4 plugs of DI water (∼25 μL) were rinsed through the concentrator (without passing through the cartridge), into the intermediate vial, and then dispensed as a series of ∼0.5 μL droplets and dried in the same manner as described above. With this modification, a total of 96% of the eluted radioactivity was loaded onto the chip, with only 4% of eluted radioactivity found as residual activity in other parts of the system (ESI Table S4†). In stark contrast to the above method where the full volume was loaded, the CLI image taken after the droplet-by-droplet loading and drying step confirmed that all of the radioactivity was confined within the reaction zone (Fig. 3E), suggesting that it would be efficiently solvated when the precursor solution is added.
3.5 Low activity [18F]fallypride synthesis
Using the optimal [18F]fluoride concentration and transfer method, [18F]fallypride synthesis was initially performed on the integrated system with low overall starting activity to verify the functionality of the system and assess its performance (Table 6). Starting with 11–170 MBq [0.3–4.5 mCi] [18F]fluoride solution, the trapping efficiency of the micro-cartridge was consistent at 100 ± 0% (n = 6), followed by high elution of trapped activity (91 ± 7%, n = 6) from the cartridge. The concentration process followed by the “dispenser” transfer method as described above resulted in 89 ± 7% (n = 6) of overall starting activity loaded onto the chip. The fluorination efficiency was 89 ± 5% (n = 6) and the radioactivity recovery was 81 ± 9% (n = 6), resulting in a crude RCY of 72 ± 8% (n = 6). After collection of the crude product, only 7 ± 3% (n = 6) of the initially-loaded radioactivity remained stuck to the chip. The crude RCY was slightly higher than we previously reported for the droplet-based synthesis using passive transport chips with low starting activity,14 i.e. 64 ± 6% (n = 4). The previously reported fluorination efficiency and radioactivity recovery were 76 ± 4% (n = 4) and 84 ± 4% (n = 4),14 respectively, suggesting that the current synthesis protocol with increased salt concentration and optimized precursor volume resulted in substantial improvement in fluorination efficiency. The integrated system had slightly lower radioactivity recovery due to the ∼6% activity loss from the concentration step.| Radionuclide concentration steps | |
|---|---|
| Starting activity (MBq [mCi]) | 11–170 [0.3–4.5] |
| Trapping efficiency (%) | 100 ± 0 |
| Elution efficiency (%) (relative to trapped activity) | 91 ± 7 |
| Waste vial (%) | 0 ± 0 |
| Activity on cartridge after elution (%) | 6 ± 6 |
| Activity on chip after elution (%) | 89 ± 7 |
| Activity in the intermediate vial after dispensing (%) | 2 ± 1 |
| Droplet radiosynthesis steps | |
|---|---|
| Fluorination efficiency (%) | 89 ± 5 |
| Radioactivity recovery (%) | 81 ± 9 |
| Crude RCY (%) | 72 ± 8 |
| Residual activity on chip after collection (%) | 7 ± 3 |
3.6 High activity [18F]fallypride synthesis
Next, syntheses were performed starting with higher activities (ranging from 3.7 GBq to 41 GBq [0.10 Ci to 1.1 Ci]). While the synthesis was successful at all scales, we observed the crude RCY to decrease from 65% to 25% as the overall starting activity increased (Fig. 5A). To better understand the effect, we looked at the performance of different factors individually. In the radionuclide concentration module, the elution efficiency of [18F]fluoride was high and consistent (96 ± 4%, n = 11) across all experimental runs, but the overall performance was adversely impacted by the trapping efficiency, which decreased from ∼94% to ∼63% as the starting activity was increased (Fig. 5B). The decreased trapping performance of the micro-cartridge suggests the capacity of the cartridge was insufficient to trap all of the [18F]fluoride. This was surprising as several reports have indicated trapping of high amounts of [18F]fluoride (up to 110 GBq [3 Ci]) using cartridges packed with only ∼2 mg of resin.4,8 After some investigation, we discovered there may be impurities in our source of [18F]fluoride that reduce the trapping efficiency far below the capacity as measured by spiking KF solutions with [18F]fluoride (see ESI, Sections 9 and 10†). Further studies, such as investigation of cartridges with higher resin mass, could potentially improve the trapping performance at high activity levels.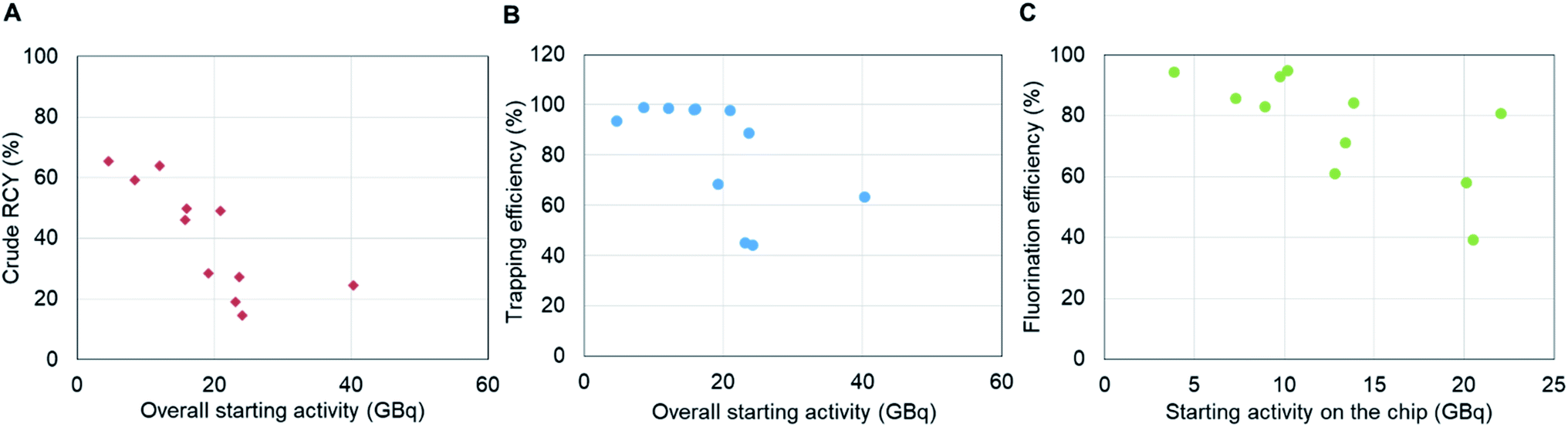 | ||
| Fig. 5 Performance of synthesis on integrated system at higher activity levels (3.7–41 GBq [0.10–1.1 Ci]). (A) Overall crude RCY (including radionuclide concentration and crude synthesis) as a function of starting activity. (B) Trapping efficiency within the cartridge as a function of starting activity. The elution efficiency remained consistent (96 ± 4%, n = 11) across all experimental runs and is not shown here. (C) Fluorination efficiency as a function of concentrated activity loaded onto the chip. | ||
Looking at the performance of the droplet synthesis process, we observed that the fluorination efficiency decreased from around 90% to 40% as the amount of starting activity on the chip increased (Fig. 5C). One potential explanation is that the reaction conditions may move out of the optimal range as the precursor to fluoride ratio decreased due to increased starting activity. Based on the molar activity of [18F]fluoride at EOB previously reported as 740 GBq μmol−1 (20 Ci μmol−1),17 the molar ratio of precursor to fluoride at low starting activity (11 to 170 MBq) ranged from 41000 to 2700 while the ratio of that at high starting activity (3.7 to 41 GBq) ranged from only 100 to 11. Potentially at the higher activity levels the reduced excess of precursor adversely impacts the yield during the short fluorination reaction. Further studies at high activity scales, e.g. using different amounts of precursor, could help to determine whether this is a factor. The decrease in fluorination efficiency when using high activities might also be due to radiolysis during the fluorination step. At the beginning of the reaction, the activity concentration in the reaction droplet is quite high, ranging from 460 to 5100 GBq mL−1 [13 to 140 Ci mL−1] (assuming 8 μL precursor solution is added) and increasing somewhat during the fluorination reaction as the reaction solvent partially evaporates. Although EtOH was preloaded in the collection vial to prevent radiolysis after the collection step (by dilution and because EtOH acts as a radical scavenger), some radiofluorinated impurities were observed in the HPLC chromatograms of the crude products when starting activity was higher than 8.0 GBq [0.22 Ci] (ESI Fig. S6†) and the number and quantity of radioactive impurities increased with increased starting activity. In contrast, the HPLC chromatogram from a batch starting with 0.20 GBq [5.4 mCi] of activity showed only two peaks, [18F]fluoride and [18F]fallypride. Further studies would be needed to confirm exactly when radiolysis is happening, which would guide potential methods to reduce it such as addition of radical scavengers at other stages of the synthesis, or attempting to change the droplet geometry (i.e. make it flatter) to reduce radiolysis.21 Another potential approach could be to divide the activity into a few smaller batches (e.g., <8 GBq [0.22 Ci], where no radiolysis was evident in chromatograms), performing several smaller-scale syntheses in parallel, and then combining the batches in the presence of a radiolysis quenching agent. At <8 GBq [0.22 Ci] activity level, the crude RCY was still ∼60%, not far from the value of 72% at low activities, potentially enabling overall conversion of 60% for much larger batch sizes.
Even though the crude RCY was only 25% with 41 GBq [1.1 Ci] starting activity, a total of 7.2 GBq [0.19 Ci] [18F]fallypride product (not decay corrected) was produced after 35 min synthesis, 5 min purification via analytical-scale HPLC, and 10 min formulation. The resulting amount of [18F]fallypride could easily supply multiple human doses (each needing ∼0.37 GBq [10 mCi] at the time of injection), even if they were scheduled throughout the day (i.e. product loss due to radioactive decay).
The overall crude synthesis took ∼35 min. This is 15 min longer than our previously reported microdroplet synthesis method,14 due to the extra time needed for concentration of [18F]fluoride (∼10 min) and transfer, loading, and drying of the [18F]fluoride onto the chip (∼6 min), compared to only ∼1 min in our previous synthesis method.14 However, the previous method was limited to using only ∼74 MBq [2 mCi] of activity (2 μL; assuming 37 GBq mL−1 [1 Ci mL−1]), and scaling up by the methods reported therein would have required sequential loading and drying of 1000 μL to load 37 GBq [1 Ci] of activity, a process that would have taken ∼170 min.
The molar activity (81–270 GBq μmol−1 [2.2–7.3 Ci μmol−1], at the end of formulation) of all experimental runs carried out on the integrated system was up to 5 times higher than previously reported molar activites for [18F]fallypride synthesis in the macroscale (15–78 GBq μmol−1 [0.4–2.1 Ci μmol−1]).22 Although 140–192 GBq μmol−1 [3.8–5.2 Ci μmol−1] was reported by Moon et al., relatively high starting activity (8.1–26 GBq [0.22–0.70 Ci]) was needed, while similar molar activity could be produced on the integrated platform using 3.7 GBq (0.1 Ci) starting activity.
3.7 Quality control tests
The resulting formulated [18F]fallypride from three separate production trials passed all QC testing (see ESI Table S5†). Thus, not only can the [18F]fallypride produced on the integrated system be produced in sufficient quantities for multiple patient doses, but these tests establish the suitability for use in patients.4 Conclusions
In this paper we successfully integrated an automated [18F]fluoride concentrator with a microdroplet-based radiosynthesizer. We presented a thorough characterization and optimization of the concentration parameters, the transfer of concentrated [18F]fluoride between the two components, and the synthesis of [18F]fallypride as a model compound. Integration of the two platforms followed by complete automation of the overall process enabled fast, safe, reliable, and high-yielding radiosynthesis of [18F]fallypride of clinical quality.With the optimized transfer method including vial rinsing and small-droplet loading approach, we were able to load 96% of activity on the chip and localize it at the reaction site. Repeatable and reliable concentration of [18F]fluoride followed by radiosynthesis of [18F]fallypride was performed with starting activities ranging from 11 MBq to 41 GBq [0.3 mCi to 1.1 Ci]. Complete concentration and synthesis could be performed in 35 min. For “low activity” syntheses starting with 11–170 MBq [0.3–4.6 mCi], fluorination efficiency and crude RCY were 89 ± 5% (n = 6) and 72 ± 8% (n = 6), respectively. As starting activity was increased (4.5–41 GBq [0.12–1.1 Ci]), the overall crude RCY dropped significantly, primarily due to a decreasing in the trapping efficiency of [18F]fluoride during the concentration process, which can likely be addressed through further cartridge optimization. We also observed some reduction in fluorination efficiency as the starting activity was increased, potentially due to mismatched stoichiometry or radiolysis effects at higher activities. Future studies will further investigate these factors and potential solutions.
This study shows that despite a small reaction volume (2–8 μL here), it is possible to load significant quantities of the radionuclide into microdroplet reactors. Starting activity was scaled up to 41 GBq [1.1 Ci], limited only by the capacity of our cyclotron facility. We also found the chips to be compatible with the high activity levels and no disruption to the droplet-based processes was observed.
This integrated platform enables production of clinical grade PET tracers in large quantities to enable imaging of several patients or imaging over several radionuclide half-lives. Production is reliable and can be completed in a short time enabling ease of use within research facilities and radio-pharmacies. We are currently exploring scaling up the synthesis of additional 18F-labeled tracers (e.g. [18F]FDOPA, [18F]FET, etc.), and molecules labeled with different isotopes. In fact, with small modifications of the concentrator module, we believe that other radioisotopes (e.g. radiometals) could be concentrated and used for production of a wide variety of radiopharmaceuticals (for imaging or targeted radionuclide therapy) at clinically-relevant scales on the integrated platform. It may also be possible to perform concentration of 18F-labeled prosthetic groups, to enable reagent-efficient, small-volume labeling of biomolecules such as peptides and proteins.23,24
Conflicts of interest
The Regents of the University of California have licensed technology to Sofie, Inc. that was invented by Dr van Dam, and have taken equity in Sofie, Inc. as part of the licensing transaction. Dr van Dam is a founder and consultant of Sofie, Inc. The remaining authors declare no conflicts of interest.Acknowledgements
The authors thank the staff of the UCLA Biomedical Cyclotron Facility for providing [18F]fluoride for these studies and for assisting with quality control testing, and Greg Khitrov of the UCLA Molecular Instrumentation Center for help with GCMS measurements. This work was supported in part by the National Cancer Institute (R21 CA212718), the National Institute on Aging (R21 AG049918), the National Institute of Mental Health (R44 MH097271), and the UCLA Foundation from a donation made by Ralph and Marjorie Crump for the UCLA Crump Institute for Molecular Imaging.References
- A. M. Elizarov, Lab Chip, 2009, 9, 1326–1333 RSC.
- S. Chen, M. R. Javed, H.-K. Kim, J. Lei, M. Lazari, G. J. Shah, M. van Dam, P. Y. Keng and C.-J. Kim, Lab Chip, 2014, 14, 902–910 RSC.
- G. Pascali, G. Mazzone, G. Saccomanni, C. Manera and P. A. Salvadori, Nucl. Med. Biol., 2010, 37, 547–555 CrossRef CAS PubMed.
- A. M. Elizarov, R. M. van Dam, Y. S. Shin, H. C. Kolb, H. C. Padgett, D. Stout, J. Shu, J. Huang, A. Daridon and J. R. Heath, J. Nucl. Med., 2010, 51, 282–287 CrossRef CAS PubMed.
- X. Zhang, F. Liu, K.-A. Knapp, M. L. Nickels, H. C. Manning and L. M. Bellan, Lab Chip, 2018, 18, 1369–1377 RSC.
- J. M. Gillies, C. Prenant, G. N. Chimon, G. J. Smethurst, W. Perrie, I. Hamblett, B. Dekker and J. Zweit, Appl. Radiat. Isot., 2006, 64, 325–332 CrossRef CAS PubMed.
- R. Bejot, A. M. Elizarov, E. Ball, J. Zhang, R. Miraghaie, H. C. Kolb and V. Gouverneur, J. Labelled Compd. Radiopharm., 2011, 54, 117–122 CrossRef CAS.
- A. Lebedev, R. Miraghaie, K. Kotta, C. E. Ball, J. Zhang, M. S. Buchsbaum, H. C. Kolb and A. Elizarov, Lab Chip, 2012, 13, 136–145 RSC.
- M.-Q. Zheng, L. Collier, F. Bois, O. J. Kelada, K. Hammond, J. Ropchan, M. R. Akula, D. J. Carlson, G. W. Kabalka and Y. Huang, Nucl. Med. Biol., 2015, 42, 578–584 CrossRef CAS PubMed.
- S. H. Liang, D. L. Yokell, M. D. Normandin, P. A. Rice, R. N. Jackson, T. M. Shoup, T. J. Brady, G. El Fakhri, T. L. Collier and N. Vasdev, Mol. Imaging, 2014, 13, 1–5 CrossRef CAS.
- M. R. Javed, S. Chen, J. Lei, J. Collins, M. Sergeev, H.-K. Kim, C.-J. Kim, R. M. van Dam and P. Y. Keng, Chem. Commun., 2014, 50, 1192–1194 RSC.
- P. Y. Keng, S. Chen, H. Ding, S. Sadeghi, G. J. Shah, A. Dooraghi, M. E. Phelps, N. Satyamurthy, A. F. Chatziioannou, C.-J. Kim and R. M. van Dam, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 690–695 CrossRef CAS PubMed.
- P. Y. Keng and R. M. van Dam, Mol. Imaging, 2015, 14, 579–594 CrossRef CAS PubMed.
- J. Wang, P. H. Chao, S. Hanet and R. M. van Dam, Lab Chip, 2017, 17, 4342–4355 RSC.
- S. A. Fiel, H. Yang, P. Schaffer, S. Weng, J. A. H. Inkster, M. C. K. Wong and P. C. H. Li, ACS Appl. Mater. Interfaces, 2015, 7, 12923–12929 CrossRef CAS PubMed.
- J. Wang, P. H. Chao and R. M. van Dam, Lab Chip, 2019, 2415–2424 RSC.
- M. Sergeev, M. Lazari, F. Morgia, J. Collins, M. R. Javed, O. Sergeeva, J. Jones, M. E. Phelps, J. T. Lee, P. Y. Keng and R. M. van Dam, Commun. Chem., 2018, 1, 10 CrossRef.
- K. Lisova, M. Sergeev, S. Evans-Axelsson, A. D. Stuparu, S. Beykan, J. Collins, J. Jones, M. Lassmann, K. Herrmann, D. Perrin, J. T. Lee, R. Slavik and R. M. van Dam, Nucl. Med. Biol., 2018, 61, 36–44 CrossRef CAS PubMed.
- P. H. Chao, M. Lazari, S. Hanet, M. K. Narayanam, J. M. Murphy and R. M. van Dam, Appl. Radiat. Isot., 2018, 141, 138–148 CrossRef CAS PubMed.
- A. A. Dooraghi, P. Y. Keng, S. Chen, M. R. Javed, C.-J. Kim, A. F. Chatziioannou and R. M. van Dam, Analyst, 2013, 138, 5654–5664 RSC.
- C. Rensch, B. Waengler, A. Yaroshenko, V. Samper, M. Baller, N. Heumesser, J. Ulin, S. Riese and G. Reischl, Appl. Radiat. Isot., 2012, 70, 1691–1697 CrossRef CAS PubMed.
- M. Lazari, J. Collins, B. Shen, M. Farhoud, D. Yeh, B. Maraglia, F. T. Chin, D. A. Nathanson, M. Moore and R. M. van Dam, J. Nucl. Med. Technol., 2014, 42, 203–210 CrossRef PubMed.
- S. Richter and F. Wuest, Molecules, 2014, 19, 20536–20556 CrossRef PubMed.
- W.-T. K. Tsai and A. M. Wu, J. Label. Compd. Radiopharm., 2018, 61, 693–714 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01212b |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |