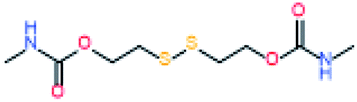DOI:
10.1039/D0RA01394C
(Review Article)
RSC Adv., 2020,
10, 13766-13782
Advances in intrinsic self-healing polyurethanes and related composites
Received
13th February 2020
, Accepted 20th March 2020
First published on 5th April 2020
Abstract
Fascinating and challenging, the development of repairable materials with long-lasting, sustainable and high-performance properties is a key-parameter to provide new advanced materials. To date, the concept of self-healing includes capsule-based healing systems, vascular healing systems, and intrinsic healing systems. Polyurethanes have emerged as a promising class of polymeric materials in this context due to their ease of synthesis and their outstanding properties. This review thereby focuses on the current research and developments in intrinsic self-healing polyurethanes and related composites. The chronological development of such advanced materials as well as the different strategies employed to confer living-like healing properties are discussed. Particular attention will be paid on chemical reactions utilized for self-healing purposes. Potential applications, challenges and future prospects in self-healing polyurethane fields are also provided.
I. Healing concepts and mechanisms
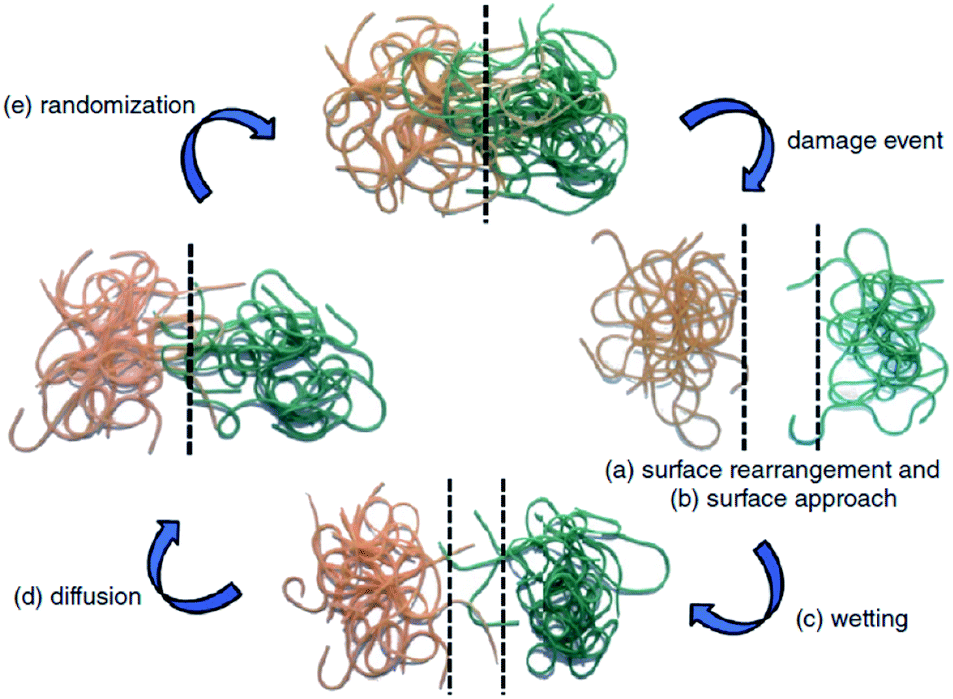
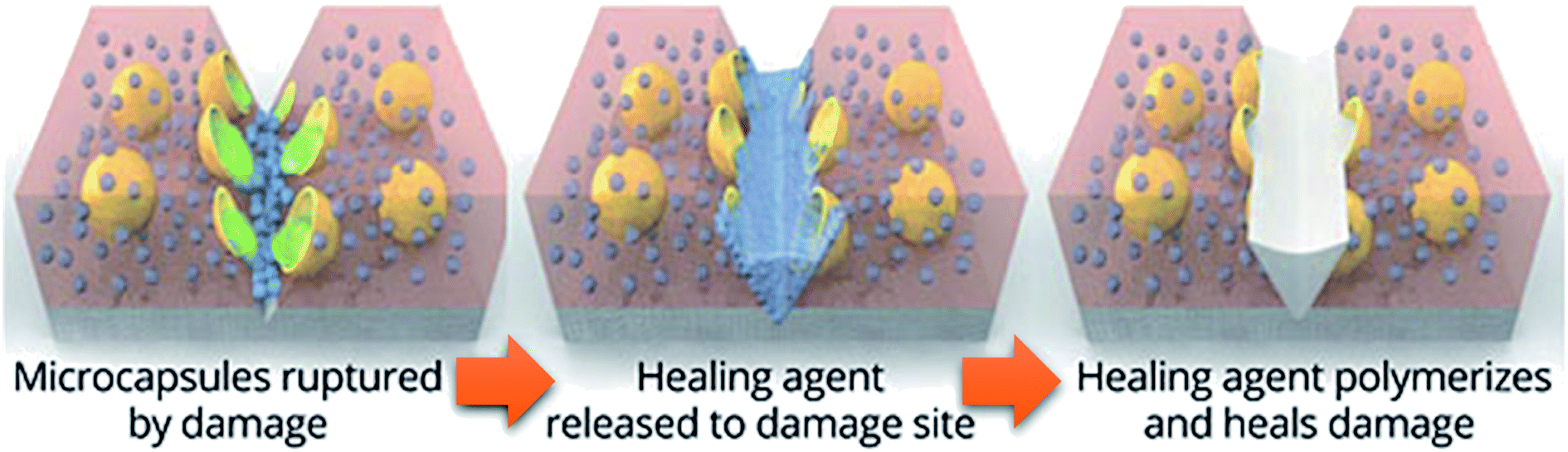
The first traditional approaches to repair polymer materials after severe damage were welding, patching and gluing.1 Despite the economic and practical aspects, these techniques are not very sustainable as they require external addition of fresh materials, and they are not applicable to heal damaged pieces of object where maintenance is dangerous and expensive (aeronautic, soft robotic, building, underwater). In the 1980s, it was noticed that a range of ionomers (i.e. polymers containing less than 15 mol% of ionic groups along the polymer backbone2) based on poly(ethylene-co-methacrylic acid) could be repaired after ballistic puncture (commercialized as Surlyn® and Nucrel® by Dupont).3 The healing mechanism of ionomers relies on concomitant elastic and plastic deformations of polymer chains after ballistic puncture and follows by a subsequent elastic recovery, local melting (heat produced by the impact) and ionic group rearrangement. Still under debate, the role of these clusters during healing is supposed to be ascribed to the increase in mechanical properties, facilitating the elastic recovery after bullet impact. Later in 1981, a self-healing mechanism based on a physical model was proposed for polymers by Wool, O'Connor et al.4 This simple and universal mechanism, applicable for nearly all self-healing concepts, is based on five stages successively occurring after a damage-to-strength recovery of ruptured polymer/polymer interfaces (Fig. 1). In this mechanism, the healing proceeds through surface rearrangement, surface approach, wetting, diffusion and randomization.5 This process is strongly related to molecular interdiffusion at (or above) the glass transition temperature (Tg) at which polymer segments are mobile enough for efficient self-repairing. In the 1990s, Dry et al. made important researches on self-repairing fiber reinforced composites and on the development of polymeric smart materials.6–9 Inspired by Dry's approach, self-healing polymers were revisited, with the pioneer works of Sottos, White et al. on microencapsulation of healing agent embedded in polymer matrix.10 In this concept, microcontainers, such as microcapsules, hollow fibers or microvascular network, containing reactive monomers (healing agent) and appropriate catalyst are dispersed into the polymer matrix. When a damage occurs, the reactive monomers, released from broken containers, fill the crack by capillary forces and subsequently crosslink, restoring extrinsically the damaged area (Fig. 2).11 During the last decade, research on self-healing polymers has grown constantly and many studies focused on the incorporation of self-healing features directly in the polymer backbone by introducing reversible bonds that can break and reform in a perpetual manner upon appropriate stimuli. This approach, called intrinsic self-healing, utilizes either dynamic covalent12–19 or non-covalent reversible3,20–24 bonds triggered by temperature, electromagnetic radiations, pH, humidity or ionic strength changes. Depending on the final targeted application of the self-healing polymers, one must wisely choose the chemistry involved in the process in terms of efficiency, reversibility conditions and feasible chemical synthesis. One key-perspective in the development of more advanced self-healing materials would be the combination of both covalent and non-covalent dynamic reactions within the same material, as reported in a few research papers.25–27 Based on the last decades of development in self-healing polymers, it is important to classify them in an unambiguous way. Beside the classification upon extrinsic or intrinsic aspects, self-healing polymers can also be classified as autonomous, if healing occurs autonomously (without external trigger), or non-autonomous if external stimulus is required to achieve healing.
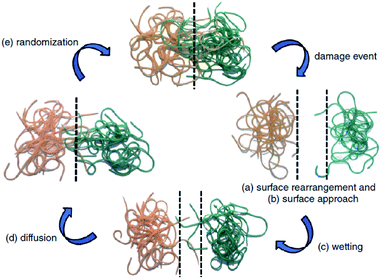 |
| | Fig. 1 Stages of self-healing mechanism for polymeric segments of random walk character: (a) surface rearrangement, (b) surface approach, (c) wetting, (d) diffusion and (e) randomization.4,5 | |
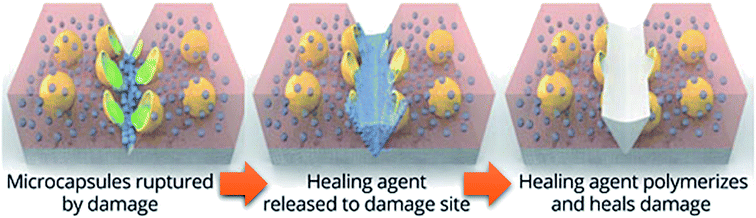 |
| | Fig. 2 Concept of self-healing based on microencapsulated healing agent (in yellow) embedded in a matrix (in red) containing a catalyst (in blue) capable of polymerizing the healing agent (in green).10,11 | |
II. Damage-repair cycles
During their utilization, polymers are subjected to several environmental aggressions, which alter their initial mechanical properties and greatly affect their reliability over time. Photo-thermal, chemical and mechanical stresses are the major external causes of material failure. When mechanical stress is considered, cracks within the material will form and propagate whether the energy released during the mechanical stress (impact or cyclic loads) is equal or larger to the energy required to generate new surfaces within the polymer.4,28 From a molecular viewpoint, mechanical damage in polymers will result in chain cleavage and/or slippage (Fig. 3).29 This results in the formation of reactive end-groups with or without the ability to proceed through conformational change. Those reactive end-groups are of, for example, free radicals and functional groups, such as COOH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO, OH, NH2, Si–O, SH, S–S or B–O. Depending on the stability of generated reactive chain ends, they will tend to reassemble at the damage site or oxidize further to form more stable products. Simultaneously, macromolecular chain segments (displaced or cleaved) will undergo conformational change or diffusion, resulting in network rearrangements. Finally, assuming that these chemico-physical processes are independent of each other or not spatially synchronized, chemical reactions favoring bonds reformation take place and the network is repaired (as it is the same for physical aspect).
C, CO, OH, NH2, Si–O, SH, S–S or B–O. Depending on the stability of generated reactive chain ends, they will tend to reassemble at the damage site or oxidize further to form more stable products. Simultaneously, macromolecular chain segments (displaced or cleaved) will undergo conformational change or diffusion, resulting in network rearrangements. Finally, assuming that these chemico-physical processes are independent of each other or not spatially synchronized, chemical reactions favoring bonds reformation take place and the network is repaired (as it is the same for physical aspect).
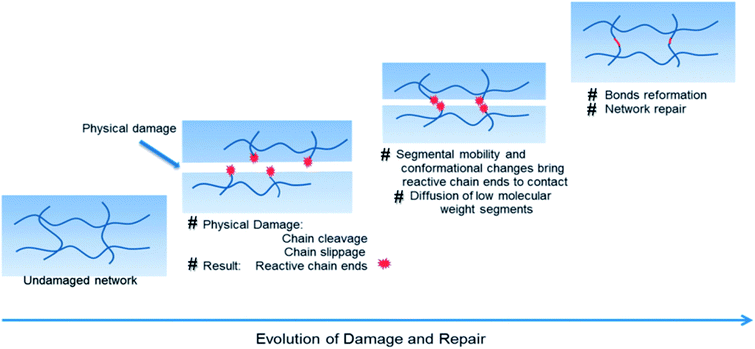 |
| | Fig. 3 Schematic representation of damage–repair cycle in polymers.29,30 | |
III. Self-healing polyurethanes
Polyurethanes (PUs) are obtained by polyaddition reactions between polyols and polyisocyanates. Herein, simply adjusting parameters such as the polyol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polyisocyanate ratio, the number of reactive end-groups or the aliphatic vs. aromatic, the crystalline vs. amorphous and the hydrophobic vs. hydrophilic composition allow readily tuning their structural, thermal and mechanical properties. PUs are thereby used in a wide range of applications such as packaging, textile, construction, automobile and electronics. Regardless the final application, PU-based materials are subjected to photo, thermal and mechanical aggressions during its utilization, which limit their performance, lifetime and reliability. Based on the economic interests that represent the polyurethane market, developing healable PUs is a key-factor for more reliable, safe, durable, sustainable, reprocessable and ecological polyurethanes. Though, an important (but not exclusive) factor to get healable polymers is the high chain mobility to fill cracks and allow reactive groups to re-associate. In polyurethane, the diisocyanate, the dihydroxy telechelic prepolymer and the chain extender dictate the final properties of the polyurethanes and their ability or not to self-heal. Other parameters such as the side-reactions during PUs synthesis and the presence of catalysts may also play an important role for the capacity of the resulting polyurethane to self-repair.
:polyisocyanate ratio, the number of reactive end-groups or the aliphatic vs. aromatic, the crystalline vs. amorphous and the hydrophobic vs. hydrophilic composition allow readily tuning their structural, thermal and mechanical properties. PUs are thereby used in a wide range of applications such as packaging, textile, construction, automobile and electronics. Regardless the final application, PU-based materials are subjected to photo, thermal and mechanical aggressions during its utilization, which limit their performance, lifetime and reliability. Based on the economic interests that represent the polyurethane market, developing healable PUs is a key-factor for more reliable, safe, durable, sustainable, reprocessable and ecological polyurethanes. Though, an important (but not exclusive) factor to get healable polymers is the high chain mobility to fill cracks and allow reactive groups to re-associate. In polyurethane, the diisocyanate, the dihydroxy telechelic prepolymer and the chain extender dictate the final properties of the polyurethanes and their ability or not to self-heal. Other parameters such as the side-reactions during PUs synthesis and the presence of catalysts may also play an important role for the capacity of the resulting polyurethane to self-repair.
(a). Role of isocyanate
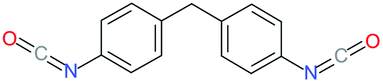
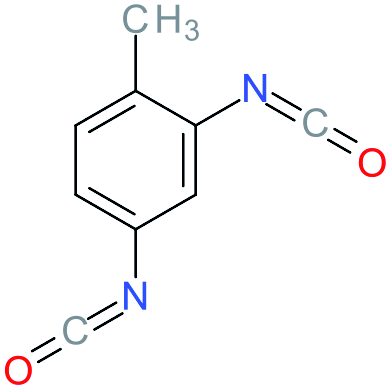
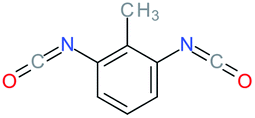
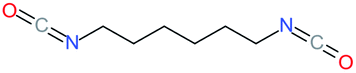
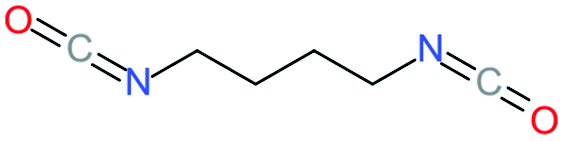
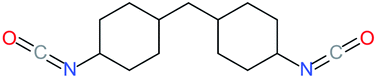
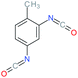
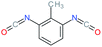
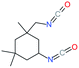
Used as coupling agent, the chemical structure of isocyanate plays an important role in the resulting thermal and mechanical properties of PUs. Introducing rigid aromatic diisocyanates will enhance rigidity and tensile strength but reduce the chain mobility compared to their flexible aliphatic counterparts. Beside this aspect, the symmetry of the isocyanate facilitates the structured organization of polymer chains. This organization is favored by hydrogen bonds between urethane groups and has a great impact on the thermo-mechanical properties of the resulting polyurethane. Finally, the reactivity of the isocyanate R–NCO is driven by the nature of R while reactivity is enhanced as R increases the electrophilicity of the isocyanate. Therefore, the reactivity decreases with the sequence: R = phenyl > benzyl > n-alkyl > cyclohexyl. With these aspects taken into consideration, one has to wisely choose the nature of diisocyanate to fulfill the desired requirement in the design of self-healing polyurethane. Table 1 shows the most commonly used commercially available diisocyanates.31
Table 1 Most commonly used commercially available diisocyanates31
| |
Name |
Chemical structure |
| MDI |
4,4′-Diphenylmethanediisocyanate |
 |
| 2,4-TDI |
Toluene-2,4-diisocyanate |
 |
| 2,6-TDI |
Toluene-2,6-diisocyanate |
 |
| 1,6-HMDI |
1,6-Hexamethylenediisocyanate |
 |
| 1,4-BDI |
1,4-Butanediisocyanate |
 |
| H12MDI |
4,4′-Dicyclohexylmethanediisocyanate |
 |
| IPDI |
Isophorone diisocyanate |
 |
(b). Role of dihydroxy telechelic prepolymer
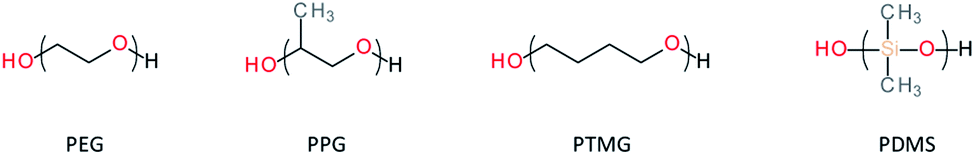
The choice of the diol-terminated prepolymer is crucial as it dictates parameters such as the crystallinity/amorphous and the hydrophobic/hydrophilic composition, the Tg as well as the molecular weight of the resulting PUs. In the context of self-healing, amorphous and flexible soft segments are preferred over crystalline and rigid ones. Low molecular weight oligomers (Mn < 5000 g mol−1) are preferred to avoid physical entanglement while facilitating chain mobility. Amorphous polyethylene glycol (PEG, Mn < 1000 g mol−1), polypropylene glycol (PPG), amorphous polytetrahydrofuran (PTHF, Mn < 1000 g mol−1) and polydimethylsiloxane (PDMS) are good candidates for making self-healing PUs (Fig. 4).
 |
| | Fig. 4 Examples of dihydroxy-terminated oligomers for the design of self-healing polyurethanes. | |
(c). Role of chain extender
Chain extender is used to confer rigidity and toughness to the resulting PUs. In conventional PUs, low molecular weight aliphatic or aromatic diols (Mn < 500 g mol−1) are used. These chain extenders give the possibility to the PU to crystallize or to introduce heterogeneity. Advantageously, the chain extenders are also used to give dynamic features to the PUs by introducing reversible chemistries within the diol structure, introducing chemical capabilities to PUs in addition to the physical aspect described hereabove.
(d). Side-reactions and presence of catalyst
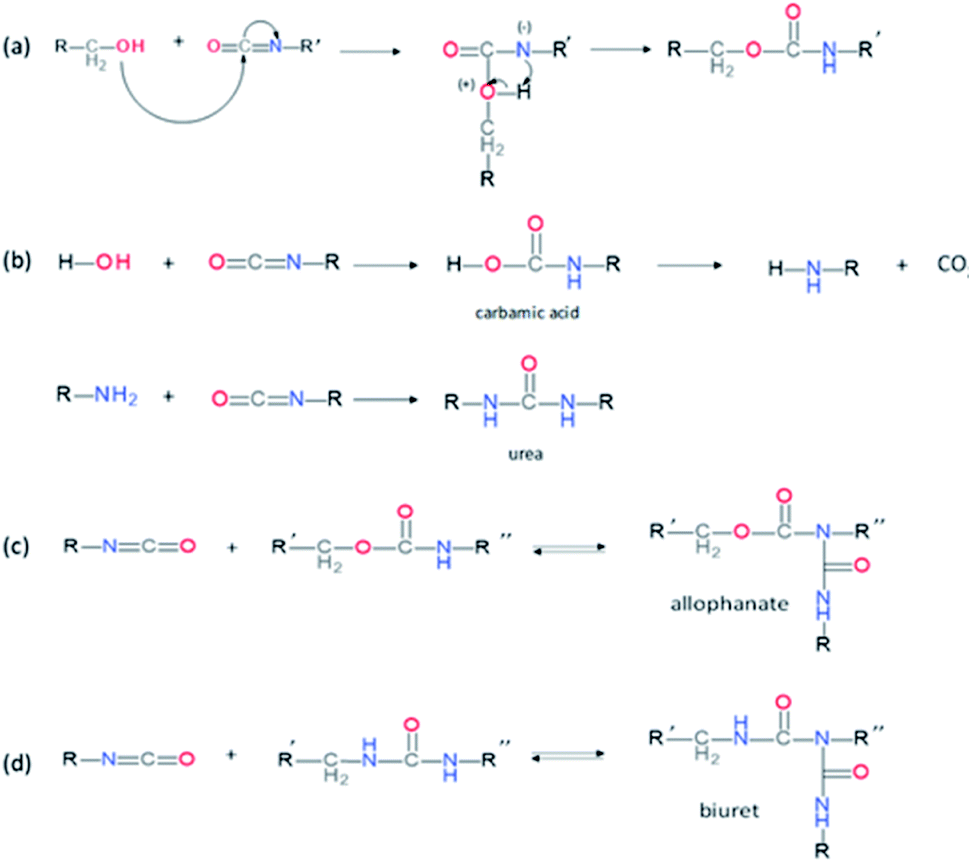
PUs synthesis must be carried out under dry and inert atmosphere. The mechanism of isocyanate formation with a primary alcohol in absence of water is presented on Fig. 5a. In the presence of water, however, side-reactions occur which lead to the formation of urea as depicted on Fig. 5b. Indeed, as shown in Table 2, labile hydrogen of water is as reactive as primary alcohol, and its presence in the reactive media can influence the chemical structure and the molecular parameters of the resulting PU. As a result, additional hydrogen/urea bonds are added within the polymer structure, modifying the targeted properties, and in some extent affecting the capacity of the material to self-heal due to restricted mobility. It is worth mentioning that side-reactions may be taken advantageous in the context of autonomous extrinsic self-healing polymers. During PU synthesis, the ratio of NCO and OH groups is usually kept equimolar. When the NCO:OH ratio is superior to one, formation of allophanate occurs by reaction of urethane and the isocyanate excess (Fig. 5c). As such, undesired crosslinking occurs within the linear polymer, therefore impacting the desired healing properties. Moreover, if NCO are exposed to water and are present in excess compared to the OH groups, the generated urea can react with excess of isocyanate, forming biuret covalent cross-linking (Fig. 5d). Besides, different catalysts used during the PU synthesis can be classified either as basic or acidic. Basic tertiary amines enhance the nucleophilicity of the diols while Lewis acid such as alkyl tin carboxylates, oxides and mercaptide oxides are supposed to raise the electrophilicity of isocyanates. Hence, the isocyanate reactivity increases with electrophilicity. Regarding self-healing abilities of PUs, catalyst can play indirectly a role as their loading will modify the molecular parameters. For instance, recent studies have demonstrated the important role of catalysts in the healing mechanism of glucose-modified PUs.32,33 Herein, Urban et al. developed different carbohydrate-based PU networks. It was demonstrated that, using the appropriate catalyst, the damaged network could be repaired in completely different fashions. Crosslinked PUs were obtained by reacting methyl-a-D-glucopyranoside (MGP) with polyethylene glycol (PEG) and hexamethylene diisocyanate trimer (HDI) and loaded with different catalysts. When loaded with dibutyltin dilaurate (DBTDL), chemical species generated upon damage react, under ambient conditions, with atmospheric water and carbon dioxide to form new urethanes and carbonate linkages.33 When loaded with zinc acetate (Zn(OAc)2), amines generated by urethane cleavage react with urethane-functionalized MGP in a urethane–urea conversion at elevated temperature (75 °C), recreating network on two separated pieces.32
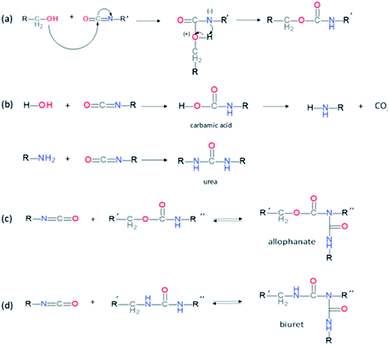 |
| | Fig. 5 Mechanism of urethane formation with isocyanate and primary alcohol (a), formation of urea by reaction of water and isocyanate (b) and formation of allophanate(c) or biuret (d) by reaction of urethane with excess of isocyanate. | |
Table 2 Reactivity of isocyanate with labile hydrogen of different groups58
| Name |
Structure |
Reactivity at 25 °C (without catalyst) |
| Aliphatic primary amine |
R–NH2 |
100000 |
| Aliphatic secondary amine |
R2–NH |
20000–25000 |
| Aromatic primary amine |
Ar–NH2 |
200–300 |
| Primary alcohol |
R–CH2–OH |
100 |
| Water |
H2O |
100 |
| Carboxylic acid |
R–C(O)OH |
40 |
| Secondary alcohol |
R2–CHOH |
30 |
| Urea |
R–NH–C(O)–NH–R′ |
15 |
| Tertiary alcohol |
R3–C(O)–H |
0.5 |
| Urethane |
R–NH–C(O)OR |
0.3 |
| Amide |
R–C(O)–NH2 |
0.1 |
IV. Self-healing polyurethanes through dynamic covalent chemistry
Dynamic covalent chemistry is commonly employed to confer healable properties to polymers.34 This chemistry can be classified based on the reactive functional groups involved in the reversible process: COOH, CC, NH2, OH, SH, Si–O, S–S, CO and other bonds can be considered, such as the formation of reversible cyclic structure. Advantages and disadvantages linked to parameters such as the energy input, the stimuli and the efficiency of the reversible reaction as well as the easiness of synthesis and bond stability, can be cited for each reactive system. PUs containing dynamic covalent bonds are of great interest in this context to lead to self-repairable materials. However, based on the PU precursors (i.e. alcohol and isocyanate), all the chemistries known for self-healing polymers in general cannot be applied. This section thereby deals with the different reversible covalent chemistries employed in the context of self-healing PUs as well as the synthetic strategies for such materials.
(a). Thermo-responsive self-healing PUs
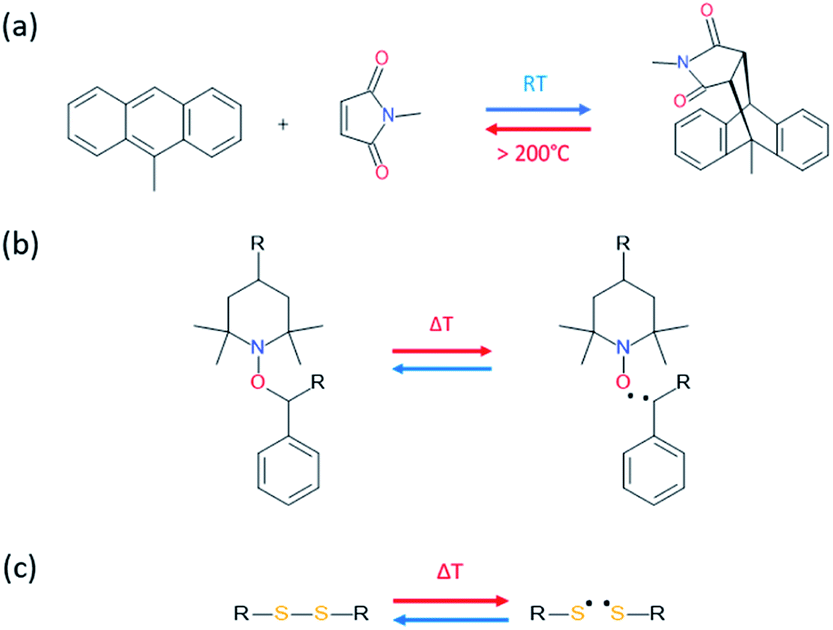
Temperature is by far one of the most available sources of energy, along with the electromagnetic radiations, and has been used extensively to trigger chemical change in dynamic materials. It is important to note that in some cases the external energy used for a material to self-heal is a combination on both thermal and electromagnetic radiation, making the differentiation between both contributions difficult. For this reason, the use of discrete energy sources as a minimal intervention is of particular of interest.29 Herein, [4+2] Diels–Alder reactions between furan and maleimide entities are known for decades and have allowed the design of a plethora of different thermo-responsive PUs. Upon thermal activation, both moieties form, at low temperature (TDA < 50 °C), a cyclo-adduct which reversibly regenerates the initial furan and maleimide groups at higher temperature (TrDA > 120 °C). This reversible bonding-debonding permits the microflow of the PU network to fill the damaged region and subsequent rebonding, enabling mechanical restoration of the damaged materials. The two most commonly used strategies to implement Diels–Alder moieties into a self-repairable polyurethane backbone are summed up in Fig. 6, namely: (i) adding diol containing maleimide-furan Diels–Alder adduct into the reaction between commercially available polyol and polyisocyanate for designing reprocessable, healable and shape-memory PU-based thermosets35–38 (Fig. 6a) and (ii) end chain-addition (Fig. 6b) or pendant-addition (Fig. 6c) of thermo-responsive moieties during the first PU synthetic step (either as maleimide/furfuryl alcohol,39–47 amine48–51 and thiol52 or maleimide/furan isocyanate53–56) and subsequent addition of complementary thermo-responsive crosslinker (multi-maleimide or multifuran), either in linear or crosslinked PU. Herein, Willocq et al. introduce single-component self-healing polyurethanes based on reversible maleimide/furan Diels–Alder reactions with good physical integrity on healing.57 Despite furan/maleimide is one of the most popular couples with respect to its low retro-scission temperature, other Diels–Alder-based moieties have been utilized for the design of self-healing PUs. For example, anthracene/maleimide moieties react by [4+2] reactions while the retroscission temperature is above 200 °C (Fig. 7a).59 These moieties have been used in a thermo-responsive diol for the design of healable PUs and composites under extreme temperature conditions. In other works, photo-thermal dimerization of anthracene was employed.60 Alkoxyamine thermo-reversible reaction was reported as another effective way for polymer network to self-heal thermally. This reaction involved the reversible thermal fission-recombination of the C–ON bond as depicted on Fig. 7b. The first use of alkoxyamine was reported for stiff alkoxyamine-based polystyrene, healing microcracks even above Tg.61 The use of this reversible chemistry was recently described for PUs, enabling healing at 80 °C.16,62 Thermal free radical fission-recombination of the S–S bonds has also been explored for self-healing materials inducing network reshuffling (Fig. 7c).15 Recovery of the PU properties after damage was possible using appropriate choice of disulfide diols and heating at 80 °C.63–65 Comparatively, diselenide –Se–Se– was used similarly to disulfide bonds in PUs and showed better healing efficiently than its sulfide counterpart thank to the faster reaction exchange of selenium thermo-reversible bond.66
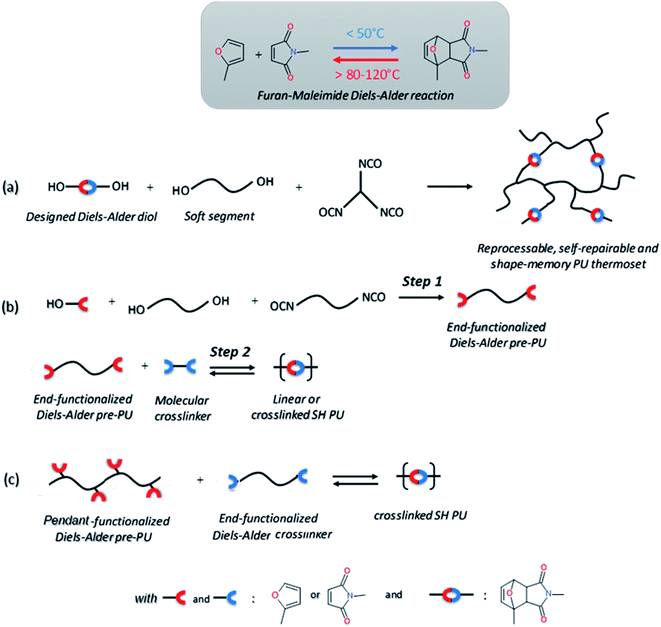 |
| | Fig. 6 Common strategies for the synthesis of Diels–Alder based self-healing PUs: incorporation of Diels–Alder adduct (a), end-chain-addition (b) and pendant-addition (c) of Diels–Alder moieties during the first PU synthetic step and subsequent addition of complementary Diels–Alder crosslinker. | |
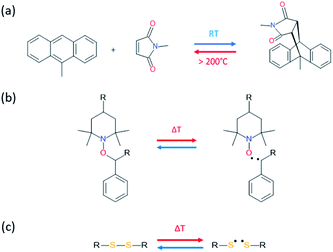 |
| | Fig. 7 [4+2] Diels–Alder reaction between maleimide and anthracyl moieties (a), reversible thermal fission-recombination of alkoxyamine based structure (b) and disulfide bond (c). | |
(b). Photo-responsive self-healing PUs
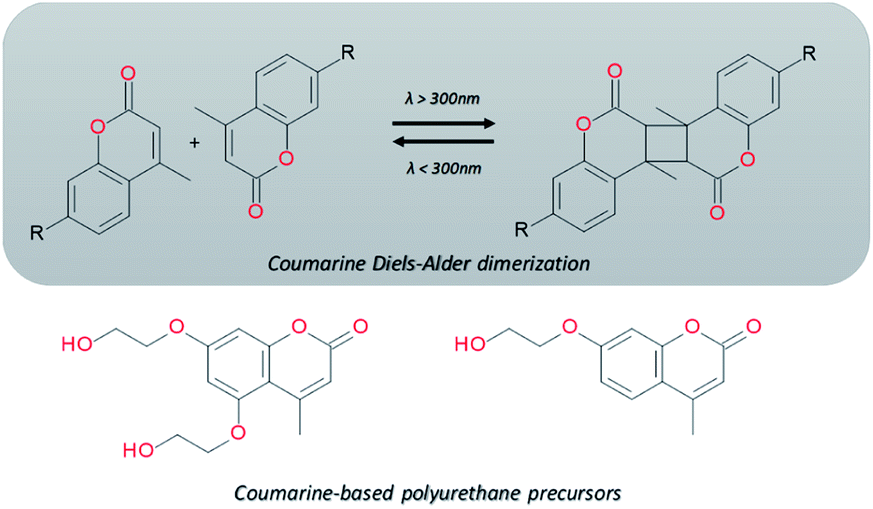
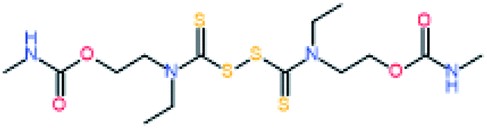
Electromagnetic radiation (visible sunlight, UV, infrared) is a convenient, available and unlimited source of energy to heal materials when considering applications as protective coating and outdoor bulk materials. For decades, light has been explored as healing trigger in different polymer matrices.67 Healable PUs with UV light were first based on [2+2] Diels–Alder moieties. Like the thermal [4+2] Diels–Alder reactions, the [2+2] cycloaddition reversibly induces formation and retro-scission of the cycloadducts when exposed to the adequate radiation wavelength. Coumarine, cinnamate and anthracene derivatives are the most reported moieties promoting Diels–Alder dimerization reaction.68 Coumarine-based polyurethanes have been developed using different synthetic coumarine-based diol or monoalcohol structures and appropriate flexible soft segment and isocyanate coupling agent (Fig. 8).69–72 Insertion of coumarine into PUs leads to the remendability of the PU network. UV-curing have also been employed for supramolecular chemistry in PUs. As an example, quadrupole hydrogen bonds moieties (ureido-pyrimidone, UPy) were incorporated into PU matrices.73 UV irradiation of hydrogen bonded UPy leads to the conversion of light into heat by absorption of UPy conjugated ring, and consequently by the dissociation of the supramolecular interaction. Despite the common use of UV radiation as healing trigger, it requires high input of energy while leading to photodegradation of the PUs.74 Interestingly, different research groups have implemented visible-light sensitive reshufflable chemistries into PUs. Xu et al. developed visible-light healable PU using diselenide chemistry.19 The dynamic properties of diselenide bond was highlighted while a wave length above 600 nm is sufficient to initiate the reshuffling by metathesis reaction (Table 3a).75 The diselenide bonds were introduced using a diselenide-based diol in linear PUs containing polytetramethylene glycol soft segments and aromatic toluene diisocyanate. Following similar track, Matyjaszewski and coworkers developed covalently crosslinked self-healing PUs under air and visible light using thiuram disulfide moieties reshuffling (Table 3b).76 Under visible light irradiation, thiuram disulfide groups rearrange by two mechanisms. On one hand, radical transfer reaction of generated S-based radical reacts with another thiuram disulfide forming a new unit. As a cascade transfer reaction, the newly generated S-based radical reacts with another thiuram disulfide groups, ensuring the dynamic of the network. On the other hand, a simple free radical crossover reaction can occur in a simpler manner. The chemical stability of such thiuram radicals has been reported to be around two weeks.77 By this approach, visible light healable PU network could be prepared in the presence of thiuram disulfide based diol, tetraethylene (soft segment), hexamethylene diisocyanate (coupling agent) and triethanolamine (crosslinking nod). Finally, Zhang et al. designed sunlight healable PUs using aliphatic disulfide groups (Table 3c).78 In their work, aliphatic disulfide were used instead of aromatic counterparts to ensure the presence of electro-donating groups that weakened the –S–S– bonds (allowing the bond cleavage using visible light).79 Aliphatic disulfides bring flexibility and resistance to yellowing during the sunlight exposure. Moreover, the presence of hydrogen bonds in the polyurethane design ensures a close contact between disulfide groups, facilitating the molecular rearrangement in the transparent PUs.
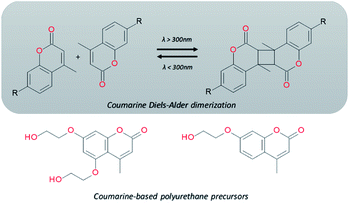 |
| | Fig. 8 Coumarine Diels–Alder light-induced dimerization and coumarine-based polyurethanes precursors. | |
Table 3 Visible-light-triggered chemistries for PUs
| Visible-light-triggered chemistrie for PURs |
Reshuffling process |
Chemical structure in the PUs |
| (a) Diselenide |
Radical metathesis |
 |
| (b) Thiuram disulfide |
Radical transfer and radical cross–over reaction |
 |
| (c) Aliphatic disulfide |
Radical metathesis |
 |
(c). Chemo-responsive self-healing PUs
Beside temperature and electromagnetic, other triggers can be employed under less drastic conditions. Recently, variation of pH or humidity have attracted the attention of the self-healing community as new and convenient ways to activate adaptive responses. These two triggers are water-based and one can easily imagine the important potentiality of repairable materials when exposed to humidity variation or acidic precipitation. Despite the increasing number of publications related to humidity or pH-induced self-healing polymers,80–82 PUs with these properties are not well documented. When PUs are exposed to humidity, it is reported that water molecules can act as plasticizers, lowering its Tg and enabling higher mobility.83,84 However, this process is not considered as self-healing mechanism as it can be applied to any water-plasticizable polymers. Therefore, to ensure that moisture induced mechanism is not due to plasticizing, novel design of the PU would confer non-hygroscopic and hydrophobic character by choosing the appropriate soft segment, diisocyanate and chain extender. Liu and coworker developed zwitterionic pyridine-based PU showing healing capabilities when exposed to humidity.85 Authors attribute the healing feature to the breakage of electrostatic forces and subsequent migration of charged species with water molecules by hydration. After drying, electrostatic interactions are reformed, and the material is healed. Rapid dynamic exchanges between urea-based moieties and moisture have been recently proposed by Willocq et al. as an alternative to promote local and spontaneous healing responses to damage using atmospheric moisture as an external stimulus.86 No example of self-healing PUs healable with pH change are reported in the literature. Interestingly, Urban and coworkers developed different PU networks generating reactive functions and readily repairing the material with reaction of urethane bonds.13,32,33,87,88 Among those works, it was demonstrated that introducing glucose ring (methyl-a-D-glucopyranoside, MGP) in the PU network endow the materials with self-healing while the mechanism of repair is highly dependent on the catalyst. When damage occurs, they assumed that urethane linkage can break either by the N–C (1) or the C–O (2) bond generating free radicals (Fig. 9a). In the first situation (1), the generated free radicals are converted into amine and methyl group by CO2 removal and hydrogen subtraction. In the second situation (2), the generated free radical reacts with a molecule of water, eliminating CO2. It is worth mentioning that in both situations, amines are formed (as demonstrated by infrared and RAMAN spectroscopy).32 The as-formed amine can then react with urethane to form urea by urethane–urea conversion if the urethane is reactive enough. Urethanes on glucose ring are more reactive because of the strain energy of the ring. Using an appropriate catalyst (e.g. zinc acetate), urethane–urea conversion could be performed with the damage-induced amine and the urethanes on glucose structure at moderated temperature (75 °C) (Fig. 9b). Formation of urea at the damage area induces healing by covalent rebonding. Beside the chemical aspect of healing, authors demonstrated the importance of physical factors after healing. For instance, the PU network exhibits elastic recovery, which ensures the gap closure and surface contact before chemical event takes place. Note that the healing mechanism is highly dependent of the catalyst used. When dibutyltin dilaurate (DBTDL) was used, the mechanism relies on the reactive species created by urethane or polyethylene glycol chain breakage. With appropriate stoichiometry, DBTDL is inserted with atmospheric water and carbon dioxide on glucose ring and interacts with urethane bond. Insertion of CO2 enables the formation of carbonate and subsequent reactions with damage generated species as amine, urethane or hydroxyl groups. This unique mechanism allows healing mechanism at room temperature and atmospheric humidity (Fig. 10).33 These examples are among the few reported where the damage itself is used to generate reactive species that are involved in the healing process. However, the drawbacks of this approach are that the healing efficiency is limited as the induced damage is generated at the extreme surface and therefore limited in quantity. Moreover, the process is not reversible while the healing mechanism will rapidly loss in its efficiency if the same damaged area is considered in a new healing process. Beside these examples, deliberately modifying the stability to introduce dynamic in urethane or urea functions is another way to propose simple polyurethane systems with healing capabilities. Dynamic urea bonds were discovered by Cheng et al., offering the simple design of self-healing polyurea-urethane at ambient conditions.18 Amide groups, widely present in biological systems like proteins, are very stable due to conjugation of π-electrons of the carbonyl function and the lone pair of the nitrogen atom and reversibility (amidolysis) required under extreme conditions. It was demonstrated that introducing bulky substituents on the amide nitrogen atom weakens the amide bond and results in possible amidolysis under mild conditions. The bulkiness of the substituents disturbs orbital co-planarity and weakens carbonyl–amine interaction. The products resulting of aminolysis is a bulky amine and ketene, too reactive for a convenient dynamic system. The dissociated carbonyl structure should be stable under ambient conditions but reactive toward amines. Isocyanate groups fulfill these requirements and can form urea with hindered amine. Molecular model reactions confirm the reversibility under mild conditions in the case of different hindered amines. Ethylene diamine with tertiobutyl substituent on nitrogen atoms was found to be the best candidate for self-healing. Indeed, the resulting urea is not too dynamic and not too stable. PU networks were obtained by mixing triethanolamine, hexamethylene diisocyanate, tetraethylene glycol (soft segment) and hindered amine. The obtained polyurea-urethane exhibits self-healing under ambient conditions by permanent dynamic reactions between bulky urea, the starting isocyanate and bulky amine. Drawbacks of this approach is the inherent toxicity of isocyanate and its reactivity toward water (ca. moisture), limiting the healing capability over time. Still, it remains a great challenge to integrate multi-stimuli responsive functions into a single polymeric material. Here, Yang et al. recently reported the synthesis of multi-responsive self-healing PUs through the use of dynamic thiol-Michael adduct as crosslinker, making the chemically crosslinked PUs healable and recyclable at an elevated temperature as well as stimuli-responsive to heat, pH and ammonia gas. Besides, the incorporation of UPy motifs into the PU backbone chains enabled the formation of dense H-bonding assemblies and endowed the material with enhanced mechanical properties.89
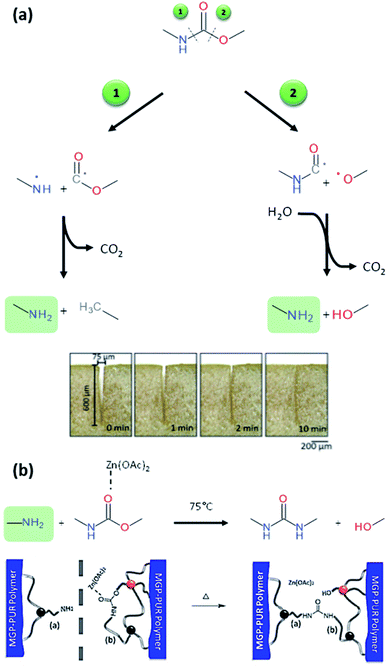 |
| | Fig. 9 Mechanism of amine formation upon urethane breakage and related gap closure after elastic recovery of the PU network (a) and urethane–urea conversion catalyzed with zinc acetate in presence of damage-induced amine and its related healing mechanism (b).32 | |
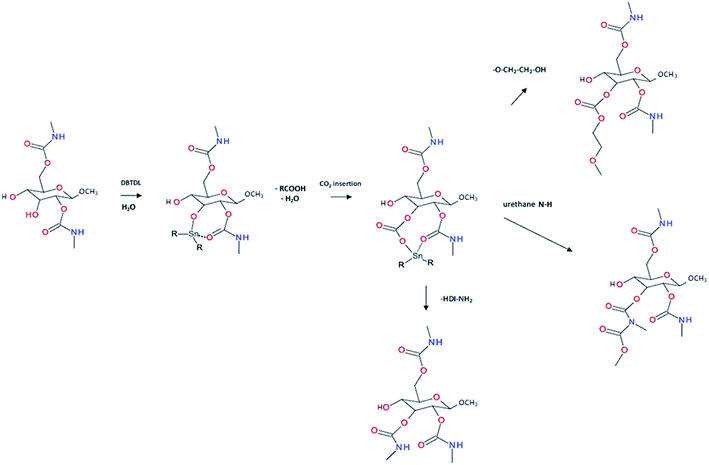 |
| | Fig. 10 Insertion mechanism of DBTDL on glucose structure and formation of reactive carbonate reacting with damage-induced reactive species.33 | |
V. Self-healing polyurethanes through supramolecular chemistry
Non-covalent interactions can be used for multiple self-repairing process such as hydrogen bonds, ionic interactions, π–π interactions, metal coordination, or host–guest interactions. Compared to covalent interactions, supramolecular interactions are fast, specific (directional and sensitive to complementary moieties) and allow multiple healing cycles with limited energy input. As such, this section deals with the different supramolecular chemistries employed in the context of self-healing PUs including hydrogen bonding, metal–ligand coordination, π-stacking and ionic interaction.
(a). Hydrogen bonding
Hydrogen-bonds are the most commonly used supramolecular bonds for self-healing. Bond strength varies from 2 to 40 kcal mol−1 depending on the nature of donor and acceptor.90,91 Though, the term “hydrogen bonded supramolecular self-healing” is ambiguous when PUs are considered. Due to the presence of hydrogen bonds from urethane linkages, hydrogen bonded physical networks are obtained even for linear PUs. Based on the hydrogen bond strengths and the PU composition, some non-functionalized PUs can undergo repairing after damage due to reassociation of hydrogen bonds.92 This reassociation can be triggered, as previously described, by temperature, light or physical reattachment (e.g. electrostatic interaction). The following section thereby focus on supramolecular self-healing PUs when hydrogen bonds are originated by other chemical structures than the urethane linkages or when urethanes are modified in an original way to confer peculiar features to the PU. Leibler et al. developed hydrogen-bonded self-healing rubber by reacting fatty acid, carboxylic acid groups and urea to form polymer structures that strongly interact through hydrogen bonds.21 The obtained supramolecular network is transparent, glassy, has “rubber like” behavior upon 90 °C and self-repairs under ambient conditions. Others reported the use of thioureas in a simple low molecular weight polymer structure, which combines mechanical robustness with self-repair abilities. In this concept, thioureas adopt a zigzag hydrogen bonds association, enabling dense hydrogen bond networks without crystallization.93 Healable hydrogels were reached by combining PU and UPy functionalized polymethacrylate polymers.94 In this approach, the hydrogels were obtained in a two-step method forming in the first step the PU with isophorane diisocyanate, polyethylene glycol and subsequent addition of methacrylate mono-alcohol. The end-capped methacrylate PU was then reacted with UPY-modified methacrylate. The resulting PU showed excellent capabilities due to the presence of quadruple associative bonds while not requiring any external stimuli for repair. However, this approach remains limited to soft hydrogels.
(b). Metal–ligand coordination
Metal–ligand coordination is a convenient way to endow the materials with self-healing capabilities through thermal or photo-thermal stimulus. Introducing metal–ligand crosslinking within the PU matrix can be achieved by choosing the appropriate monomer. As an example, self-healing PUs were obtained by adding pyridine derivative during the PU synthesis (using 2,6-diaminopyridine).95 Along the linear polyurethane chain, pyridines have the possibility to interact with different metal forming networks. Based on the selected metal, bi- (Fe and Zn) or tridentate (Tb) complexes are formed. The dynamic properties of the as-formed PU networks can be tuned as the strength of interaction will be dependent on the metallic center. For example, Zn–pyridine interaction is strong while not dynamic as an opposition to the Fe–pyridine and Tb–pyridine interactions which are used for self-healing. The limitation of this approach is the toxicity of pyridine derivative and the stability of the metal used toward oxidation.
(c). Π-stacking
Healable supramolecular PU blends based on aromatic electron deficient and rich entities that form π–π interactions was reported.97 In this system, pyrenyl-functionalized telechelic PU was blended with polydiimide forming π–π interactions by chain folding and complemented with hydrogen bonds of the PU backbone. Another approach consists in the introduction of methoxyphenyl and nitrophenyl groups, self-complementary in π–π interactions.96 These functions were introduced in a telechelic PU to endow healing under mild conditions (ca. at 45 °C) (Fig. 11).
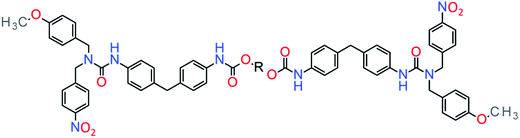 |
| | Fig. 11 Example of self-healable PU through π-stacking.96 | |
(d). Ionic interaction
Electrostatic clusters obtained by introducing sulfonate98,99 or carboxylate100 groups were also reported for the promotion of self-healing through ionic interactions in PUs. Once the matrix undergoes damage, the clusters dissociate and reassociate leading to self-healing abilities.
VI. Self-healing polyurethane composites
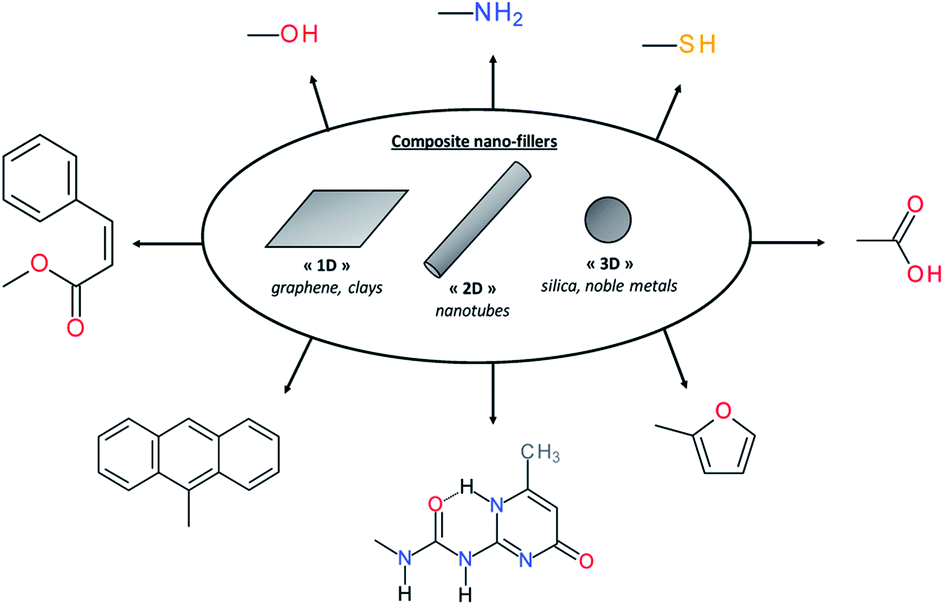
In material science, it is known for decades that polymers loaded with (nano)fillers possess improved mechanical properties if the dispersion and interfacial adhesion of filler/matrix are controlled. Depending on their dimensions, rigid (nano)fillers can be 3D-particles, rods, tubes, sheets or fibers. However, in the context of self-healing polymers composites, the introduction of rigid particles is counter-intuitive and leads mostly to the loss of healing properties due to restricted mobility in the composite. To solve this limitation, surface chemical functionalization of (nano)fillers is a promising alternative to use (nano)fillers as part of the self-healing networks. When their surfaces are modified with chemical functions as carboxylic acid,101 hydroxyl,102 sulfonate,103 amine,104 UPy,105 furfuryl,106 thiol,107 cinnamoyl,108 anthracene109 or pyrenyl110 groups, (nano)fillers can react with the matrix and interfacial healing is possible using one of the reversible chemistries (either covalent or supramolecular) previously described (Fig. 12). This approach is of interest to develop mechanically reinforced and functional self-healing PU composites. However, nanoparticle/polyurethane interface should be controlled to ensure good adhesion and dispersion of the nanofiller, resulting in improved rigidity without compromising the strain at break. This aspect can be achieved by functionalizing nanofillers with chemical groups that are compatible with the polymer matrix. In the context of self-healing nanocomposites, this surface modification is crucial to have healable interface as the introduction of rigid particles within the polymer matrix will restrict the chain mobility. Beside the improvement of mechanical properties, the real interest of nanoparticles in self-healing composites is the addition of functional properties associated to the intrinsic properties of nanoparticles such as electrical, magnetic, moisture-sensing, piezoelectric, damage-sensing or optical properties. Moreover, the possibility to activate healing in a “remote-control” fashion is highly sought.111 The following sections thereby report different examples of functional self-healing PU composites.
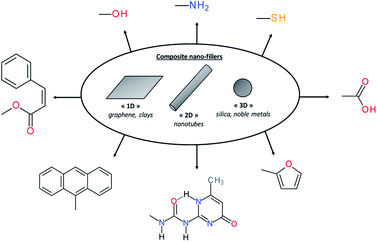 |
| | Fig. 12 Different nanofillers based on dimensional aspect and examples of chemical modifications of surface nanofillers for self-healing composites. | |
(a). Electric self-healing PU composites
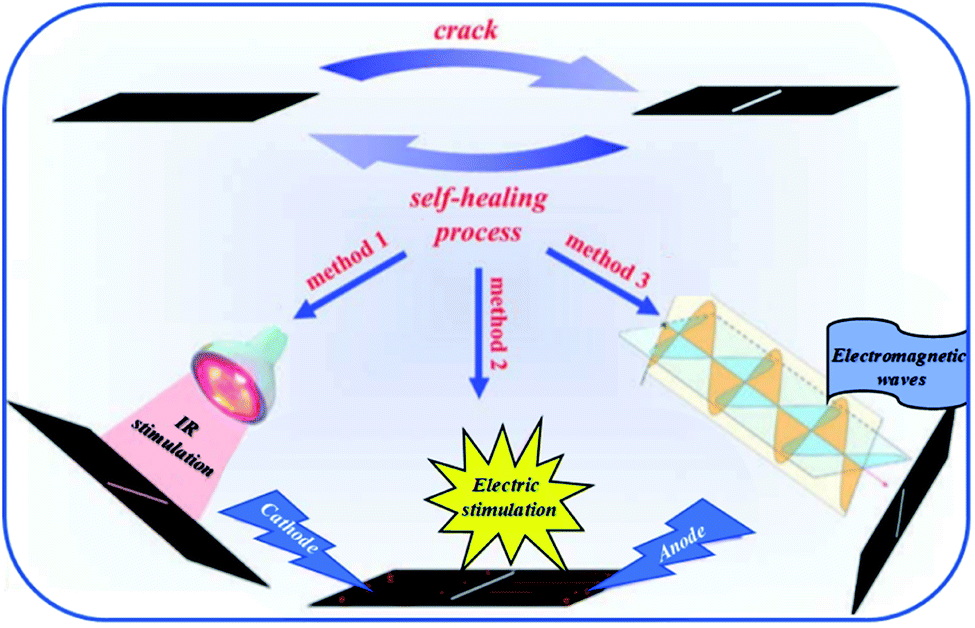
Allotropic forms of carbon such as carbon black, carbon nanotubes or graphene have been extensively used as reinforcing agents.112–114 In addition, these particles confer electrical and thermal properties to the resulting composites if a percolation network is achieved. At least 1 to 5 %wt of carbon nanotubes is required to endow the composite with sufficient electrical properties. Carbon nanotubes have been reported for different systems of self-healing PU composites. For instance, Gu et al. mixed multimaleimide and multifuran moieties with modified multi-walled carbon nanotubes to obtain heat-resistant, electrostatic dissipative and thermally stable reversible composites.117 In this work, carbon nanotubes, not participating to self-healing process, were modified with carboxylic acid to enhance the compatibility between the PU matrix and the nanofillers through hydrogen bonds. Similar investigations were conducted with hydroxyl-functionalized carbon nanotubes. Despite a restoration of thermal conductivity, and thus a certain network percolation after healing, mechanical performances were still reduced as compared to original ones due to restricted motions of carbon nanotubes.118 Graphene is widely used in organic electronic devices as its high electrical performances allow designing self-healing electrically conductive PUs. Hence, Chen et al. reported multichannel and repeatable healing of graphene–polyurethane composites by dispersing few layer of graphene (as obtained by arc discharge method) into commercial thermoplastic polyurethane (TPU) (Fig. 13).115 In addition to electrical conductivity and due to π-conjugated electrons, graphene sheets offer the possibility to heal the PUs by conversion of different stimuli into heat. When exposed to infrared irradiation, more efficient healing capabilities are observed compared to pristine matrix due to heat conversion induced by graphene. This feature is of interest when polymer films are exposed to sunlight. A second stimulus is based on electrical conductivity of graphene when finely dispersed and forming a percolating network. During electrical activation, current is converted into heat by Joule heating. At scratch tips, electrical resistance is higher and thermal energy locally release is sufficient to get repairs. Herein, Willocq et al. demonstrated the possibility to heal local macroscopic damage by a confined temperature increase arising from the Joule effect on thermo-reversible and electrically conductive poly(ester-urethane)/carbon nanotube nanocomposites.119 Finally, microwave irradiation was demonstrated as a third stimulus for healing. In this case, the microwaves are absorbed and converted into heat due to dipole distortion of graphene sheets that act as nanoscale heater. Besides, healing of graphene–PU composites using near infrared stimulus was possible thanks to the fine dispersion achieved after graphene surface functionalization. For this purpose, graphene oxide was chemically modified with phenylisocyanate forming urethane bonds on graphene surface and enhancing the compatibility with the matrix.120 Remarkably, in another work, improved mechanical properties could be obtained with 0.1%wt of graphene without compromising healing capabilities.121 This could be possible by in situ polymerization of PU chains from graphene oxide reactive groups. After addition of appropriate diisocyanate and soft segment, the grafted chains were end-capped with furfuryl groups. Subsequent addition of dimaleimide coupling agent leads to the formation of thermally remendable and highly reinforced composites. A similar strategy was reported for Diels–Alder modified graphene–PU composites healable with infrared laser.122 Ultrafast healing was obtained due to excellent thermal conduction of graphene opening a promising way for flexible electronic devices repair. Lignin-modified graphene healable PU nanocomposites were prepared in a facile, aqueous-based and green method.123 As lignin was non-covalently bonded to graphene, the sp2 structure of graphene was preserved ensuring good electrical conductivity and UV resistance due to the presence of lignin acting as radical scavenger. Beside the use of carbon-based nanofillers for electrical induced self-healing, copper nanowires mixed with PUs to produce self-healing flexible transparent conductor were reported.124 The nanocomposite, obtained after nanowire deposition and subsequent drop casting of polyurethane solution, showed conductive properties and repeatable healing without compromising the electrical conductivity.
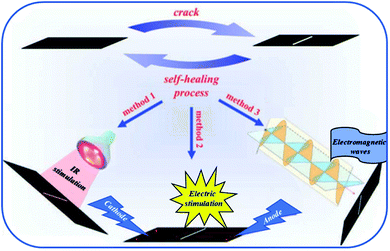 |
| | Fig. 13 Graphene–polyurethane composite healed under infrared, electric and electromagnetic stimulus.115 | |
(b). Magnetic self-healing PU composites
Comparatively to electrical particles, magnetic particles such as paramagnetic iron particles can be used to generate heat and subsequent healing. γ-Fe2O3 or Fe3O4 superparamagnetic nanoparticles were used to generate local melt flow under oscillating magnetic field.125,126 Under oscillation, the particles produce local heat increasing the polymer chains mobility and offering the possibility of multiple healing with non-invasive magnetic waves. For instance, introduction of Fe2O3 particles into PU fibers showed effective inductive heating under alternative magnetic field while no associated healing properties were reported so far.127 Other studies using similar concepts have been reported.128 For example, the use of noble metal such as silver was investigated to generate local heating by plasmonic effect (collective excitation of electrons, converting electromagnetic radiations into heat). When exposed to visible light, silver nanoparticles dispersed in thermoplastic produce a plasmonic effect, offering the possibility to heal material through photo-induced healing.129 As last example, polyurethane matrices containing dispersed graphene layers have the possibility to self-heal under microwave irradiation absorption.115
(c). Bio-based self-healing PU composites
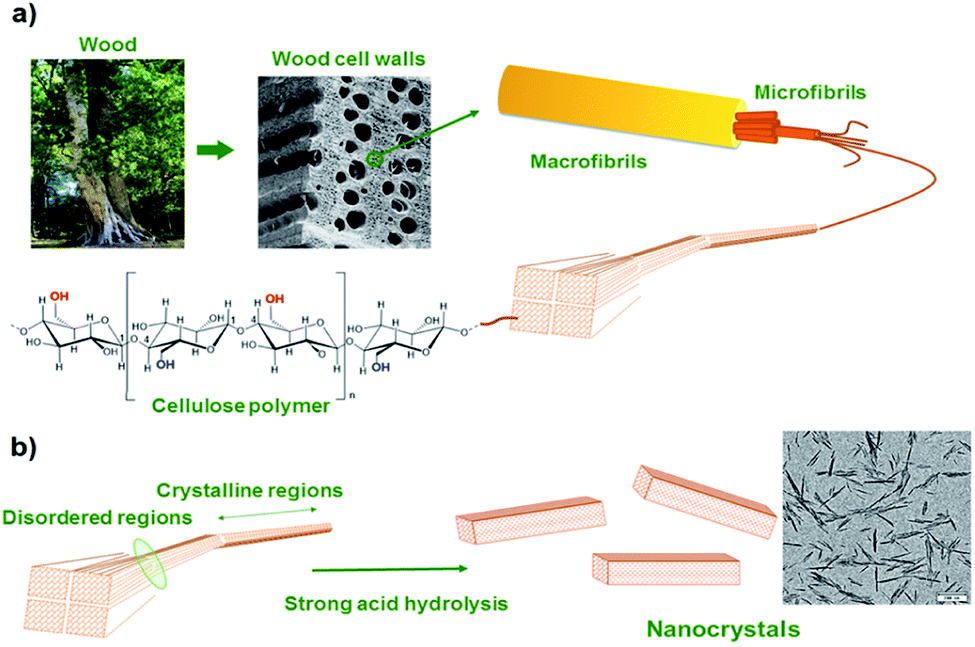
To comply with growing ecological challenges, the development of sustainable materials and by extension sustainable self-healing composite materials is of great importance. Recently, bio-based cellulose nanocrystals have demonstrated its potential for reinforced, bio-based and optically active composites.130 Cellulose nanocrystals (CNCs) are rigid and crystalline nanoparticles obtained by acidic hydrolysis of cellulose from plant (ramie fiber, cotton) or animals (tunicate). Such nanofillers, having glucose-based structures, exhibits a large number of hydroxyl groups on its surface, offering the possibility of surface modification and better nanofiller-matrix compatibilization (Fig. 14). Based on this possible surface modification, Weder et al. have developed light healable CNC–PU composites.105 CNCs hydroxyl groups were reacted with isocyanate containing UPy moieties and further dispersed in UPy-telechelic linear PU, building a network through UPy–UPy hydrogen bond interactions. Upon UV irradiation, hydrogen bonds dissociated enabling enhanced mobility and network healing. CNCs were modified similarly with reversible mechanophore diarylbibenzofuranone providing PU composites with self-healing and damage-probing abilities.133 Despite their non bio-based origin, attapulgites are natural hydrated magnesium–aluminium silicate clays characterized as randomly oriented networks of packed rods. Similar to CNCs, attapulgite surfaces are rich in hydroxyl groups. Favored hydrogen bond interactions of hydroxyl groups with PU matrix allow for a good filler dispersion and enhanced mechanical properties. Attapulgites were therefore dispersed in disulfide-based PUs and self-healing nanocomposites with shape memory effect were reported.134
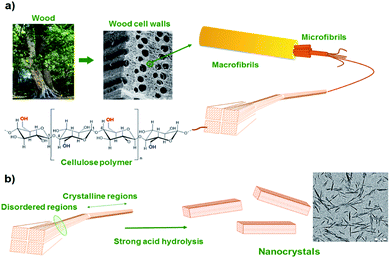 |
| | Fig. 14 From macroscopic to microscopic composition of cellulose (a) and acidic treatment for cellulose nanocrystal synthesis (b).116 | |
VII. Future directions
This section deals with the future directions in the context of self-healing PUs including shape-memory assisted self-healing PUs, self-healing non-isocyanate PUs and mechanochromic self-healing PUs.
(a). Shape-memory assisted self-healing PUs
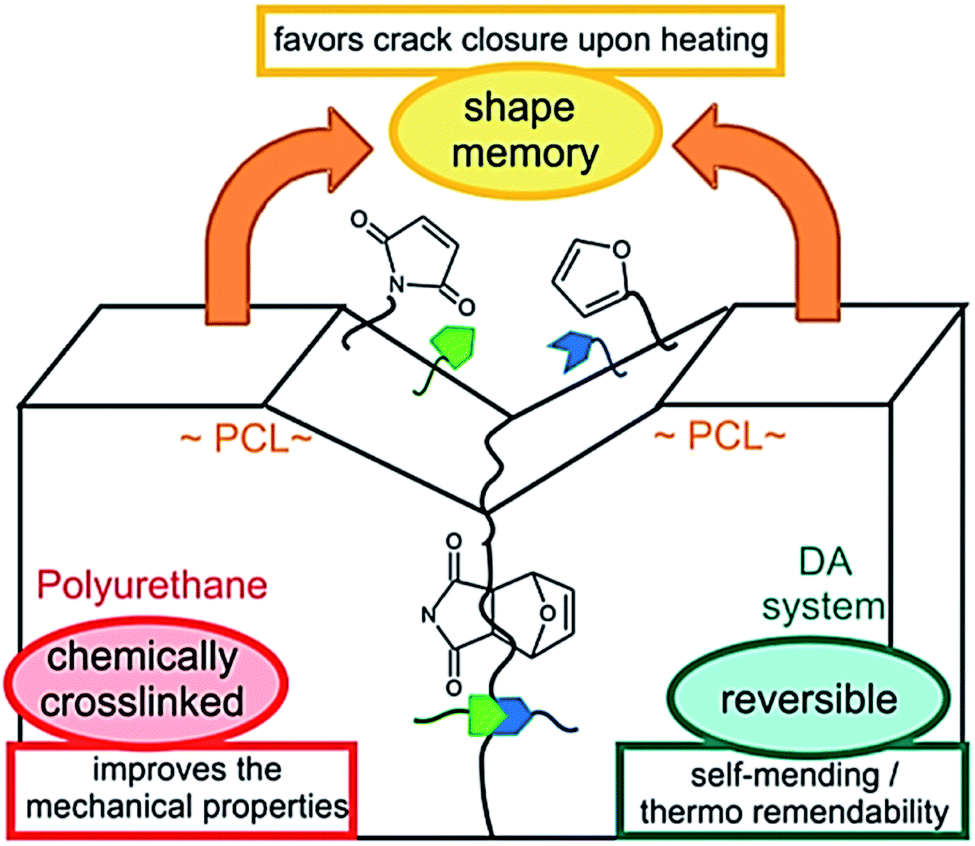
Note that chemical and physical aspects are equally important to provide efficient self-healing. Except few examples, self-healing materials, so far, are limited to micrometer sized scratch healing or require physical reattachment of damaged parts to ensure the healing process. To address this problem, the concept of shape-memory assisted self-healing polymers was introduced in 2013.131,132 Shape memory polymers that are, similarly to self-healing materials, stimuli adaptive materials have been extensively studied.136 Shape memory polymers are characterized by their ability to store (or memorize) a temporary shape and return to their initial permanent shape upon stimuli. Crystallization, glass transition temperature or reversible crosslinking can be used to fix the temporary shape while entanglement, physical or chemical crosslinking form the permanent shape.136 PUs are polymers of choice for shape–memory properties as physical permanent networks are intrinsic of this polymer class thanks to the presence of hydrogen bonds from the urethanes linkages or from chain entanglements. On the other hand, switchable domains are obtained after introducing e.g. crystalline soft segments, reversible crosslinking point or a combination of both to the obtained multi-stimuli responsive shape memory PUs.137 Hence, Mather et al. used cleverly shape-memory concept to provide shape-memory systems with self-healing capabilities. Herein, linear and crosslinkable polycaprolactone (PCL) were mixed with fibers as-obtained by electrospinning technique. This association led to thermally healable polymer combining the shape recovery of the crosslinked PCL matrix bringing surfaces into contact and the melting/flowing of linear PCL chains rebonding the fibers at cracks. In this example, shape memory is a way to heal macroscopic damaged (millimeter sized, puncture) (Fig. 15). Du Prez et al. developed thermo-remendable shape-memory PUs in a one-pot synthesis using Diels–Alder reactions in PCL-based PUs.42 In this system, authors demonstrated that the induced crack favored breaking of Diels–Alder cycloadduct, and shape memory at temperature above PCL melting (i.e. 50 °C) permits the gap closure and subsequent reassociation of Diels–Alder moieties for healing (Fig. 16). On similar track, Zhu et al. reported the design of cyclic PCL-based PUs using the furan-maleimide chemistry for shape-memory assisted self-healing PUs.36 These recent studies opened new horizons in self-healing concept through the possibility of healing macroscopically damages which will certainly be a major topic in coming years.
 |
| | Fig. 15 Shape memory assisted self-healing of macroscopic damaged using thermally healable crosslinked/linear PCL fibers.131,132 | |
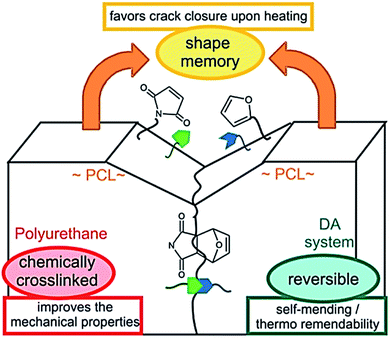 |
| | Fig. 16 Schematic representation of shape-memory assisted self-healing process in a Diels–Alder based PCL–polyurethane.42 | |
(b). Self-healing non-isocyanate PUs
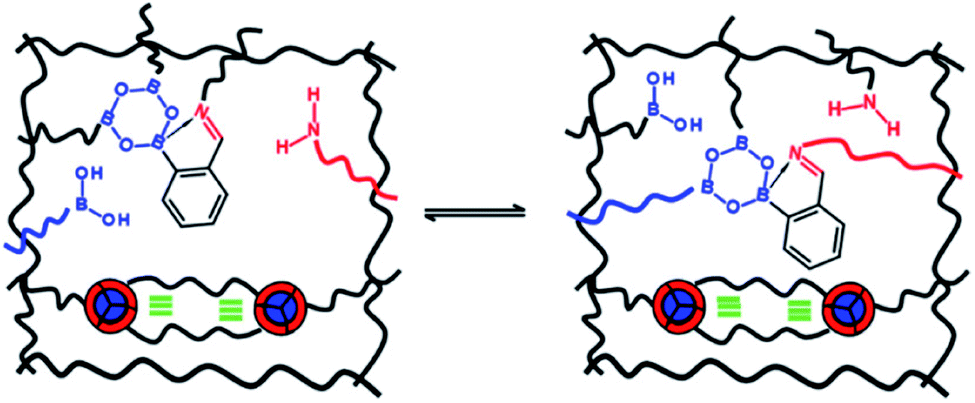
Introducing reversibility in PUs is of great interest to extend their lifetime by healing and reprocessability. So far, all the developed self-healing PUs remains with an unsolved problematic, which is the use of isocyanate as building block. Despite their intensive use in industry, isocyanate synthesis and handling require special care and are not the best option from a sustainable viewpoint (e.g. toxicity of isocyanate during production, safety for customers). Therefore, a new trend is to develop non-isocyanate PUs (so-called polyhydroxyurethane) currently synthesized from 5-membered ring cyclocarbonates and amines. Cyclocarbonates are produced with epoxy and carbon dioxide, valorizing green-house effect gas into high-performance polymers. As a promising example, Caillol et al. have developed thermoreversible and reprocessable non-isocyanate based PUs. Furfuryl glycidyl ether was therein converted into cyclocarbonate derivative and mixed with dimaleimide-terminated polypropylene glycol and a diamine, forming a thermomendable networks.140,141 Delpierre et al. herein described a simple approach to design a nonisocyanate-based PU network featuring multi-responsiveness to humidity and temperature and outstanding healing properties by combining iminoboronate and boroxine chemistry (Fig. 17).135 Recently, Zheng et al. revealed that classical thermoset PUs are readily capable of permanent reshaping after a topological network rearrangement that is induced by transcarbamoylation.142 Later, Chen et al. further discovered that the presence of a catalyst, i.e. dimethylaminopyridine, in polyhydroxyurethane leads to dynamic and reprocessable networks through transcarbamoylation exchange reactions and reversible cyclic carbonate aminolysis (Fig. 18).143 This offers the possibility of designing multi-recyclable isocyanate-free PU networks and is a source of inspiration for the self-healing community.
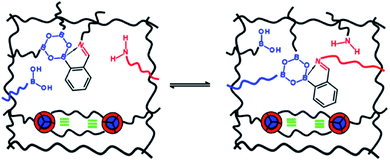 |
| | Fig. 17 Design of self-healing nonisocyanate-based PUs combining iminoboronate and boroxine chemistry.135 | |
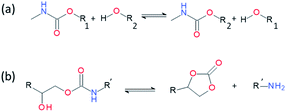 |
| | Fig. 18 Transcarbamoylation (a) and reversible cyclic carbonate aminolysis (b) catalyzed by dimethylaminopyridine. | |
(c). Mechanochromic self-healing PUs
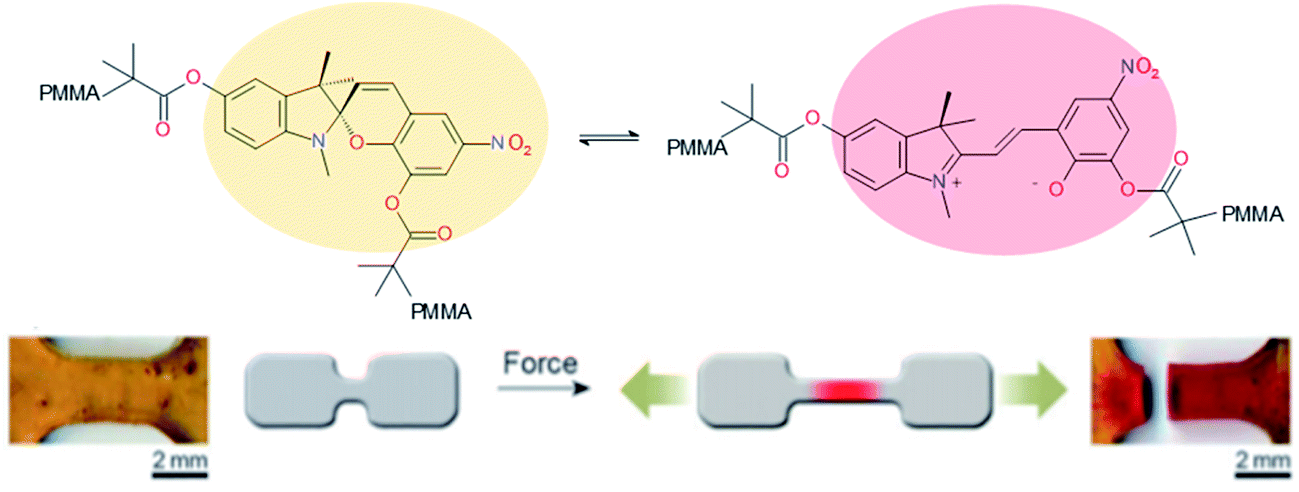
Ideal self-healing materials should have the ability to manage crack/damage in a real-time response as well as the possibility to provide visual inspection of damage when microcracks are not easily visible (i.e. as a preventive method). Mechanochemistry refers to chemical reactions induced by the input of mechanical energy (e.g. grinding, compression, scratch) and mechanochromic molecules possess the ability to undergo conformational changes inducing color changes (i.e. change of conjugation degree) under mechanical solicitations.144 One of the first examples of damage probing using mechanochromic molecules in polymer networks was reported by Sotto et al. in 2009.138 Spiropyran-based polymethylmethacrylate networks show color change (from colorless to purple red) when submitted to mechanical stress (Fig. 19). This was attributed to mechanically-induced conformational changes of spiropyran molecules (i.e. rearrangement to merocyanine).139 This concept using spyropyran as visual probe of damages was transferred to PU networks by designing spiropyran diol monomers. Weng et al. finally reported dual cross-linked elastomers with spiropyran probes and UPy moieties presenting healing and damage probing properties.145 The same group extended this concept to dual spiropyran/metal–ligand self-healing Pus, showing healing ability with solvent and damage-induced color changes.146 Other molecular designs were reported for mechanochromic self-healing PUs such as diarylbibenzofuranone enabling healing by forming stable free radicals. Moreover, due to aromatic character, the formation of free radical species modified the absorbance of diarylbibenzofuranone and generated purple colors. Diarylbibenzofuranone groups were also introduced in linear PU matrices by designing the appropriate diol monomer and mixing it with polypropylene glycol, 4,4′-methylenebisphenyl isocyanate and butanediol as chain extender.17,147 Under stress, the color of the elastomer changes in a reversible way (i.e. disappearance of purple color after several times following the mechanical stress, Fig. 20). It is clear that mechanochromic effect is of great interest when considering the probing of damage in PU-based materials. In addition, the presence of dual specific bonds within the material is a promising feature to get on time healable and detectable damage.
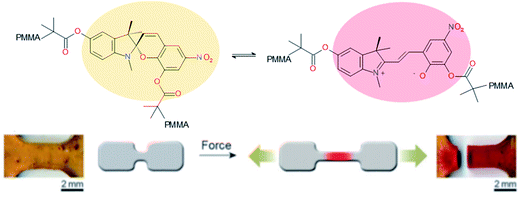 |
| | Fig. 19 Mechanochromic spyropyran-based PMMA network.138,139 | |
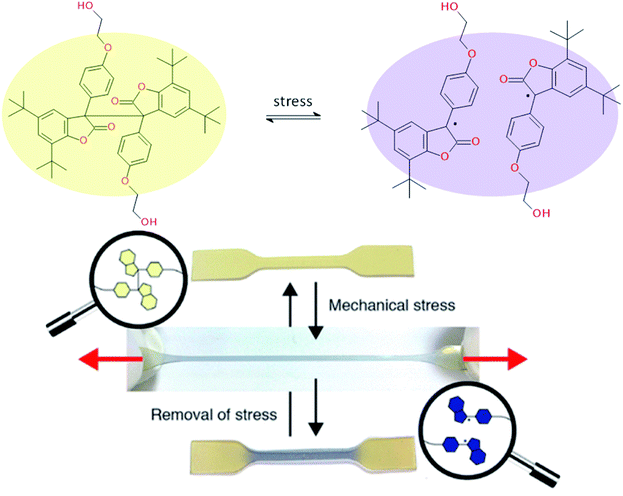 |
| | Fig. 20 Diarylbibenzofuranone mechanochromic diol for PU synthesis and the resulting PU elastomers under mechanical stretching.17,147 | |
VIII. Conclusions and challenges
PU materials are highly used in our daily life while finding approaches to extend their lifetime which dictate their use in future applications, requiring highly advanced materials. Herein, self-healing PUs provide opportunities for designing novel materials with high performance, lifetime and reliability. Although the number of publications reporting self-healing PUs has grown exponentially the past decade, so far, their presence on the market is still limited. Non-exhaustive list of drawbacks limiting their commercialization are the following: (i) no practical up-scaling synthesis: multi-step synthesis, low yield, production price, use of solvent; (ii) limited healing recovery: in terms of reactive functions (e.g. damage-induced self-healing PUs), cycles (e.g. hindered urea) or mobility (e.g. self-healing PU composites); (iii) high energy input required for self-healing: thermal or UV irradiations tend to be replaced by softer stimuli as humidity or visible light; (iv) healing limited to microscopic damages: limited to micrometer sized scratch or requiring physical reattachment of cut material; (v) non-functional: only applicable to commodity applications (unless addition of functional nanoparticles is considered) and (vi) no detection of damage in PUs (as a prevention): up to now, only mechanochromic PUs provide damage probing feature. PUs addressing these issues will be considered as the perfect self-healing PUs. To achieve such non-utopist challenges, combination of the different concepts described in the present review should open future directions in this challenging but fascinating quest. Developing chemical or physical approaches separately will always be less efficient than their combination in a single material. Finally, and equally important, the development of spectroscopic techniques and knowledges allowing to highlight healing mechanism in PUs still remain a key-factor for designing efficient systems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge support from the Belgian Federal Government Office of Science Policy (SSTC-PAI 6/27), the European Commission/Walloon Region (FSE and FEDER) as well as the Belgian National Fund for Scientific Research (FNRS). B. W. and J. M. R. are F. R. S.-F. N. R. S. research fellows.
References
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- A. Eisenberg and M. Rinaudo, Polym. Bull., 1990, 24, 671 CrossRef CAS.
- S. J. Kalista, T. C. Ward and Z. Oyetunji, Mech. Adv. Mater. Struct., 2007, 14, 391–397 CrossRef CAS.
- R. P. Wool and K. M. O'Connor, J. Appl. Phys., 1981, 52, 5953–5963 CrossRef CAS.
- D. Döhler, P. Michael and W. Binder, in Self-Healing Polymers, 2013, ch. 1, pp. 5–60, DOI:10.1002/9783527670185.ch1.
- D. Carolyn, Smart Mater. Struct., 1994, 3, 118 CrossRef.
- C. Dry, Composite Structures, 1996, 35, 263–269 CrossRef.
- C. Dry, Design of self-growing, self-sensing, and self-repairing materials for engineering applications, SPIE, 2001 Search PubMed.
- C. M. Dry and N. R. Sottos, Passive smart self-repair in polymer matrix composite materials, SPIE, 1993 Search PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794 CrossRef CAS PubMed.
- S. H. Cho, S. R. White and P. V. Braun, Adv. Mater., 2009, 21, 645–649 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- B. Ghosh, K. V. Chellappan and M. W. Urban, J. Mater. Chem., 2011, 21, 14473–14486 RSC.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- C. e. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Adv. Mater., 2015, 27, 7740–7745 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467 CrossRef CAS PubMed.
- R. Araya-Hermosilla, G. M. R. Lima, P. Raffa, G. Fortunato, A. Pucci, M. E. Flores, I. Moreno-Villoslada, A. A. Broekhuis and F. Picchioni, Eur. Polym. J., 2016, 81, 186–197 CrossRef CAS.
- R. Martin, A. Rekondo, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, J. Mater. Chem. A, 2014, 2, 5710–5715 RSC.
- C. Bao, Y.-J. Jiang, H. Zhang, X. Lu and J. Sun, Adv. Funct. Mater., 2018, 28, 1800560 CrossRef.
- T. A. Osswald and G. Menges, in Materials Science of Polymers for Engineers, Carl Hanser Verlag GmbH & Co. KG, 2012, pp. 423–487, DOI:10.3139/9781569905241.010.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- A. Södergård and M. Stolt, in Poly(Lactic Acid), John Wiley & Sons, Inc., 2010, pp. 27–41, DOI:10.1002/9780470649848.ch3.
- Y. Yang and M. W. Urban, Polym. Chem., 2017, 8, 303–309 RSC.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 12142–12147 CrossRef CAS PubMed.
- S. Billiet, X. K. D. Hillewaere, R. F. A. Teixeira and F. E. Du Prez, Macromol. Rapid Commun., 2013, 34, 290–309 CrossRef CAS PubMed.
- S. Chen, F. Wang, Y. Peng, T. Chen, Q. Wu and P. Sun, Macromol. Rapid Commun., 2015, 36, 1687–1692 CrossRef CAS PubMed.
- W. Chen, Y. Zhou, Y. Li, J. Sun, X. Pan, Q. Yu, N. Zhou, Z. Zhang and X. Zhu, Polym. Chem., 2016, 7, 6789–6797 RSC.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- A. K.-L. Mohammad, H. M. Mohammad and A. S. Henry, Smart Mater. Struct., 2017, 26, 055003 CrossRef.
- P. Du, X. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, RSC Adv., 2013, 3, 15475–15482 RSC.
- L. Irusta, M. J. Fernández-Berridi and J. Aizpurua, J. Appl. Polym. Sci., 2017, 134, 44543 CrossRef.
- N. Okhay, N. Mignard, C. Jegat and M. Taha, Des. Monomers Polym., 2013, 16, 475–487 CrossRef CAS.
- G. Rivero, L.-T. T. Nguyen, X. K. D. Hillewaere and F. E. Du Prez, Macromolecules, 2014, 47, 2010–2018 CrossRef CAS.
- Y. S. Ryu, K. W. Oh and S. H. Kim, Macromol. Res., 2016, 24, 874–880 CrossRef CAS.
- Y. Wei and X. Ma, Adv. Polym. Technol., 2018, 37, 1987–1993 CrossRef CAS.
- R. Zhang, S. Yu, S. Chen, Q. Wu, T. Chen, P. Sun, B. Li and D. Ding, J. Phys. Chem. B, 2014, 118, 1126–1137 CrossRef CAS PubMed.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, Polym. Chem., 2016, 7, 7278–7286 RSC.
- S. Yu, R. Zhang, Q. Wu, T. Chen and P. Sun, Adv. Mater., 2013, 25, 4912–4917 CrossRef CAS PubMed.
- P. Du, M. Wu, X. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, J. Appl. Polym. Sci., 2014, 131, 40234 Search PubMed.
- L. Feng, Z. Yu, Y. Bian, J. Lu, X. Shi and C. Chai, Polymer, 2017, 124, 48–59 CrossRef CAS.
- X. Liu, P. Du, L. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, Polym. Bull., 2013, 70, 2319–2335 CrossRef CAS.
- Y. Zhong, X. Wang, Z. Zheng and P. Du, J. Appl. Polym. Sci., 2015, 132, 41944 Search PubMed.
- L.-T. T. Nguyen, H. T. Nguyen and T. T. Truong, J. Polym. Res., 2015, 22, 186 CrossRef.
- C. Gaina, O. Ursache and V. Gaina, Polym.-Plast. Technol. Eng., 2011, 50, 712–718 CrossRef CAS.
- V. Gaina, O. Ursache, C. Gaina and E. Buruiana, Des. Monomers Polym., 2012, 15, 63–73 CrossRef CAS.
- C.-D. Varganici, O. Ursache, C. Gaina, V. Gaina and B. C. Simionescu, J. Therm. Anal. Calorim., 2013, 111, 1561–1570 CrossRef CAS.
- C.-D. Varganici, O. Ursache, C. Gaina, V. Gaina, D. Rosu and B. C. Simionescu, Ind. Eng. Chem. Res., 2013, 52, 5287–5295 CrossRef CAS.
- B. Willocq, F. Khelifa, J. Brancart, G. Van Assche, P. Dubois and J. M. Raquez, RSC Adv., 2017, 7, 48047–48053 RSC.
- R. Herrington and K. Hock, Flexible Polyurethane Foams, The Dow Chemical Co., 1997 Search PubMed.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- Y. Fang, X. Du, Z. Du, H. Wang and X. Cheng, J. Mater. Chem. A, 2017, 5, 8010–8017 RSC.
- C. e. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. e. Yuan, Polym. Chem., 2013, 4, 4648–4654 RSC.
- T. Wan and D. Chen, J. Mater. Sci., 2017, 52, 197–207 CrossRef CAS.
- Y. Xu and D. Chen, Macromol. Chem. Phys., 2016, 217, 1191–1196 CrossRef CAS.
- Y. Yang, X. Lu and W. Wang, Mater. Des., 2017, 127, 30–36 CrossRef CAS.
- S. Delpierre, B. Willocq, J. De Winter, P. Dubois, P. Gerbaux and J.-M. Raquez, Chem.–Eur. J., 2017, 23, 6730–6735 CrossRef CAS PubMed.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- C. Cardenas-Daw, A. Kroeger, W. Schaertl, P. Froimowicz and K. Landfester, Macromol. Chem. Phys., 2012, 213, 144–156 CrossRef CAS.
- R. H. Aguirresarobe, L. Martin, N. Aramburu, L. Irusta and M. J. Fernandez-Berridi, Prog. Org. Coat., 2016, 99, 314–321 CrossRef CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2011, 21, 18373–18380 RSC.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- J. Ling, M.-z. Rong and M.-q. Zhang, Chin. J. Polym. Sci., 2014, 32, 1286–1297 CrossRef CAS.
- R. Liu, X. Yang, Y. Yuan, J. Liu and X. Liu, Prog. Org. Coat., 2016, 101, 122–129 CrossRef CAS.
- D. Rosu, L. Rosu and C. N. Cascaval, Polym. Degrad. Stab., 2009, 94, 591–596 CrossRef CAS.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- L. M. García-Con, M. J. Whitcombe, E. V. Piletska and S. A. Piletsky, Angew. Chem., 2010, 122, 4169–4172 CrossRef.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683–10690 RSC.
- J. M. Matxain, J. M. Asua and F. Ruiperez, Phys. Chem. Chem. Phys., 2016, 18, 1758–1770 RSC.
- J. J. Wilker, Nat. Mater., 2014, 13, 849 CrossRef CAS PubMed.
- N. N. Xia, X. M. Xiong, J. Wang, M. Z. Rong and M. Q. Zhang, Chem. Sci., 2016, 7, 2736–2742 RSC.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867 CrossRef CAS PubMed.
- P. Pissis, L. Apekis, C. Christodoulides, M. Niaounakis, A. Kyritsis and J. Nedbal, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1529–1539 CrossRef CAS.
- Y. Ya-Jen, H. Keith, S. W. Thomas and J. M. Duncan, Smart Mater. Struct., 2011, 20, 085010 CrossRef PubMed.
- H. Wen, S. Chen, Z. Ge, H. Zhuo, J. Ling and Q. Liu, RSC Adv., 2017, 7, 31525–31534 RSC.
- B. Willocq, F. Khelifa, J. Odent, V. Lemaur, Y. Yang, P. Leclère, J. Cornil, P. Dubois, M. W. Urban and J.-M. Raquez, ACS Appl. Mater. Interfaces, 2019, 11, 46176–46182 CrossRef CAS PubMed.
- B. Ghosh, K. V. Chellappan and M. W. Urban, J. Mater. Chem., 2012, 22, 16104–16113 RSC.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- Z. Yang, F. Wang, C. Zhang, J. Li, R. Zhang, Q. Wu, T. Chen and P. Sun, Polym. Chem., 2019, 10, 3362–3370 RSC.
- J. W. Larson and T. B. McMahon, Inorg. Chem., 1984, 23, 2029–2033 CrossRef CAS.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC.
- Y. J. Kim, P. H. Huh and B. K. Kim, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 468–474 CrossRef CAS.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- Y. Lin and G. Li, J. Mater. Chem. B, 2014, 2, 6878–6885 RSC.
- Z. Wang, C. Xie, C. Yu, G. Fei, Z. Wang and H. Xia, Macromol. Rapid Commun., 2018, 1700678 CrossRef PubMed.
- A. Feula, A. Pethybridge, I. Giannakopoulos, X. Tang, A. Chippindale, C. R. Siviour, C. P. Buckley, I. W. Hamley and W. Hayes, Macromolecules, 2015, 48, 6132–6141 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- Y. Xiao, H. Huang and X. Peng, RSC Adv., 2017, 7, 20093–20100 RSC.
- S. Chen, F. Mo, Y. Yang, F. J. Stadler, S. Chen, H. Yang and Z. Ge, J. Mater. Chem. A, 2015, 3, 2924–2933 RSC.
- M. Comí, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Eur. Polym. J., 2017, 91, 408–419 CrossRef.
- C. Wang, N. Liu, R. Allen, J. B. H. Tok, Y. Wu, F. Zhang, Y. Chen and Z. Bao, Adv. Mater., 2013, 25, 5785–5790 CrossRef CAS PubMed.
- S. K. Yadav and J. W. Cho, Appl. Surf. Sci., 2013, 266, 360–367 CrossRef CAS.
- J. Odent, J. M. Raquez, P. Dubois and E. P. Giannelis, J. Mater. Chem. A, 2017, 5, 13357–13363 RSC.
- M. Yoonessi, Y. Shi, D. A. Scheiman, M. Lebron-Colon, D. M. Tigelaar, R. A. Weiss and M. A. Meador, ACS Nano, 2012, 6, 7644–7655 CrossRef CAS PubMed.
- M. V. Biyani, E. J. Foster and C. Weder, ACS Macro Lett., 2013, 2, 236–240 CrossRef CAS.
- T. Engel and G. Kickelbick, Polym. Int., 2014, 63, 915–923 CrossRef CAS.
- V. Artel, R. Cohen, I. Aped, M. Ronen, D. Gerber and C. N. Sukenik, Langmuir, 2013, 29, 191–198 CrossRef CAS PubMed.
- P. Froimowicz, D. Klinger and K. Landfester, Chem.–Eur. J., 2011, 17, 12465–12475 CrossRef CAS PubMed.
- A. R. Smith and D. F. Watson, Chem. Mater., 2010, 22, 294–304 CrossRef CAS.
- R. Vaiyapuri, B. W. Greenland, S. J. Rowan, H. M. Colquhoun, J. M. Elliott and W. Hayes, Macromolecules, 2012, 45, 5567–5574 CrossRef CAS.
- S. Ahmad and M. S. Annette, Smart Mater. Struct., 2016, 25, 084018 CrossRef.
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- I. Balberg, Carbon, 2002, 40, 139–143 CrossRef CAS.
- L. Huang, N. Yi, Y. Wu, Y. Zhang, Q. Zhang, Y. Huang, Y. Ma and Y. Chen, Adv. Mater., 2013, 25, 2224–2228 CrossRef CAS PubMed.
- M. Kaushik, C. Fraschini, G. Chauve, J.-L. Putaux and A. Moores, in The Transmission Electron Microscope – Theory and Applications, InTech, 2015 Search PubMed.
- G. Fu, L. Yuan, G. Liang and A. Gu, J. Mater. Chem. A, 2016, 4, 4232–4241 RSC.
- P. Szatkowski, K. Pielichowska and S. Blazewicz, J. Mater. Sci., 2017, 52, 12221–12234 CrossRef CAS.
- B. Willocq, R. K. Bose, F. Khelifa, S. J. Garcia, P. Dubois and J. M. Raquez, J. Mater. Chem. A, 2016, 4, 4089–4097 RSC.
- J. T. Kim, B. K. Kim, E. Y. Kim, S. H. Kwon and H. M. Jeong, Eur. Polym. J., 2013, 49, 3889–3896 CrossRef CAS.
- J. Li, G. Zhang, L. Deng, S. Zhao, Y. Gao, K. Jiang, R. Sun and C. Wong, J. Mater. Chem. A, 2014, 2, 20642–20649 RSC.
- A. V. Menon, G. Madras and S. Bose, ACS Omega, 2018, 3, 1137–1146 CrossRef CAS PubMed.
- S. I. Seyed Shahabadi, J. Kong and X. Lu, ACS Sustainable Chem. Eng., 2017, 5, 3148–3157 CrossRef CAS.
- X. Zhang, Z. Tang, D. Tian, K. Liu and W. Wu, Mater. Res. Bull., 2017, 90, 175–181 CrossRef CAS.
- C. C. Corten and M. W. Urban, Adv. Mater., 2009, 21, 5011–5015 CrossRef CAS PubMed.
- W. Post, R. Bose, S. García and S. van der Zwaag, Polymers, 2016, 8, 436 CrossRef PubMed.
- C.-H. Park, S.-J. Kang, L. D. Tijing, H. R. Pant and C. S. Kim, Ceram. Int., 2013, 39, 9785–9790 CrossRef CAS.
- N. Zhong and W. Post, Composites, Part A, 2015, 69, 226–239 CrossRef CAS.
- I. Alessandri, Small, 2010, 6, 1679–1685 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- X. Luo and P. T. Mather, ACS Macro Lett., 2013, 2, 152–156 CrossRef CAS.
- E. D. Rodriguez, X. Luo and P. T. Mather, ACS Appl. Mater. Interfaces, 2011, 3, 152–161 CrossRef CAS PubMed.
- K. Imato, J. C. Natterodt, J. Sapkota, R. Goseki, C. Weder, A. Takahara and H. Otsuka, Polym. Chem., 2017, 8, 2115–2122 RSC.
- Y. Xu and D. Chen, Mater. Chem. Phys., 2017, 195, 40–48 CrossRef CAS.
- S. Delpierre, B. Willocq, G. Manini, V. Lemaur, J. Goole, P. Gerbaux, J. Cornil, P. Dubois and J.-M. Raquez, Chem. Mater., 2019, 31, 3736–3744 CrossRef CAS.
- F. Pilate, A. Toncheva, P. Dubois and J.-M. Raquez, Eur. Polym. J., 2016, 80, 268–294 CrossRef CAS.
- F. Pilate, R. Mincheva, J. De Winter, P. Gerbaux, L. Wu, R. Todd, J.-M. Raquez and P. Dubois, Chem. Mater., 2014, 26, 5860–5867 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- E. Dolci, V. Froidevaux, G. Michaud, F. Simon, R. Auvergne, S. Fouquay and S. Caillol, J. Appl. Polym. Sci., 2017, 134, 44408 CrossRef.
- D. Elena, M. Guillaume, S. Frédéric, F. Stéphane, A. Rémi and C. Sylvain, Green Mater., 2016, 4, 160–170 CrossRef.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- H. Zhang, Y. Chen, Y. Lin, X. Fang, Y. Xu, Y. Ruan and W. Weng, Macromolecules, 2014, 47, 6783–6790 CrossRef CAS.
- G. Hong, H. Zhang, Y. Lin, Y. Chen, Y. Xu, W. Weng and H. Xia, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, ACS Macro Lett., 2015, 4, 1307–1311 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jérémy Odent†
,
Jérémy Odent†