
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate controlled, regioselective carbopalladation for the one-pot synthesis of C4-substituted tetrahydroisoquinoline analogues†
Singarajanahalli Mundarinti Krishna Reddya,
Pavithira Suresha,
Subbiah Thamotharan b,
Jagadeesh Babu Nanuboluc,
Surisetti Suresh*d and
Subramaniapillai Selva Ganesan*a
b,
Jagadeesh Babu Nanuboluc,
Surisetti Suresh*d and
Subramaniapillai Selva Ganesan*a
aDepartment of Chemistry, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur-613401, Tamil Nadu, India. E-mail: selva@biotech.sastra.edu
bBiomolecular Crystallography Laboratory, Department of Bioinformatics, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur 613401, India
cCentre for X-ray Crystallography, CSIR-Indian Institute of Chemical Technology, Room No 150, Hyderabad-500007, India
dOrganic Synthesis & Process Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, India
First published on 21st April 2020
Abstract
6-Exo-trig cyclization reaction through regioselective carbopalladation was demonstrated with N-(2-halobenzyl)-N-allylamines to furnish the corresponding C4-substituted tetrahydroisoquinoline derivatives. The scope of the reaction was extended to the synthesis of C4-quaternary tetrahydroisoquinoline derivatives also. The nature of the substituent on the olefin moiety dictates the course of the carbopalladation sequence. Regioselective carbopalladation is substantiated by performing the reaction with unsymmetrical diallylated amine substrates.
Introduction
Tetrahydroisoquinolines are one of the key nitrogen heterocycles with innumerable biological activities.1a–i Noscapine, salsolinol, gigantine are representative examples of tetrahydroisoquinolines with anticancer, antihistaminic and hallucinogenic activities.2 A representative list of bioactive tetrahydroisoquinoline derivatives is given in Fig. 1. | ||
| Fig. 1 Selected bioactive tetrahydroisoquinoline analogues. | ||
Among tetrahydroisoquinolines, C4-substituted analogues are well-acclaimed subset due to their highly desirable pharmacologically relevant properties. Nomifensine is a C4-substituted isoquinoline analogue used as antidepressant agent without any sedative side effect.3 C4-substituted analogues are also used as serotonin reuptake inhibitors with histamine H3 antagonist activity4 and function as 4-hydroxytamoxifen analogues.5 Cherylline is a C4-substituted tetrahydroisoquinoline isolated from the natural sources.6
Despite several significances, only a limited number of examples are available for the one-pot synthesis of C4-substituted tetrahydroisoquinoline derivatives. These were prepared by the palladium catalysed [4+2] annulation reaction7a or superacid-catalyzed Pictet–Spengler cyclization reactions (Chart 1).7b Hu et al., reported palladium mediated aminoalkenylation of alkenes for the synthesis of C4-substituted tetrahydroisoquinoline derivatives.7c Nandakumar et al., reported the synthesis of similar tetrahydroquinoline by the intramolecular cyclization of more reactive alkynes.7d Broggini and co-workers reported palladium catalysed synthesis of C4-spiroannulated tetrahydroisoquinoline derivatives.8 However, the scope of the reaction was largely confined to N-allyl derivative.
 | ||
| Chart 1 Methods for the synthesis of C4-substituted tetrahydroisoquinolines and our methodology. | ||
Unlike N-allyl derivatives, 6-exo-trig cyclization of N-cinnamyl/N-crotyl derivatives is challenging due to the plausible formation of a mixture of isomers due to the regio- and stereochemical scrambling (vide infra). Since the generation of a convenient method to access these valuable pharmacophores is highly appreciated, it was decided to perform 6-exo-trig cyclization of terminal carbon substituted acyclic N-allylic amine derivatives with palladium catalysis (Chart 1).
Results and discussion
To obtain the highest level of stereocontrol in palladium catalysis, directing functional groups needs to be anchored on the alkene or the use of Ag or Tl salts is required.9a,b In the absence of any directing group or metal salt additive, it is difficult to secure a single isomeric product from the mixture of stereo-/regio-isomers (Scheme 1). | ||
| Scheme 1 Plausible products in Pd catalysed cyclization of 1a–l. | ||
Apart from the formation of a mixture of isomers as shown in Scheme 1, the competitive deallylation reaction of the substrate 1a-1l is yet another challenge.10 To suppress deallylation, allyl moiety needs to be tethered to a heterocyclic ring11 or reaction must be performed with N-allyl amides.12a,b These constraints limit the synthetic utility of the 6-exo-trig cyclization for accessing C4-substituted tetrahydroisoquinoline analogues.
It was previously reported in the literature that the nature of the substituent on the double bond plays a pronounced role in determining the type of the product formed in the reaction.13 Likewise, the use of alkyl amine would also facilitate the cyclization reaction by suppressing the undesired deallylation reaction. Hence, tertiary amine derivative 1a was designed to have a cinnamyl group anchored on the aliphatic amine. Reaction conditions were optimized with substrate 1a (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Temp. (°C) | Solvent | Time (h) | Yield (%) 3ab |
| a Unless otherwise mentioned, all the reactions were performed with aryl halide 1a (0.1 mmol), Pd catalyst (0.01 mmol) and base (0.2 mmol) in 3 mL of solvent.b Isolated Yield.c Presence of unreacted 1a was observed if the reaction mixture was quenched before 24 h.d PPh3 (0.02 mmol) was used as ligand. | ||||||
| 1 | Pd(dppf)Cl2 | Cs2CO3 | 100 | NMP | 26 | 60 |
| 2c | Pd2(dba)3 | Cs2CO3 | 110 | Toluene | 24 | 90 |
| 3 | Pd2(dba)3 | Cs2CO3 | 120 | DMF | 26 | 74 |
| 4 | Pd2(dba)3 | Cs2CO3 | 120 | DMSO | 24 | 65 |
| 5 | Pd2(dba)3 | Cs2CO3 | 130 | Xylene | 24 | 75 |
| 6 | Pd2(dba)3 | K2CO3 | 110 | Toluene | 24 | 60 |
| 7 | Pd2(dba)3 | Cs2CO3 | 80 | Toluene | 30 | 60 |
| 8d | Pd(OAc)2 | Cs2CO3 | 110 | Toluene | 24 | 95 |

Reaction performed with commercially available, pre-functionalized palladium phosphine complexes such as Pd(dppf)Cl2 gave the corresponding tetrahydroisoquinoline product 3a in 60% yield (Table 1, entry 1). It is of interest to mention here that the exclusive formation of the isomer 3a with an exocyclic double bond was observed in the reaction medium. The migration of exocyclic double bond to the corresponding stable endocyclic double bond through the reinsertion of the palladium followed by second elimination was not observed. Encouraged by this result, we decided to investigate further the role of palladium on the 6-exo-trig cyclization reaction. Gratifyingly, changing of palladium complex from Pd(dppf)Cl2 to Pd2(dba)3 (Pd2+ to Pd0) improved the product yield to 90% (Table 1, entry 2). Replacing the non-polar solvent toluene with polar aprotic solvents such as DMF or DMSO reduced the yield of the product 3a (Table 1, entries 3 and 4). Changing of base to K2CO3 or reducing the reaction temperature drastically affected the yield of the product 3a (Table 1, entries 6 and 7). Interestingly, replacing pre-functionalized palladium complexes with in situ generated palladium phosphine complexes using Pd(OAc)2/PPh3 gave the desired product 3a with 95% yield (Table 1, entry 8). Hence, it was decided to perform the reaction with Pd(OAc)2/PPh3. After optimizing the reaction conditions, the scope of the reaction was screened with diverse alkyl/aryl substituents.
The carbopalladation/cyclization reaction was found to be general for both N-(n-butyl) and N-ethyl amine derivatives (Fig. 2). The products 3a and 3b were obtained in appreciable yields. The replacement of N-ethyl substituent on the nitrogen atom with sterically more demanding tert-butyl substituent didn't show much effect on the yield of the product [3b (87%) vs. 3c (78%)]. These results show that the nature of the aliphatic substituent on the nitrogen atom doesn't influence much on the yield of the isoquinoline products 3.
 | ||
| Fig. 2 Substrate scope for 6-exo-trig cyclization reaction. | ||
Replacing the phenyl group on the terminal olefin with a relatively electron richer 4-methoxyphenyl group also did not affect the carbopalladation/cyclization sequence. The products 3d–f were obtained in high yields. In the case of benzylamine derivatives, the presence of methoxy substituent on the olefin aromatic moiety or amine did not alter the yield of the products 3g–h. The structure of the product 3g was confirmed unequivocally by single crystal XRD and it revealed that the product 3g adopts (Z)-configuration (Fig. 2). As speculated, the replacement of aliphatic substituent on the nitrogen atom with an aromatic moiety resulted in more amounts of unreacted substrate 1i in the reaction medium. An increase in the reaction time or reaction temperature did not improve the yield of the product 3i. Interestingly, the reaction carried out with N-aryl-N-allyl derivative gave the product 3j in 80% yield. Reaction performed with N,N-diallylated substrate gave the product 3k in 75% yield. Likewise, lipophilic N-octyl-N-allyl derivative also yielded the product 3l in good yields. In the synthesis of both 3k and 3l, cyclization proceeded with substituent-free allyl moiety itself. The formation of deallylated product was not observed even in the absence of any substituent at the terminal double bond. To demonstrate the practical utility of this method as a synthetic tool, we have performed a larger scale synthesis of 3c with 10 mmol of substrate 1c. It is imperative to mention that the desired product 3c was formed in 76% yield in this scale-up reaction. In addition, 3c synthesis performed with Pd(PPh3)4 also gave the product in 70% yields. This shows that Pd(0) plays a crucial role in this cyclization sequence.
Mechanistic investigations and quantum chemical calculations
It was previously reported in the literature that the facial selectivity of the olefin and the subsequent carbopalladation plays a crucial role in determining the product selectivity.14 Oxidative addition of palladium onto the aryl bromide could result in the formation of aryl palladium complex (Fig. 3). For the 6-exo-trig cyclization, intramolecular coordination of palladium with olefin moiety is necessary. We presume, during the carbopalladation, the phenyl substituent on the olefin tends to move away from the sterically more hindered triphenylphosphine ligand tethered palladium. This could have been resulted in the preferential formation of (Z)-isomer over (E)-isomer (Fig. 3). A detailed plausible mechanism is given in Scheme 3. | ||
| Fig. 3 Plausible cyclization pathway. | ||
If steric factor plays a crucial role in the carbopalladation, then the use of unsymmetrical diallylated substrate 1m–p or 1s would lead to regioselective carbopalladation at sterically less hindered olefin moiety. To check this hypothesis, acyclic amine substrates 1m–p were designed to have both substituted allyl and cinnamyl moieties tethered on to the nitrogen atom (Scheme 2(i) and (ii)). In the case of substrates 1m/1n, cyclization prefers sterically less hindered allyl moiety rather than relatively more steric cinnamyl moiety and the products 3m/3n are formed in good yields. Likewise, for the substrates 1o/1p, cyclization proceeds through sterically less hindered cinnamyl/allyl moiety than sterically more crowded 2,3-dimethylallyl moiety. Hence, products 3o/3p form in good yields. In scheme (i) and (ii), the formation of products 3m′/3n′ and 3o′/3p′ are not observed. The carbopalladation at 2,3-dimethyl allyl derivative 1q is challenging due to (i) the presence of sterically more demanding methyl groups, (ii) lack of β-hydrogen on the ring (Scheme 2(iii)). Interestingly, reaction performed with 1q gave the corresponding C4-quaternary isoquinoline derivative 3q. Thus the proposed method offers a facile method for the synthesis of C4-quaternary derivatives. However, the reaction performed with 1r failed to yield the desired product 3r. Lack of product selectivity was observed if the substrate (1s) possesses two cinnamyl groups with similar steric influence but varies in electronic effect (Scheme 2(iv)).
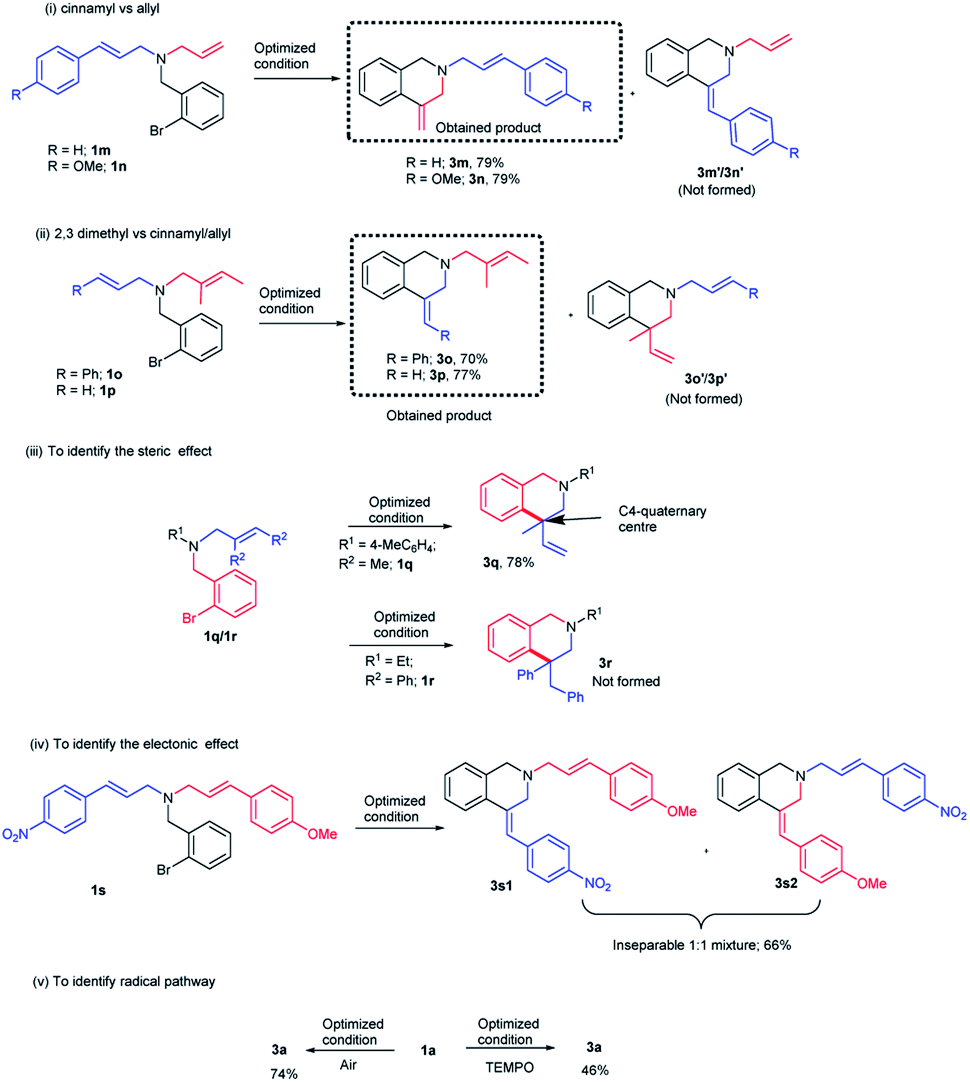 | ||
| Scheme 2 Regioselective carbopalladation and mechanistic studies. | ||
The carbopalladation/cyclization reaction performed under open-air reaction conditions also gave the product 3a in 74% yield (Scheme 2(v)). To further confirm the absence of radical pathway, the reaction was performed in the presence of radical quencher TEMPO (Scheme 2(v)). As predicted, TEMPO did not interfere with the course of the reaction and product 3a was obtained in 46% yield.
Quantum chemical calculations were performed using Gaussian09 program15 with B3LYP/6-311++G(d,p) level of theory for 1m and 1o. The result revealed that the electron density of the highest occupied molecular orbitals (HOMO) is localized over allyl and cinnamyl moieties and they are comparable (Fig. 4). Hence, the products 3o and 3m formed from 1o and 1m are largely driven by the steric factor. Studies performed on both (E) and (Z)-isomers of 3g revealed that the (Z)-isomer is 2.6 kcal mol−1 more stable form than the corresponding (E)-isomer (Fig. S1, ESI†). Based on the results obtained from mechanistic studies and quantum chemical calculations, we propose a plausible mechanism for the formation of the product.
 | ||
| Fig. 4 (I) HOMO of 1o; (II) HOMO of 1m. | ||
The addition of both Pd(OAc)2 and PPh3 could result in the formation of anionic palladium complex A (Scheme 3).16 Oxidative addition of anionic palladium on to the aryl halide followed by the coordination with olefin would result in the formation of palladium complex B1 or B2.
 | ||
| Scheme 3 Plausible mechanism for Pd-catalysed regioselective 6-exo-trig cyclization. | ||
The nature of the substituent on the olefin dictates this crucial carbopalladation step.13 If R = Me, the carbopalladation might proceed through sterically less hindered cinnamyl derivative via cycle I to form B1. In cycle I, the migratory insertion of the olefin on to the aryl palladium would result in the formation of complex C1. Likewise, if R = H, the reaction could proceed through cycle II and C2 is formed. The syn coplanar arrangement of the metal center and the β-hydrogen atom is required for the β-hydride elimination to take place in C1 and C2.17 The β-hydride elimination of the complex C1 and C2 could result in the formation of the product 3m and 3o respectively. Interestingly, the re-insertion of the palladium on to the product was not observed under the reaction conditions. Hence the formation of the product with endocyclic double bond was not found.
Conclusion
In conclusion, we have developed an intramolecular sequential carbopalladation and cyclization methodology for the synthesis of highly biologically relevant C4-substituted tetrahydroisoquinoline analogues. The prime advantages of this transformation are being the exclusive formation of (Z)-exo olefin group containing tetrahydroisoquinoline derivatives. The possibility for the generation tetrahydroisoquinolines with all carbon quaternary stereogenic centers at C4 carbon atom is also demonstrated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Council of Scientific and Industrial Research, CSIR (80(0085)/16/EMR-II) is gratefully acknowledged by S. S. G. The authors thank the SASTRA Deemed University for providing lab space and the NMR facility. We thank Director, CSIR-IICT for the support (IICT/Pubs./2019/315).References
- (a) J. D. Scott and R. M. Williams, Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed; (b) S. M. Devine, C. Yong, D. Amenuvegbe, L. Aurelio, D. Muthaih, C. Pouton, R. Callaghan, B. Capuano and P. J. Scammells, Synthesis and pharmacological evaluation of noscapine-inspired 5-substituted tetrahydroisoquinolines as cytotoxic agents, J. Med. Chem., 2018, 61, 8444–8456 CrossRef CAS PubMed; (c) N. Oku, S. Matsunaga, R. W. M. Vansoest and N. Fusetani, Renieramycin J, a highly cytotoxic tetrahydroisoquinoline alkaloid, from a marine sponge neopetrosia sp., J. Nat. Prod., 2003, 66, 1136–1139 CrossRef CAS PubMed; (d) Y. Fang, H. Zhou, Q. Gu and J. Xu, Synthesis and evaluation of tetrahydroisoquinoline–benzimidazole hybrids as multifunctional agents for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2019, 167, 133–145 CrossRef CAS PubMed; (e) A. S. Capilla, R. Soucek, L. Grau, M. Romero, J. R. Martnez, D. H. Caignard and M. D. Pujol, Substituted tetrahydroisoquinolines: synthesis, characterization, antitumor activity and other biological properties, Eur. J. Med. Chem., 2018, 145, 51–63 CrossRef CAS PubMed; (f) M. Kono, T. Oda, M. Tawada, T. Imada, Y. Banno, N. Taya, T. Kawamoto, H. Tokuhara, Y. Tomata, N. Ishii, A. Ochida, Y. Fukase, T. Yukawa, S. Fukumoto, H. Watanabe, K. Uga, A. Shibata, H. Nakagawa, M. Shirasaki, Y. Fujitani, M. Yamasaki, J. Shirai and S. Yamamoto, Discovery of orally efficacious RORγt inverse agonists. part 2: design, synthesis, and biological evaluation of novel tetrahydroisoquinoline derivatives, Bioorg. Med. Chem., 2018, 26, 470–482 CrossRef CAS PubMed; (g) M. Kairuki, Q. Qiu, M. Pan, Q. Li, J. Zhou, H. Ghaleb, W. Huang, H. Qian and C. Jiang, Designed P-glycoprotein inhibitors with triazol-tetrahydroisoquinoline-core increase doxorubicin-induced mortality in multidrug resistant K562/A02 cells, Bioorg. Med. Chem., 2019, 27, 3347–3357 CrossRef CAS PubMed; (h) Y. S. Li, X. Y. Liu, D. S. Zhao, Y. X. Liao, L. H. Zhang, F. Z. Zhang, G. P. Song and Z. N. Cui, Tetrahydroquinoline and tetrahydroisoquinoline derivatives as potential selective PDE4B inhibitors, Bioorg. Med. Chem. Lett., 2018, 28, 3271–3275 CrossRef CAS PubMed; (i) H. Watanabe, K. Fukui, Y. Shimizu, Y. Idoko, Y. Nakamoto, K. Togashi, H. Saji and M. Ono, Synthesis and biological evaluation of F-18 labeled tetrahydroisoquinoline derivatives targeting orexin 1 receptor, Bioorg. Med. Chem. Lett., 2019, 29, 1620–1623 CrossRef CAS PubMed.
- I. P. Singh and P. Shah, Tetrahydroisoquinolines in therapeutics: a patent review (2010–2015), Expert Opin. Ther. Pat., 2017, 27, 17–36 CrossRef CAS PubMed and references cited therein.
- Z. L. Zhao, Q. L. Xu, Q. Gu, X. Y. Wu and S. L. You, Enantioselective synthesis of 4-substituted tetrahydroisoquinolines via palladium-catalyzed intramolecular Friedel–Crafts type allylic alkylation of phenols, Org. Biomol. Chem., 2015, 13, 3086–3092 RSC and references cited therein.
- X. Deng, J. T. Liang, J. Liu, H. McAllister, C. Schubert and N. S. Mani, A practical synthesis of enantiopure 7-alkoxy-4-aryl-tetrahydroisoquinoline, a dual serotonin reuptake inhibitor/histamine H3 antagonist, Org. Process Res. Dev., 2007, 11, 1043–1050 CrossRef CAS.
- M. Kihara, M. Ikeuchi, A. Yamauchi, M. Nukatsuka, H. Matsumoto and T. Toko, Synthesis and biological evaluation of 7-hydroxy-3,4-diphenyl-1,2-dihydroisoquinolines as new 4-hydroxytamoxifen analogues, Chem. Pharm. Bull., 1997, 45, 939–943 CrossRef CAS PubMed.
- A. Brossi, G. Grethe, S. Teitel, W. C. Wildman and D. T. Bailey, Cherylline, a 4-phenyl-1,2,3,4-tetrahydroisoquinoline alkaloid, J. Org. Chem., 1970, 35, 1100–1104 CrossRef CAS.
- (a) X. Vidal, J. L. Mascarenas and M. Gulias, Palladium-catalyzed, enantioselective formal cycloaddition between benzyltriflamides and allenes: straightforward access to enantioenriched isoquinolines, J. Am. Chem. Soc., 2019, 141, 1862–1866 CrossRef CAS PubMed; (b) S. Nakamura, M. Tanaka, T. Taniguchi, M. Uchiyama and T. Ohwada, Stereoselectivity of superacid-catalyzed Pictet-Spengler cyclization reactions, Org. Lett., 2003, 5, 2087–2090 CrossRef CAS PubMed; (c) Y. Hu, Y. Xie, Z. Shen and H. Huang, Palladium-catalyzed ring-forming aminoalkenylation of alkenes with aldehydes initiated by intramolecular aminopalladation, Angew. Chem., Int. Ed., 2017, 56, 2473–2477 CrossRef CAS PubMed; (d) A. Nandakumar, D. Muralidharan and P. T. Perumal, Synthesis of functionalized tetrahydroisoquinolines via palladium-catalyzed 6-exo-dig carbocyclization of 2-bromo-N-propargylbenzylamines, Tetrahedron Lett., 2011, 52, 1644–1648 CrossRef CAS.
- E. M. Beccalli, G. Broggini, M. Martinelli, N. Masciocchi and S. Sottocornola, New 4-spiroannulated tetrahydroisoquinolines by a one-pot sequential procedure. Isolation and characterization of σ-alkylpalladium Heck intermediates, Org. Lett., 2006, 8, 4521–4524 CrossRef CAS PubMed.
- (a) K. Karabelas, C. Westerlund and A. Hallberg, The effect of added silver nitrate on the palladium-catalyzed arylation of allyltrimethylsilanes, J. Org. Chem., 1985, 50, 3896–3900 CrossRef CAS; (b) R. Grigg, V. Loganathan, V. Sridharan, P. Stevenson, S. Sukirthalingam and T. Worakun, Palladium catalysed tandem cyclisation – anion capture processes. Part 2. Cyclisation onto alkynes or allenes with hydride capture, Tetrahedron, 1996, 52, 11479–11502 CrossRef CAS.
- F. G. Helion, A. Merzouk and F. Guibe, Mild and selective palladium(0)-catalyzed deallylation of allylic amines. Allylamine and diallylamine as very convenient ammonia equivalents for the synthesis of primary amines, J. Org. Chem., 1993, 58, 6109–6113 CrossRef.
- S. Nandi, S. Samanta, S. Jana and J. K. Ray, Synthesis of substituted benzimidazo[2,1-a]isoquinolines and its condensed analogues using Pd(0)-catalyzed cyclization/C–H activation, Tetrahedron Lett., 2010, 51, 5294–5297 CrossRef CAS.
- (a) R. D. Heeten, L. J. P. van der Boon, D. L. J. Broere, E. Janssen, F. J. J. de Kanter, E. Ruijter and R. V. A. Orru, Concise synthesis of highly substituted benzo[a]quinolizines by a multicomponent reaction/allylation/Heck reaction sequence, Eur. J. Org. Chem., 2012, 275–280 CrossRef; (b) A. Dhami, M. F. Mahon, M. D. Lloyd and M. D. Threadgill, 4-Substituted 5-nitroisoquinolin-1-ones from intramolecular Pd-catalysed reaction of N-(2-alkenyl)-2-halo-3-nitrobenzamides, Tetrahedron, 2009, 65, 4751–4765 CrossRef CAS.
- U. M. Estaibalez, O. G. Calvo, V. O. de-Elguea, N. Sotomayor and E. Lete, Intramolecular Mizoroki–Heck reaction in the regioselective synthesis of 4-alkylidene-tetrahydroquinolines, Eur. J. Org. Chem., 2013, 3013–3022 CrossRef.
- D. A. Petrone, H. Yoon, H. Weinstabl and M. Lautens, Additive effects in the palladium-catalyzed carboiodination of chiral N-allyl carboxamides, Angew. Chem., Int. Ed., 2014, 53, 7908–7912 CrossRef CAS PubMed and references cited therein.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- C. Amatore and A. Jutand, Anionic Pd(0) and Pd(II) intermediates in palladium-catalyzed Heck and cross-coupling reactions, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed.
- T. M. Gogsig, New Discoveries on the β-Hydride Elimination, Springer, Berlin Heidelberg, 2012, DOI:10.1007/978-3-642-32099-6.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1912875. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01539c |
| This journal is © The Royal Society of Chemistry 2020 |