
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultidimensional assembly using layer-by-layer deposition for synchronized cardiac macro tissues†
Yongjun
Jang
 a,
Da Jung
Jung
b,
Seung-Cheol
Choi
c,
Do-Sun
Lim
c,
Jong-Hoon
Kim
d,
Gi Seok
Jeoung
*b,
Jongseong
Kim
*a and
Yongdoo
Park
*a
a,
Da Jung
Jung
b,
Seung-Cheol
Choi
c,
Do-Sun
Lim
c,
Jong-Hoon
Kim
d,
Gi Seok
Jeoung
*b,
Jongseong
Kim
*a and
Yongdoo
Park
*a
aDepartment of Biomedical Sciences, College of Medicine, Korea University, Seoul, Korea. E-mail: envokim72@korea.ac.kr; ydpark67@korea.ac.kr
bBiomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea. E-mail: gsjeong@amc.seoul.kr
cDepartments of Cardiology, Cardiovascular Center, Korea University Anam Hospital, Seoul, Korea
dDepartment of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul, Korea
First published on 18th May 2020
Abstract
The fabrication of biomimetic structures for tissues and organs is emerging in the fields of biomedical engineering and precision medicine. While current progress in biomedical research provides a number of biofabrication methods, the construction of multi-dimensional cardiac tissue is highly challenging due to difficulties in the maturation and synchronization of cardiomyocytes (CMs) in conjunction with other types of cells, such as myofibroblasts and endothelial cells. Here, we show a simple fabrication methodology to construct multi-dimensional cardiac macro tissue (mCMT) by layer-by-layer (LBL) deposition of cells on micro patterned PDMS. mCMTs formed by LBL deposition of pluripotent stem cell (PSC)-derived cardiomyocytes and cardiac fibroblasts formed 3D patterned structures with synchronized beating characteristics. We also demonstrate that cardiac maturation factors such as the gene expression of MLC2v and cTNI and formation of sarcomeres in mCMTs were significantly enhanced by LBL deposition and growth factors during the maturation process. Fabrication of matured mCMTs with synchronized beating enables providing an efficient platform for evaluating the efficacy and toxicity of drug candidates. These results have important implications because mCMTs are applicable to diverse in vitro studies and drug screening methods that require tissue-like structures and functions in a physiological environment.
1 Introduction
The fabrication of laboratory-grown macro tissues consisting of cells, a matrix, and functional biomolecules has been in high demand due to their great potential as a core platform for laboratory and clinical applications.1–5 Assembling and organizing tissue-like structures from individual cells, matrices, and biomolecules require morphogenic guidance by biochemical and/or biophysical cues to be successful in the formation of biologically functional mimetics. In particular, morphogenic guidance by macroscale geometry confinement and microscale cell–cell interactions is quintessential for the assembly of biologically functional macro tissues, which requires fabrication methodology in multiple dimensions.6–9 For example, a magnetic alginate microfiber was used as a template to assemble cells to fabricate microvascular-like structures.10 The macroscale anisotropic alignment of cardiac cells affected the formation of cell–cell junctions.11 The cellular capability of self-organization was enhanced by filament-shaped micropatterning.12As regenerative agents and drug screening platforms, cardiac micro tissues from pluripotent stem cell (PSC)-derived cardiomyocytes (CMs) have been extensively developed but also show limitations for their use in heart disease and cardiotoxicity screening. To fabricate biologically functional cardiac tissues, myocardial cells were transformed into various patterns designed by the use of two-dimensional (2D) topography, resulting in three-dimensional (3D) cardiac tissue.13–23 Although such 2D patterning facilitated the formation of cardiac tissue, the anisotropic alignment of CMs, which is quintessential for cardiac tissue, was hampered by a lack of 3D structural information. Therefore, a 3D matrix was utilized to overcome the limitation of the 2D scaffolds.24–30 For example, electrospinning and collagen patterning allowed for guiding cell–cell interactions and thereby the 3D alignment of CMs.31–35 A cardiac biowire was used as a platform for alignment, maturation and drug assessment using surgical sutures and collagen type I.36 A myocardial patch from epicardiac cells was used to induce the alignment of the myocardium.11 The use of a nanofiber enabled the anisotropic alignment of myocardial cells with a scaffold.37
To achieve biologically functional cardiac tissues from stem cell-derived CMs, there are two essential features: the maturation of embryonic stem cells to an adult phenotype38,39 and the synchronization of contractile cardiac tissue through cell–cell junctions to avoid the arrhythmogenic potential of asynchronous stem cells.40,41 In particular, cell-to-cell interactions and mechanical coupling between CMs and stromal tissue play key roles not only in maturation but also in synchronized cardiac beating under the influence of their microenvironment.42–47 Since cardiac tissue regulates beating by mediating the mechanical and electrical signals between them, enhancing the spatial networks among cardiac cells attenuates the beating fluctuation. For instance, the mechanical connection of fibroblasts to CMs allowed the regulation of electrical signals and beating.48–51 Therefore, the fabrication of a biomimetic architecture with biophysical integrity is important for constructing in vitro cardiac tissues with synchronized beating properties.
In this study, we developed accordion-like honeycomb (ALH) patterned multi-dimensional cardiac macro tissues (mCMTs) by layer-by-layer (LBL) deposition and centrifugation, which yielded cardiac tissues with synchronized beating.37,52,53 Our finding showed that the addition of fibroblast growth factor 4 (FGF4) and ascorbic acid facilitated cellular maturation and contractility in the mCMT. We also demonstrated that mCMTs prepared by LBL and growth factors could be used to evaluate the efficacy and toxicity of drug candidates. Our findings have important implications for exploring the use of biomimetic mCMTs in biomedical engineering and precision medicine.
2 Methods
2.1 Fabrication of ALH-patterned substrates
To generate the ALH micropattern, a PDMS (Sylgard® 184, Dow Chemical Co., Midland, MI, USA) microchannel was replicated from a master mold. The master mold was made using conventional soft lithography on a silicon wafer (SU-8, MicroChem, Westborough, MA, USA). A mixture of a PDMS prepolymer containing a PDMS precursor and curing agent in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was decanted into the SU-8 mold and polymerized in a drying oven (at 60 °C) for 2 h. For construction of the round shaped channel, PDMS prepolymer was poured on the fabricated PDMS master mold and then removed the poured PDMS from the master mold. Then, the master mold was cured again using a drying oven (at 60 °C for 2 h).54,55 Finally, a PDMS microstructure (8 × 10 × 5 mm) containing 196 patterns (14 × 14) arrayed in an accordion-like channel (200 × 300 μm, width and depth) was fabricated (Fig. S1†).
:1 ratio was decanted into the SU-8 mold and polymerized in a drying oven (at 60 °C) for 2 h. For construction of the round shaped channel, PDMS prepolymer was poured on the fabricated PDMS master mold and then removed the poured PDMS from the master mold. Then, the master mold was cured again using a drying oven (at 60 °C for 2 h).54,55 Finally, a PDMS microstructure (8 × 10 × 5 mm) containing 196 patterns (14 × 14) arrayed in an accordion-like channel (200 × 300 μm, width and depth) was fabricated (Fig. S1†).
2.2 hPSC culture and differentiation into CMs
Human embryonic stem cells (hESC) (H9, WiCell Research Institute, Madison, WI, USA) were maintained on Matrigel (BD Biosciences, San Jose, CA, USA)-coated plates in Essential 8™ medium (Gibco, Grand Island, NY, USA). hESCs were dissociated into single cells with Accutase (Sigma-Aldrich, Saint Louis, MO, USA) at 37 °C for 3 min and then seeded onto a Matrigel-coated 6-well plate at 200000 cells per cm2 in Essential 8™ medium (Gibco) supplemented with 2 μM thiazovivin (Sigma-Aldrich) for 24 h. Thiazovivin was used to inhibit Rho kinase activity due to their role in maintaining stem cells.56 Our cardiac differentiation protocol is based on temporal modulation of canonical Wnt signalling method.57 At day 0, cells were treated with 6 μM CHIR99021 (Sigma-Aldrich) in RPMI 1640 medium (Gibco) with B-27™ supplement (without insulin, Gibco) for 2 days. At day 2, cells were treated with 2 μM of IWP2 (Tocris, Bristol, UK) in RPMI 1640 medium with B-27™ supplement (without insulin) for 2 days. At day 4, the medium was changed daily. At day 7, the medium was changed to RPMI 1640 medium with B-27™ supplement (without vitamin A, Gibco), following the medium change at every 2 days. The hESCs were harvested at day 12 for mCMT fabrication. Human induced pluripotent stem cells (hiPSCs)-derived CMs (Cardiosight-S®, NEXEL Co., Ltd, Seoul, South Korea) were cultured on Matrigel-coated plates in RPMI 1640 medium with B-27™ supplement (without vitamin A) for 4 days by changing the medium at every 2 days and then harvested for mCMT construction.
2.3 Multi-dimensional cardiac macro tissue (mCMT) fabrication
To fabricate mCMT, we utilized hESC-derived CMs, hiPSCs-drived CMs, and cardiac fibroblasts (NHCF-V, Normal Human Ventricular Cardiac Fibroblast, LONZA, MD, USA) that were cultured in FBM™ basal medium with FGM™-3 SingleQuot Supplements (Lonza). The solution for gelation (collagen concentration, 2 mg ml−1) was prepared by mixing collagen type I (rat tail, BD Biosciences) with phosphate-buffered saline (PBS) supplemented with phenol red, and the pH was adjusted to 7.4 using 0.5 N NaOH. For construction of mixture cardiac patches, the harvested CM pellet and the CFB pellet were mixed with 400 μl of type I collagen solution. Next, the mixture of 1 × 106 CMs and 2 × 105 CFBs were loaded into the ALH-patterned mold and centrifuged for 1 min (190 rcf). For the LBL cardiac patch, the CM pellet and the CFB pellet were mixed with 300 μl and 100 μl of type I collagen solution, respectively. Next, 1 × 106 cells/300 μl of CMs mixture were loaded into the ALH-patterned mold and centrifuged for 1 min (190 rcf). Sequentially, 2 × 105 cells/100 μl of CFBs mixture were loaded into the ALH-patterned mold on top of the CM-loaded gel followed by centrifugation for 1 min (190 rcf). After gelation in a humidified 5% CO2 incubator at 37 °C for 40 min, RPMI 1640 medium (Gibco) with or without 20 ng ml−1 FGF4 (Sigma-Aldrich) and 200 μg ml−1L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma-Aldrich) were added and placed back in the incubator for maturation. The mCMT was cultured for 6 days after construction of each patch by changing the medium every 2 days. The mCMT was cultured in PDMS mold for first 2 days and then incubated in 35 mm cell culture dish for following 4 days.2.4 Immunofluorescence staining and image analysis
mCMTs were washed with PBS (Gibco) and fixed with 4% paraformaldehyde (PFA) solution (Sigma-Aldrich) for 30 min at room temperature. The patches were permeabilized with 0.4% Triton X-100 for 20 min and blocked with 5% bovine serum albumin (BSA, Sigma-Aldrich) for 45 min at room temperature. After overnight incubation at 4 °C with primary antibodies, which included cardiac troponin T mouse (Thermo Scientific, Waltham, MA, USA), MLC2v rabbit (MYL2, Proteintech, Rosemont, IL, USA), or FSP1 rabbit (fibroblast-specific protein-1, Millipore, Billerica, MA, USA) at dilutions of 1:300, 1:200 and 1:200 in 5% BSA solution, respectively, each patch was washed five times with PBST for 15 min each. Alexa Fluor 488 goat anti-mouse IgG (H+L) for cardiac troponin T and Alexa Fluor 594 goat anti-rabbit IgG (H+L) (Life Technologies, Carlsbad, CA, USA) for MLC2v or FSP1 were added as secondary antibodies at dilutions of 1:1000, 1:500 and 1:600 in 5% BSA solution, respectively, and then incubated for 3 h at room temperature. 4′,6-Diamidino-2-phenylindole, dihydrochloride (DAPI) (1:2000; Molecular Probes, Carlsbad, CA, USA) was also added to each sample for nuclear staining. After staining, the samples were washed again five times with PBST for 15 min each. Samples were then visualized using an LSM 800 (Carl Zeiss, Oberkochen, Germany) confocal fluorescence microscope. ImageJ software was used for image analysis. Co-localization of cTNT and MLC2v was evaluated by utilizing ‘coloc2’ function in the imageJ software. Coloc2 illustrates the degree of correlation through the pixel intensity as a Pearson's value.
2.5 Quantitative PCR
Total RNA was extracted from each mCMT patch on day 6 using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), with slight modifications of the manufacturer's recommendations. In short, each sample was homogenized by pipetting 1 ml of TRIzol reagent for 5 min and then mixed with 0.2 ml of chloroform (Sigma), followed by vertexing it for 15 s. The resulting mixture was centrifuged at 15000 rpm for 15 min at 4 °C and then the colorless supernatant was transferred to a new tube which was gently mixed with the same amount of isopropanol (Sigma). The supernatant mixture was centrifuged at 15000 rpm for 10 min at 4 °C, yielding a RNA pellet. After removing the supernatant, the RNA pellet was washed with 500 μl of 75% ethanol and centrifuged at 15000 rpm for 10 min at 4 °C. This step was repeated twice. To remove the ethanol completely, the pellet was dried in a dry oven (56 °C) for 5 min. The dried RNA pellet was dissolved in nuclease free duplex buffer (Integrated DNA Technologies, Coralville, IA, USA). The concentration of extracted mRNA was measured by using a NanoDrop 2000c (Thermo Fisher Scientific). Extracted RNA (∼500 ng) was reverse transcribed into complementary DNA (cDNA) using PrimeScript™ (cDNA synthesis kit; Takara, Shiga, Japan). PCR was performed with iQSYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) using a MyiQ 2 real-time PCR detection system (Bio-Rad). The relevant primers are listed in ESI Table 1,† and β-actin served as the reference gene. The expression level of each gene was normalized to that of the control group by the ddCt method.
2.6 Beating analysis using a captured movie
The contractile properties of CMTs were estimated by analyzing the movies taken with a microscope (Nikon, Ti2-E, Tokyo, Japan) at 40× magnification. Movies were captured at 50 frames per second and then analyzed by NIS software (Nikon), which counts the variation in light intensity in a selected region during 15 s. The beating kinetics of each sample were estimated to identify beating regularity and homogeneity.2.7 Drug treatment and evaluation studies
Treatment of drugs was performed on LBL GF+ mCMT patches inside incubator at day 6 after seeding. Fluo-4 solution was prepared by adding 50 μl of pluronic F-127 (20%) solution in DMSO (Invitrogen) into 50 μg of fluo-4, AM (Invitrogen) and used to measure calcium transient of mCMTs. mCMT patches were moved to 12 well plate. 1250 μl of RPMI 1640 medium with B-27™ supplement (without vitamin A) with calcium chloride (Sigma) solution to make 2 mM of Ca2+ and 15 μl of Fluo-4 solution was added to each patch well prior to drug dose experiments. A dilution of the drugs in media was added in 250 μl doses. Sample were incubated and recorded at 0 h, 0.5 h and 2 h time points. The final concentration of the drug used was 10 μM for isoproterenol (Sigma), 30 μM for ruthenium red (Abcam, Cambridge, UK), and 10 μM for yoda1 (Sigma).2.8 Statistical analysis
Statistical analyses were performed to evaluate the differences between the experimental groups using one-way or two-way ANOVA (SPSS, IBM Corporation, NY, USA). Levene's test was performed to determine variance homogeneity. If the variance is equal, Scheffe post hoc test was performed. Dunnett's T3 post hoc test was used if the variances were not equal.3 Results
3.1 Fabrication of multi-dimensional cardiac macro tissues (mCMTs)
To prepare mCMT, we selected human pluripotent stem cell (hPSC)-derived CMs and human cardiac fibroblasts (hCFBs) due to the synergetic role of hCFBs on CM maturation.58 A square frame on the ALH patterned PDMS mold was utilized to fabricate mCMT by placing both hPSC-CM and hCFB cells through simple centrifugation of the cell–collagen mixture (Fig. 1A and B).52 The LBL method followed by the sequential centrifugation of hPSC-CM-collagen type 1 and hCFB-collagen type I was performed, resulting in biphasic mCMT, with each layer composed of a dominant layer of hPSC-CMs and hCFBs, respectively. Alternatively, a mixture of hPSC-CMs, hCFBs, and collagen type I was centrifugally precipitated into the ALH scaffold to form mixed CMT (Fig. 1C). After incubation in culture media, the mCMT imbedded in the collagen gel lifted up from the ALH scaffold, yielding a patterned patch-like structure surrounded by a frame. In the use of drug molecules, the mCMT patch was utilized to measure beating characteristics and calcium transient (Fig. 1A and B).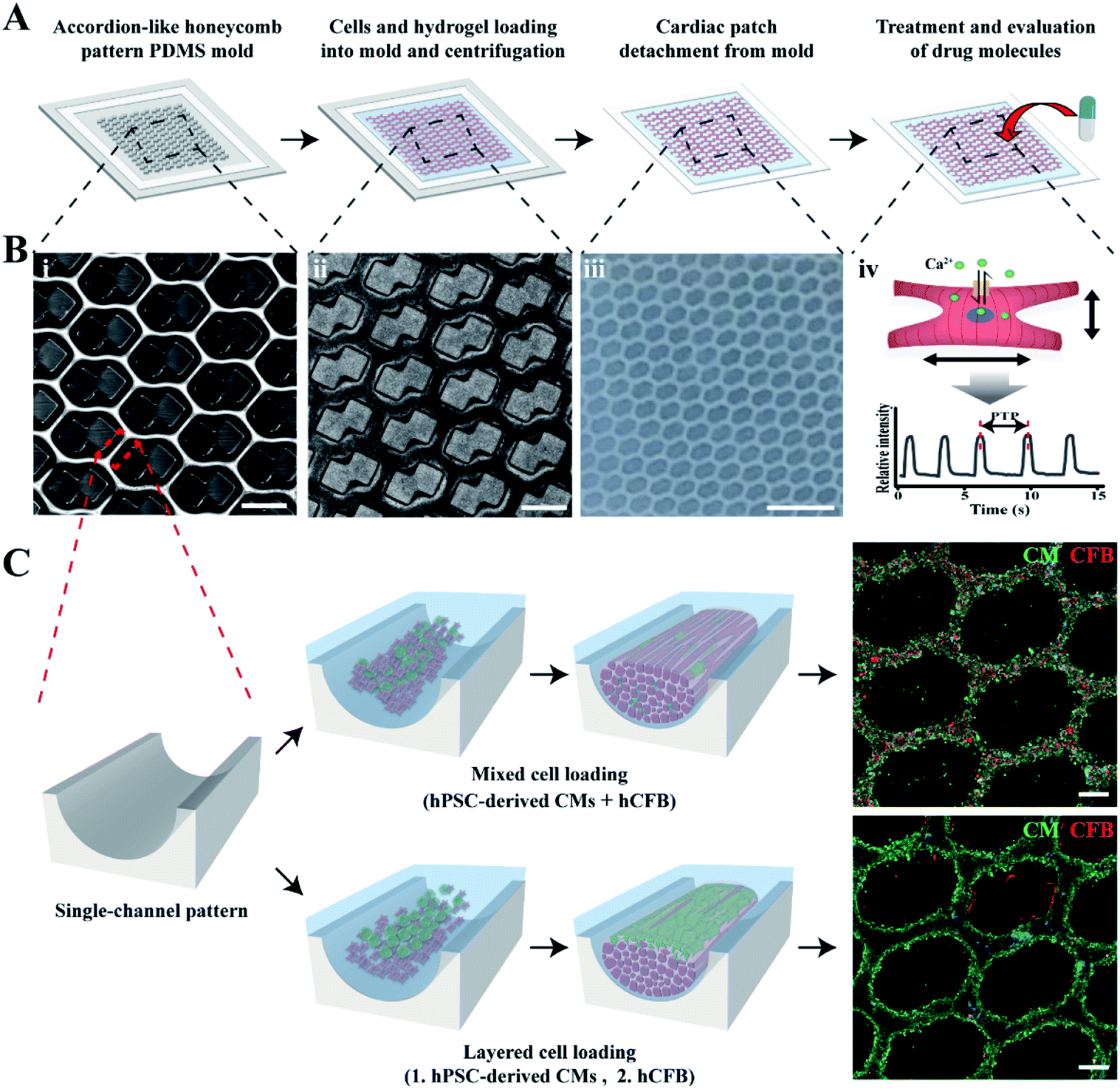 | ||
| Fig. 1 Fabrication of cardiac microtissues by LBL or the mixture deposition of cells in an accordion-like honeycomb (ALH) pattern. A representative scheme to prepare cardiac patches for drug test (A) and image of the ALH pattern mold (B(i)), ALH pattern mold with patterned cell image (B ii), cardiac patch detached from the ALH pattern mold (B(iii)) and concept of drug evaluation (B(iv)). (C) Schematic image of different cell deposition according to cell loading method to make LBL and mixture cardiac patches. Immunofluorescent images represent differences in cell deposition. Scale bars: 500 μm in (B(i and ii)), 2.5 mm in (B(iii)) and 200 μm in (C). | ||
The maturation of mCMT was monitored to examine the effect of FGF4 and ascorbic acid and used for cell deposition by either LBL or simple mixture gelation (Fig. 2). The microscopic images of mCMT revealed quite well-patterned patches not only in the poly(dimethylsiloxane) (PDMS) mold (days 2 and 3) but also in detached gels (day 6), which suggests that our method by the use of the ALH scaffold and centrifugation is suitable for the preparation of mCMT and its in situ maturation (Fig. S2†). Notably, all four groups processed by the mixture or LBL gelation with or without treatment with FGF4 and ascorbic acid maintained the integrity of the ALH-patterned structure.
 | ||
| Fig. 2 Bright field microscope image of cardiac patches in the mold on day 2 and detached cardiac patches on day 6. Scale bar: 500 μm. | ||
3.2 Cell distribution of mCMTs in the ALH patterned mold
Confocal fluorescence microscopy was used to identify a specific cell type at a location within the ALH-patterned mCMT and thereby to validate the cell deposition methods by LBL and the mixture. To this end, we stained CMs and fibroblasts with cardiac troponin T (cTNT; green) which is a specific marker present in the actin site of CMs and fibroblast-specific marker 1 (FSP1; red) for CFBs, respectively (Fig. 3). For the quantification, the ratio of FSP1/cTNT area in each bottom layer was normalized as 1 and then the relative ratio of FSP1/cTNT area in each corresponding top layer was estimated to that of the bottom layer. In the mixture, at day 1, both cTNT and FSP1 coexisted in the bottom and top layers of the mCMT, indicating that CMs and hCFBs formed entangled mCMT (Fig. 3). However, the LBL sample at day 1 showed that cTNT was dominant in the bottom layers, while FSP1 was populated only in the top layer (Fig. 3). This result suggests that the LBL method is suitable for constructing mCMT with specific cell types. After 6 days of further maturation, the mixture CMT maintained its initial characteristics, with a slight change in the cell population. Thus, CMs and hCFBs coexisted in the bottom and top regions of the CMT mixture (Fig. 3 and S3†). The LBL CMT, each CM and hCFB dominant layers existed in the bottom and top regions, respectively (Fig. 3 and S3†).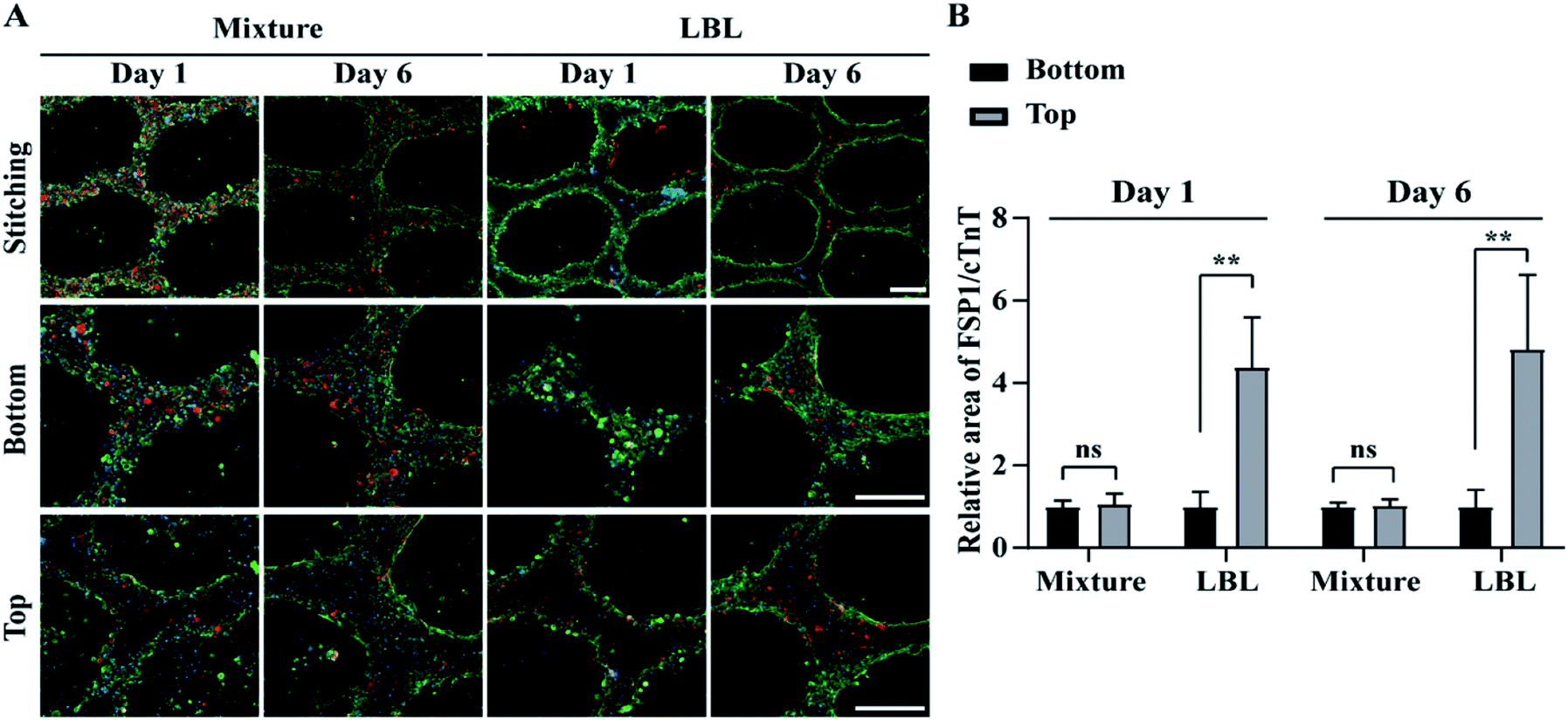 | ||
| Fig. 3 Distribution of hPSC-derived CMs and CFBs in mCMT by LBL and mixture deposition at day 1 and 6, using confocal fluorescence microscopy. (A) Cardiac troponin T (cTNT; green), fibroblast-specific protein 1 (FSP1; red) and nucleus (blue) were immunohistochemically stained, respectively. (B) Quantitative analysis of FSP1 area relative to cTNT in bottom and top of mCMTs. Scale bars are 200 μm. *P < 0.05, **P < 0.01, ***P < 0.001, unpaired two-tailed Student's t-test. Note that N = 4 per each group for graph B and the error bars were estimated by the standard deviation. | ||
3.3 Beating properties of CMTs
Spontaneous beating of mCMTs were observed by inverted optical microscopy as early as 1–2 days after cell deposition. Representative beating patterns of each group, which were analysed by real-time images of mCMTs beating, are shown in Fig. 4 (from day 2 to day 6). mCMTs with no FGF4 and ascorbic acid treatment (GF−) revealed that the beating frequency was not synchronized in either the mixture or the LBL groups at days 2 and 6, respectively (Fig. 4A and B). For mCMTs with FGF4 and ascorbic acid treatment (GF+), however, the frequency was well synchronized in the LBL group but not in the mixture group after 6 days of incubation, although synchronized beating was not observed in any sample at day 2. We further analysed mCMT behaviour (contraction, relaxation, and peak-to-peak duration) at four time points: days 2, 3, 5, and 6. The contraction of mCMTs was increased until day 5 and then decreased at day 6. The relaxation and peak-to-peak duration of mCMTs showed a similar trend, with a variation in beating spots in the LBL GF+ group being significantly lower than that of all other groups after 6 days of incubation. Therefore, FGF4 and ascorbic acid enhanced the beating regularity of CM, which was synergistically increased in the LBL mCMTs. This result suggests that the LBL deposition method with chemical supplements improves the synchronization of mCMTs beating.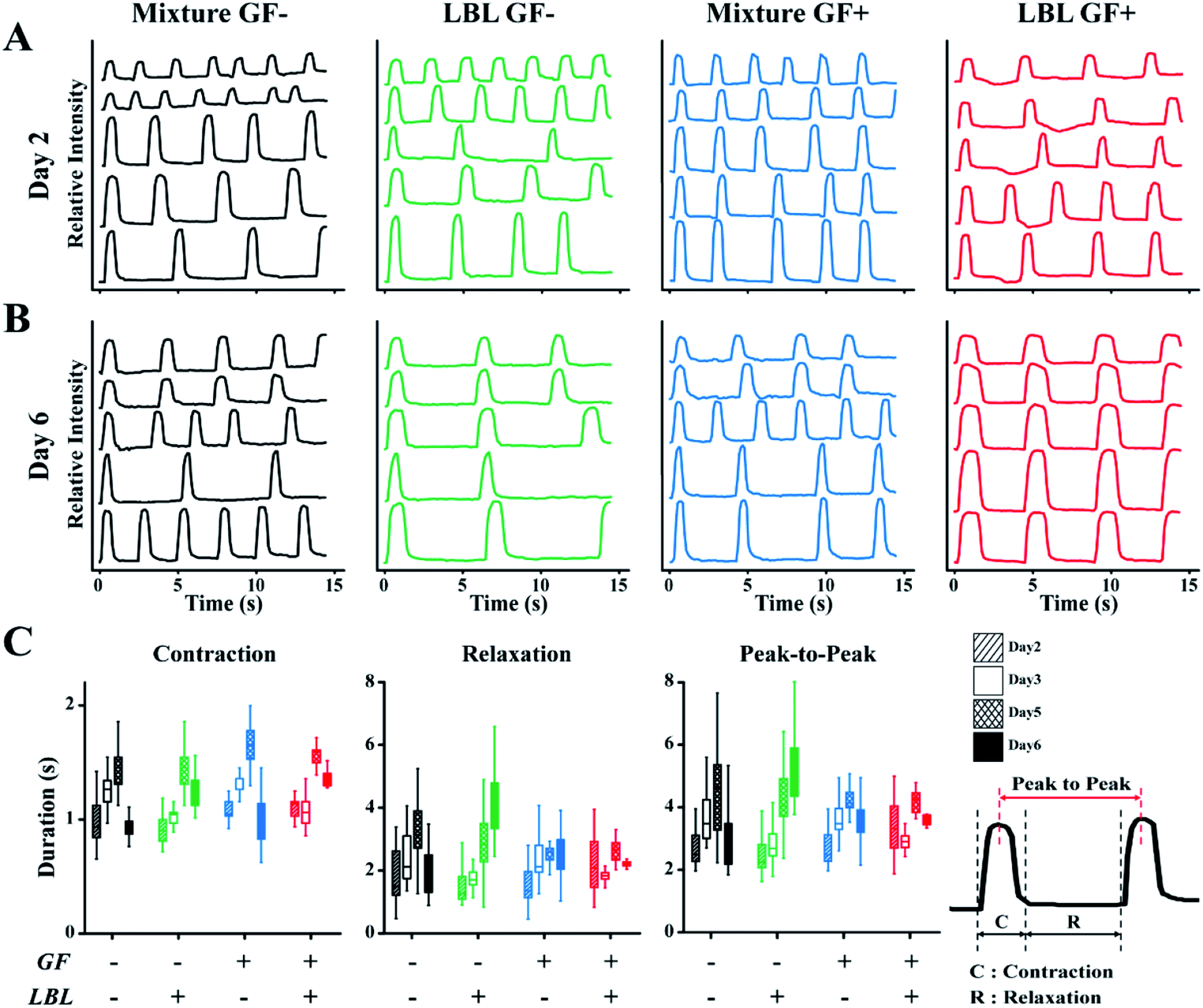 | ||
| Fig. 4 Characterization of mCMTs beating. Beating characteristics at day 2 (A) and day 6 (B) are represented by monitoring pixel intensity in regions of interest over a period of 15 s. The wave in the each graph represents change in light intensity of the different area, respectively. (C) Comparison of contraction, relaxation and peak-to-peak duration (PTP) among prepared groups. Note that the statistical analysis by one-way or two-way ANOVA is shown in Fig. S4† and Tables S1–S3,†N = 5 per each group for beating analysis. | ||
3.4 Gene expression levels of maturation markers in mCMTs
We also evaluated the gene expression profile of mCMTs associated with CM maturation and electromechanical function at day 6. The GF+ groups exhibited increased maturation factors (ventricular myosin light chain-2 (MLC2v) and cardiac troponin I (cTNI)) (Fig. 5A and B), whereas there was no significant difference in cardiac troponin T (cTNT) or gap junction alpha-1 (GJA1) between the groups (Fig. 5C and F). In particular, MLC2v in the GF+ groups was approximately 20-fold higher than that in the GF− groups (Fig. 5A). Therefore, FGF4 and ascorbic acid positively influence the differentiation of immature CMs into ventricular-specific CMs.59 In addition, pre-Mir21 (MicroRNA 21), a hypertrophy marker, was slightly increased in the LBL GF+ CMT compared with the other groups. Interestingly, the mechanosensitive ion channel Piezo1 was also slightly increased with LBL and GF.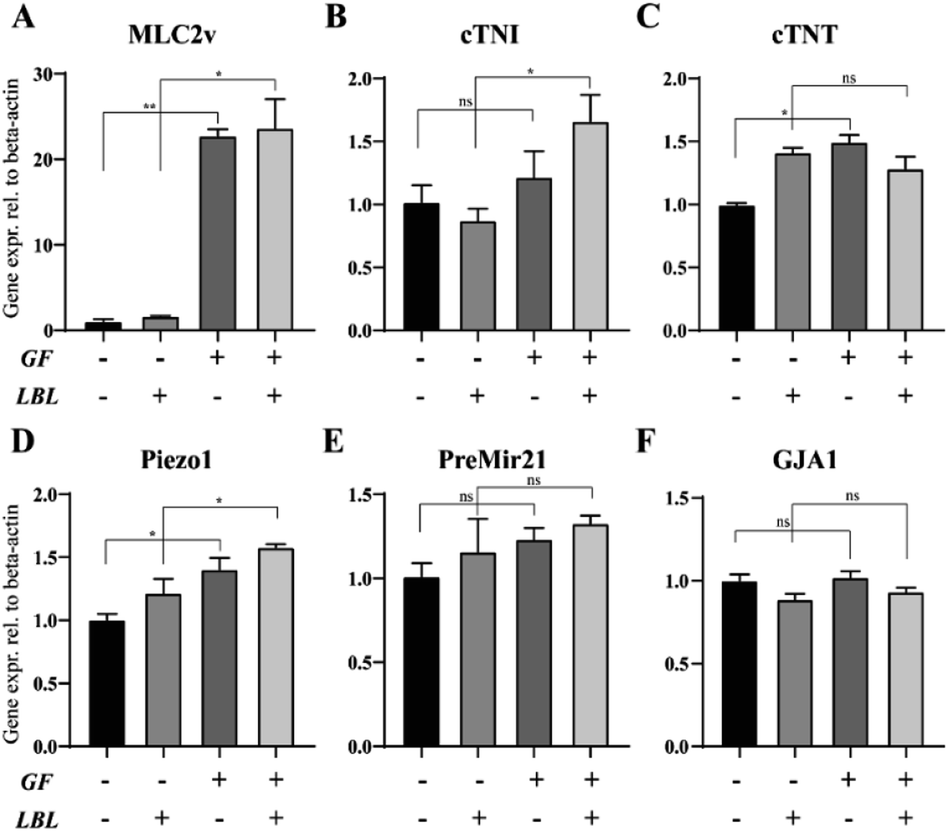 | ||
| Fig. 5 Effect of growth factors and the LBL method on the gene expression of mCMTs by real-time PCR analysis (A–F). Shown are the expression levels of the cardiac-specific markers (cTNT, cTNI, GJA1), ventricle-specific marker (MLC2v), hypertrophy marker (PreMir21) and mechanosensitive ion channel (piezo1). Expression values were normalized to β-actin. Statistically significant values were defined as *P < 0.05, **P < 0.01. P values were determined according to the unpaired two-tailed Student's t-test. Note that the experiment was triplicated, and the error bars were estimated by the standard deviation. | ||
3.5 Identification of MLC2v and sarcomere alignment in mCMTs
In the next step, we performed confocal fluorescence microscopy by immunostaining with cTNT which is located at the actin of CMs and MLC2v which is present in the thick filaments of the ventricle type CMs to evaluate CMs alignment and maturation at the level of protein expression, respectively. In the GF+ mCMT, sarcomeric actin was formed by cTNT. In particular, the LBL mCMT appears to have better actin alignment than the mixed mCMT (Fig. 6A). Although the expression of MLC2v in the LBL GF+ mCMT was approximately less than twice as high as that of the mixture GF+ mCMT (Fig. 6B), the LBL GF+ mCMT was well colocalized with cTNT and MLC2v in the sarcomeric actin, while the GF+ mixture mCMT showed cytoplasmic staining rather than actin in MLC2v staining (Fig. 6C). Therefore, the alignment and maturation of CMs in the LBL GF+ mCMT is better than that in the other group (Fig. S5†).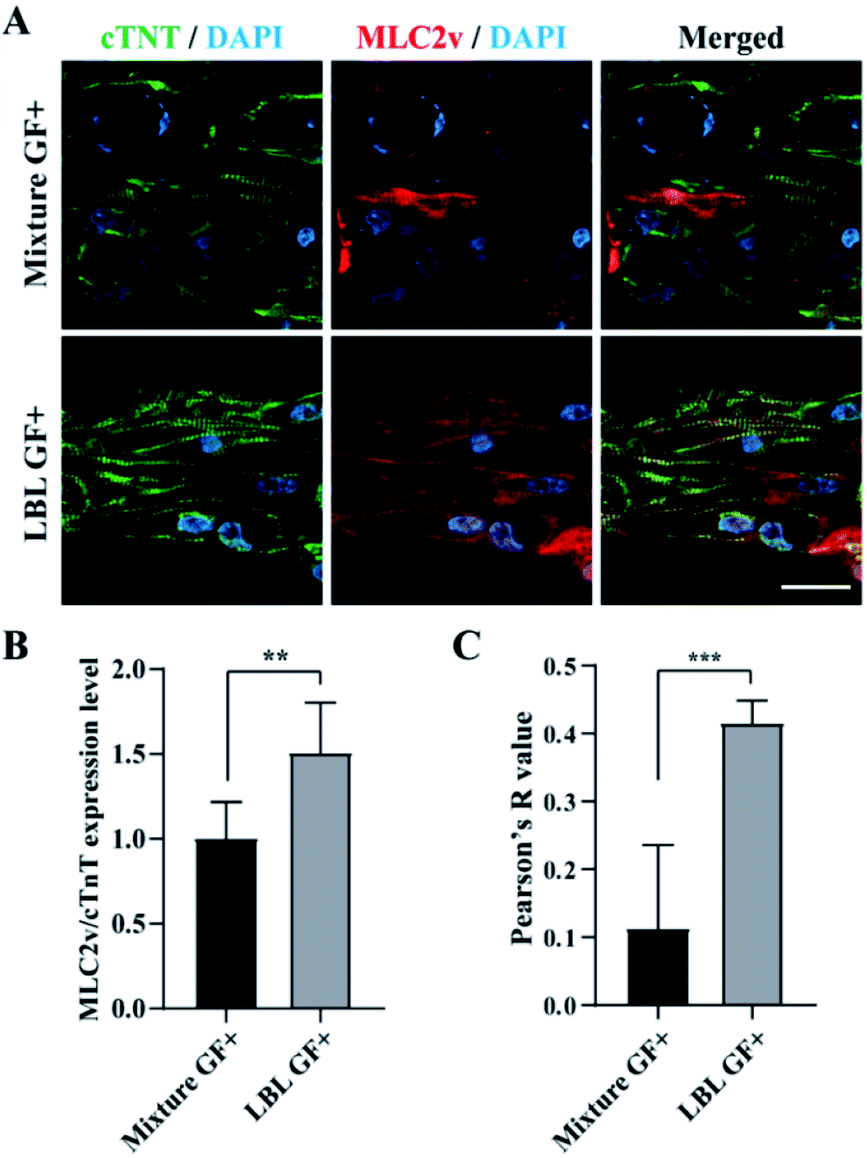 | ||
| Fig. 6 Identification of MLC2v expression and sarcomere alignment in cardiac patches using confocal fluorescence microscopy at the day 6. (A) cTNT (green), MLC2v (red), and DAPI (blue) staining images in the mixture and LBL groups, respectively. (B) Normalized expression level of MLC2v over cTNT of mCMTs. (C) Colocalization of MLC2v and cTNT analyzed by Pearson's R value. Scale bars: 10 μm. Statistically significant value was defined as *P < 0.05, **P < 0.01, ***P < 0.001 and unpaired two-tailed Student's t-test. Note that the experiment was N = 8 for B and N = 5 for C, respectively and the error bars were estimated by the standard deviation. | ||
3.6 Evaluation of mCMTs using agonist and antagonist
The method of constructing mCMT by the LBL deposition and centrifuge enables preparation of biomimetic mCMT to evaluate the efficacy and toxicity of drug molecules. Since LBL GF+ mCMT exists in the form of a tiny patch with synchronized beating behaviour, the mCMT is applicable to evaluate the effect of agonists and antagonists for the function of CMs. Therefore, we prepared the mCMTs to measure their beating property and calcium transient by adding agonists, Yoda1 and isoproterenol and antagonist, ruthenium red (Fig. 7). Yoda1 is a agonist of mechanoresponsive piezo1 ion channel, which increases both calcium influx through the channel and beating rate of cardiac cells.60 Ruthenium red is known to block cationic channels, which influences on the beating of cardiomyocytes.61 Isoproterenol is a beta-adrenergic agonist that increases the beating rate of cardiomyocytes.62 The control group proceeded together for comparison as a group with no drugs added. To characterize the cardiac beating and calcium transient of the mCMTs, we analysed peak-to-peak duration of mCMTs, which was shown in Fig. 4 (Fig. 7A). In our experiments, isoproterenol and Yoda1 treated mCMTs began to beat faster than that of untreated mCMTs, while ruthenium red treated mCMTs were slower in their beating at the time point of 0.5 h and 2 h after the treatment (Fig. 7B). Similarly, calcium transients of the mCMTs were faster for isoproterenol and Yoda1 treated groups, but slower for the ruthenium red in comparison to the untreated control group (Fig. 7C). Therefore, the mCMTs prepared by LBL and GF+ were successfully used to evaluate cardiac beating and calcium transient upon addition of agonist and antagonist. We also found that the effect of isoproterenol and ruthenium red on the beating and calcium transient of mCMTs were attenuated for 2 hours, while Yoda1 keeps the effect for the same time.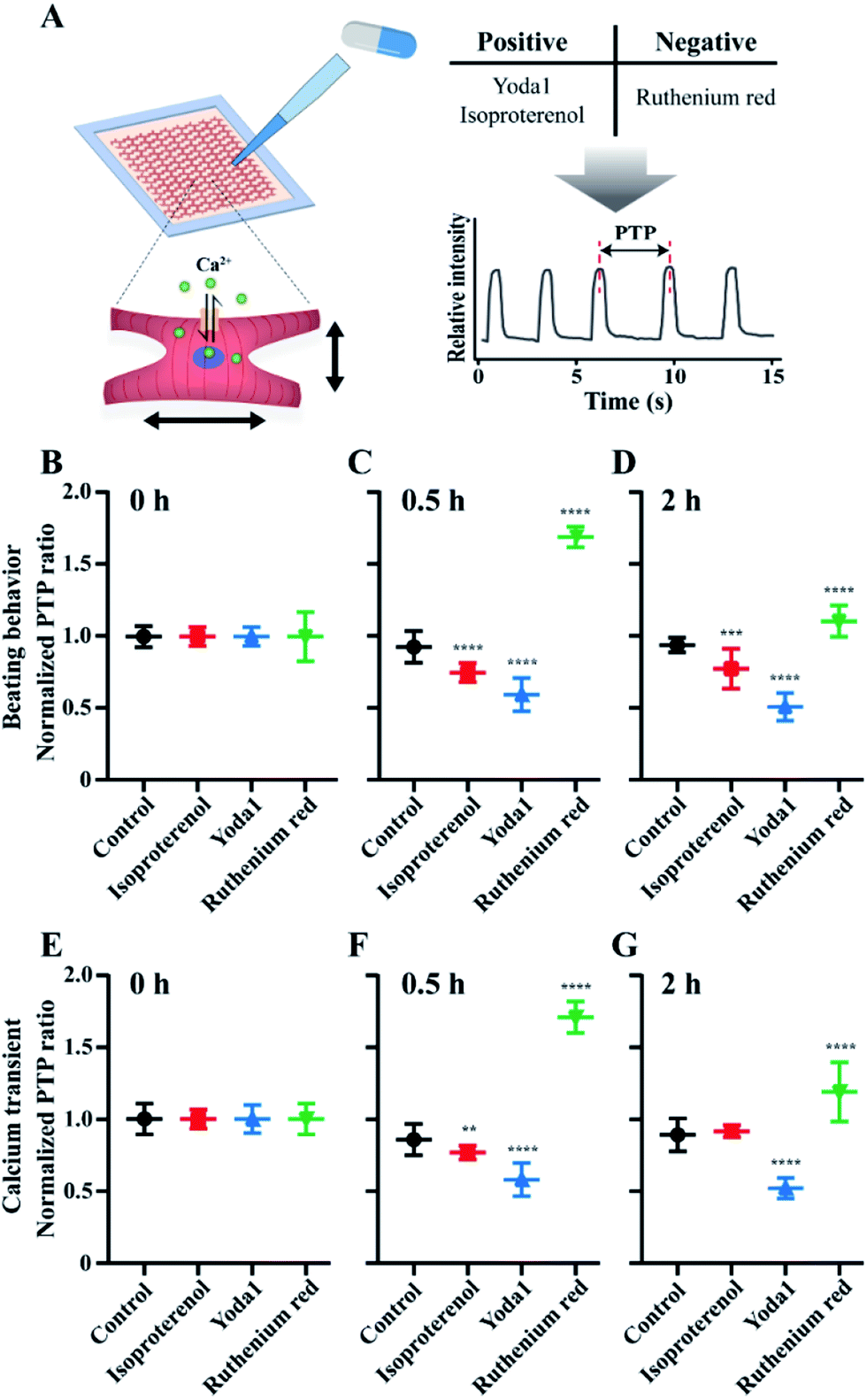 | ||
| Fig. 7 Evaluation of beating and calcium transient of mCMTs upon addition of agonist and antagonist. (A) A scheme of drug evaluation process. After treatment of agonist and antagonist, the PTP was measured by beating (B, C, D) and calcium transient (E, F, G), respectively, and normalized to estimate PTP ratio at 0 h (B, E), 0.5 h (C, F) and 2 h (D, G), respectively. Statistical significance was estimated as *P < 0.05, **P < 0.01, ***P < 0.001 and unpaired two-tailed Student's t-test. Note that the number of beatings is N = 16 for control group, N = 15 for isoproterenol group, N = 15 for Yoda1 group and N = 10 for ruthenium red group, respectively and the error bars were estimated by the standard deviation. | ||
4 Discussions
Recent emerging technologies in biomaterials and tissue engineering enable the development of a complex structure for regeneration and precision medicine.11,63 Despite such advancement, the fabrication of precisely organized and aligned cell–matrix structures mimicking real tissues has been challenging. Fabricating complex structure requires complex technologies, which makes the barrier higher. LBL based on cells was achieved by using other polymer layers for cell stacking or using thermo-responsive coated plate to harvest cell layers and stacking the cell layers.64,65 In this study, simple centrifugation of cells–hydrogel mixture on the PDMS molds enabled the LBL deposition of different types of cells for the fabrication of complex structure (Fig. 1). The structural integrity of LBL based mCMT were kept for 6 days of the maturation process (Fig. 3), which suggests the fidelity of our method. Demolding of the cardiac structure, as well as its fabrication, is another technical challenge to fabricate the integrated mCMTs. Our results show that round bottomed PDMS molds allow to retrieve the intact mCMTs from PDMS molds with no surface treatment. Technology developed in this study by the combination of PDMS based round bottom mold with centrifugation of cell–matrix by LBL deposition allows to fabricate patterned mCMTs for investigating the synchronization and maturation of cardiac tissues.Synchronized beating of mCMTs is essential for their use in tissue engineering and drug screening.38,66 For instance, the irregular beating of cardiac tissues is corresponding to the abnormal heartbeats that is a symptom of an heart disease, called arrhythmia.31,67 Our findings show that synchronization of the mCMTs is significantly enhanced by both LBL deposition of cardiac cells with fibroblasts and growth factors (Fig. 4). During the assembly process of mCMTs, cell–cell connections are important for cardiac bundle formation. In the LBL mCMTs with fibroblast layer, our observation suggests that CMs are fairly aligned with unidirectional sarcomere formation (Fig. 6). In contrast, CMs of the mixture mCMTs with fibroblasts are assembled with no directionality of sarcomeres, hinting at a potential role of fibroblasts acting as interrupting CMs connections (Fig. 6). It is also worthwhile to note that fibroblasts act as a activator of contractility and firing of CMs by lowering membrane potentials.43,68 Thus, our findings in the enhanced synchronization of LBL mCMTs could be explained by the fact that CMs were interconnected each other without the physical intercalation of CFs, but with their activating effects in proximity. This is also supported by our analysis that the cardiac maturation marker, MLC2v and cTNI, was upregulated in a RNA level (Fig. 5A and B) and the sarcomere structure was better formed in a protein level for the LBL mCMTs with growth factors (Fig. 6).
Controlling cell assembly in 3D structure is challenging, but prerequisite to utilize mCMT in a number of applications including drug screening. In the previous study, we demonstrated the development of cardiac patch to regulate the interplay between CMs and CFBs through LBL deposition. However, there was a caveat to control cellular aggregation and uniform distribution of cells in the whole area of mCMT. Such limitation made us difficult to prepare reproducible mCMTs with high fidelity, which was critical to evaluate the efficacy and toxicity of drug molecules using mCMTs. In the current study, we employed one of the well-known patterns, i.e. accordion-like honeycomb,52 to introduce a geometrical guidance cue into construction of mCMT. The use of the pattern allowed us to prepare quite reproducible mCMTs by regulating aggregation and distribution of cells in the whole area (Fig. S6 and S7†), which also facilitated the alignment and assembly of mCMTs. Thus, we were able to use the mCMTs for evaluation of the beating and calcium transient upon addition of drug molecules.
Assembly of mCMTs by a LBL deposition and centrifuge has proposed a potential route to develop biomimetic mCMT in an easy and convenient way.9,69 Since not only a functional cardiac tissue assembled by hPSC-derived cardiomyocytes could mimic the beating behaviour of human cardiac cells, but also the failure in cardiac beating is known for most common causes of drug rejection,70 the mCMT could be used to screen drug molecules for the efficacy and toxicity. In this study, we utilized agonist and antagonist of CM to demonstrate that cardiac beating and calcium transient of mCMTs could be used for the functional assay of CM properties. When exposed to each agonist, i.e. isoproterenol and Yoda1, and antagonist, i.e. ruthenium red, we were able to see the change in beating property and calcium transient of mCMT. It is also interesting to note that Yoda1 increases the beating and calcium transient by ∼2 times compared with those of control group, while ruthenium red adversely results in decrease of the properties. Since Yoda1 and ruthenium red are activator and inhibitor for mechanoresponsive Piezo1 ion channel, respectively, our result suggests that Piezo1-mediated calcium transient could be an important factor in beating of CMs.71 Our observation could shed current obstacles of drug screening using mCMT, providing innovative, yet easy biofabrication technologies for many desired applications.
5 Conclusions
We report the development of mCMT, which enabled us to control the maturation and synchronization of cardiac tissues and the multicellular structure and dimension. The formation of mCMT by LBL deposition and centrifugation represents an advancement in the ability to fabricate cardiac tissues on demand. The characterization of cardiac beating and gene expression for maturation sheds light on the sufficiency of our method to fabricate cardiac tissues for basic research, biomedical engineering, and healthcare industries. Evaluation of mCMT function using drug molecules could be further enhanced by employing active mechanical stimuli and signalling modulation. The multicellular structure and synchronized beating aspects of this approach should facilitate its application to solve problems in basic development and cardiac disorders including arrhythmias, cardiomyopathy, and cardiac hypertrophy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the grant from the National Research Foundation of Korea, Republic of Korea (Grant No. 2016-M3A9B6947892) and a Korea University Grant.Notes and references
- X. Y. Zhang, Y. Yanagi, Z. Sheng, K. Nagata, K. Nakayama and T. Taguchi, Biomaterials, 2018, 167, 1–14 CrossRef CAS PubMed.
- Y. Wang, X. Yuan, K. Yu, H. Meng, Y. Zheng, J. Peng, S. Lu, X. Liu, Y. Xie and K. Qiao, Biomaterials, 2018, 171, 118–132 CrossRef CAS PubMed.
- C. P. Jackman, A. M. Ganapathi, H. Asfour, Y. Qian, B. W. Allen, Y. Li and N. Bursac, Biomaterials, 2018, 159, 48–58 CrossRef CAS PubMed.
- M. Shah, P. Kc and G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 23893–23900 CrossRef CAS PubMed.
- L. R. Sewanan, J. Schwan, J. Kluger, J. Park, D. L. Jacoby, Y. Qyang and S. G. Campbell, JACC Basic Transl. Sci., 2019, 4, 495–505 CrossRef PubMed.
- J. Foolen, S. L. Wunderli, S. Loerakker and J. G. Snedeker, Matrix Biol., 2018, 65, 14–29 CrossRef CAS PubMed.
- P. Andersen, E. Tampakakis, D. V. Jimenez, S. Kannan, M. Miyamoto, H. K. Shin, A. Saberi, S. Murphy, E. Sulistio, S. P. Chelko and C. Kwon, Nat. Commun., 2018, 9, 3140, DOI:10.1038/s41467-018-05604-8.
- P. Hoang, J. Wang, B. R. Conklin, K. E. Healy and Z. Ma, Nat. Protoc., 2018, 13, 723–737 CrossRef CAS PubMed.
- A. J. Hughes, H. Miyazaki, M. C. Coyle, J. Zhang, M. T. Laurie, D. Chu, Z. Vavrusova, R. A. Schneider, O. D. Klein and Z. J. Gartner, Dev. Cell, 2018, 44, 165–178 CrossRef CAS PubMed.
- T. Sun, Q. Shi, Q. Huang, H. Wang, X. Xiong, C. Hu and T. Fukuda, Acta Biomater., 2018, 66, 272–281 CrossRef CAS PubMed.
- W. Bian, N. Badie, H. D. t. Himel and N. Bursac, Biomaterials, 2014, 35, 3819–3828 CrossRef CAS PubMed.
- M. A. Lancaster, N. S. Corsini, S. Wolfinger, E. H. Gustafson, A. W. Phillips, T. R. Burkard, T. Otani, F. J. Livesey and J. A. Knoblich, Nat. Biotechnol., 2017, 35, 659–666 CrossRef CAS PubMed.
- P. P. S. S. Abadi, J. C. Garbern, S. Behzadi, M. J. Hill, J. S. Tresback, T. Heydari, M. R. Ejtehadi, N. Ahmed, E. Copley, H. Aghaverdi, R. T. Lee, O. C. Farokhzad and M. Mahmoudi, Adv. Funct. Mater., 2018, 28, 1707378, DOI:10.1002/adfm.201707378.
- N. Annabi, K. Tsang, S. M. Mithieux, M. Nikkhah, A. Ameri, A. Khademhosseini and A. S. Weiss, Adv. Funct. Mater., 2013, 23, 4950–4959, DOI:10.1002/adfm.201300570.
- A. Bettadapur, G. C. Suh, N. A. Geisse, E. R. Wang, C. Hua, H. A. Huber, A. A. Viscio, J. Y. Kim, J. B. Strickland and M. L. McCain, Sci. Rep., 2016, 6, 28855, DOI:10.1038/srep28855.
- C. W. Hsiao, M. Y. Bai, Y. Chang, M. F. Chung, T. Y. Lee, C. T. Wu, B. Maiti, Z. X. Liao, R. K. Li and H. W. Sung, Biomaterials, 2013, 34, 1063–1072 CrossRef CAS PubMed.
- H. Takahashi, T. Shimizu, M. Nakayama, M. Yamato and T. Okano, Biomaterials, 2013, 34, 7372–7380 CrossRef CAS PubMed.
- S. P. Sheehy, F. Pasqualini, A. Grosberg, S. J. Park, Y. Aratyn-Schaus and K. K. Parker, Stem Cell Rep., 2014, 2, 282–294 CrossRef CAS PubMed.
- K. M. Tsang, N. Annabi, F. Ercole, K. Zhou, D. Karst, F. Li, J. M. Haynes, R. A. Evans, H. Thissen, A. Khademhosseini and J. S. Forsythe, Adv. Funct. Mater., 2015, 25, 977–986 CrossRef CAS PubMed.
- D. Carson, M. Hnilova, X. Yang, C. L. Nemeth, J. H. Tsui, A. S. Smith, A. Jiao, M. Regnier, C. E. Murry, C. Tamerler and D. H. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 21923–21932 CrossRef CAS PubMed.
- J. Han, Q. Wu, Y. Xia, M. B. Wagner and C. Xu, Stem Cell Res., 2016, 16, 740–750 CrossRef CAS PubMed.
- N. Penland, E. Choi, M. Perla, J. Park and D. H. Kim, Nanotechnology, 2017, 28, 075103, DOI:10.1088/1361-6528/aa55e0.
- N. P. Williams, M. Rhodehamel, C. Yan, A. S. T. Smith, A. Jiao, C. E. Murry, M. Scatena and D. H. Kim, Biomaterials, 2020, 240, 119856, DOI:10.1016/j.biomaterials.2020.119856.
- S. Fleischer, A. Shapira, R. Feiner and T. Dvir, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1898–1903 CrossRef CAS PubMed.
- A. Tijore, S. A. Irvine, U. Sarig, P. Mhaisalkar, V. Baisane and S. Venkatraman, Biofabrication, 2018, 10, 025003, DOI:10.1088/1758-5090/aaa15d.
- Y. S. Zhang, A. Arneri, S. Bersini, S. R. Shin, K. Zhu, Z. Goli-Malekabadi, J. Aleman, C. Colosi, F. Busignani, V. Dell'Erba, C. Bishop, T. Shupe, D. Demarchi, M. Moretti, M. Rasponi, M. R. Dokmeci, A. Atala and A. Khademhosseini, Biomaterials, 2016, 110, 45–59 CrossRef CAS PubMed.
- J. Jang, H. J. Park, S. W. Kim, H. Kim, J. Y. Park, S. J. Na, H. J. Kim, M. N. Park, S. H. Choi, S. H. Park, S. W. Kim, S. M. Kwon, P. J. Kim and D. W. Cho, Biomaterials, 2017, 112, 264–274 CrossRef CAS PubMed.
- R. A. Li, W. Keung, T. J. Cashman, P. C. Backeris, B. V. Johnson, E. S. Bardot, A. O. T. Wong, P. K. W. Chan, C. W. Y. Chan and K. D. Costa, Biomaterials, 2018, 163, 116–127 CrossRef CAS PubMed.
- J. U. Lind, T. A. Busbee, A. D. Valentine, F. S. Pasqualini, H. Yuan, M. Yadid, S. J. Park, A. Kotikian, A. P. Nesmith, P. H. Campbell, J. J. Vlassak, J. A. Lewis and K. K. Parker, Nat. Mater., 2017, 16, 303–308 CrossRef CAS PubMed.
- M. Kapnisi, C. Mansfield, C. Marijon, A. G. Guex, F. Perbellini, I. Bardi, E. J. Humphrey, J. L. Puetzer, D. Mawad, D. C. Koutsogeorgis, D. J. Stuckey, C. M. Terracciano, S. E. Harding and M. M. Stevens, Adv. Funct. Mater., 2018, 28, 1800618, DOI:10.1002/adfm.201800618.
- J. Li, I. Minami, M. Shiozaki, L. Yu, S. Yajima, S. Miyagawa, Y. Shiba, N. Morone, S. Fukushima, M. Yoshioka, S. Li, J. Qiao, X. Li, L. Wang, H. Kotera, N. Nakatsuji, Y. Sawa, Y. Chen and L. Liu, Stem Cell Rep., 2017, 9, 1546–1559 CrossRef CAS PubMed.
- L. A. MacQueen, S. P. Sheehy, C. O. Chantre, J. F. Zimmerman, F. S. Pasqualini, X. Liu, J. A. Goss, P. H. Campbell, G. M. Gonzalez, S.-J. Park, A. K. Capulli, J. P. Ferrier, T. F. Kosar, L. Mahadevan, W. T. Pu and K. K. Parker, Nat. Biomed. Eng., 2018, 2, 930–941 CrossRef CAS PubMed.
- V. Y. Sidorov, P. C. Samson, T. N. Sidorova, J. M. Davidson, C. C. Lim and J. P. Wikswo, Acta Biomater., 2017, 48, 68–78 CrossRef CAS PubMed.
- W. Zhang, C. W. Kong, M. H. Tong, W. H. Chooi, N. Huang, R. A. Li and B. P. Chan, Acta Biomater., 2017, 49, 204–217 CrossRef CAS PubMed.
- B. M. Ulmer, A. Stoehr, M. L. Schulze, S. Patel, M. Gucek, I. Mannhardt, S. Funcke, E. Murphy, T. Eschenhagen and A. Hansen, Stem Cell Rep., 2018, 10, 834–847 CrossRef CAS PubMed.
- S. S. Nunes, J. W. Miklas, J. Liu, R. Aschar-Sobbi, Y. Xiao, B. Zhang, J. Jiang, S. Masse, M. Gagliardi, A. Hsieh, N. Thavandiran, M. A. Laflamme, K. Nanthakumar, G. J. Gross, P. H. Backx, G. Keller and M. Radisic, Nat. Methods, 2013, 10, 781–787 CrossRef CAS PubMed.
- Y. Liu, G. Xu, J. Wei, Q. Wu and X. Li, Mater. Sci. Eng., C, 2017, 81, 500–510 CrossRef CAS PubMed.
- K. Ronaldson-Bouchard, S. P. Ma, K. Yeager, T. Chen, L. Song, D. Sirabella, K. Morikawa, D. Teles, M. Yazawa and G. Vunjak-Novakovic, Nature, 2018, 556, 239–243 CrossRef CAS PubMed.
- H. Uosaki, P. Cahan, D. I. Lee, S. Wang, M. Miyamoto, L. Fernandez, D. A. Kass and C. Kwon, Cell Rep., 2015, 13, 1705–1716 CrossRef CAS PubMed.
- I. Kehat, L. Khimovich, O. Caspi, A. Gepstein, R. Shofti, G. Arbel, I. Huber, J. Satin, J. Itskovitz-Eldor and L. Gepstein, Nat. Biotechnol., 2004, 22, 1282–1289 CrossRef CAS PubMed.
- K. Kojima, T. Kaneko and K. Yasuda, Biochem. Biophys. Res. Commun., 2006, 351, 209–215 CrossRef CAS PubMed.
- I. Nitsan, S. Drori, Y. E. Lewis, S. Cohen and S. Tzlil, Nat. Phys., 2016, 12, 472–477 Search PubMed.
- H. Saini, A. Navaei, A. Van Putten and M. Nikkhah, Adv. Healthcare Mater., 2015, 4, 1961–1971 Search PubMed.
- B. Liau, C. P. Jackman, Y. Li and N. Bursac, Sci. Rep., 2017, 7, 42290, DOI:10.1038/srep42290.
- J. Rother, C. Richter, L. Turco, F. Knoch, I. Mey, S. Luther, A. Janshoff, E. Bodenschatz and M. Tarantola, Open Biol., 2015, 5, 150038, DOI:10.1098/rsob.150038.
- J. Bargehr, L. P. Ong, M. Colzani, H. Davaapil, P. Hofsteen, S. Bhandari, L. Gambardella, N. Le Novere, D. Iyer, F. Sampaziotis, F. Weinberger, A. Bertero, A. Leonard, W. G. Bernard, A. Martinson, N. Figg, M. Regnier, M. R. Bennett, C. E. Murry and S. Sinha, Nat. Biotechnol., 2019, 37, 895–906 CrossRef CAS PubMed.
- F. Varzideh, E. Mahmoudi and S. Pahlavan, J. Cell. Biochem., 2019, 120, 16681–16691 CrossRef CAS PubMed.
- T. Kaneko, F. Nomura and K. Yasuda, J. Nanobiotechnol., 2011, 9, 21, DOI:10.1186/1477-3155-9-21.
- L. McArthur, L. Chilton, G. L. Smith and S. A. Nicklin, Biochem. Soc. Trans., 2015, 43, 513–518 CrossRef CAS PubMed.
- J. Trieschmann, D. Bettin, M. Haustein, A. Koster, M. Molcanyi, M. Halbach, M. Hanna, M. Fouad, K. Brockmeier, J. Hescheler, K. Pfannkuche and T. Hannes, Stem Cells Int., 2016, 2016, 2936126, DOI:10.1155/2016/2936126.
- T. Y. Kim, C. M. Kofron, M. E. King, A. R. Markes, A. O. Okundaye, Z. Qu, U. Mende and B. R. Choi, PLoS One, 2018, 13, e0196714, DOI:10.1371/journal.pone.0196714.
- G. C. Engelmayr Jr, M. Cheng, C. J. Bettinger, J. T. Borenstein, R. Langer and L. E. Freed, Nat. Mater., 2008, 7, 1003–1010 CrossRef PubMed.
- B. Zhang, M. Montgomery, L. Davenport-Huyer, A. Korolj and M. Radisic, Sci. Adv., 2015, 1, e1500423, DOI:10.1126/sciadv.1500423.
- G. S. Jeong, J. H. Song, A. R. Kang, Y. Jun, J. H. Kim, J. Y. Chang and S. H. Lee, Adv. Healthcare Mater., 2013, 2, 119–125 CrossRef CAS PubMed.
- G. S. Jeong and S.-H. Lee, Tissue Eng. Regener. Med., 2014, 11, 291–296 CrossRef.
- S. Park, D. Kim, Y. G. Jung and S. Roh, Anim. Reprod. Sci., 2015, 161, 47–57 CrossRef CAS PubMed.
- C. H. Xiaojun Lian, G. Wilson, K. Zhu, L. B. Hazeltine, S. M. Azarin, K. K. Raval, T. J. K. Jianhua Zhang and S. P. Palecek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1848–1857 Search PubMed.
- J. E. Cartledge, C. Kane, P. Dias, M. Tesfom, L. Clarke, B. McKee, S. Al Ayoubi, A. Chester, M. H. Yacoub, P. Camelliti and C. M. Terracciano, Cardiovasc. Res., 2015, 105, 260–270 CrossRef CAS PubMed.
- Y. Jang, S. C. Choi, D. S. Lim, J. H. Kim, J. Kim and Y. Park, Soft Matter, 2020, 16, 428–434 RSC.
- R. Syeda, J. Xu, A. E. Dubin, B. Coste, J. Mathur, T. Huynh, J. Matzen, J. Lao, D. C. Tully, I. H. Engels, H. M. Petrassi, A. M. Schumacher, M. Montal, M. Bandell and A. Patapoutian, Elife, 2015, 4, e07369, DOI:10.7554/eLife.07369.
- X. S. Lingjia Qian, H. Ren, J. Gong and S. Cheng, Cell Stress Chaperones, 2004, 9, 281–293 CrossRef PubMed.
- I. Goldfracht, Y. Efraim, R. Shinnawi, E. Kovalev, I. Huber, A. Gepstein, G. Arbel, N. Shaheen, M. Tiburcy, W. H. Zimmermann, M. Machluf and L. Gepstein, Acta Biomater., 2019, 92, 145–159 CrossRef CAS PubMed.
- G. S. Jeong, Y. No da, J. Lee, J. Yoon, S. Chung and S. H. Lee, Nat. Commun., 2016, 7, 11269, DOI:10.1038/ncomms11269.
- S. Ryu, J. Yoo, J. Han, S. Kang, Y. Jang, H. J. Han, K. Char and B.-S. Kim, Chem. Mater., 2017, 29, 5134–5147 CrossRef CAS.
- Y. Amano, A. Nishiguchi, M. Matsusaki, H. Iseoka, S. Miyagawa, Y. Sawa, M. Seo, T. Yamaguchi and M. Akashi, Acta Biomater., 2016, 33, 110–121 CrossRef CAS PubMed.
- H. Zeng, B. Balasubramanian, A. Lagrutta and F. Sannajust, J. Pharmacol. Toxicol. Methods, 2018, 91, 18–26 CrossRef CAS PubMed.
- D. Schocken, J. Stohlman, J. Vicente, D. Chan, D. Patel, M. K. Matta, V. Patel, M. Brock, D. Millard, J. Ross, D. G. Strauss and K. Blinova, J. Pharmacol. Toxicol. Methods, 2018, 90, 39–47 CrossRef CAS PubMed.
- Y. Zhao, N. Rafatian, E. Y. Wang, N. T. Feric, B. F. L. Lai, E. J. Knee-Walden, P. H. Backx and M. Radisic, Matrix Biol., 2019, 189–204, DOI:10.1016/j.matbio.2019.04.001.
- K. Kuribayashi-Shigetomi, H. Onoe and S. Takeuchi, PLoS One, 2012, 7, e51085, DOI:10.1371/journal.pone.0051085.
- A. Eder, I. Vollert, A. Hansen and T. Eschenhagen, Adv. Drug Delivery Rev., 2016, 96, 214–224 CrossRef CAS PubMed.
- T. Y. Wong, W. C. Juang, C. T. Tsai, C. J. Tseng, W. H. Lee, S. N. Chang and P. W. Cheng, J. Clin. Med., 2018, 7, 410, DOI:10.3390/jcm7110410.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01577f |
| This journal is © The Royal Society of Chemistry 2020 |