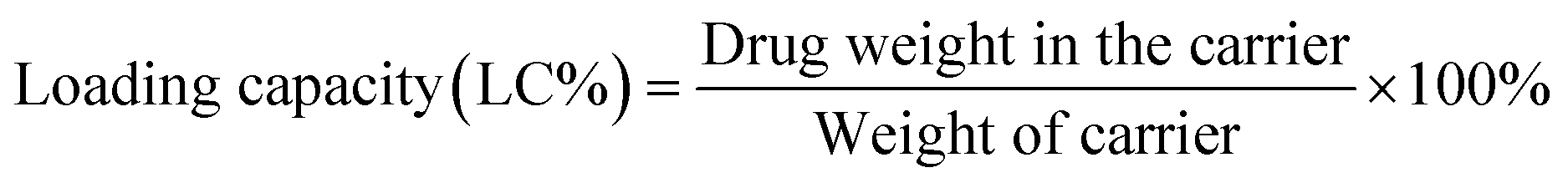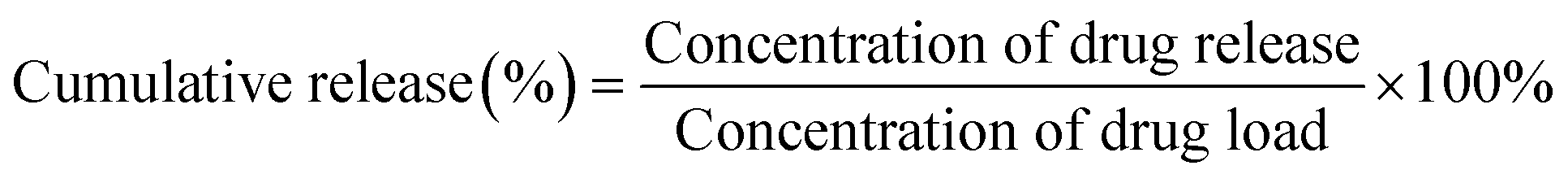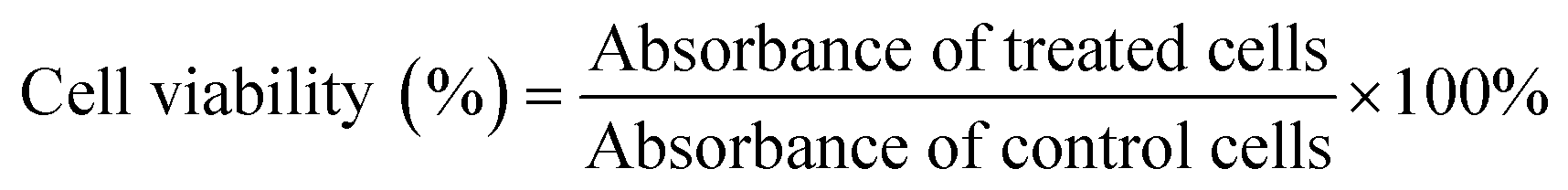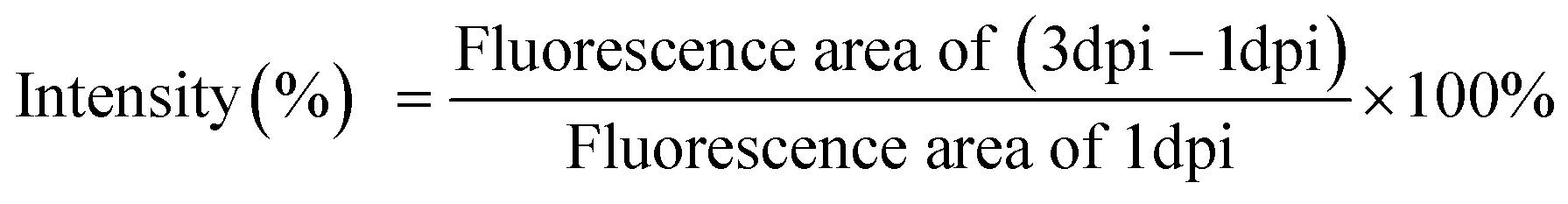DOI:
10.1039/D0RA01589J
(Paper)
RSC Adv., 2020,
10, 20682-20690
Amino acid-modified PAMAM dendritic nanocarriers as effective chemotherapeutic drug vehicles in cancer treatment: a study using zebrafish as a cancer model
Received
19th February 2020
, Accepted 27th April 2020
First published on 1st June 2020
Abstract
The use of nanomaterials for drug delivery offers many advantages including the targeted delivery of drugs and their controlled release. Nonetheless, entry into the target cells remains a challenge for many nanomaterials used for drug delivery. Moreover, cellular uptake limits the therapeutic efficiency of many anticancer drugs. An important goal is to increase the specific accumulation of these nanoparticles (NPs) at the desired cancerous tissues. Notably, cancer cells show a high demand for some amino acids and we have used this knowledge to develop novel carrier systems. In this study, drug carriers were produced by the conjugation of multiple amino acids such as L-histidine (H) and L-cysteine (C) or single amino acids such as only H with the G4.5 dendrimers (G) to produce GHC aggregates and GH NP carriers, respectively. Doxorubicin was loaded into the G4.5, GH, and GHC dendrimers (G/DOX, GH/DOX and GHC/DOX, respectively) and the release mechanism was demonstrated at pH 7.4 and pH 5.0. GH/DOX and GHC/DOX showed better stability under physiological conditions than the dendrimer alone (G/DOX). GH/DOX and GHC/DOX exhibited higher inhibition of HeLa cell proliferation in in vitro and in vivo studies in zebrafish, confirming the early release of DOX by disrupting the endosomal membrane and triggering the destabilization of carriers at a lower pH of 5.0.
1. Introduction
Chemotherapy is the most general treatment in cancer therapy, and the common chemotherapeutic drug is doxorubicin (DOX), which is one of the most important anthracyclines drug. The mechanism of DOX involves interaction with the DNA to block topoisomerase II and then prevent DNA replication and cell division.1,2 However, the critical limitations of chemotherapy are high systemic cytotoxicity, short half-life, and non-site specific action.3 Therefore, specific drug delivery systems with targeting ability are capable of reducing the harmful side effects while optimizing the therapeutic potency for cancer therapy.4 Recently, polymeric carriers as promising vehicles have been used to regulate the number of drugs safely released in malignant tissues.5,6 Among these nanocarrier systems, liposome, micelle, and dendrimer systems are potential platforms for constructing a variety of versatile biocompatible systems for drug delivery applications. Dendrimers are core–shell nanostructures having a precise architecture with monodispersity. The dendrimers are synthesized in a generation-by-generation fashion around a core unit, resulting in branching points and surface functionalities.7 The possibility to modify the surface of dendrimers makes them ideal carriers for guest molecule encapsulation, allowing the dissolution of hydrophobic foreign materials in water. Comparison to PAMAM G4.5, lower-molecular-weight or lower branched dendrimers (e.g., PAMAM G1.5) are easily excreted from the body, which has been proved in many studies. The G4.5 PAMAM dendrimer is a highly branched, monodispersed, and biocompatible dendritic polymer and exhibits pH-dependent conformational changes.8–10 Thus, researchers have widely used these unique dendrimer systems for drug delivery and biomedical applications.
For delivering therapeutic agents into the target cells,11 the first challenging step for most of the nanocarriers is the cell uptake process.12,13 The major uptake of nanoparticle-mediated carriers into the cells is generally via the endocytic pathway (non-specific internalization or receptor-mediated uptake).5,14 Nevertheless, the effect of cellular uptake limits the therapeutic efficacy of many anticancer drugs.15 Therefore, an ideal nano-drug delivery system is capable of increasing the targeted internalization with specific high-dose drug accumulation in cancerous tissues.16,17 Briefly, the targeting efficiency is dependent on the type of the ligand and the biostability of the designed ligand in the carrier system.18 By binding NPs with cell-penetrating ligands, such as peptides, antibodies, and folic acid, the uptake of NPs would be dramatically enhanced through receptor-mediated endocytosis, which is also called the active targeting of cancer cells.19 Meanwhile, amino acids are commonly used as the targeting group for improving the uptake of nanoparticles in the cancer cells. In fact, glucose and amino acids are common substances that exhibit high metabolic requirements of cancer cells, resulting in increase in the demands of these substrates in the tumor cells when compared to that observed for normal cells.20–22 Many studies have reported that amino acids are also used for tumor detection and assessment before the therapeutic treatment.23 The unique structural characteristics of each amino acid result in different physicochemical performances and therefore, amino acids play an irreplaceable role in biochemistry and biological performance inside the cells.
Among these amino acids, L-histidine as an essential amino acid and L-cysteine as a nonessential amino acid can improve the cell uptake for drug delivery, facilitating high cellular endocytic behavior and early release of the payload into the nucleus via the protonation effect. Histidine is the most active and versatile amino acid and plays multiple roles in protein interactions.24 Furthermore, the imidazole ring of histidine has a lone pair of electrons on the unsaturated nitrogen, endowing it with amphoteric nature via protonation–deprotonation (pKa value of 6).25 Imidazole groups ionize at acidic conditions, thereby triggering osmotic swelling and inducing the destabilization of polymeric NPs, which lead to the release of the incorporated drug.26 Researchers have reported that the imidazole-containing polymer demonstrates improved efficiency of gene transfection27,28 and high drug delivery efficiency. In addition, the sequence of cysteine and histidine residues incorporated into the trans-activator of transcription (TAT) structure promotes the gene transfection efficiency by up to 7000-fold when compared with the original Tat peptide.29 The PHEA-g-C18-His drug delivery system can increase the buffering ability and improve the inhibition of cell proliferation.11 In addition, histidine-enriched multifunctional peptides can promote cellular uptake,26 while the conjugation of cysteine with the TAT peptide can enhance the cellular uptake of cargos in cancer cells (Gd3+, 4.7 times higher than normal cells).30 Overall, the histidine and cysteine-containing peptides have been confirmed to increase the cellular uptake. Due to cancer cell rapid proliferation and increased antioxidant demands,20,22 amino acids facilitate improvements in cellular uptake. Among all other various natural amino acids, histidine is the most active and versatile member; it plays multiple roles in protein interactions and is often the key residue in enzyme catalytic reactions.31 In addition, the SH group of cysteine molecules readily interacts with the carbonyl group of many proteins.32 In our study, we used PAMAM-conjugated histidine and cysteine (GH and GHC) NPs to induce enhanced protein interactions, resulting in the enhanced cellular uptake of NPs.
In this study, high-uptake dendrimer derivatives have been developed for potent chemotherapeutic delivery. The biocompatible G4.5 PAMAM dendrimer was selected as the nanocarrier of the system by decorating with amino acids, namely, histidine and cysteine to build single and multiple amino acid-modified dendrimer NPs (GH and GHC, respectively). In addition, the amino acid-modified dendrimers were characterized and explored to understand if the existence of amino acids on dendrimers could enhance the cellular uptake and trigger an increased amount of drug to induce cell inhibition. In this regard, the therapeutic efficiency and cell viability were further studied in vitro against HeLa cells and in vivo in a zebrafish model.
2. Results and discussion
2.1. Synthesis and characterization of GH nanoparticles and GHC aggregates
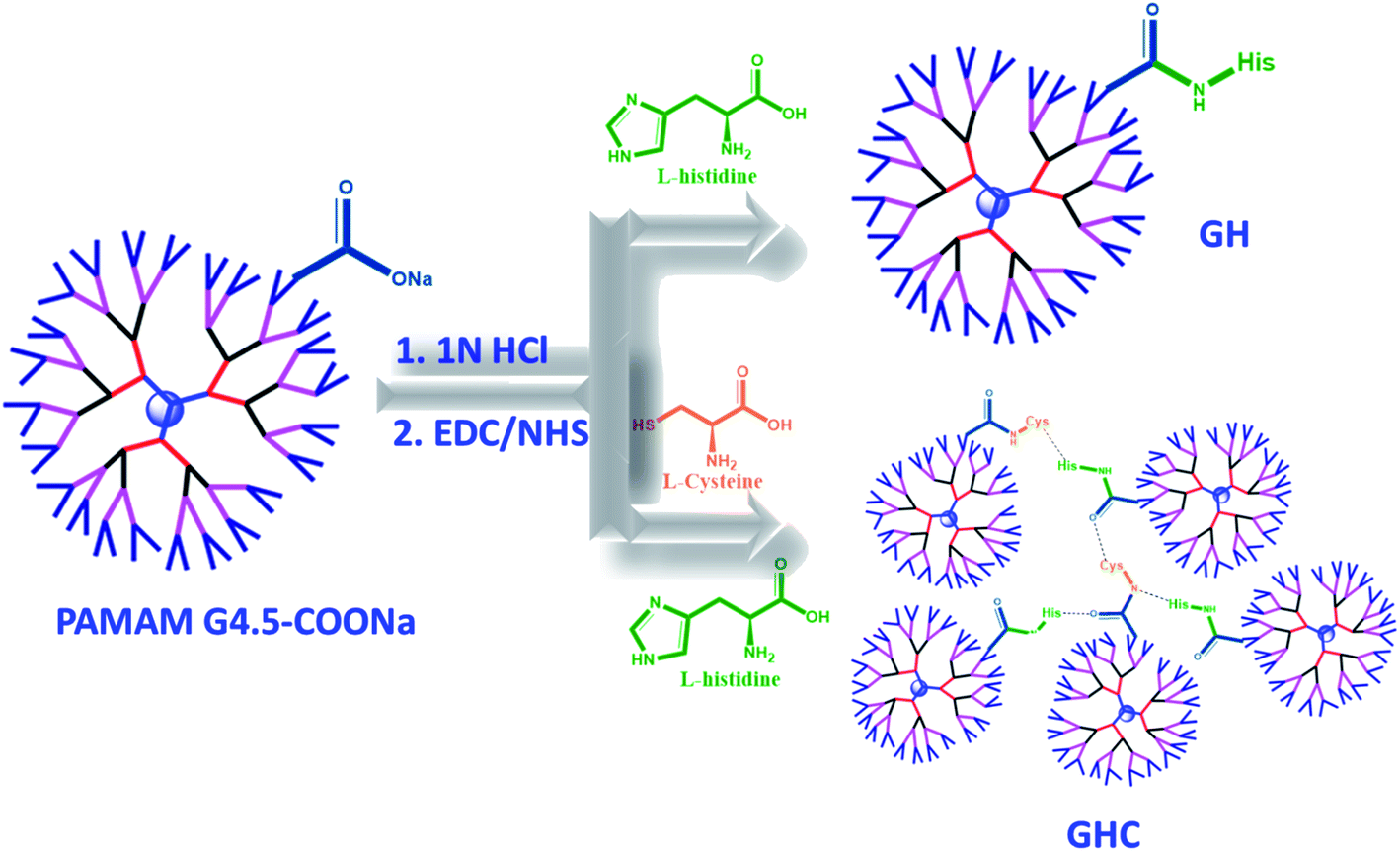
In the present study, we developed nanocarrier systems composed of one amino acid and two amino acids decorated with G4.5 dendrimers and evaluated the efficacy of unmodified and amino acid-modified G4.5 dendrimers as drug delivery vehicles. The main aim of our study was to improve the cellular uptake and bioactivity of anticancer drugs. The G4.5 dendrimers were coupled with either single L-histidine (H) or multiple amino acids such as L-histidine and L-cysteine (H and C) using the EDC/NHS agents to form stable amide bonds.33 As shown in Scheme 1, the G4.5 dendrimers were conjugated with L-His to form GH NPs. On the other hand, the G4.5 dendrimers were conjugated with both H and C to form GHC aggregates through disulfide bonding. Afterwards, DOX will be encapsulated in the G4.5 dendrimers, GH NPs, and GHC aggregates to obtain anticancer drug delivery systems (Scheme 1).
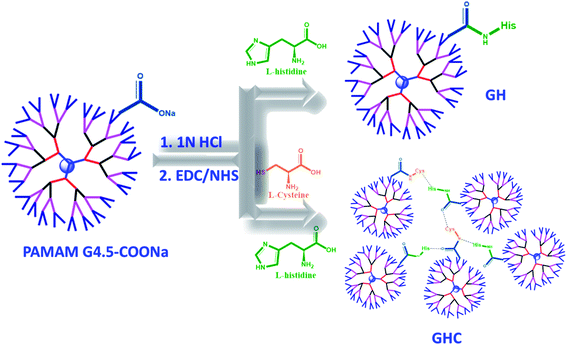 |
| | Scheme 1 The conjugation of the G4.5 dendrimers with L-histidine and L-cysteine (GH and GHC NPs) and loading the chemotherapy drug (DOX). | |
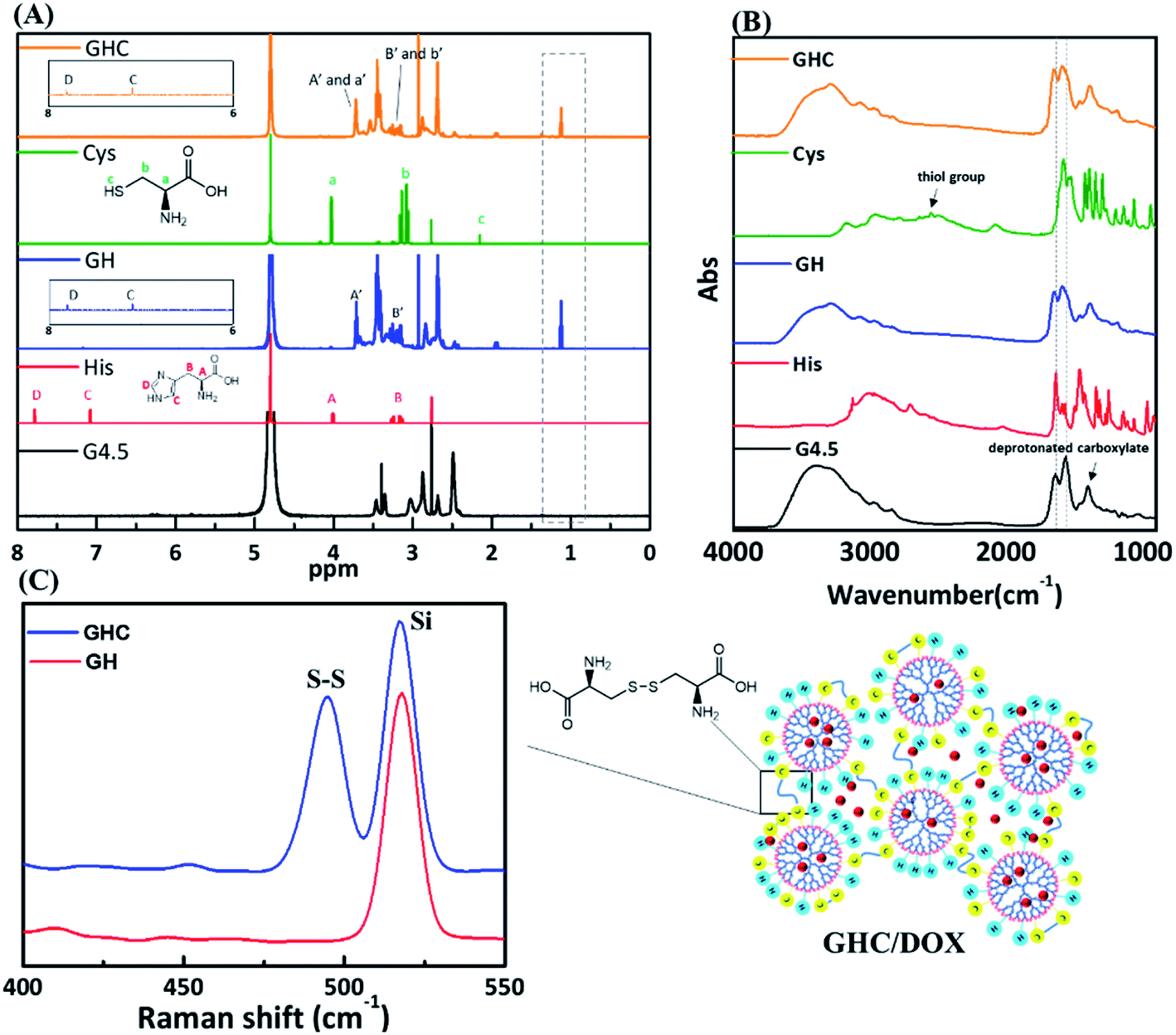
The as-synthesized GH NPs and GHC aggregates were characterized using 1H NMR, FT-IR, and Raman spectroscopy. Fig. 1 shows the 1H NMR and FT-IR spectra of the G4.5 dendrimers, GH, and GHC NPs. For both cases, a new amide proton peak is located at 1.1 ppm, confirming that the primary amino groups successfully reacted with the carboxylic groups of the G4.5 dendrimer. Moreover, in the NMR spectrum of GHC, the proton signal nearby nitrogen atom (i.e. CHN) in the L-Cys signal is observed to be slightly shifted, which is consistent with a previous report for chemical conjugation. In both the spectra, the characteristic peak of the imidazole ring of L-His appeared at 7.0–7.8 ppm11 (Fig. 1A). The successful conjugation of amino acids to the dendrimer was also demonstrated by FT-IR spectroscopy. The spectra obtained for the G4.5 dendrimer exhibit characteristic peaks of amide I NH stretching at 3278 cm−1 and NH bending at 1638 cm−1, amide II C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1561 cm−1, and broad OH stretching vibrations at 3400 cm−1.34,35 Histidine, cysteine and the G4.5 dendrimer comprise amine groups and carboxylic acids. For GH and GHC (containing an additional amide bond), although the spectra are similar to that observed for the unmodified dendrimer, the amide I and II peaks shift slightly (Fig. 1B). Notably, the GHC aggregates of the thiol group of L-Cys36 are absent (Fig. 1B), indicating disulfide linkage formation.37 Subsequently, the GHC aggregates exhibit a strong sharp band of the disulfide bonds at 495 cm−1 in the Raman spectra (Fig. 1C), which is in agreement with a previous report.38
O stretching at 1561 cm−1, and broad OH stretching vibrations at 3400 cm−1.34,35 Histidine, cysteine and the G4.5 dendrimer comprise amine groups and carboxylic acids. For GH and GHC (containing an additional amide bond), although the spectra are similar to that observed for the unmodified dendrimer, the amide I and II peaks shift slightly (Fig. 1B). Notably, the GHC aggregates of the thiol group of L-Cys36 are absent (Fig. 1B), indicating disulfide linkage formation.37 Subsequently, the GHC aggregates exhibit a strong sharp band of the disulfide bonds at 495 cm−1 in the Raman spectra (Fig. 1C), which is in agreement with a previous report.38
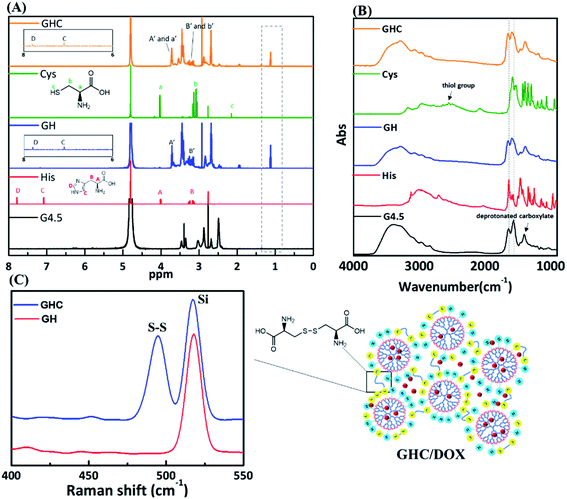 |
| | Fig. 1 (A) 1H NMR, (B) FT-IR, and (C) Raman spectra of the amino acid-modified G4.5 dendrimers GH and GHC. | |
2.2. Drug loading and releasing efficiency
After the validation of successful synthesis, DOX was loaded into the G4.5, GH, and GHC dendrimer carriers. Table 1 summarizes the DOX encapsulation efficiency and loading capacity of the G4.5 dendrimer and dendrimer derivatives. The results showed that DOX was successfully loaded into all the dendrimer carriers. However, drug loading increased in the amino acid-modified dendrimers compared to that in the unmodified dendrimer (LC, 2–3% increase; EE, 14–16% increase). Following drug loading, the unique imidazole structure of histidine in the interior sections of the dendrimer derivatives facilitates favorable interactions with DOX through (1) π–π stacking23 and (2) hydrogen bonding,3 which might be one of the reasons for enhanced drug loading in the dendrimer derivatives. Because the imidazole structure of histidine comprises a conjugative π–plane, favorable π–π stacking interactions with the aromatic rings of DOX are facilitated. In addition, weak hydrogen bonding interactions can occur between the hydrogen atoms in imidazole/histidine and oxygen atoms in the carbonyl groups of DOX and also between the oxygen atom in the carbonyl groups of histidine and the glycosidic amine of DOX.10 These additional interactions between DOX and dendrimer-derivative carriers may account for the observed enhanced drug loading (in both GH/DOX and GHC/DOX). Moreover, the aggregation of NPs through the disulfide network of GHC is conducive to maximum drug loading. Thus, the DOX contents of the histidine and cysteine amino acid-modified dendrimers significantly increased.
Table 1 Encapsulation efficiency (EE) and loading capacity (LC) of dendrimer (G4.5) and amino acid-modified dendrimers (GH and GHC) (mole ratio of carriers to DOX: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9)
:9)
| Name |
LC (%) |
EE (%) |
| G4.5/DOX |
6.2 ± 1.9 |
33.2 ± 10.8 |
| GH/DOX |
8.7 ± 0.5 |
47.5 ± 3.5 |
| GHC/DOX |
9.0 ± 0.9 |
49.0 ± 4.9 |
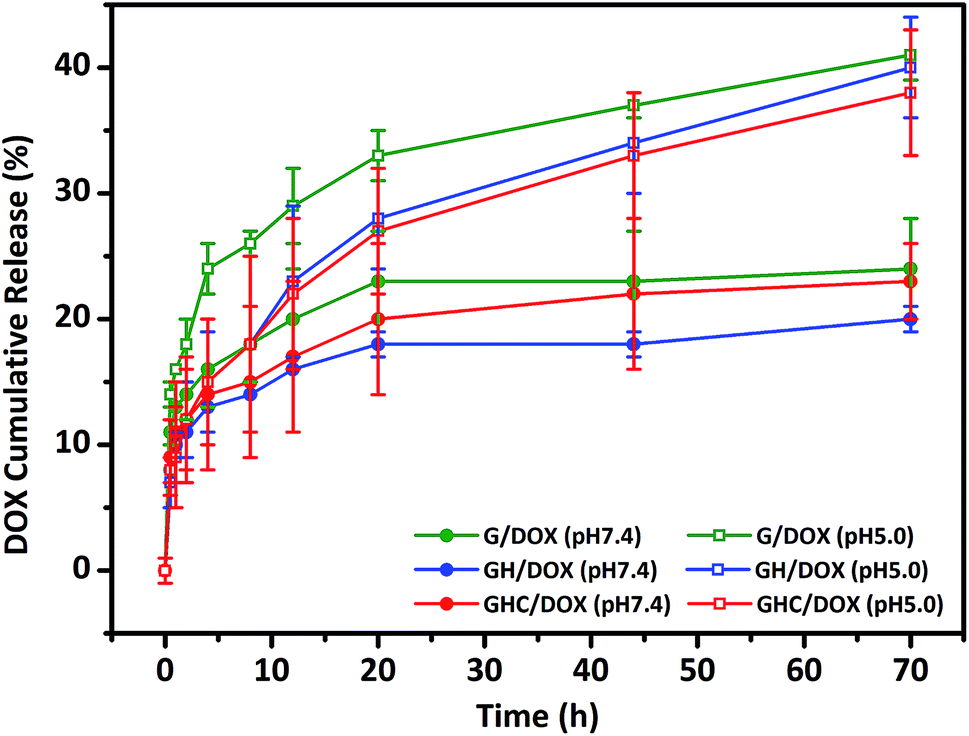
GH and GHC comprise ionized imidazole groups, which become protonated upon acidic pH (internal stimulus) stimulation.10,23 We evaluated the impact of these imidazole groups on the release behavior in the amino acid derived-dendrimers (G4.5/DOX, GH/DOX and GHC/DOX) at different pH values (7.4 and 5.0). As shown in Fig. 2, under all conditions, the DOX release follows a biphasic pattern; an initial rapid release is followed by long-term sustained release, which is in agreement with a previous report.35 At pH 7.4, only a small percentage of DOX was released from the carriers (about 20% over 72 h). Notably, both cysteine and histidine-modified dendrimer complexes (GH/DOX and GHC/DOX) conferred enhanced drug release at acidic conditions (pH 5.0), which was higher than that at physiological conditions (pH 7.4) in the G, GH, and GHC carriers (increased by 17%, 20%, and 15%, respectively), confirming the pH sensitivity of the dendrimer-based drug carriers.36 Under acidic conditions, the drug release rate of G4.5 remained the highest during the 72 h release profile. Because there were no amino acids on the surface of the unmodified dendrimer, an acidic buffer could easily protonate the interior of the dendrimer, leading to a conformational change and enhanced drug release. GH is the most sensitive to the microenvironment (demonstrating an increase of 20% in drug release) since imidazole protonation triggers the destabilization of carriers.10,24,37 In contrast, GHC was the most stable, with the minimum increase in drug release upon pH stimulation, and this was attributed to the stable microenvironment within the aggregates. It is possible that the amino acids (imidazole and disulfide) on the exterior surface might serve as a barrier to protect the interior of the dendrimer. Therefore, these results indicate that GH/DOX might yield a promising delivery system, releasing a high dose on the target due to the micro-environmental differences between healthy tissues and cancer cells.
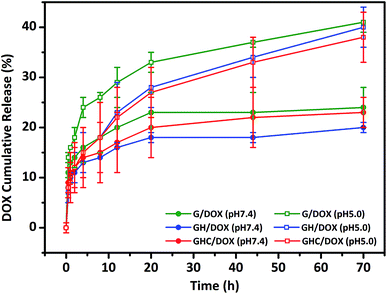 |
| | Fig. 2 In vitro DOX release study from unmodified and amino acid-modified G4.5 dendrimers at different pH values. | |
2.3. Cellular uptake and distribution of dendrimer–drug complexes
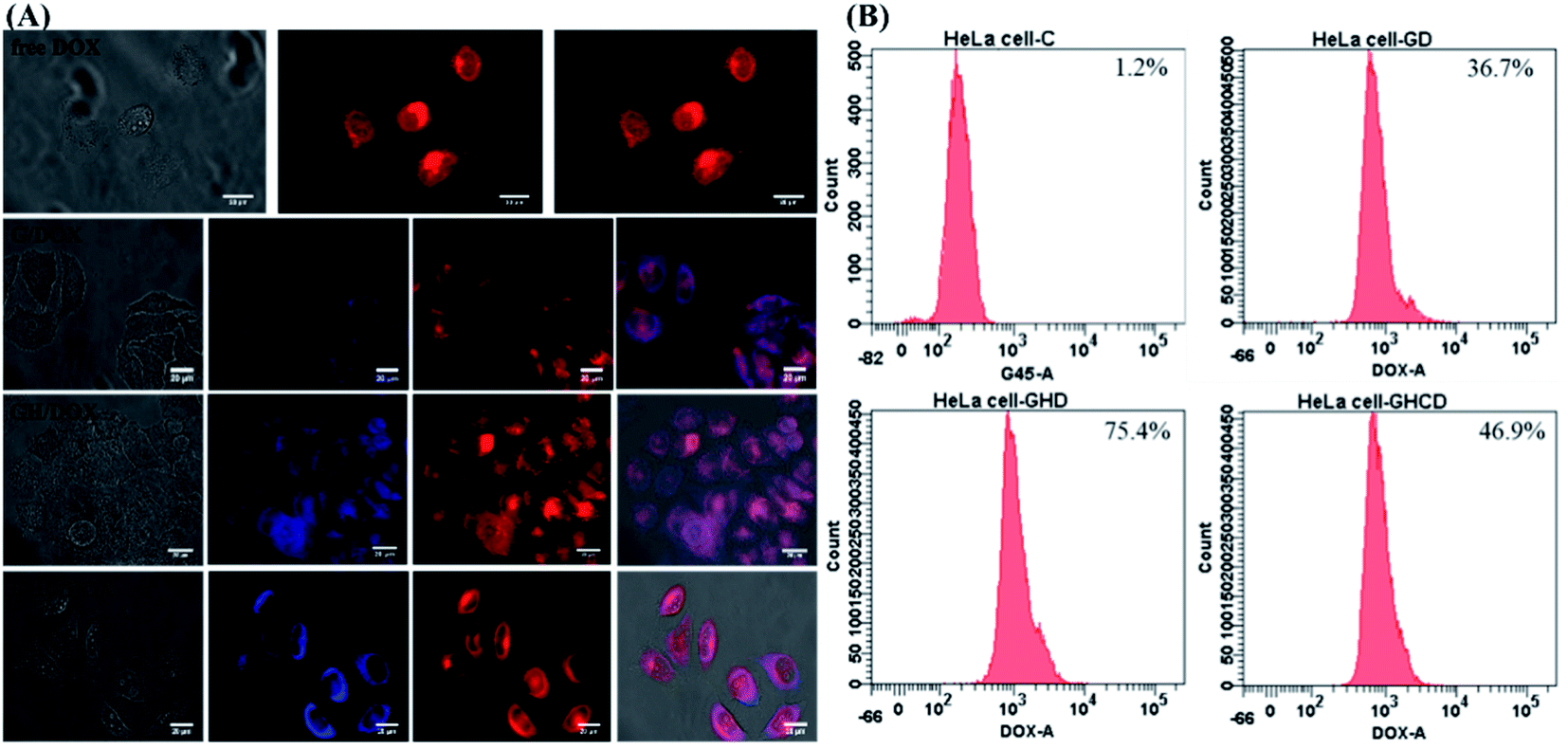
Intracellular morphological observations were obtained using the iRiS™ Digital Cell Imaging System. Fig. 3A shows the successful uptake of amino acid-modified dendrimer carriers by HeLa cells and the subsequent drug transfer to the cell nuclei. After 8 h incubation with free DOX, a strong DOX fluorescence signal was observed in the cytoplasm and nucleus, indicating that the quick diffusion into the cells through the cell membrane was followed by transfer to the nucleus and consequent blocking of DNA replication, as previously reported.39 In contrast, the nanoparticle-mediated entry into the cells via the endocytic pathway is a relatively slow process.5 Although a relatively weak fluorescence signal was observed because DOX was located in the dendrimer–drug system, the drugs accumulated within the target cells. Moreover, amino acid modification altered the pattern of cellular distribution and slowed down the amount of the drug delivered to the cells. Indeed, less DOX was observed in the nucleus with G4.5 compared to that for GH and GHC. One of the reasons for this observation is that imidazole endows pH-dependent amphoteric properties (pKa ∼ 6) and endosomolytic activity14,40,41 Therefore, the histidine-rich dendrimer promoted the disruption of the endosomal membrane and early release of entrapped molecules in the amino acid-modified dendrimer systems.
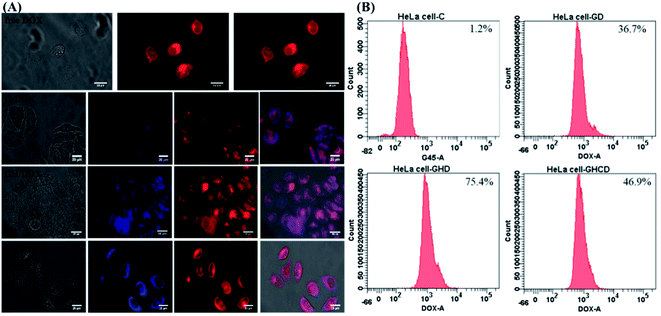 |
| | Fig. 3 HeLa cells treated with G/DOX, GH/DOX, and GHC/DOX: (A) fluorescence microscopy images. G4.5 dendrimers (blue); DOX (red) and (B) flow cytometry analysis. | |
Another issue is the cellular uptake capacity of the dendrimer-based complexes in HeLa cells. Studies have reported that amino acids can facilitate cancer drug delivery in other nanotechnological applications.11,20,25,41 Flow cytometry was used to quantify the cellular uptake from the DOX-loaded G4.5 dendrimer derivatives in 12 h treatment. The control revealed a lack of fluorescence, whereas G/DOX, GH/DOX, and GHC/DOX showed enhanced cellular fluorescence. Higher fluorescence was observed in GH/DOX and GHC/DOX compared to that in the G4.5 dendrimer, confirming that amino acid modification improved the cellular uptake capacity. These observations are attributed to the over-expression of some amino acid transporters in immortalized cells since amino acids are necessary for the rapid proliferation and increased antioxidant demands of cancer cells.21,42 The next step was to demonstrate that the observed increase in the efficiency of DOX release from the amino acid-modified dendrimers translated into the inhibition of HeLa cell proliferation in vitro and in vivo.
2.4. In vitro inhibition efficiency of cancer cell proliferation
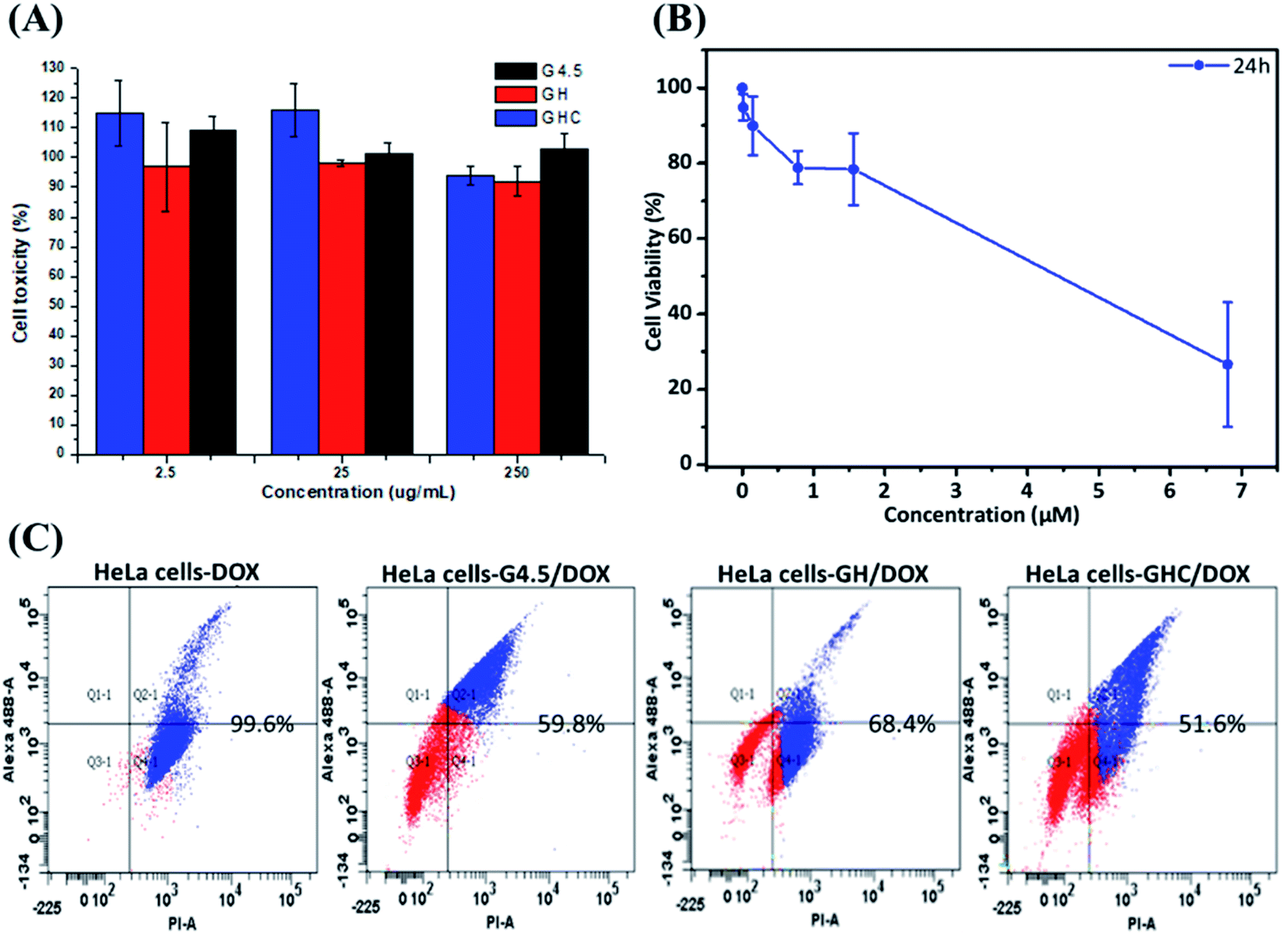
Generally, the primary critical consideration for biomaterials is cytotoxicity; PAMAM dendrimers have demonstrated biocompatibility with animal physiology.43,44 The high survival rate of HeLa cells after 24 h exposure to the dendrimer G4.5 and both dendrimer derivatives (GH and GHC) confirmed their compatibility and suitability for drug delivery (Fig. 4A). The dose-dependent cytotoxicity of free DOX with HeLa cells was evaluated using an MTT assay. Fig. 4B shows the proportional reduction in the HeLa cell viability with an increased concentration of free DOX (which is capable of efficiently inhibiting cell proliferation). Subsequently, cell death was assessed using flow cytometry and annexin V–propidium iodide (PI) staining. Fig. 4C indicates the lower cytotoxicity of the DOX-loaded dendrimer derivatives compared to that of free DOX (a consequence of the different uptake routes and release mechanisms). Efficient nanocarriers enhance the local drug delivery in the desired cancerous tissues and reduce the off-target tissue effects. The inhibition capacities of G4.5/DOX, GH/DOX, and GHC/DOX treatments were 59.8%, 68.4%, and 51.6%, respectively (Fig. 4C). Despite the fact that the highest release was observed with G4.5/DOX after 12 h (Fig. 2), the inhibition of cell reproduction was greater in GH/DOX and was almost as high as that of G/DOX. These results confirmed the high affinity of the amino acid-modified dendrimers for HeLa cells (Fig. 3), resulting in an accumulation of drugs within the cells. The amino acid-modified dendrimers rapidly released drugs within the cytoplasm following the protonation of histidine. In addition, the disulfide linkages between the aggregates of GHC were reduced by glutathione (GSH) in the tumor tissues, exposing more NPs to the cellular environment. Taken together, the results indicate that the DOX-loaded amino acid dendrimers demonstrate excellent efficiency for the inhibition of HeLa cell proliferation. The results suggest that these drug delivery systems show great promise.
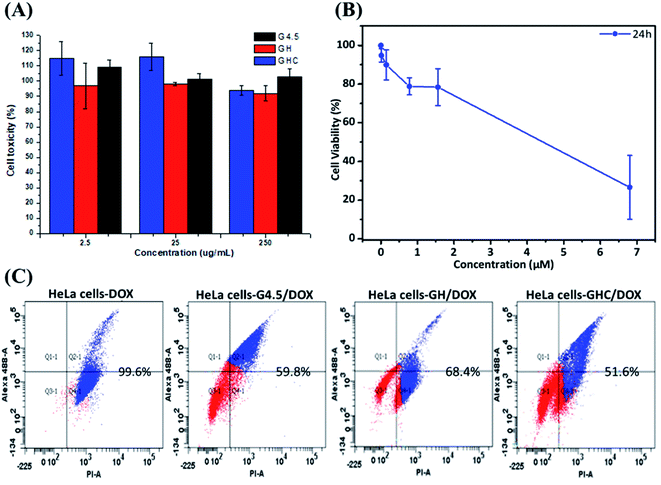 |
| | Fig. 4 In vitro cytotoxicity of HeLa cells. MTT assay after treatment for 24 h with (A) G4.5 dendrimer, GH NPs and GHC aggregates; (B) different concentrations of free DOX, (C) apoptosis and necrosis in cell populations were determined using flow cytometry with annexin V/propidium iodide staining (treated with G/DOX, GH/DOX and GHC/DOX for 12 h). | |
2.5. In vivo zebrafish experiments
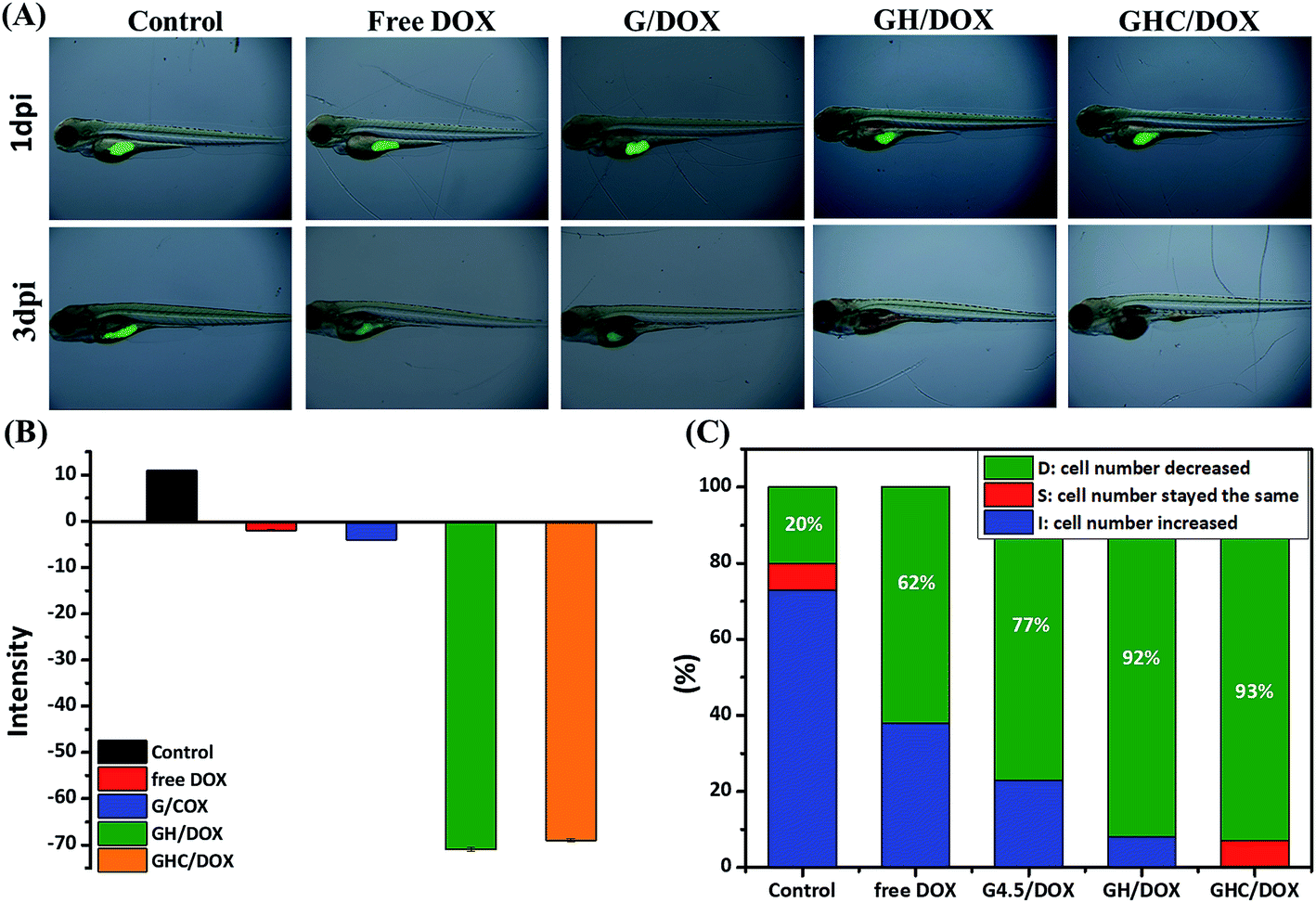
Zebrafish models have also been used in preclinical research for the evaluation of novel anticancer nanomedicines.45 HeLa cell proliferation in zebrafish embryos was monitored with noninvasive fluorescence imaging. Prior to the implantation of cells into the yolkspace of zebrafish embryos, the cells were labeled by carboxyfluorescein succinimidyl ester (CFSE) (2bf). Then, the zebrafish embryos (2 dbf) were treated with the DOX-loaded dendrimer complexes for 48 h and the intensities of the fluorescent markers were detected at 1 dpi (1 day post imaging) and 3 dpi. CFSE fluorescence was used to monitor the HeLa cell proliferation and differentiation46–48 in the cell viability assays. Fig. 5A–C show the survival and morphological appearance of the embryos after DOX treatment. As a result, the fluorescence in zebrafish was significantly attenuated after treatments with free DOX, G/DOX, GH/DOX, and GHC/DOX (Fig. 5A). Moreover, the observed decrease in the fluorescence intensity (negative value, Fig. 5B) of the free DOX and DOX-loaded dendrimer complexes was considered as an indication of cell death compared to that for the control group, confirming the drug-induced cytotoxicity for HeLa cells. The negative value has been calculated from eqn (5) in the Experimental section. The statistics of the HeLa cell viability were recorded by the CFSE fluorescence intensity (Fig. 5C). In GH/DOX and GHC/DOX, a remarkable decrease in the HeLa cell number demonstrated the high potency to suppress the cell proliferation (up to 90%). Therefore, the findings of this study suggest that GH/DOX and GHC/DOX can substantially enhance the antitumor effect during in vivo activity.
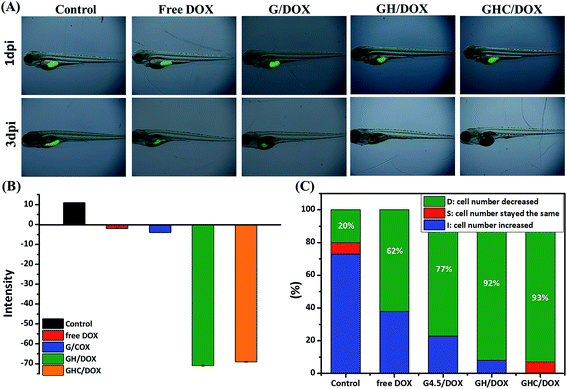 |
| | Fig. 5 In vivo zebrafish study. HeLa cells were labeled with CFSE in the embryo and treated with PBS, free DOX, G/DOX, GH/DOX, and GHC/DOX for 48 h. Proliferation inhibition was evaluated by comparison of the intensity of CFSE-labeled cells at 1 dpi and 3 dpi. (A) CFSE fluorescence images were recorded at 0 dpi and 3 dpi; (B) average CFSE fluorescence; (C) survival rate of HeLa cells within zebrafish (each group contained 15 zebrafish). | |
3. Conclusion
In this study, a novel drug delivery system was developed through the modification of G4.5 dendrimers with either L-histidine (H) alone or L-histidine (H) and L-cysteine (C) together (GH nanoparticles and GHC aggregates, respectively). Biocompatible amino acid modification of the dendrimers increased the drug loading capacity but retained the pH-dependent DOX release behavior. GH and GHC demonstrated high affinity for HeLa cells and enhanced stability at non-physiological conditions (compared to the G4.5 dendrimers). Therefore, these NPs enhanced the specific accumulation at the desired cancerous tissues. An in vitro drug release study demonstrated a lower release rate and HeLa cell proliferation was effectively inhibited by GH and GHC. Similar results were obtained in an in vivo zebrafish animal model. At a lower pH, the ionized imidazole groups of histidine disrupted the endosomal membrane and triggered the destabilization of carriers, resulting in an early release of the entrapped anticancer drug DOX towards the cancer cell nucleus. In summary, the amino acid-modified PAMAM dendrimers have potential as local delivery systems for various biomolecules in the future.
4. Experimental section
4.1. Synthesis and characterization of amino acid-modified dendrimers
The synthetic amide bond formation methodology was adopted for the preparation of histidine and cysteine-modified PAMAM G4.5 dendrimers and schematically presented in Scheme 1. The G4.5–COO–Na+ solutions were acidified with 1 N HCl solution up to a pH of 5 and dialyzed. Afterward, the carboxylic groups of the G4.5 dendrimer were activated by using an excess amount of NHS and EDC overnight. The activated dendrimer solution was reacted with L-His (mole ratio of 1:320) for 24 h. Finally, the purification was dialyzed (molecular weight [MWCO]: 6–8 kDa) against ultra-distilled water to remove the unbound molecules and lyophilized overnight for the further confirmation of the GH and GHC nanocarriers. The obtained modified dendrimers were confirmed through FT-IR (PerkinElmer) and NMR (Bruker Avance III HD600-MHz NMR with D2O as the solvent) spectroscopy. A similar procedure was followed to conjugate both cysteine and histidine (mole ratio of 1:160:160).
4.2. Drug loading and in vitro release study
DOX loading within the G4.5, GH, and GHC dendrimers was carried out, as previously reported.38 Neutralized doxorubicin hydrochloride (DOX–HCl) dissolved in dimethyl sulfoxide (DMSO) was slowly dropped into a diluted dendrimer solution. The resulting solution was continuously stirred at room temperature overnight and then dialyzed against PBS for 24 h to remove the unloaded drug. The dendrimer–drug interaction and free DOX calibration curve were determined using a UV-vis spectrophotometer at 500 nm (Jasco V-730 UV-vis spectrophotometer).4,49 Loading efficiency and encapsulation efficiency (EE%) were obtained using eqn (1) and (2), respectively:50| |
 | (1) |
| |
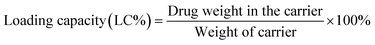 | (2) |
The drug release profiles from the DOX-loaded dendrimers (G/DOX, GH/DOX, and GHC/DOX) were evaluated at different pH using the dialysis method. Briefly, solutions containing the complexes were transferred into a dialysis membrane (MWCO 1 kDa) and immersed in phosphate buffer (pH 7.4) or citrate buffer (pH 5.0) at 37 °C under constant stirring. The external buffer solution (2 mL) was collected at a predetermined time interval and the concentration of the drugs was measured by UV-vis spectrophotometry. Furthermore, the external buffer system was maintained at a constant volume and pH by replacement with a corresponding fresh volume of PBS.6,51 The experiment was performed in triplicate and cumulative release was calculated using eqn (3):
| |
 | (3) |
4.3. In vitro cellular uptake
Human cervical carcinoma (HeLa) cells were grown in Dulbecco's modified Eagle's medium and supplemented with 10% fetal bovine serum and 1% sodium pyruvate at 37 °C and 5% CO2. Then, the cells (1 × 106 cells per well) were incubated with free DOX, G/DOX, GH/DOX and GHC/DOX (DOX concentration: 5 μg mL−1) for 8 h, washed with PBS, and captured through fluorescence microscopy (iRiSTMDigital Cell Imaging System).
4.4. Cell biological evaluation
The toxicity of biomaterials and the anticancer drug was evaluated by MTT assays using previously reported protocols.49,51 HeLa cells were seeded in 96-well plates at a density of 5000 cells per well and treated with different concentrations of free DOX and amino acid-modified dendrimers (GH NPs and GHC aggregates) for 24 h. After incubation, the medium was replaced with an MTT solution (0.5 mg mL−1) for 4 h and then, DMSO was substituted. Finally, absorbance (570 nm) was measured by an Enzyme-Linked ImmunoSorbent Assay (ELISA). Cell viability was calculated as per the following formula (4):| |
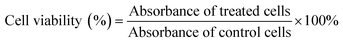 | (4) |
The inhibiting cell growth effect and quantitative cellular uptake were conducted through flow cytometry (Becton Dickinson FACS Area III cell sorter). Here, 11 μM of free DOX, G/DOX, GH/DOX and GHC/DOX were treated with HeLa cells (1 × 106 cells per well) incubated in T25 dishes for 12 h. After that, the cells were washed, collected and resuspended in 500 μl of 1× annexin-binding buffer for analysis. Alexa Fluor@ 488 annexin V and PI were added following the manufacturer's recommendation.
4.5. In vivo zebrafish tumor-xenograft study
Zebrafish (National Health Research Institutes) as an animal model was used to evaluate the effects of in vivo chemotherapy. HeLa cell lines (200 cells) labeled with CFSE were implanted into the yolk space of 2 dpf zebrafish embryos (embryos at 0–3 d post-fertilization [dpf]) based on previous protocols.52,53 After 1 day post-implantation, the 3 dpf blastulas were immersed in 2 μM of free DOX and payload G4.5, GH and GHC solutions for 48 h. Subsequently, the images were recorded at 3 days post-implantation and 5 days post-fertilization (3 dpi or 5 dpf) (as shown in Fig. 6); the intensity was calculated as follows (5):| |
 | (5) |
 |
| | Fig. 6 In vivo zebrafish tumor-xenograft design for the evaluation of the therapeutic efficacy of drug carriers. | |
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of National Health Research Institutes (NHRI) and approved by the Institution Animal Care and Use Committee (IACUC) of the NHRI, protocol number NHRI-IACUC-107058.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to thank the Ministry of Science and Technology of the Republic of China (Taiwan) (Grant No. MOST 108-2211-E-182-005), National Taiwan University of Science and Technology, National Taiwan University and Taipei Medical University Joint Research Program (Grant No. TMU-NTUST-104-10 and, TMU-NTUST-105-07 and TMU-NTUST-106-06), Lo-Hsu Medical Foundation, LotungPoh-Ai Hospital (Funding Number: 10908) and Hi-Q Marine Biotech International Ltd., New Taipei City, Taiwan for providing the financial support.
References
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- M. A. Deli, N.-T. Chen, C.-Y. Wu, C.-Y. Chung, Y. Hwu, S.-H. Cheng, C.-Y. Mou and L.-W. Lo, PloS One, 2012, 7, e44947 CrossRef PubMed.
- Y. Liu, K. Li, Y. Wu, J. Ma, P. Tang, Y. Liu and D. Wu, Biomater. Sci., 2019, 7, 3662–3674 RSC.
- F. Fu, Y. Wu, J. Zhu, S. Wen, M. Shen and X. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 16416–16425 CrossRef CAS PubMed.
- Y. Liu, W. Wang, J. Yang, C. Zhou and J. Sun, Asian J. Pharm. Sci., 2013, 8, 159–167 CrossRef.
- P. S. Lai, P. J. Lou, C. L. Peng, C. L. Pai, W. N. Yen, M. Y. Huang, T. H. Young and M. J. Shieh, J. Controlled Release, 2007, 122, 39–46 CrossRef CAS PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS.
- E. V. Munsell, N. L. Ross and M. O. Sullivan, Curr. Pharm. Des., 2016, 9, 1227–1244 CrossRef PubMed.
- N. Oddone, N. Lecot, M. Fernandez, A. Rodriguez-Haralambides, P. Cabral, H. Cerecetto and J. C. Benech, J. Nanobiotechnol., 2016, 14, 45 CrossRef PubMed.
- M. Markowicz, P. Szymánski, M. Ciszewski, A. Kłys and E. z. Mikiciuk-Olasik, Biol. Phys., 2012, 38, 637–656 CrossRef CAS PubMed.
- S. R. Yang, H. J. Lee and J. D. Kim, J. Controlled Release, 2006, 114, 60–68 CrossRef CAS PubMed.
- V. V. Munsell, N. L. Ross and M. O. Sullivan, Curr. Pharm. Des., 2016, 22, 1227–1244 CrossRef PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- F. Fekri, J. Abousawan, S. Bautista, L. Orofiamma, R. M. Dayam, C. N. Antonescu and R. Karshafian, Sci. Rep., 2019, 9, 17768 CrossRef PubMed.
- F. Alexis, J. W. Rhee, J. P. Richie, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Urol. Oncol., 2008, 26, 74–85 CrossRef CAS PubMed.
- R. Misra, S. Acharya and S. K. Sahoo, Drug Discov. Today, 2010, 15, 842–850 CrossRef CAS PubMed.
- H.-Q. Yin, D.-S. Mai, F. Gan and X.-J. Chen, RSC Adv., 2014, 4, 9078 RSC.
- Y. Wei, T. Tang and H. B. Pang, Nat. Commun., 2019, 10, 3646 CrossRef PubMed.
- H. Nazari, K. Ashtari, M. Soleimani, K. Bagherzadeh, S. Safari and B. Mehravi, Iran. J. Pharmacol. Ther., 2018, 16, 1–8 Search PubMed.
- B. Mehravi, M. S. Ardestani, M. Damercheli, H. Soltanghoraee, N. Ghanaldarlaki, A. M. Alizadeh, M. A. Oghabian, M. S. Shirazi, S. Mahernia and M. Amanlou, Mol. Imag. Biol., 2014, 16, 519–528 CrossRef PubMed.
- C. Munoz-Pinedo, N. El Mjiyad and J. E. Ricci, Cell Death Dis., 2012, 3, e248 CrossRef CAS PubMed.
- T. Singhal, T. K. Narayanan, V. Jain, J. Mukherjee and J. Mantil, Mol. Imag. Biol., 2008, 10, 1–18 CrossRef PubMed.
- S. M. Liao, Q. S. Du, J. Z. Meng, Z. W. Pang and R. B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- E. S. Lee, H. J. Shin, K. Na and Y. H. Bae, J. Controlled Release, 2003, 90, 363–374 CrossRef CAS PubMed.
- Z. Meng, L. Luan, Z. Kang, S. Feng, Q. Meng and K. Liu, J. Mater. Chem. B, 2017, 5, 74–84 RSC.
- D. W. Pack, D. Putnam and R. Langer, Biotechnol. Bioeng., 2000, 67, 217–223 CrossRef CAS PubMed.
- M. E. Davis, Mol. Pharm., 2009, 6, 659–668 CrossRef CAS PubMed.
- S. L. Lo and S. Wang, Biomater. Sci., 2008, 29, 2408e2414 Search PubMed.
- H. Nazari, K. Ashtari, M. Soleimani, K. Bagherzadeh, S. Safari and B. Mehravi, Iran. J. Pharmacol. Ther., 2018, 16, 1–8 Search PubMed.
- S.-M. Liao, Q.-S. Du, J.-Z. Meng, Z.-W. Pang and R.-B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- D. Pal and P. Chakrabarti, J. Biomol. Struct. Dyn., 1998, 15, 1059–1072 CrossRef CAS PubMed.
- S. L. Mekuria and H. C. Tsai, Colloids Surf. B Biointerfaces, 2015, 135, 253–260 CrossRef CAS PubMed.
- M. J. Prieto, N. E. del Rio Zabala, C. H. Marotta, H. Carreno Gutierrez, R. Arevalo Arevalo, N. S. Chiaramoni and S. del Valle Alonso, PLoS One, 2014, 9, e90393 CrossRef PubMed.
- T. W. Mekonnen, Y. S. Birhan, A. T. Andrgie, E. Y. Hanurry, H. F. Darge, H. Y. Chou, J. Y. Lai, H. C. Tsai, J. M. Yang and Y. H. Chang, Colloids Surf. B Biointerfaces, 2019, 184, 110531 CrossRef CAS PubMed.
- Q. Z. Li Li, Y. Ding, X. Cai, S. Gu and Z. Cao, Anal. Methods, 2014, 6, 2715 RSC.
- G. L. Wang, Y. M. Dong, H. X. Yang and Z. J. Li, Talanta, 2011, 83, 943–947 CrossRef CAS PubMed.
- S. Y. Wu, H. Y. Chou, C. H. Yuh, S. L. Mekuria, Y. C. Kao and H. C. Tsai, Adv. Sci., 2018, 5, 1700339 CrossRef PubMed.
- L. Jiang, Z.-m. Gao, L. Ye, A.-y. Zhang and Z.-g. Feng, Biomater. Sci., 2013, 1, 1282 RSC.
- N. Ferrer-Miralles, J. L. Corchero, P. Kumar, J. A. Cedano, K. C. Gupta, A. Villaverde and E. Vazquez, Microb. Cell Factories, 2011, 10, 101 CrossRef CAS PubMed.
- N. N. Sheveleva, D. A. Markelov, M. A. Vovk, I. I. Tarasenko, M. E. Mikhailova, M. Y. Ilyash, I. M. Neelov and E. Lahderanta, Molecules, 2019, 24, 2481 CrossRef CAS PubMed.
- M. Lo, Y. Z. Wang and P. W. Gout, J. Cell. Physiol., 2008, 215, 593–602 CrossRef CAS PubMed.
- M. Saad, O. B. Garbuzenko, E. Ber, P. Chandna, J. J. Khandare, V. P. Pozharov and T. Minko, J. Controlled Release, 2008, 130, 107–114 CrossRef CAS PubMed.
- A. M. Krause-Heuer, A. M. P. Grant, A. N. Orkey and J. R. Aldrich-Wright, Aust. J. Chem., 2008, 61, 675–681 CrossRef CAS.
- C. Gutierrez-Lovera, A. J. Vazquez-Rios, J. Guerra-Varela, L. Sanchez and M. de la Fuente, Genes, 2017, 8, 349 CrossRef PubMed.
- F. Fenaroli, D. Westmoreland, J. Benjaminsen, T. Kolstad, F. M. Skjeldal, A. H. Meijer, M. v. d. Vaart, L. Ulanova, N. Roos, B. Nystro, J. Hildahl and G. Griffiths, ACS Nano, 2014, 8, 7014–7026 CrossRef CAS PubMed.
- B. J. Quah, H. S. Warren and C. R. Parish, Nat. Protoc., 2007, 2, 2049–2056 CrossRef CAS PubMed.
- G. Bocharov, T. Luzyanina, J. Cupovic and B. Ludewig, Front. Immunol., 2013, 4, 264 Search PubMed.
- M. Zhang, R. Guo, M. Keri, I. Banyai, Y. Zheng, M. Cao, X. Cao and X. Shi, J. Phys. Chem. B, 2014, 118, 1696–1706 CrossRef CAS PubMed.
- S. Chandra, S. Dietrich, H. Lang and D. Bahadur, J. Mater. Chem., 2011, 21, 5729 RSC.
- Y. Wang, X. Cao, R. Guo, M. Shen, M. Zhang, M. Zhu and X. Shi, Polym. Chem., 2011, 2, 1754 RSC.
- K. D. Addisu, W.-H. Hsu, B. Z. Hailemeskel, A. T. Andrgie, H.-Y. Chou, C.-H. Yuh, J.-Y. Lai and H.-C. Tsai, ACS Biomater. Sci. Eng., 2019, 5, 5453–5469 CrossRef CAS.
- H. S. Lin, Y. L. Huang, Y. S. Wang, E. Hsiao, T. A. Hsu, H. Y. Shiao, W. T. Jiaang, B. P. Sampurna, K. H. Lin, M. S. Wu, G. M. Lai and C. H. Yuh, Cancers, 2019, 11, 739 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *fg,
Rajeshkumar Anbazhagan*fg,
Chiou-Hwa Yuhhij,
Jen Ming Yang*kl and
Yen-Hsiang Changl
*fg,
Rajeshkumar Anbazhagan*fg,
Chiou-Hwa Yuhhij,
Jen Ming Yang*kl and
Yen-Hsiang Changl