
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized polyhedral oligosilsesquioxane (POSS) based composites for bone tissue engineering: synthesis, computational and biological studies†
Laura Legnania,
Daniela Iannazzob,
Alessandro Pistoneb,
Consuelo Celestib,
Salvatore Giofrè c,
Roberto Romeoc,
Angela Di Pietrod,
Giuseppa Visallid,
Monica Frestaa,
Paola Bottinoa,
Ignazio Blancoe and
Maria Assunta Chiacchio*a
c,
Roberto Romeoc,
Angela Di Pietrod,
Giuseppa Visallid,
Monica Frestaa,
Paola Bottinoa,
Ignazio Blancoe and
Maria Assunta Chiacchio*a
aDipartimento di Scienze del Farmaco, Università di Catania, Viale A. Doria 6, 95125 Catania, Italy. E-mail: ma.chiacchio@unict.it
bDipartimento di Ingegneria, Università di Messina, Contrada Di Dio, 98166 Messina, Italy
cDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università di Messina, Via S.S. Annunziata, 98168 Messina, Italy
dDipartimento di Scienze Biomediche, Odontoiatriche e delle Immagini Morfologiche e Funzionali, Via Consolare Valeria 1, 98100, Messina, Italy
eDipartimento di Ingegneria Civile e Architettura, Università di Catania, Viale A. Doria 6, 95125 Catania, Italy
First published on 19th March 2020
Abstract
Functionalized polyhedral oligosilsesquioxanes (POSS) containing an isoxazolidine nucleus have been synthesized by microwave assisted 1,3-dipolar cycloaddition of N-methyl-C-alkoxycarbonyl nitrone 1 with POSS containing olefin moieties. The results of cycloaddition processes were rationalized by computational studies at the DFT level. The covalent conjugation of chitosan with the cycloadduct 3a leads to composite material CS-POSS 7 which was gelified using genipin as cross linking agent. The suitability of the system for bone tissue engineering purposes was evaluated by in vitro drug release studies using ketoprofen as a model drug and cytotoxicity assays performed on human fetal osteoblastic cells. The preliminary biological tests showed the lack of cytotoxicity of the hybrid material and suggest its potential role in bone tissue engineering applications.
1. Introduction
Biopolymer-based composites containing inorganic materials with inherent bioactive properties and cytocompatibility have attracted significant interest in tissue engineering, leading to the development of bone scaffolds to be used both as bone grafts and drug delivery systems.1,2 These biomaterials have shown the ability to promote damaged tissue repair and regeneration by intrinsic osteoinductive properties and/or by triggering stimulating effects on bone cells and tissues, such as pH, ionic strength, temperature, light and magnetic fields.3 The main advantage in using hybrid materials lies in their ability to mimic the composite structure of natural bone and teeth which are mainly constituted of an organic phase, collagen type 1, and an inorganic component, hydroxyapatite.4 Ideal composites for tissue engineering applications should be biocompatible, able to mimic and interact with the natural environment of extracellular matrix (ECM), and possess suitable mechanical properties to provide support for the newly formed tissue and also to withstand external forces during bone tissue regeneration.5Several fabrication techniques have been used to develop bone tissue engineering scaffolds, using a wide range of biomaterials whose choice mainly depends on the site of implantation for which the scaffold is designed. Three-dimensional scaffolds constituted by natural polymeric biomaterials including proteins and polysaccharides, have shown to provide ideal structural support and to allow a suitable environment for cell adhesion, migration, proliferation and differentiation.6
Among the different polysaccharide-based 3D scaffolds, the naturally derived polymers such as chitin, chitosan, collagen, gelatin, alginate, hyaluronic acid and their composites, due to their high biocompatibility, biodegradability, low cost, ease of handling, and possibility of gelation in situ, represent resourceful biomaterials for bone and soft tissue engineering applications.7 Moreover, these biopolymers can be cross-linked, allowing the incorporation of cells, growth factors and the controlled release of bioactive molecules.8 Among the different investigated biopolymers, the natural polysaccharide, chitosan (CS), obtained from the deacetylation of chitin, due to its good biocompatibility, biodegradability, ease of chemical modifications, antibacterial properties and high affinity in vivo with macromolecules represent an ideal organic material for the development of biopolymer based scaffold for organ and tissue regeneration.9,10 However, besides its outstanding chemical, physical and biological features, the applications of scaffolds based on this natural polysaccharide show some limitations mainly related to its low mechanical strength, quick hydrolysis and burst drug release which limit its use, as single component, in bone tissue engineering formulations.11,12
In order to improve the mechanical behavior and biological stability of chitosan based scaffolds the 3D hydrogels have been reinforced with bioactive ceramic materials such as hydroxyapatite (HA), bioglass ceramic, silica nanoparticles, titanium dioxide and zirconium oxide.13–15 Among the different inorganic compounds, silica nanoparticles have shown to improve mechanical properties of polymers by providing enhancement in their structure and increasing the bioactivity of the related composites. Moreover, a recent in vitro study demonstrated the ability of silicate-based bioactive glasses to induce calcium hydroxy-carbonate apatite on the silica surface, with a close composition to bone mineral composition.16
The polyhedral oligomeric silsesquioxanes (POSS) are silicon/oxygen cage structures with RSiO3/2 repeat units and size range of 1–3 nm. These compounds are characterized by a hybrid chemical composition, intermediate between that of inorganic materials (SiO2) and organic silicone polymers (R2SiO).17 This structural peculiarity confers to POSS, “hybrid” properties and then the ability to be inert and thermally stable due to the presence of the inorganic Si–O–Si fragment and also the reactivity and ability to be readily modified because of the presence of R–Si fragment. Due to their nanoscale dimensions and the possibility to form stable covalent and non covalent interactions with organic polymers, several POSS containing composites have been developed for applications in different materials areas such as the development of bio-polymers, high-temperature composites, liquid crystals, and coatings for spacecraft.17,18 In particular, POSS silica cages, due to their biocompatibility and physico-chemical properties, able to enhance the mechanical and rheological properties of biopolymers, have shown to be suitable nanofillers for a wide range of composites for biomedical applications, thus leading to the development of biomedical devices, tissue engineering scaffolds, drug delivery systems, and biosensors.19–21 In addition, it was proved that these nanostructured materials can stimulate important biological responses at nanoscopic dimensions, such as cell adhesion and apatite formation.22,23
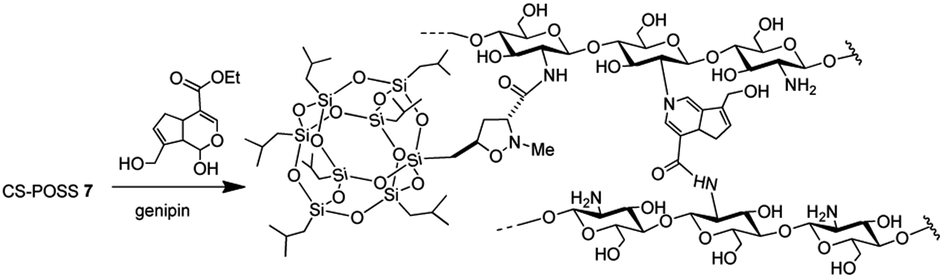
In this study, we exploited the useful functionalization of cubic silsesquioxanes with (RSiO3/2)8 formula in order to afford POSS substrates containing suitable functional groups able to form a covalent bond with the chitosan structure. The insertion of a reactive olefin moiety24 at the organic side chain (group R) of heptaisobutyl-POSS, allowed the effective microwave assisted 1,3-dipolar cycloaddition reaction with the N-methyl-C-alkoxycarbonyl nitrone 1, affording the corresponding isoxazole derivatives 3a,b, 4a,b and 5b, containing an ethoxycarbonyl group at the C-3 position of heterocyclic nucleus. The further amidation reaction between the ethoxycarbonyl group of the isomer formed in the greater amount 3a and the amino group of chitosan allowed the formation of the POSS-chitosan hybrid material which was gelified using the cross-linking agent genipin, a natural compound endowed with very low toxicity.25 The synthesized hydrogel scaffolds were investigated for their drug release abilities using ketoprofen as model drug which was included in the scaffold during the gelation procedure. The results of in vitro drug release studies and the preliminary biological tests performed on osteoblasts culture have shown the lack of cytotoxicity of the hybrid material, thus revealing the CS-POSS 7 as a potential candidate for bone tissue engineering applications.
2. Results and discussion
2.1. Synthesis of isoxazolidinyl-POSS 3a,b, 4a,b and 5b
The 1,3-dipolar cycloaddition reactions of allyl-2a and vinyl-heptaisobutyl-POSS 2b with N-methyl-C-ethoxycarbonylnitrone 1, were performed under microwave irradiation at the temperature of 100–110 °C in 90 min, following a previously reported procedure.26 The pericyclic reaction with allyl-heptaisobutyl-POSS 2a leads to the formation of two cycloadducts: 3a and 4a, with no formation of other regioisomers 5a and 6a. The reaction showed a very good control of regio- and stereoselectivity with the trans isomer 3a as the main adduct with a relative ratio trans/cis 3a/4a of 5.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 1).
:1 (Scheme 1).
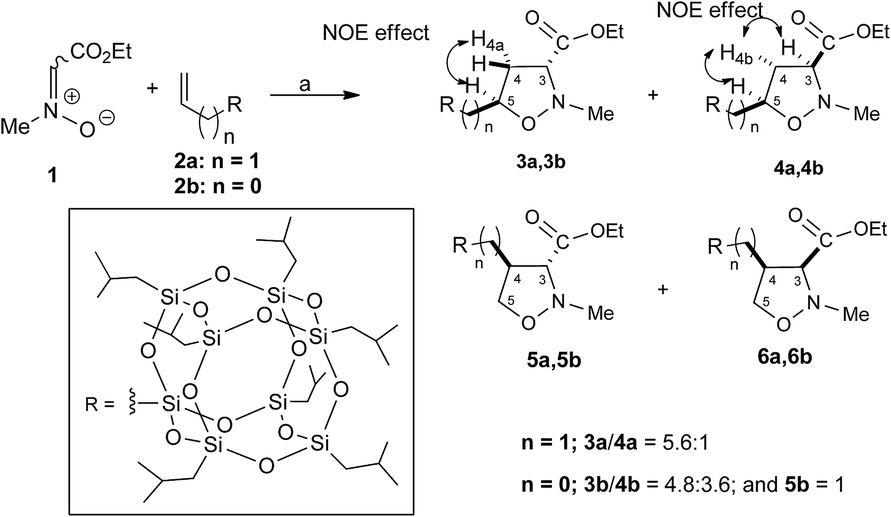 | ||
| Scheme 1 Synthesis of isoxazolidinyl-POSS 3a, 4a, 3b, 4b and 5b. Reagents and conditions: toluene, 100 W, 110 °C, 90 min. [(3a: 85%; 4a: 15% yield); (3b: 48.0%, 4b: 36.0% and 5b: 10.0% yield)]. | ||
The structure and regiochemistry of the obtained compounds were assigned on the basis of the spectroscopic data. In particular, the 1H NMR spectra of 3a shows the presence of the diagnostic methine proton at C-5 as multiplet at 4.27–4.21 ppm, the H-3 signal as a doublet of doublet centered at 3.27 ppm, while the H-4 protons resonate as doublet of triplet at 2.58 ppm and as doublets of doublets of doublets at 2.15 ppm. The stereoisomer 4a shows the H-5 proton as a doublet of triplet of doublets centered at 4.39 ppm, the H-3 proton as triplet centered at 3.51 δ, and the H-4 protons at 2.68 and 2.22 ppm as doublets of doublets of doublets. The 29Si NMR spectrum shows the signal of the O–Si–CH2–isoxazolidine group at −67.68 ppm and the signal of –O–Si–CH2CH(CH3)2 units at −67.97 ppm (see ESI†).
The stereochemical assignment of compounds 3a and 4a were obtained by NOE experiments. A positive NOE effect was registered for the trans compound 3a between the H5 and the upfield H4a protons while for the compound 4a the positive NOE effect between H5, H3 and H4b protons are indicative of a topological cis relationship.
The 1,3-dipolar cycloaddition reaction of vinyl-heptaisobutyl-POSS 2b performed in the same reaction conditions used for 2a afforded a mixture of 3b, 4b and 5b in a 1.5:1.4:1 relative ratio with no formation of the 6b (Scheme 1). The appearance of the regioisomer 5b in the reaction mixture between 1 and 2b was easily deduced from the 1H NMR spectrum which presents the H-5 protons as doublets of doublets at 4.11 and 3.99 ppm, respectively.
The stereochemical assignment of compounds 3b, 4b and 5b were obtained by NOE experiments. Thus, a positive NOE effect was registered for the trans compound 3b between the H-5 and the upfield H4a protons while for the compound 4b the positive NOE effect between H-5, H-3 and H4b protons are indicative of a topological cis relationship. For the compound 5b, no NOE effect was registered between H-3 and H-4 protons indicating a relative topological trans relationship.
2.2. Theoretical calculations
In order to rationalize the regio- and stereoselectivity of the reaction, considering the different dipolarophile reactivity of the POSS-substituted olefin 2a,b with nitrone 1, a mechanistic study of the cycloaddition process was done using DFT calculations.27Calculations were performed using the Gaussian 16 program package.28 All the structures were optimized at the B3LYP/6-31G(d) level. Vibrational frequencies were computed at the same level of theory to define the optimized structures as minima or transition states.
Starting from the nitrone 1 E or Z and taking into account the possible exo or endo approach of the dipolarophiles for the two possible regioisomers, eight different transition states (TSs) were supposed. In Fig. 2 are reported the possible TSs, giving, for each nitrone's isomer, the stereoisomeric products 3–6, with the groups POSS and –CO2Et in cis or trans, respectively.
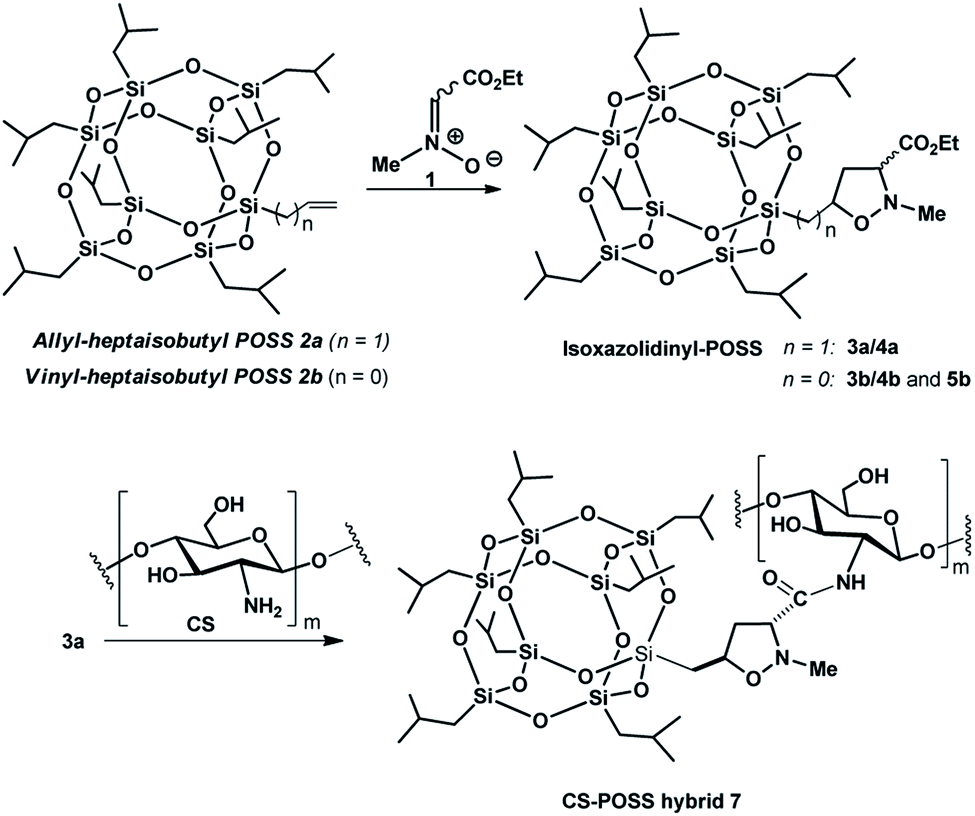 | ||
| Fig. 1 Synthesis of CS-POSS hybrid 7. | ||
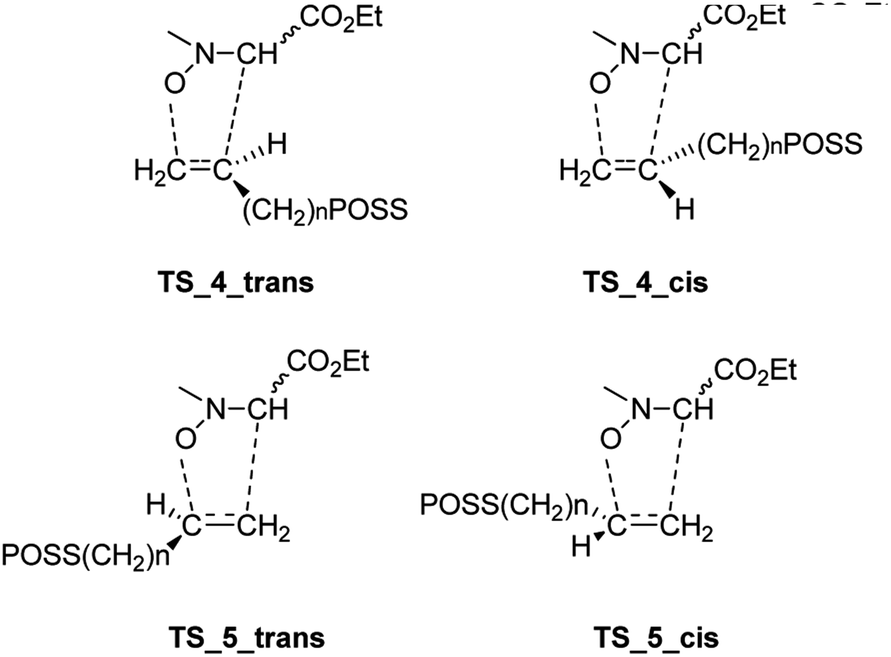 | ||
| Fig. 2 Possible TSs of the cycloaddition of nitrone 1 with dipolarophiles 2a,b. | ||
Considering the cycloaddition, involving the heptaisobutyl-1-allyl-POSS 2a, in Fig. 3 are reported the three-dimensional plots of the TSs leading to the cycloadducts 3a and 4a (TSs of compounds 5a and 6a are reported in ESI†). The activation energy and free energy values at 383 K are given in Table 1. The TS 3a_E/Z are favorites with ΔG‡ of 26.20 and 28.16 kcal mol−1, which produce 95% yield of trans adduct 3a. The TS 4a_E/Z (ΔG‡ 29.60/30.64 kcal mol−1), is higher in energy and gives 3.1% yield of the cis derivative 4a. As regard to the regioisomer 5a, the lower TS (TS_5a_E) for its formation has a ΔG‡ of 31.40 kcal mol−1 and leads to the formation of 2% yield of this cycloadduct but, having an energy barrier more than 30 kcal mol−1, the cycloaddition does not occur. Furthermore, the TS energies of 6a (TS_6a_E and TS_6a_Z) are too high, thus, this compound can't be obtained in this cycloaddition process.
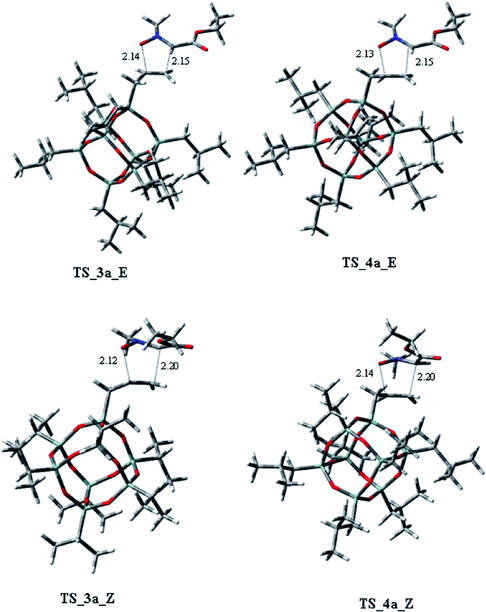 | ||
| Fig. 3 Transition states for 2a with 1 leading to stereoisomers 3a, 4a. Displacement vectors for TS imaginary frequencies are shown as grey arrows and the values of the forming bond lengths are reported in angstroms. | ||
| TSs | ΔE‡ (kcal mol−1) | ΔG‡ (kcal mol−1) | % (T = 383 K) |
|---|---|---|---|
| TS_3a_E | 13.14 | 26.20 | 78.7 |
| TS_4a_E | 15.15 | 29.60 | 5.5 |
| TS_5a_E | 15.51 | 31.40 | 3.4 |
| TS_6a_E | 17.03 | 34.23 | 0.5 |
| TS_3a_Z | 14.38 | 28.16 | 11.2 |
| TS_4a_Z | 16.92 | 30.64 | 0.4 |
| TS_5a_Z | 17.97 | 33.48 | 0.1 |
| TS_6a_Z | 17.75 | 34.15 | 0.1 |
Both C–C and C–O bonds are formed in a concerted albeit asynchronous way, as evident by distances between atoms involved in the forming bonds (Å) reported in the 3D plots. The pathway leading to regioisomers 5a and 6a (see ESI†) resulted to be more asynchronous respect to that leading to regioisomers 3a and 4a; anyway, both processes are concerted, as confirmed by IRC analysis.
On the other hand, considering the cycloaddition of 2b with 1 in Fig. 4 and 5 are reported the three-dimensional plots of the transition states to the regioisomeric cycloadducts 3b–6b and in Table 2 the activation energy, the free energy values and the percentages at 383 K.
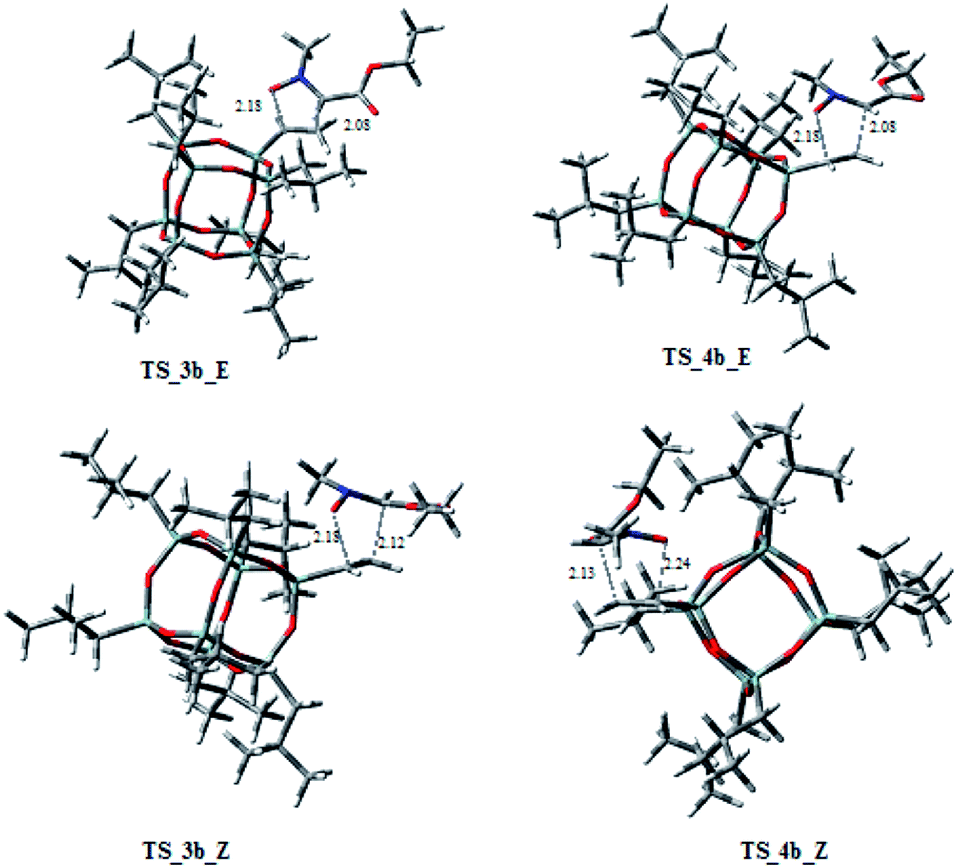 | ||
| Fig. 4 Transition states for the reaction of 2b with 1 leading to stereoisomers 3b and 4b. Displacement vectors for TS imaginary frequencies are shown as grey arrows and the values of the forming bond lengths are reported in angstroms. | ||
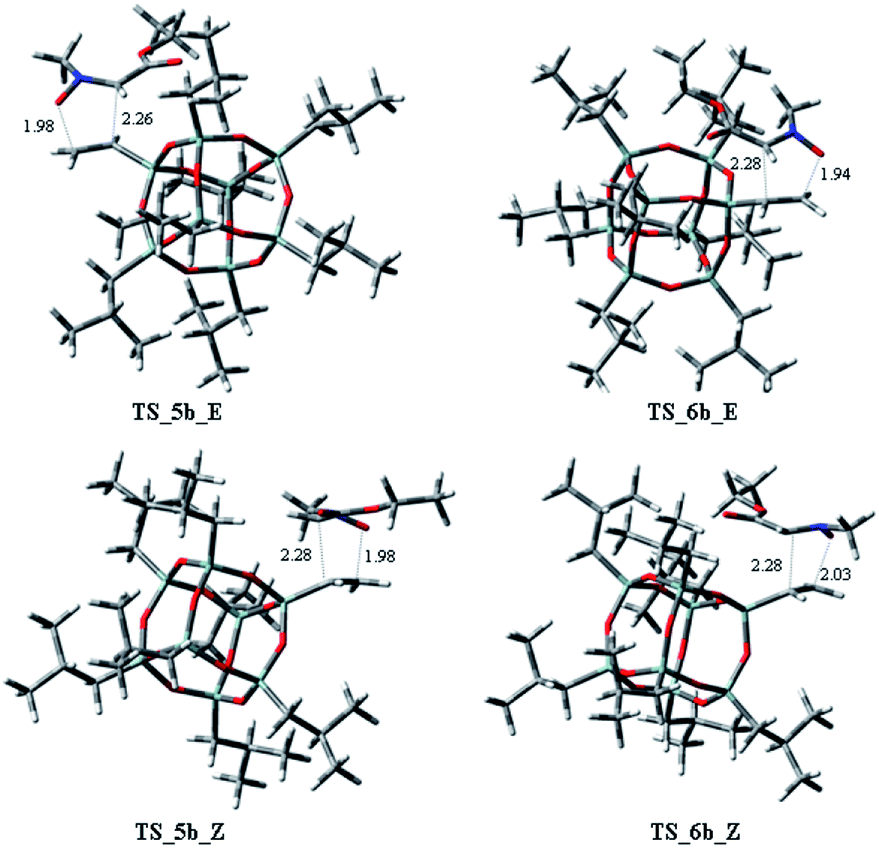 | ||
| Fig. 5 Transition states for the reaction of 2b with 1 leading to stereoisomers 5b and 6b. Displacement vectors for TS imaginary frequencies are shown as grey arrows and the values of the forming bond lengths are reported in angstroms. | ||
| TSs | ΔE‡ (kcal mol−1) | ΔG‡ (kcal mol−1) | % (T = 383 K) |
|---|---|---|---|
| TS_3b_E | 12.23 | 27.86 | 36.1 |
| TS_4b_E | 12.86 | 28.36 | 15.7 |
| TS_5b_E | 12.95 | 28.73 | 0.4 |
| TS_6b_E | 12.60 | 30.84 | 1.8 |
| TS_3b_Z | 15.40 | 31.64 | 13.9 |
| TS_4b_Z | 14.26 | 28.96 | 22.2 |
| TS_5b_Z | 13.01 | 29.94 | 9.4 |
| TS_6b_Z | 15.27 | 32.86 | 0.5 |
In this case, the ratio of the two regioisomers 3b/4b vs. 5b is 8.4:1, according with experimental data. The trans/cis ratio in 3b vs. 4b is 4.8:3.6. It is worth to note that the TSs 5b_E, and 5b_Z, leading to 5b have ΔG‡ of 28.73 and 29.94 kcal mol−1, clearly lower of the corresponding regioisomer 5a (5a_E, 5a_Z: 31.40/33.48 kcal mol−1). Thus, these results could justify the preferred formation of 5b with respect to 5a. As observed for the cycloaddition reaction of 2a, also for 2b, the IRC analyses allowed us to verify that the reaction is concerted, albeit slightly asynchronous (Fig. 4 and 5). In conclusion, the modeling study, according to the experimental results, rationalized that the 1,3-dipolar cycloaddition of 1 with 2a or 2b is regio- and stereo-selective.
2.3. Synthesis of CS-POSS hybrid hydrogels
The main isomer 3a obtained from the 1,3-dipolar cycloaddition of 2a with nitrone 1 was used for the tert-butoxide-assisted amidation reaction with the free nucleophilic amino group of chitosan.29 The reaction led to the formation of the hybrid sample CS-POSS 7 (Fig. 1). In order to achieve the complete amidation of 3a, the reaction was performed using an excess of biopolymer (see Experimental section). The effectiveness of the coupling reaction between 3a and chitosan was investigated by FTIR spectroscopy and TGA analysis. The FTIR spectra of CS-POSS 7, compared with those of precursors chitosan (CS) and of POSS cycloadduct 3a is reported in Fig. 6. The spectrum of CS shows a strong band at 3150–3600 cm−1 corresponding to the stretching of N–H and O–H bonds, the peaks at 2850 and 2920 cm−1 due to the aliphatic C–H stretching, a band at 1575 cm−1 ascribable to the N–H bending, a peak at 1030 cm−1 corresponding to the C–O stretching and a peak at 1647 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the primary amide of residual N-acetyl groups. Compound 3a shows the diagnostic signals at 1111 cm−1 ascribable to the Si–O–Si stretching of silica cage, the aliphatic C–H stretching of isobutyl substituents at 2954 cm−1 and 2871 cm−1 and the presence of a peak at 1747 cm−1 due to the CO stretching of the ester group present at C-3 position of the isoxazolidine nucleus. The hybrid sample CS-POSS 7 shows the presence of the diagnostic peak at 1680 cm−1 attributable to the CO stretching of the newly formed amide bond, the presence of a peak at 1550 cm−1 ascribable to the N–O stretching and a peak at 1250 cm−1 due to the absorbance of the C–N bond; moreover a large band at 3100–3600 cm−1 lower in intensity with respect to CS sample, can be observed.
O stretching of the primary amide of residual N-acetyl groups. Compound 3a shows the diagnostic signals at 1111 cm−1 ascribable to the Si–O–Si stretching of silica cage, the aliphatic C–H stretching of isobutyl substituents at 2954 cm−1 and 2871 cm−1 and the presence of a peak at 1747 cm−1 due to the CO stretching of the ester group present at C-3 position of the isoxazolidine nucleus. The hybrid sample CS-POSS 7 shows the presence of the diagnostic peak at 1680 cm−1 attributable to the CO stretching of the newly formed amide bond, the presence of a peak at 1550 cm−1 ascribable to the N–O stretching and a peak at 1250 cm−1 due to the absorbance of the C–N bond; moreover a large band at 3100–3600 cm−1 lower in intensity with respect to CS sample, can be observed.
 | ||
| Fig. 6 FTIR spectra of samples 3a, CS, and CS-POSS 7. | ||
The covalent conjugation of chitosan with 3a that leads to CS-POSS 7 was also confirmed by thermogravimetric analysis (TGA) performed under nitrogen environment (Fig. 7). The thermal degradation of pure chitosan shows two stages in the range of 50–400 °C. The first (range 50–100 °C) is due to the loss of water with a weight loss of about 9%, while the second stage (range 250–400 °C) shows a complete degradation of the sample with a maximum decomposition temperature at 300 °C and a total weight loss of about 70%. The POSS cycloadduct 3a shows the initial thermal decomposition temperature at 235 °C, a maximum decomposition temperature at 293 °C and a complete thermal degradation at 600 °C. The TGA curve of the hybrid material CS-POSS 7 shows a different profile with respect to precursors chitosan and 3a. For this sample, three different degradation stages can be observed: the first at 50–100 °C attributable to the loss of water, the second at 190–300 °C with a maximum decomposition at 254 °C and the third at 300–400 °C with a maximum decomposition temperature at 371 °C and a complete degradation of the sample with a total weight loss of about 75%. The different degradation temperatures and the different shape of TGA curves are indicative of the formation of a new composite material with a different thermal behavior with respect to precursors CS and 3a.
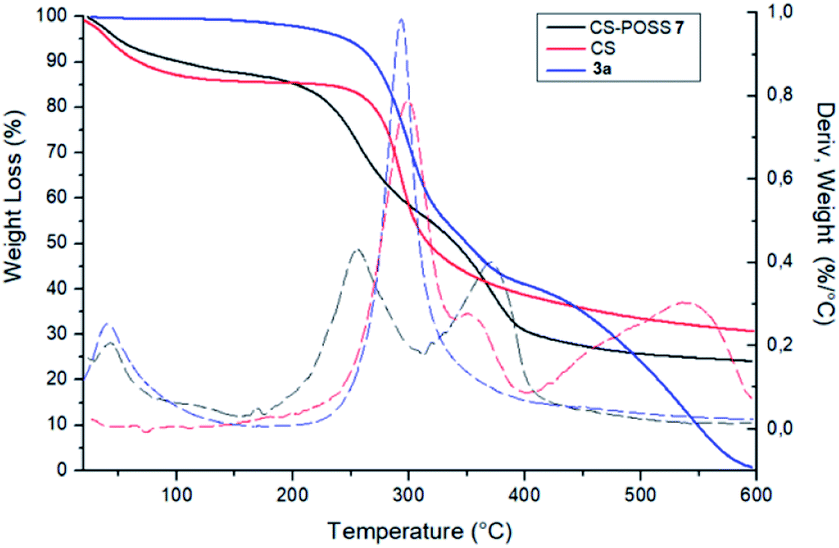 | ||
| Fig. 7 TGA thermograms of CS-POSS 7, CS and of compound 3a performed under nitrogen atmosphere. | ||
Furthermore, the 29Si NMR spectrum of CS-POSS 7 performed in CDCl3 (see ESI†) confirms that under our reaction conditions, the cage of the POSS remains intact. In fact, only two signals appeared at −67.65 and −67.96, corresponding to Si–CH2–isoxazolidine and to Si–CH2–CH(CH3)2, respectively.
The synthesized CS-POSS 7 and pure chitosan were gelified using the natural and biocompatible cross-linking agent genipin in order to compare the drug release properties of the formed hydrogels. The gelification process was performed in the presence of an aqueous solution of the anti-inflammatory drug ketoprofen (80 mg mL−1) at the concentration of 4 wt% with respect to the weight of biopolymers-based samples. Since genipin cross-links macromolecules by binding two amine groups between neighboring chains, the CS-POSS 7, because of the steric hindrance caused by POSS molecule bonded to chitosan chains, showed a more delayed gelation time with respect to the hydrogel obtained from unmodified chitosan (Scheme 2).
 | ||
| Scheme 2 Cross-linking reaction of CS-POSS 7 with genipin (see Experimental details). | ||
Compared to the CS hydrogel, the introduction of POSS molecule in the CS-POSS 7 hydrogel, have shown to decrease the water penetration, due probably to the different crosslinking ability and to the presence of hydrophobic components. In fact, the amount of water, evaluated as difference between the weight of composite before and after drying, was found to be of 94 wt% and 78 wt% for CS and CS-POSS 7 hydrogels, respectively.
The rheological properties of the synthesized chitosan-based hydrogels were evaluated by measuring the storage modulus (G′) and the loss modulus (G′′), as a function of frequency range from 10−1 Hz to 10 Hz (See ESI†). The results of these studies demonstrated, for both hydrogels, G′ values higher than the G′′ value, thus indicating an elastic solid behaviour. Moreover, higher G′ and G′′ values have been found for CS-POSS 7 sample, revealing for this hybrid material, an increased stiffness with respect to the pure CS hydrogel. These results are in complete agreement to what reported in literature for hydrogels containing POSS molecules.18,21
2.4. Drug release studies
The drug release behaviour was evaluated from the hydrogels obtained from CS and CS-POSS 7, in order to investigate the effect of the presence of the silica cage on the drug release properties of the chitosan-based matrices. The study was performed at the normal body temperature of 37 °C, in buffer phosphate solution (pH = 7.4) using the dialysis bag diffusion technique and in an orbital shaker rotating at 30 rpm. The amount of drug released was quantified by UV-Vis absorption spectra, by measuring the drug absorbance at 260 nm (Fig. 8). The hydrogel obtained from CS shows a burst drug release (about 65%) within 1 hour that reached 100% of drug released within 1 day. For the hydrogel obtained from CS-POSS 7 the release curve showed also a fast release of great amounts of ketoprofen (45%) in the first hour but a more controlled release in the next two days, reaching a plateau at about 78% of ketoprofen loaded. The results on this study suggest the presence of specific intermolecular interactions between the functionalized silica cage and the drug and open new possibilities for a long term sustained release of drugs entrapped inside the polymeric matrix during its in vivo biodegradation.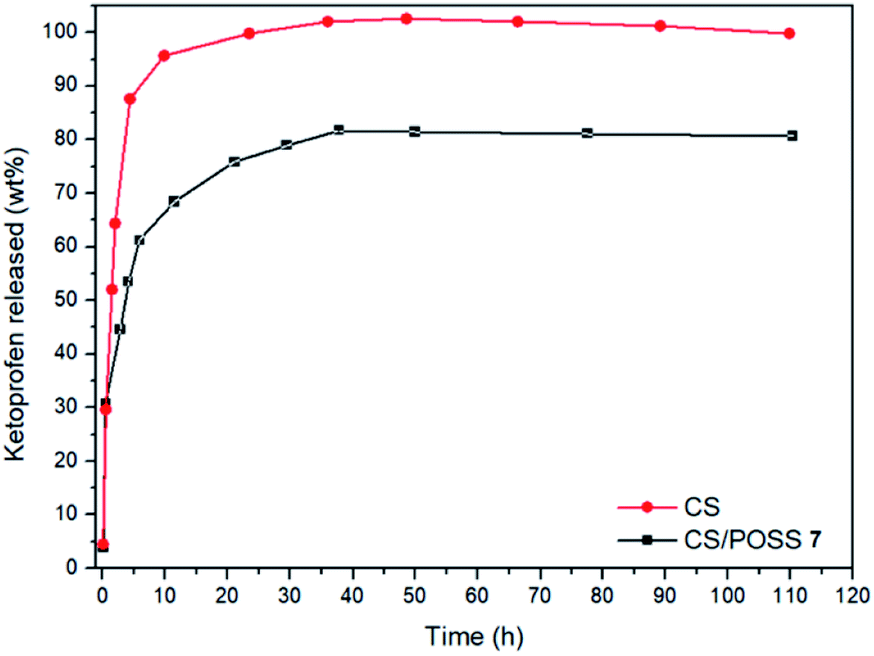 | ||
| Fig. 8 Ketoprofen release from CS and CS-POSS 7 hydrogels at 37 °C, in PBS (pH 7.4). | ||
2.5. Cell cultures and biological assays
Human fetal osteoblastic cells (hFOB 1.19) were used as in vitro model to evaluate the cytotoxicity of the synthesized CS and CS-POSS 7 hydrogels. Both samples were gelified at room temperature and the cells were plated and grown on the formed hydrogels for 48 h at 34 °C in an atmosphere of 5% CO2 in order to simulate the natural living tissue environment. Cells cultured in their specific medium, were used as negative controls.Cytotoxicity studies have shown that the investigated hydrogels, as evaluated using the DNA intercalating probe propidium iodine (PI) and by fluorimetric analysis (λexc 493–λem 636 nm) were not cytotoxic to osteoblasts. At the lowest dose tested, the percentages of dead cells were superimposable with control cells (5% ± 0.3 vs. 5.2 ± 0.4 and 5.4 ± 0.6 CS and CS-POSS 7 hydrogels respectively). As shown in Fig. 9, a very limited increase in the mortality rates was observed at the highest doses, highlighting the low cytotoxicity of both hydrogels. In particular, compared to highly biocompatible CS, the hybrid material increased the percentages of dead cells on average by 12%.
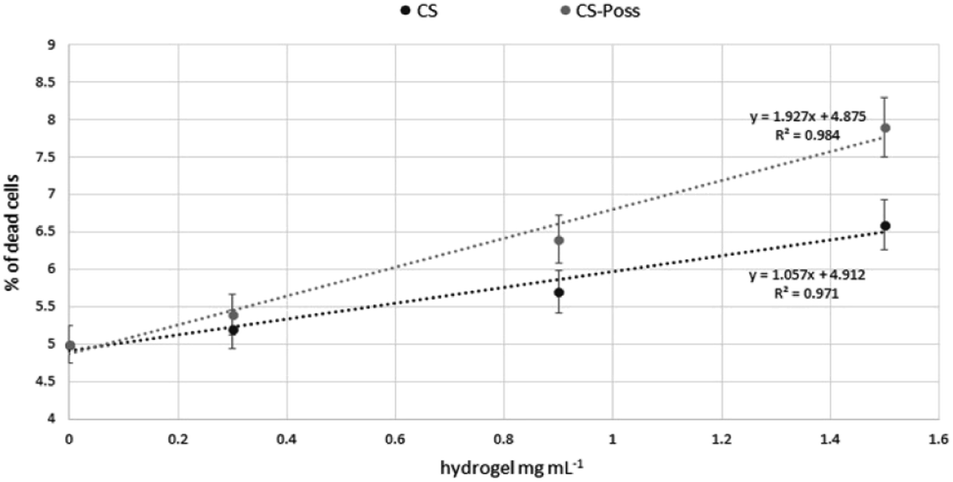 | ||
| Fig. 9 Percentages of dead cells grown in presence of CS and CS-POSS 7 hydrogels. | ||
In addition, the use of acridine orange (AO), a cell-permeable, nucleic acid-selective fluorescent cationic dye, allowed the analysis of cell compartments. The Confocal Laser Scanning Microscope, (CLSM) images confirmed that the cells grown in presence of the hydrogels were morphologically analogous to controls (Fig. 10).
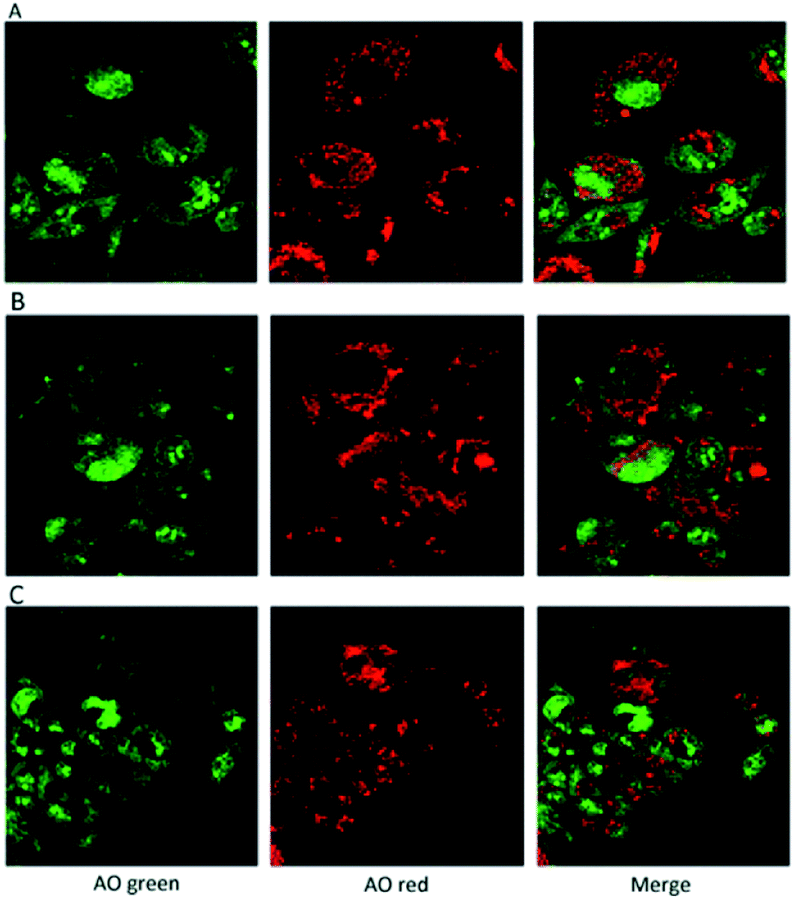 | ||
| Fig. 10 CLSM images of hFOB 1.19 cells labelled with the metachromatic fluorophore AO. (A) Control cells; (B) osteoblastic cells grown in presence of CS hydrogel; (C) osteoblastic cells grown in presence of CS-POSS 7 hydrogel. In the nucleus the AO, intercalating with DNA emits a green florescence while in the cytosol, the fluorophore is sequestered in the intact lysosomes where, due to the low pH, it becomes protonated and emits a red florescence. In presence of acid compartment damage only green florescence is emitted. | ||
3. Conclusions
We have exploited the useful functionalization of POSS silica cage to afford substrates containing functional groups suitable for the covalent bonding with the chitosan structure. The microwave assisted 1,3-dipolar cycloaddition of POSS derivatives containing olefin moieties, with the N-methyl-C-alkoxycarbonyl nitrone afforded the corresponding isoxazolidine derivatives containing an ethoxycarbonyl group at the C-3 position of heterocyclic nucleus. Theoretical calculations confirmed the experimental observations leading to C-5 substituted derivatives as preferred cycloadducts. Furthermore, the relevant formation of compound 5b in the case of cycloaddition of 1 with vinyl-POSS 2b can be rationalized by ΔG‡ energies of the corresponding TSs and by the shorter distance of the incoming C–O bond in the transition state with respect to what observed for the cycloaddition of allyl-POSS 2a.The amidation reaction between the amino group of chitosan and the ethoxycarbonyl group of the major isomer 3a, allowed the formation of the hybrid material CS-POSS 7 which was gelified using the cross-linking agent genipin. The synthesized hydrogel scaffold was investigated for its ability to release ketoprofen, which was included in the scaffold during the gelation procedure. In vitro drug release studies and preliminary biological tests performed on osteoblasts culture, have shown the ability of the system to release drugs in a controlled manner and the lack of cytotoxicity of the hybrid material. Thus, the results of this work highlight the sample CS-POSS 7 as a potential candidate for bone tissue engineering applications.
4. Experimental section
4.1. Materials and methods
Solvents, allyl-heptaisobutyl-POSS 2a and vinyl-heptaisobutyl POSS 2b and other chemical reagents were purchased from Sigma-Aldrich and were used without further purification. The reactions under microwave irradiations were carried out using a CEM Corporation Focused Microwave System, Model Discover. Thin-layer chromatographic separations were carried out on Merck silica gel 60-F254 precoated aluminum plates (Merck, Darmstadt, Germany). Flash chromatography was carried out using Merck silica gel (200–400 mesh). Preparative separations were carried out using a Büchi C-601 MPLC instrument (BUCHI Italia S.r.l., Milano, Italy) using Merck silica gel 0.040–0.063 mm, and the eluting solvents were delivered by a pump at the flow rate of 3.5–7.0 mL min−1. NMR spectra were recorded on a Varian Unity Inova instrument (1H at 500 MHz, 13C at 125.67 MHz, 29Si at 99.32 MHz). Chemical shifts are in ppm (δ) using TMS as internal standard. NOE difference spectra were obtained by subtracting alternatively right-off-resonance free induction decays (FIDS) from right-on-resonance-induced FIDS. Thermogravimetric analyses (TGA) were performed using a TAQ500 instrument (TA Instruments, New Castle, DE, USA) from 100 to 700 °C, with a rate of 20 °C per minute, under air atmosphere. The Infrared spectra were obtained using the Fourier-Transform Infrared (FT-IR) spectrometer VERTEX 80/80v (Bruker Optik GmbH, Ettlingen, Germany), by the method of KBr pellets in the range of 4000–500 cm−1. UV spectra have been performed by Thermo Nicolet mod, Evolution 500 spectrophotometer, measuring the drug absorbance at 260 nm. Rheological properties measurements were carried out on a rotational Rheometer (Mod. SR5, Rheometric Scientific) with parallel plate geometry at 37 °C; cylindrical samples had a diameter of 25 mm and a height of 1 mm. Storage and loss modulus were measured by using a frequency sweep mode with the range from 0.1 to 10 Hz (rad s−1). Cell line hFOB 1.19 was purchased from Izsler (Istituto Zooprofilattico “B. Ubertini” Brescia, Italia), cell culture media and fluorophores were purchased from Sigma-Aldrich. Fluorimetric analyzes were performed by a microtiter plate reader (Tecan Italia, Milan-Italy) while a TCS SP2 (Leica Microsystems, Heidelberg, Germany), equipped with Ar/Kr laser and coupled to a microscope (Leica DM IRB) was used for morphological analyses.4.2. General procedure
:20) as eluent, to afford isoxazolidines 3a, 4a from allyl-heptaisobutyl POSS 2a and isoxazolidines 3b, 4b and 5b from vinyl-heptaisobutyl-POSS 2b.
4.2.1.1. Reaction of nitrone 1 with allyl-heptaisobutyl POSS 2a. The first eluted product was ethyl(3RS,5RS)-5-(3,5,7,9,11,13,15-heptaisobutylpentacyclo[9.5.1.13,9.15,15.17,13]octasilox-1-yl)-2-methylisoxazolidine-3-carboxylate (3a) (85%, 170 mg), sticky white solid, δ 1H (500 MHz, CDCl3) 4.28–4.19 (m, 3H, O–CH2–, and H5), 3.27 (dd, 1H, J = 6.5, 9.9 Hz, H3), 2.79 (s, 1H, N–CH3), 2.58 (dt, 1H, J = 12.4, 6.5 Hz, H4b), 2.15 (ddd, 1H, J = 12.4, 9.9, 8.1 Hz, H4a), 1.93–1.78 (m, 7H, CH isobutyl), 1.29 (t, 3H, J = 7.1 Hz,
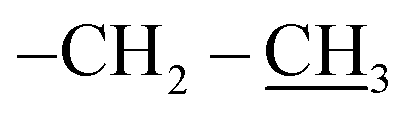 ), 0.96 (d, 21H, J = 1.4 Hz, CH3 isobutyl), 0.95 (d, 21H, J = 1.3 Hz, CH3 isobutyl), 0.94–0.81 (m, 2H, –CH2–Si), 0.60 (d, 14H, J = 7.2 Hz, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 170.87, 74.05, 70.07, 61.32, 40.64, 29.86, 25.84, 24.01, 22.62, 22.56, 18.52, 14.30. δ 29Si NMR (99.32 MHz, CDCl3) −67.68 and −66.97. Anal. calcd for C37H79NO15Si8: C, 44.32; H, 7.94; N, 1.40. Found: C, 44.30; H, 7.95; N, 1.39.
), 0.96 (d, 21H, J = 1.4 Hz, CH3 isobutyl), 0.95 (d, 21H, J = 1.3 Hz, CH3 isobutyl), 0.94–0.81 (m, 2H, –CH2–Si), 0.60 (d, 14H, J = 7.2 Hz, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 170.87, 74.05, 70.07, 61.32, 40.64, 29.86, 25.84, 24.01, 22.62, 22.56, 18.52, 14.30. δ 29Si NMR (99.32 MHz, CDCl3) −67.68 and −66.97. Anal. calcd for C37H79NO15Si8: C, 44.32; H, 7.94; N, 1.40. Found: C, 44.30; H, 7.95; N, 1.39.The second eluted product was ethyl(3SR,5RS)-5-(3,5,7,9,11,13,15-heptaisobutylpentacyclo[9.5.1.13,9.15,15.17,13]octasilox-1-yl)-2-methylisoxazolidine-3-carboxylate (4a) (15%, 30 mg), sticky white solid, δ 1H (500 MHz, CDCl3) 4.39 (dtd, 1H, J = 10.9, 7.2, 3.7 Hz, H5), 4.21 (q, 2H, J = 7.0 Hz, O–CH2–), 3.51 (t, 1H, J = 7.6 Hz, H3), 2.75 (s, 3H, N–CH3), 2.68 (ddd, 1H, J = 12.6, 8.5, 6.9 Hz, H4b), 2.22 (ddd, 1H, J = 12.6, 7.6, 6.7 Hz, H4a), 1.91–1.78 (m, 7H, CH isobutyl), 1.29 (t, 3H, J = 7.0 Hz, 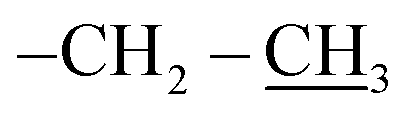 ), 1.10–1.03 (m, 2H, –CH2–Si), 0.96 (d, 21H, J = 1.3 Hz, CH3 isobutyl), 0.95 (d, 21H, J = 1.3 Hz, CH3 isobutyl), 0.60 (d, 14H, J = 7.0 Hz, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 171.34, 73.26, 61.39, 39.76, 29.85, 25.85, 25.83, 25.80, 24.02, 24.00, 22.61, 22.56, 18.60, 14.33. Anal. calcd for C37H79NO15Si8: C, 44.32; H, 7.94; N, 1.40. Found: C, 44.35; H, 7.96; N, 1.42.
), 1.10–1.03 (m, 2H, –CH2–Si), 0.96 (d, 21H, J = 1.3 Hz, CH3 isobutyl), 0.95 (d, 21H, J = 1.3 Hz, CH3 isobutyl), 0.60 (d, 14H, J = 7.0 Hz, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 171.34, 73.26, 61.39, 39.76, 29.85, 25.85, 25.83, 25.80, 24.02, 24.00, 22.61, 22.56, 18.60, 14.33. Anal. calcd for C37H79NO15Si8: C, 44.32; H, 7.94; N, 1.40. Found: C, 44.35; H, 7.96; N, 1.42.
4.2.1.2. Reaction of nitrone 1 with vinyl-heptaisobutyl POSS 2b. The first eluted product was (3RS,5SR)-ethyl 5-(3,5,7,9,11,13,15-heptaisobutyl-2,4,6,8,10,12,14,16,17,18,19,20-dodecaoxa-1,3,5,7,9,11,13,15-octasilapentacyclo[9.5.1.13,9.15,15.17,13]icosan-1-yl)-2-methylisoxazolidine-3-carboxylate (3b) (48%, 60 mg), white solid, mp: 113–115 °C. δ 1H (500 MHz, CDCl3) 4.22 (q, J = 7.1 Hz, 2H, O–CH2–), 3.66 (td, J = 9.4, 1.7 Hz, 1H, H5), 3.19 (t, J = 7.5 Hz, 1H, H3), 2.77 (s, 3H, N–CH3), 2.67–2.45 (m, 2H, H4), 2.02–1.75 (m, 7H, CH isobutyl), 1.29 (t, J = 7.1 Hz, 3H,
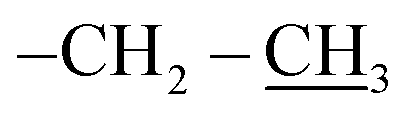 ), 0.96 (d, J = 1.4 Hz, 21H, CH3 isobutyl), 0.95 (d, J = 1.4 Hz, 21H, CH3 isobutyl), 0.61 (dd, J = 10.3, 7.1 Hz, 14H, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 171.21, 69.50, 66.37, 61.43, 36.39, 27.07, 25.85, 25.83, 24.00, 23.97, 23.96, 22.61, 22.42, 14.29. Anal. calcd for C35H75NO15Si8: C, 43.13; H, 7.76; N, 1.44. Found C, 43.16; H, 7.78; N, 1.46.
), 0.96 (d, J = 1.4 Hz, 21H, CH3 isobutyl), 0.95 (d, J = 1.4 Hz, 21H, CH3 isobutyl), 0.61 (dd, J = 10.3, 7.1 Hz, 14H, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 171.21, 69.50, 66.37, 61.43, 36.39, 27.07, 25.85, 25.83, 24.00, 23.97, 23.96, 22.61, 22.42, 14.29. Anal. calcd for C35H75NO15Si8: C, 43.13; H, 7.76; N, 1.44. Found C, 43.16; H, 7.78; N, 1.46.The second eluted product was (3SR,4RS)-ethyl 4-(3,5,7,9,11,13,15-heptaisobutyl-2,4,6,8,10,12,14,16,17,18,19,20-dodecaoxa-1,3,5,7,9,11,13,15-octasilapentacyclo[9.5.1.13,9.15,15.17,13]icosan-1-yl)-2-methylisoxazolidine-3-carboxylate (5b) (10%, 12.3 mg), white solid, mp: 93–95 °C. δ 1H (500 MHz, CDCl3) 4.27–4.13 (m, 2H, O–CH2–), 4.11 (dd, J = 9.3, 8.1 Hz, H5b), 3.99 (dd, J = 9.3, 8.1 Hz, 1H, H5a), 3.40 (d, J = 8.5 Hz, 1H, H3), 2.73 (s, 3H, N–CH3), 2.35 (td, J = 9.3, 8.5 Hz, 1H H4), 1.91–1.78 (m, 7H, CH isobutyl), 1.30 (t, J = 7.1 Hz, 3H, 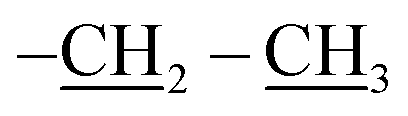 ), 0.96 (d, J = 3.0 Hz, 21H, CH3 isobutyl), 0.95 (d, J = 3.0 Hz, 21H, CH3 isobutyl), 0.65–0.58 (m, 14H, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 170.96, 70.90, 67.12, 61.58, 32.02, 27.06, 25.83, 25.82, 23.98, 22.59, 22.57, 22.47, 14.27. Anal. calcd for C35H75NO15Si8: C, 43.13; H, 7.76; N, 1.44. Found C, 43.14; H, 7.77; N, 1.46.
), 0.96 (d, J = 3.0 Hz, 21H, CH3 isobutyl), 0.95 (d, J = 3.0 Hz, 21H, CH3 isobutyl), 0.65–0.58 (m, 14H, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 170.96, 70.90, 67.12, 61.58, 32.02, 27.06, 25.83, 25.82, 23.98, 22.59, 22.57, 22.47, 14.27. Anal. calcd for C35H75NO15Si8: C, 43.13; H, 7.76; N, 1.44. Found C, 43.14; H, 7.77; N, 1.46.
The third eluted product was (3RS,5RS)-ethyl 5-(3,5,7,9,11,13,15-heptaisobutyl-2,4,6,8,10,12,14,16,17,18,19,20-dodecaoxa-1,3,5,7,9,11,13,15-octasilapentacyclo[9.5.1.13,9.15,15.17,13]icosan-1-yl)-2-methylisoxazolidine-3-carboxylate (4b) (36%, 44 mg), white solid, mp: 111–113 °C. δ 1H (500 MHz, CDCl3) 4.19 (q, J = 7.1 Hz, 2H, O–CH2), 3.70 (dd, J = 12.0, 6.4 Hz, 1H, H5), 3.46 (t, J = 7.7 Hz, 1H, H5), 2.68 (s, 3H, N–CH3), 2.67–2.61 (m, 1H, H4b), 2.53–2.48 (td, J = 12.1, 7.1 Hz, 1H, H4a), 1.93–1.79 (m, 7H, CH isobutyl), 1.28 (t, J = 7.1 Hz, 3H, 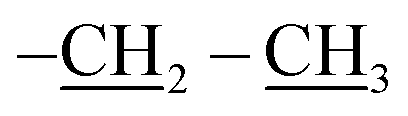 ), 0.95 (d, J = 1.0 Hz, 21H, CH3 isobutyl), 0.94 (d, J = 1.0 Hz, 21H, CH3 isobutyl), 0.67–0.56 (m, 14H, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 171.63, 69.42, 63.17, 61.35, 46.73, 36.85, 25.83, 25.82, 25.78, 23.99, 23.96, 23.91, 22.61, 22.36, 14.34. Anal. calcd for C35H75NO15Si8: C, 43.13; H, 7.76; N, 1.44. Found C, 43.16; H, 7.78; N, 1.45.
), 0.95 (d, J = 1.0 Hz, 21H, CH3 isobutyl), 0.94 (d, J = 1.0 Hz, 21H, CH3 isobutyl), 0.67–0.56 (m, 14H, CH2 isobutyl). δ 13C (125 MHz, CDCl3) 171.63, 69.42, 63.17, 61.35, 46.73, 36.85, 25.83, 25.82, 25.78, 23.99, 23.96, 23.91, 22.61, 22.36, 14.34. Anal. calcd for C35H75NO15Si8: C, 43.13; H, 7.76; N, 1.44. Found C, 43.16; H, 7.78; N, 1.45.
000 Da) for 8 h. After removal of water under vacuum at 50 °C, the mixture was dried at 50 °C for 24 h at the vacuum drying pressure of 65 mbar. The sample was characterized by FTIR and TGA analysis and used for the subsequent reactions without further purification.:1 mixture of Dulbecco's Modified Eagle's Medium/Nutrient Mixture F-12 Ham with L-glutamine (2.5 mM), sodium pyruvate (0.5 mM), sodium bicarbonate (1.2 g L−1), G418 (0.3 mg mL−1) and fetal calf serum (10%). We used exposure concentrations ranging from 0.3 to 1.5 mg mL−1 for CS hydrogel and CS-POSS 7 hydrogel (see ESI†) that were stratified in a 12 multi-well plate, forming drops which were gelified at room temperature. In each well the surface covered by the nanosystems was calculated by means the Image J software. Hence, the plates were sterilized through a 24 h UV radiation treatment and then the cells were plated (approximately 3 × 105 cells per well) and grown for 48 h at 34 °C in an atmosphere of 5% CO2. In each experiment, we included monolayers grown in the absence of compounds as negative controls (i.e. control cells). Since the interference of compounds did not make it possible to perform the formazan-based colorimetric viability test, we labelled hFOB 1.19 cells with the DNA intercalating probe propidium iodine (PI) in order to evaluate cytotoxicity. Briefly, after the growth of the monolayers for the experimental times with and without the examined compounds, suspensions formed by detached cells were loaded with propidium iodine (3 mg mL−1) and counted. The mortality rate was assessed by fluorimetric analysis (λexc 493–λem 636 nm). The emission values recorded in treated cells were compared with control cells whose cell mortality rate was 5%. To quantify the viable cells, we fixed and permeabilized the adherent cells with cold 70% ethanol for 30 min at 4 °C, and then we stained with propidium iodine (3 mg mL−1). By using the same wavelengths above reported, the values were recorded by using a microtiter plate reader (Tecan Italia, Milan-Italy). The biological tests were all performed in triplicate, and the replicates were calculated as means ± SD comparing the obtained values in exposed cells with those obtained by the negative control. In addition, to evaluate cell morphology and the lysosomal compartment, the adherent treated and untreated cells were also labelled with the metachromatic fluorophore Acridine Orange (AO) and confocal observations were performed (CLSM, TCS-SP2, Leica-Microsystems, Germany).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Universities of Catania and Messina, Interuniversity Research Centre on Pericyclic Reactions and Synthesis of Hetero and Carbocycles Systems and the Interuniversity Consortium for Innovative Methodologies and Processes for Synthesis (CINMPIS) for partial financial support. The authors are very grateful to Professor Pierluigi Caramella for helpful discussion and suggestions.References
- S.-B. Park, E. Lih, K.-S. Park, Y. K. Joung and D. K. Han, Prog. Polym. Sci., 2017, 68, 77–105 CrossRef CAS.
- Y. Zhang, X. Liu, L. Zeng, J. Zhang, J. Zuo, J. Zou, J. Ding and X. Chen, Adv. Funct. Mater., 2019, 29, 1903279 CrossRef.
- K. Zhang, S. Wang, C. Zhou, L. Cheng, X. Gao, X. Xie, J. Sun, H. Wang, M. D. Weir, M. A. Reynolds, N. Zhang, Y. Bai and H. H. K. Xu, Bone Res., 2018, 6, 31 CrossRef PubMed.
- K. Farbod, M. R. Nejadnik, J. A. Jansen and S. C. G. Leeuwenburgh, Tissue Eng., Part B, 2013, 20, 173–188 CrossRef PubMed.
- L. Roseti, V. Parisi, M. Petretta, C. Cavallo, G. Desando, I. Bartolotti and B. Grigolo, Mater. Sci. Eng., C, 2017, 78, 1246–1262 CrossRef CAS PubMed.
- R. Song, M. Murphy, C. Li, K. Ting, C. Soo and Z. Zheng, Drug Des., Dev. Ther., 2018, 12, 3117–3145 CrossRef CAS PubMed.
- S. Tiwari, R. Patil and P. Bahadur, Polymers, 2019, 11, 1–23 CrossRef PubMed.
- K. M. Galler, R. N. D'Souza and J. D. Hartgerink, J. Mater. Chem., 2010, 20, 8730–8746 RSC.
- M. Rodríguez-Vázquez, B. Vega-Ruiz, R. Ramos-Zúñiga, D. A. Saldaña-Koppel and L. F. Quiñones-Olvera, BioMed Res. Int., 2015, 1–15 Search PubMed.
- F. Croisier and C. Jérôme, Eur. Polym. J., 2013, 49, 780–792 CrossRef CAS.
- R. LogithKumar, A. KeshavNarayan, S. Dhivya, A. Chawla, S. Saravanan and N. Selvamurugan, Carbohydr. Polym., 2016, 151, 172–188 CrossRef CAS PubMed.
- S. Gao, G. Tang, D. Hua, R. Xiong, J. Han, S. Jiang, Q. Zhang and C. Huang, J. Mater. Chem. B, 2019, 7, 709–729 RSC.
- S. Deepthi, J. Venkatesan, S.-K. Kim, J. D. Bumgardner and R. Jayakumar, Int. J. Biol. Macromol., 2016, 93, 1338–1353 CrossRef CAS PubMed.
- K. C. Kavya, R. Jayakumar and S. K. P. Chennazhi, Int. J. Biol. Macromol., 2013, 59, 255–263 CrossRef CAS PubMed.
- (a) A. Pistone, D. Iannazzo, C. Celesti, E. Piperopoulos, D. Ashok, A. Cembran, A. Tricoli and D. Nisbet, Materials, 2019, 12, 2321 CrossRef CAS PubMed; (b) A. Pistone, D. Iannazzo, S. Galvagno, C. Espro, A. Tampieri, m. Montesi, S. Panseri and M. Sandri, Engineering, 2017, 3(1), 55–59 CrossRef CAS.
- Y. Yu, Z. Bacsik and M. Edén, Materials, 2018, 11, 1–18 CrossRef CAS PubMed.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- S. Tamburaci and F. Tihminlioglu, J. Mater. Sci.: Mater. Med., 2018, 29, 1–15 CrossRef CAS PubMed.
- S. L. Chew, K. Wang, S. P. Chai and K. L. Goh, J. Mater. Sci.: Mater. Med., 2011, 22, 1365–1374 CrossRef CAS PubMed.
- Y. Zhang, M. Chen, J. Tian, P. Gu, H. Cao, X. Fan and W. Zhang, Biomater. Sci., 2019, 7, 3266–3276 RSC.
- H. Ghanbari, B. G. Cousins and A. M. Seifalian, Macromol. Rapid Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed.
- S. L. Chew, K. Wang, S. P. Chai and K. L. Goh, J. Mater. Sci.: Mater. Med., 2011, 22, 1365–1374 CrossRef CAS PubMed.
- Y.-M. Ha, T. Amna, M.-H. Kim, H.-C. Kim, M. S. Hassan and M.-S. Khil, Colloids Surf., B, 2013, 102, 795–802 CrossRef CAS PubMed.
- (a) U. Chiacchio, A. Padwa and G. Romeo, Curr. Org. Chem., 2009, 13, 422–447 CrossRef CAS; (b) R. Romeo, C. Carnovale, S. V. Giofrè, M. A. Chiacchio, A. Garozzo, E. Amata, G. Romeo and U. Chiacchio, Beilstein J. Org. Chem., 2015, 11, 328–334 CrossRef CAS PubMed; (c) S. V. Giofrè, R. Romeo, C. Carnovale, R. Mancuso, S. Cirmi, M. Navarra, A. Garozzo and M. A. Chiacchio, Molecules, 2015, 20, 5260–5275 CrossRef PubMed; (d) F. P. Ballistreri, C. M. A. Gangemi, A. Pappalardo, G. A. Tomaselli, R. M. Toscano and G. Trusso Sfrazzetto, Int. J. Mol. Sci., 2016, 17, 1112 CrossRef PubMed; (e) A. Brandi, F. Cardona, S. Cicchi, M. Cordero and A. Goti, Org. React., 2017, 94, 1–321 CAS; (f) I. Blanco, L. Abate, P. Bottino and M. A. Chiacchio, J. Therm. Anal. Calorim., 2018, 134, 1337–1344 CrossRef CAS; (g) L. Abate, F. A. Bottino, G. Cicala, M. A. Chiacchio, G. Ognibene and I. Blanco, Polymers, 2019, 11, 1475 CrossRef CAS PubMed; (h) M. A. Chiacchio, L. Legnani, A. Campisi, P. Bottino, G. Lanza, D. Iannazzo, L. Veltri, S. Giofré and R. Romeo, Org. Biomol. Chem., 2019, 17, 4892–4905 RSC; (i) S.-I. Murashi and Y. Imada, Chem. Rev., 2019, 119, 4684–4716 CrossRef PubMed; (j) A. I. Said and T. I. El-Emary, RSC Adv., 2020, 10, 845–850 RSC.
- C. C. Tsai, R. N. Huang, H. W. Sung and H. C. Liang, J. Biomed. Mater. Res., 2000, 52, 58–65 CrossRef CAS PubMed.
- M. A. Chiacchio, l. Borrello, G. Di Pasquale, A. Pollicino, F. A. Bottino and A. Rescifina, Tetrahedron, 2005, 61, 7986–7993 CrossRef CAS.
- (a) L. Legnani, L. Toma, M. A. Chiacchio, S. Giofré, I. Delso, T. Tejero and P. Merino, J. Org. Chem., 2016, 81, 7733–7740 CrossRef CAS PubMed; (b) M. A. Chiacchio, L. Legnani, P. Caramella, T. Tejero and P. Merino, Eur. J. Org. Chem., 2017, 1952–1960 CrossRef CAS; (c) P. Merino, M. A. Chiacchio, L. Legnani, I. Delso and T. Tejero, Org. Chem. Front., 2017, 4, 1541–1554 RSC; (d) M. A. Chiacchio, L. Legnani, P. Caramella, T. Tejero and P. Merino, Tetrahedron, 2018, 74, 5627–5634 CrossRef CAS; (e) L. Legnani, R. Puglisi, A. Pappalardo, M. A. Chiacchio and G. Trusso Sfrazzetto, Chem. Commun., 2020, 56, 539–542 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- B. R. Kim, H. G. Lee, S. B. Kang, G. H. Sung, J. J. Kim, J. K. Park, S. G. Lee and Y. J. Yoon, Synthesis, 2012, 1, 42–50 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01636e |
| This journal is © The Royal Society of Chemistry 2020 |