
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt oxysulphide/hydroxide nanosheets with dual properties based on electrochromism and a charge storage mechanism†
Saran Kalasinaab,
Ketsuda Kongsawatvoragulab,
Nutthaphon Phattharasupakunab,
Phitchayapha Phattharaphutiab and
Montree Sawangphruk *ab
*ab
aDepartment of Chemical and Biomolecular Engineering, School of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand. E-mail: montree.s@vistec.ac.th
bCentre of Excellence for Energy Storage Technology (CEST), Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
First published on 7th April 2020
Abstract
The charge storage mechanism of mixed cobalt oxysulphide/hydroxide materials having electrochromic properties was investigated. The cobalt oxysulphide/hydroxide materials exhibit a dual reversible redox reaction and electrochromic properties in 1 M KOH during charging and discharging.
Pseudo-supercapacitors and batteries are normally associated with faradaic electrochemical reactions at the electrode's surface and bulk structures, respectively. Interestingly, some materials can reversibly change colour during a faradaic chemical reaction, which is known as electrochromism.1,2 An application of electrochromic materials is for example in a ‘‘smart window’’ in which the mechanism is that they can change transmittance under different potentials. To enhance the functionality of the application, the energy storage and electrochromic properties are integrated together, and in this work this is called an electrochromic energy storage device. The challenge in the search for electrochromic energy storage devices is to find ideal materials that can provide excellent charge storage performance, good cycling stability or reversibility and excellent electrochromism.
Although many metal oxides (i.e., WO3, Nb2O5, TiO2, NiO, MnO2 and V2O5) have been widely used in electrochromic energy storage devices,1,2 metal sulphides are not yet fully investigated in these devices. Cobalt sulphides exhibit higher charge storage performance than the conventional cobalt hydroxides and/or oxides.3–6 However, they are not so stable over long cycling as compared to metal oxides. They can be decomposed generating H2S, for example. On the other hand, the S-doping in cobalt oxides can improve the electronic structure and electrochemical property of the host materials maintaining their high stability.7–9 As a result, in this work we have investigated the cobalt oxysulphide/hydroxide nanosheets and used them as the electrodes of the electrochromic energy storage devices.
Basically, all the cobalt oxides and/or sulphides can occur multiple redox reactions as normally observed by the electrochemical measurement. Although the redox mechanisms of these materials i.e., Co(OH)2, Co3O4 and CoS are normally proposed via conventional reactions, a clear evidence is also essential for supporting these mechanisms.8,10 The intermediate state of the material is very important to deeply understand the mechanisms.11 Note, the intermediate state is very difficult to be detected requiring more sophisticated techniques.12,13 In this work, we then used in situ UV-visible and X-ray absorption spectroscopy (XAS) techniques to determine the intermediate state.
In this work, the cobalt oxysulphide/hydroxide film was prepared by the electrodeposition method using thiourea (CH4N2S) as a sulphur source in the deposition solution. After the synthesis, the yellow-brown film of cobalt oxysulphide/hydroxide film was obtained and then its electrochemical property was tested in 1 M KOH. The as-prepared cobalt oxysulphide/hydroxide exhibits the electrochromic property in KOH solution after applying different potentials. The film can change its colour from yellow-brown to black during the electrochemical testing as shown in Fig. 1. The mechanism of the reversible change in colour of cobalt oxysulfide/hydroxide was fundamentally investigated by in situ techniques in which its intermediate state was clearly studied.
 | ||
| Fig. 1 FESEM image (a), FESEM-EDS image (b), HRTEM image (c), and the selected area electron diffraction (SAED) patterns (d) of the cobalt oxysulphide/hydroxide nanosheets. | ||
A mixed cobalt oxysulphide/hydroxide film, which is a semiconductor material, produced by a simple electrodeposition method shows such a dual property. After the electrodeposition, the yellowish-brown film of cobalt oxysulphide/hydroxide on the fluorine-doped tin oxide (FTO) was obtained (see Fig. S1b†), and its electrochemical property was investigated in 1 M KOH (see Fig. S2†). Interestingly, the as-prepared cobalt oxysulphide/hydroxide exhibits electrochromic characteristics during charging and discharging. In another word, the film shows different colours at different applying working potentials. This is due to its reduced and oxidised states of cobalt in the oxysulphide/hydroxide material.
The morphology and structure of the as-electrodeposited cobalt oxysulphide/hydroxide film on FTO glass were investigated by field emission scanning electron microscopy (FESEM), energy dispersive X-ray spectroscopy (EDS), and high-resolution transmission electron microscopy (HRTEM) techniques. An FESEM image of cobalt oxysulphide/hydroxide displays a nanosheet structure as shown in Fig. 1a. EDX mapping in Fig. 1b confirms that the film consists of Co, O, and S elements and all the elements are homogeneously distributed within the film (see Fig. S3†). Fig. 1c displays an HRTEM image of cobalt oxysulphide/hydroxide. The d-spacing values of ca. 0.5 and 0.2 nm are due to (001) and (103) planes, respectively, (Fig. S4†). The diffraction rings of cobalt oxysulfide/hydroxide from HRTEM are shown in Fig. 1d. Four rings of α-, (001), (100), (002), and (102) planes relate to Co(OH)2. Four rings of (002), (101), (004), and (103) planes refer to CoO(OH), and two rings of (100) and (102) planes represent CoS.
The cobalt oxysulfide/hydroxide electrode was also investigated by EDX technique with the elemental mapping of Co, O and S as shown in Fig. S3.† The EDS displays the Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S ratio of 9:1. Besides, the crystalline phase as well as the chemical composition and oxidation state of the as-electrodeposited cobalt oxysulphide/hydroxide nanosheets were investigated by powder X-ray diffraction (PXRD) and X-ray photoelectron spectroscopy (XPS) as shown in Fig. 2.
:S ratio of 9:1. Besides, the crystalline phase as well as the chemical composition and oxidation state of the as-electrodeposited cobalt oxysulphide/hydroxide nanosheets were investigated by powder X-ray diffraction (PXRD) and X-ray photoelectron spectroscopy (XPS) as shown in Fig. 2.
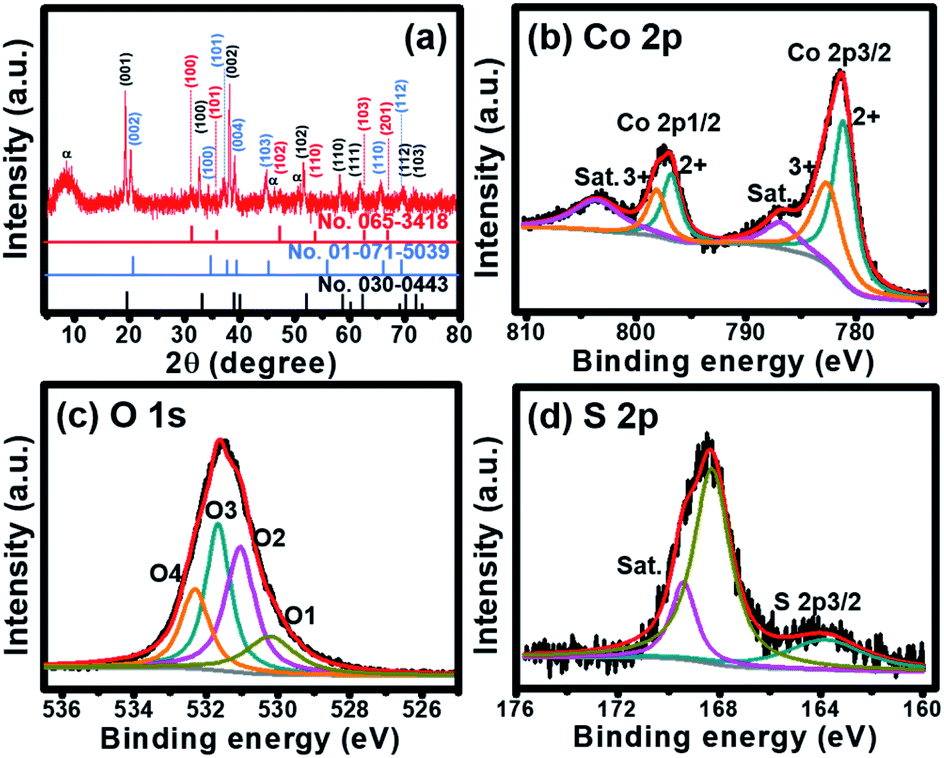 | ||
| Fig. 2 (a) XRD pattern and (b–d) XPS spectra of cobalt oxysulphide/hydroxide nanosheets; Co 2p (b), S 2p (c), and O 1s (d). | ||
The crystalline phase of the as-electrodeposited cobalt oxysulphide/hydroxide nanosheets investigated using the XRD is shown in Fig. 2a. The diffraction peaks display at 2θ = 19.5°, 33.5°, 38.5°, 51.3°, 58.5°, 62.0°, 70.5°, and 73.5° representing (001), (100), (002), (102), (110), (111), (112), and (103) planes of Co(OH)2 (JCPDS no.030-0443), respectively. The diffraction peaks at 20.3°, 35.2°, 38.0°, 40.1°, 45.3°, 65.5°, and 69.3° refer to (002), (100), (101), (004) (103), (110), and (112) planes of CoO(OH) (JCPDS no. 01-071-5039), respectively. Whilst, the diffraction peaks at 2θ = 30.9°, 36.5°, 47.2°, 53.8°, 63.2°, and 67.1° correspond to (100), (101), (102), (110), (103), and (201) planes of CoS (JCPDS no.065-3418), respectively. The XRD patterns suggest that the material consists of three crystalline planes of Co(OH)2, CoO(OH), and CoS. The mixture between Co(OH)2 and CoO(OH) has also previously been reported.14 In addition, a ratio between Co:S was calculated by wavelength dispersive X-ray fluorescence (WDXRF) supporting the XRD results. Generally, the as-prepared α-Co(OH)2 film can be obtained by the electrodeposition technique.15 The Co(OH)2 is a hydrotalcite-like structure with the hexagonal crystal phase of space group P3m1 with lattice parameters of a = b = 0.3183 nm, and c = 0.4652 nm.15,16 From the XRD pattern, Co(OH)2 is the main composition in the mixed materials. Note that the peak at 9.8° refers to the (001) plane of α-phase Co(OH)2 relating to anions (NO3−, S2− and SOx2−) intercalated within the interlayer of nanosheets (JCPDS no.051-1731).17–19 The Co(OH)2 can be converted into CoO(OH) during the oxidation process with the reduction of d-spacing value and the shift of the peak of (001) plane of Co(OH)2 to (002) plane of CoO(OH).14,16,20 The CoO(OH) is a hexagonal crystal phase of space group R3m with lattice parameters of a = b = 0.3183 nm, and c = 1.315 nm.15,16 The c-axis of CoO(OH) clearly expands and disorders when compared with the crystal structure of Co(OH)2. Meanwhile, CoS is a hexagonal crystal phase of space group P63/mmc with lattice parameters of a = b = 0.3368 nm, and c = 0.517 nm.21,22 Based on the same crystal structure, CoS can be substituted into the crystal structure of Co(OH)2. In addition, FTIR spectrum supports the existence of these peaks on the crystalline structures. The spectrum exhibits Co–S, Co![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, Co–O, and Co–OH bonds as shown in Fig. S5.†
S, Co–O, and Co–OH bonds as shown in Fig. S5.†
The chemical composition and oxidation state of Co in cobalt oxysulphide/hydroxide nanosheets were studied by X-ray photoelectron spectroscopy (XPS). There are two main peaks of the XPS spectrum at 781.2 and 796.8 eV referring to Co 2p3/2 and Co 2p1/2, respectively (Fig. 2b).23 The 2p3/2 peak can be deconvoluted into two peaks of Co2+ 2p3/2 and Co3+ 2p3/2 at 781.0 and 782.6 eV, respectively. Whilst, the Co 2p1/2 peak can also be deconvoluted at 796.6 and 798.2 eV for Co2+ 2p1/2 and Co3+ 2p1/2, respectively.24,25 The result indicates the coexistence of Co2+ and Co3+ in cobalt oxysulphide/hydroxide.26 The two main peaks of Co2+ and Co3+ represent Co–S bond.27,28 Note that the peaks also show a slight shift to a higher binding energy when compared to CoOOH (779.2, 781.4, 796.0, and 798.3 eV for Co3+ 2p3/2, Co2+ 2p3/2, Co3+2p1/2, and Co2+ 2p1/2, respectively).26 Due to the multiple electron excitation of Co2+ and Co3+ in cobalt oxysulphide/hydroxide, there are two peaks of shake-up satellite at 786.8 and 803.6 eV. The distance of the binding energy between Co 2p3/2 and Co 2p1/2 is about 15.6 eV implying that the cobalt oxysulphide/hydroxide film consists of both Co2+ and Co3+ with an average Co oxidation number of ca. 2.41. The O 1s spectrum of the cobalt oxysulphide/hydroxide is shown in Fig. 2c. The spectrum can be fitted into O1 (530.2 eV), O2 (531.1 eV), O3 (531.7 eV), and O4 (532.3 eV) components. The O1 component relates to the bonding between metal and oxygen (M–O–M). O2 refers to the oxygen of the hydroxyl groups, O3 involves with the number of defect sites with low oxygen coordination and O4 is the physisorption/chemisorption of water molecules on the surface of the cobalt oxysulphide/hydroxide.29–32 Fig. 2d displays the XPS spectrum of S 2p exhibiting two main peaks at 163.8 eV and 168.3 eV. The binding energy at 163.8 eV is owing to the bonding of Co–S in the material.33–35 The binding energy at 168.3 eV can be assigned to the oxidized sulphur group (such as –SOx–, x = 2–4) whilst a shake-up satellite at 169.4 eV can be referred to the bonding S–O.33–37
The electrochemical property of as-fabricated electrodes was firstly investigated by cyclic voltammetry (CV) with Hg/HgO and Pt as reference and counter electrodes, respectively. Cobalt oxysulphide/hydroxide was tested at a potential range of −0.5 to 0.8 V vs. Hg/HgO. The CV of cobalt oxysulphide/hydroxide at a scan rate of 25 mV s−1 is shown in Fig. 3a. It was found that the cobalt oxysulphide/hydroxide electrode changes its colour from yellowish brown to black for the forward scan and from black to yellowish brown for the backward scan in 1 M KOH electrolyte (see the video in ESI†). The oxidation processes (forward scan) start from −0.5 V to 0.8 V where two redox peaks can be observed at 0.3 and 0.6 V, respectively. The peak at 0.3 V relates to the oxidation of Co2+ to Co3+ which can be referred to the first oxidation reaction of Co(OH)2 and/or CoS by following reactions (1) and (2), respectively;8,10
| Co(OH)2 + OH− ↔ CoO(OH) + H2O + e− | (1) |
| CoS + OH− ↔ CoS(OH) + e− | (2) |
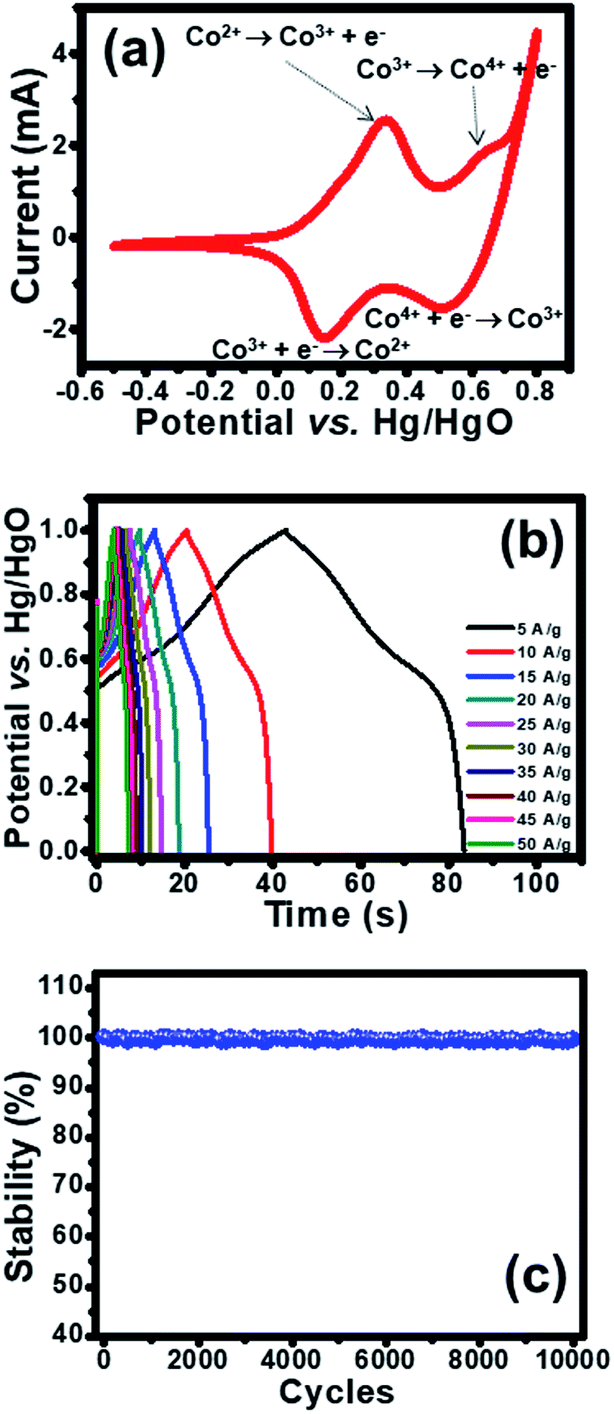 | ||
| Fig. 3 (a) CVs at 25 mV s−1 and (b) GCDs at 5–50 A g−1 (b) of half-cell electrode of cobalt oxysulphide/hydroxide nanosheets in 1 M KOH electrolyte and (c) cycling stability test from GCD over 10000 cycles at 5 A g−1. | ||
The peak at 0.6 V relates to the oxidation of Co3+ to Co4+.8,10 The second oxidation reactions of CoO(OH) and/or CoS(OH) follow reactions (3) and (4), respectively;
| CoO(OH) + OH− ↔ CoO2 + H2O + e− | (3) |
| CoS(OH) + OH− ↔ CoSO + H2O + e− | (4) |
The reduction processes (backward scan) start from 0.8 V to −0.5 V vs. Hg/HgO. There are two peaks at 0.55 and 0.15 V vs. Hg/HgO, respectively. The peak at 0.55 V vs. Hg/HgO relates to the reversible reactions (3) and (4) and the peak at 0.15 V Hg/HgO refers to the reversible reactions (1) and (2). These oxidation and reduction reactions can be clearly observed by the colour change of the electrode in 1 M KOH. In addition, the electrode was investigated by galvanostatic charge discharge (GCD) at various applied current densities (5–50 A g−1) as shown in Fig. 3b. The enlarge GCDs at 30–50 A g−1 were also shown in Fig. S8.† The GCDs were applied with a potential window of 1.0 V vs. Hg/HgO to avoid water splitting reaction. During charging, the charge storage mechanisms follow the forward oxidation reactions (1)–(4) or forward scan where the film colour changes from yellowish brown to black (see the video in ESI†). Whilst, the discharging mechanism follows the backward reduction reactions (4)–(1), the film colour returns from black to yellowish brown. The specific capacities at applied current densities of 5, 10, 15, 20, 25, 30, 35, 40, 45, and 50 A g−1 are 112.4, 106.9, 102.3, 99.6, 97.1, 95.0, 94.1, 93.1, 92.5, and 91.4 mA h g−1, respectively. In addition, the cycling performance of the cobalt oxysulphide/hydroxide electrode was tested for 10000 cycles at an applied current of 5.0 A g−1 as shown in Fig. 3d. The electrode exhibits very good capacity retention which is close to 100% maintaining the colour of the electrode like the initial colour (see Fig. S7†). Note that the cobalt oxysulphide/hydroxide is stable in KOH electrolyte.
Due to the colour change of the electrode from yellowish brown to black during the electrochemical testing in 1 KOH, in situ UV-visible spectrometer (Avantes) was utilized and the result is shown in Fig. 4a. The electrode was applied by cyclic voltammetry (CV) method from −0.5 V to 0.8 V vs. Hg/HgO in 1 M KOH. During CV testing, the UV-visible absorption was simultaneously investigated. After the CV measurement was started from −0.5 V to 0.8 V for the oxidation reaction, the absorption of the cobalt oxysulphide/hydroxide electrode increases with the highest absorption at 0.65 V vs. Hg/HgO. The absorption of the cobalt oxysulphide/hydroxide electrode decreases during the backward reduction process from 0.8 V to −0.5 V vs. Hg/HgO. At −0.5 V vs. Hg/HgO for which the absorption spectrum returns to its initial state suggesting that the redox reactions of cobalt oxysulphide/hydroxide are highly reversible.
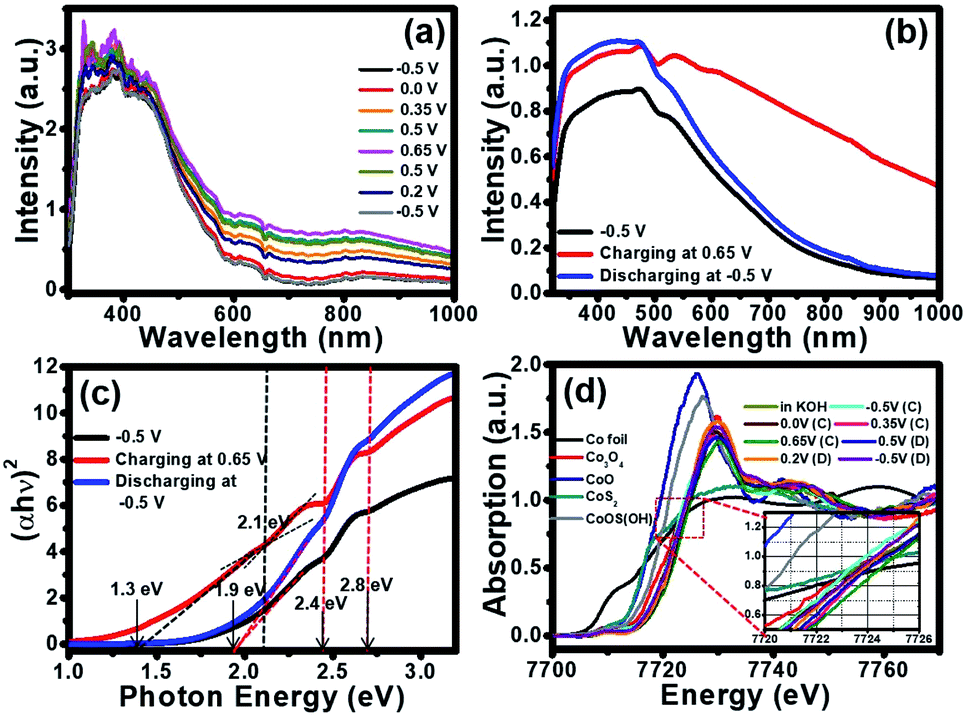 | ||
| Fig. 4 (a) In situ UV-visible spectra with Avantes UV-visible spectrometer, (b) in situ UV-visible spectra with chrono amperometry method, (c) Tauc plot of in situ UV-visible spectra, and (d) in situ XAS of cobalt oxysulphide/hydroxide nanosheets in 1 M KOH. | ||
To gain better understanding the property–function relationship of the materials, their electronic properties, the band gap energies of cobalt oxysulphide/hydroxide and their intermediates were further investigated by in situ UV-visible spectrometry together with a chronoamperometry at −0.5 V, 0.65 V, and −0.5 V vs. Hg/HgO. The absorption spectra of cobalt oxysulphide/hydroxide at different applied potentials are shown in Fig. 4b. The absorption spectrum of cobalt oxysulphide/hydroxide covers visible light region and displays the highest intensity at 470 nm. Note, the optical property of cobalt oxysulfide/hydroxide was measured under ambient condition (see details in ESI†). The absorption spectrum at −0.5 V vs. Hg/HgO shows the same result as that of the electrode without applying potential. The cobalt oxysulphide/hydroxide electrode starts to change its colour from yellowish brown to black at the applied potential of 0.65 V vs. Hg/HgO. This suggests that the cobalt oxysulphide/hydroxide can form the intermediates. The absorption spectrum of the intermediate exhibits higher intensity than that of the electrode with an applied potential of −0.5 V vs. Hg/HgO (initial state). This relates to the colour change into black colour. The electrode was then applied at −0.5 V vs. Hg/HgO back to the initial state (yellowish brown colour) in which the absorption spectrum is the same as the initial state. In addition, these absorption spectra were converted by Tauc equation to find their band gap energies at each state. The Tauc plot at each state is shown in Fig. 4c. The band gap energy at the initial state displays three values including 1.9, 2.4, and 2.8 eV. When the electrode was applied at 0.65 V vs. Hg/HgO, the gap energies at 2.4 and 2.8 eV remain constant, whilst the lowest gap energy decreases to 1.3 eV. Interestingly, the gap energy at 2.1 eV can also be observed referring to the energy level of the intermediate of cobalt oxysulphide/hydroxide. This gap energy is an intermediate energy level similar to the intermediate band for the solar cell.38–41 The more light absorption and the lower gap energy can be observed relating to the change of cobalt oxysulphide/hydroxide colour into the intermediate state (black film). After the intermediate state, the electrode was applied at −0.5 V vs. Hg/HgO, back to the initial state. The gap energy of the cobalt oxysulfide/hydroxide remains the same as the initial state. This clearly indicates that the intermediate of cobalt oxysulphide/hydroxide can be detected, and the redox reaction exhibits good reversible process. The change of the lowest gap energy and the existence of the gap energy at 2.1 eV support the energy diagram and the existence of the intermediate state.
To gain more information about the charge storage mechanism of cobalt oxysulphide/hydroxide, in situ electrochemical XAS was also performed along with the chrono amperometry. The experiment was set up in 1 M KOH electrolyte (Fig. S2†) for which the absorption energy of each electrode was measured at different applied potentials of −0.5, 0.0, 0.35, and 0.65 V vs. Hg/HgO (forward scan) and 0.5, 0.2, and −0.5 V vs. Hg/HgO (backward scan). The absorption energy of Co element was compared with the standard compounds including Co foil, CoO, Co2O3, Co3O4, and CoS2. The Co K-edge XANES spectra of cobalt oxysulphide/hydroxide and Co standard compounds in the fluorescent mode at different applied potentials are shown in Fig. 4d. The absorption edge of cobalt oxysulphide/hydroxide is shifted ca. 2 eV towards a high-energy as compared to the CoO standard indicating that the Co ions exhibit higher valence state.26,42 The average oxidation number of Co in cobalt oxysulphide/hydroxide after being immersed in the electrolyte is +2.67 (7721.8 eV), which remains unchanged after being charged at −0.5 V vs. Hg/HgO (7721.8 eV). The oxidation number of Co slightly increases to +2.94 (7722.8 eV), +3.03 (7723.1 eV) and +3.05 (7723.2 eV) with applied potentials at −0.5, 0.0, 0.35 and 0.65 V vs. Hg/HgO, respectively. This suggests that the charge storage mechanisms of cobalt oxysulphide/hydroxide occur as the proposed mechanism above. During backward scans at 0.5, 0.2, and −0.5 V vs. Hg/HgO, the oxidation number of Co continuously decreases to +3.04 (7723.2 eV), +3.01 (7723.1 eV) and +2.90 (7722.6 eV), respectively, resulting from the reduction processes of Co in cobalt oxysulphide/hydroxide (see the backward reactions (4)–(1)).
The extended X-ray absorption fine structure (EXAFS) was further analysed to obtain fundamental information about the neighbouring atoms (S and O) surrounding the cobalt chiral atoms in the crystal structure of cobalt oxysulphide/hydroxide. From EXAFS data, the R-space can be plotted from the Fourier transform EXAFS spectra with the Artemis freeware program. The peaks from the R-space analysis can be identified by comparing the R-spaces of Co standards as shown in Fig. 5a. It shows the bond length between Co and the neighbouring atoms. The bond length of the Co–S of the standard CoS2 is lower than that of the Co–O of the standard CoO. The spectra of all samples exhibit lower bond length than the standards since the ionic radius of S− is lower than that of OH−.26 The spectra of all samples exhibit lower bond length than the standards. The decreasing bond length implies the Co–S bonding and the existence of substituted S atoms in the cobalt oxysulfide/hydroxide film.
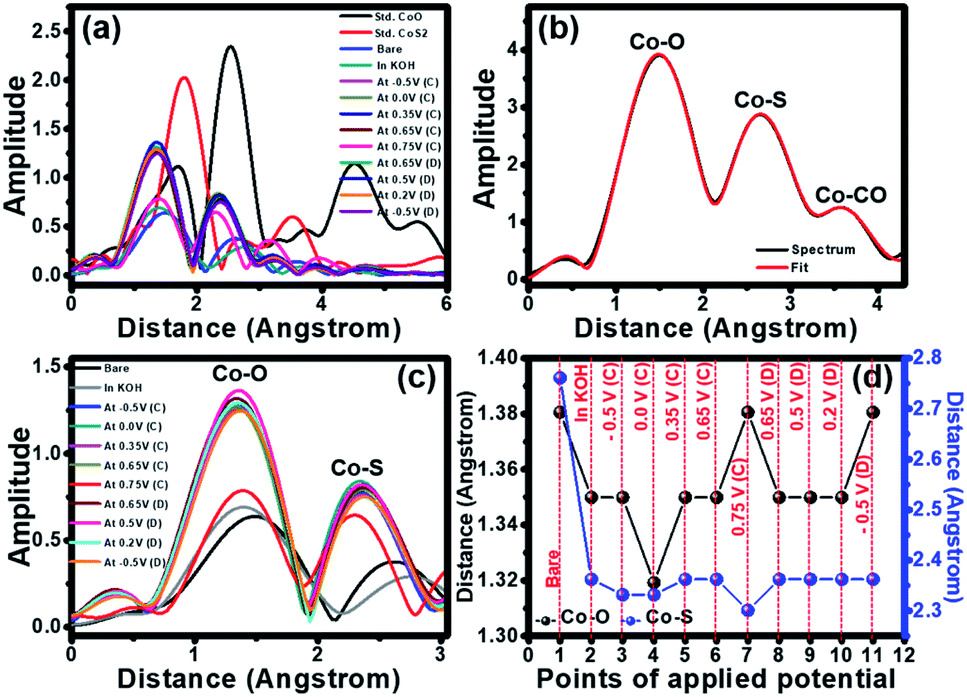 | ||
| Fig. 5 (a) EXAFS spectra of cobalt oxysulfide/hydroxide compared with standard compounds, (b) EXAFS spectrum is fitted by the standard modelling, (c) EXAFS spectra of cobalt oxysulfide/hydroxide at various applied potentials and (d) their R space applied with various potentials. | ||
The EXAFS spectrum of the as-prepared cobalt oxysulphide/hydroxide film was also fitted by using the standard modelling of CoO2 (mp-25476) and CoS2 (mp-2070) as shown in Fig. 5b. The fitted EXAFS parameters of the cobalt oxysulfide/hydroxide film are listed in Table S1.† These peaks relate to three shells of the bond between Co and neighbouring atoms. The first, second, and third shells can be assigned to Co–O, Co–S, and Co–Co bonds, respectively. Note, the fitted EXAFS data in Fig. 5b were used as the standard to compare with others (see Table S1†). The data suggests that the coordination number of Co is close to 6. The cobalt oxysulphide/hydroxide consists of some structural distortion owning to the dangling bonds in the surface Co octahedron owing to the substitution of O atoms with S atoms.42 In situ EXAFS was also measured together with the chronoamperometry method at −0.5, 0.0, 0.35, 0.65, and 0.75 V vs. Hg/HgO for the forward scan and 0.65, 0.5, 0.2, and −0.5 V vs. Hg/HgO for the backward scan. The R space plots of EXAFS spectra of cobalt oxysulphide/hydroxide are shown in Fig. 5c. It displays the movement of an amplitude and bond distance with applied potentials. The increasing amplitude relates to the increasing coordination number whilst the bond distance represents to the bonding change during applying potentials. The fitted data indicates that the coordination number of Co in the cobalt oxysulphide/hydroxide film is close to 6 and the coordination number slightly increases when the electrode was immersed in 1 M KOH. This is due to the chemical interaction between cobalt oxysulfide and oxygen atoms from OH−.23,43 When the electrode was applied with −0.5 and 0.0 V vs. Hg/HgO, the coordination number clearly increases with the highest amplitude at 0.35 V vs. Hg/HgO for the forward scan. After 0.35 V vs. Hg/HgO, the coordination number decreases to the minimum at 0.75 V vs. Hg/HgO. This suggests that the cobalt oxysulphide/hydroxide has structural distortion due to the interaction with the adsorbed OH− ions forming the intermediate state at 0.75 V.26,44
For discharge process, the potential was applied with 0.65, 0.5, 0.2, and −0.5 V vs. Hg/HgO, respectively. The coordination number increases at 0.65 V vs. Hg/HgO and gradually decreases with applied potentials of 0.5, 0.2, and −0.5 V vs. Hg/HgO, respectively. This suggests that the intermediate state returns to the initial state. Note that the coordination number after the discharging process shows higher than that before the charging process. This is a result of the chemical reaction of cobalt oxysulphide/hydroxide with the adsorbed OH− ions, leading to longer Co–O bond. The R space values of each applied potential points are shown in Fig. 5d. It clearly shows the shift of the bond distance with applied potentials. The bond distances of Co–O and Co–S for the as-electrodeposited cobalt oxysulfide film are 1.38 and 1.39 Å, respectively (point 1 in Fig. 5d). When the electrode was immersed in 1 M KOH (point 2 in Fig. 5d), the bond distances of Co–O and Co–S clearly decrease and continuously decrease with the applied potentials at −0.5 V vs. Hg/HgO (point 3 in Fig. 5d) and 0.0 V vs. Hg/HgO (point 4 in Fig. 5d) for the charging process. The bond distance of Co–O displays the lowest of all at 0.0 V and return to the highest of all at 0.75 V vs. Hg/HgO for charging (point 7 in Fig. 5d). This relates to the oxidation reaction of Co (also see the CV result). At 0.75 V vs. Hg/HgO for charging, the bond distance of Co–S exhibits the lowest distance which is opposite to the Co–O bonding. This suggests that the crystal structure of cobalt oxysulphide/hydroxide changes due to the redox reaction relating to the electrochromic property of cobalt oxysulphide/hydroxide.
The t2g orbital of Co provides high active sites for the electrochemical interaction. The valence electronic configuration (3d6 Co3+) with a high spin state consists of unpair electrons that can bind with S2− influencing the mismatch degree of Jahn–Teller distortion so-called the oxygen vacancy formation.26,45,46 The oxygen vacancy formation provides the active site for the electrochemical reactions during charging and discharging.23,42 It can be concluded here that the XANES data also suggests that the redox reaction occurs as the proposed mechanism. The EXAFS data show the changing Co–O and Co–S bonds during charging and discharging. The change can clearly be observed with applied potential at 0.75 V (point 7 in Fig. 5d). Oxygen from OH– with high electronegativity of all the reactants reacting with cobalt oxysulphide/hydroxide affects the bond distance of Co–O and Co–S. The changes of bond distance lead to the distortion of hexagonal crystal structure along c-axis. Note that all cobalt compounds are hexagonal.
In summary, the as-prepared cobalt oxysulphide/hydroxide nanosheets consist of three crystalline phases of (Co(OH)2, CoO(OH), and CoS) introducing unique optical properties including the electrochromism. In addition to clear observation for the colour change of the as-prepared material during charging and discharging by naked eyes, the charge storage performance of the as-prepared cobalt oxysulphide/hydroxide nanosheets was investigated together with in situ UV-visible and X-ray absorption spectroscopy techniques in 1 M KOH. It was found that during charging the increasing light absorption of cobalt oxysulphide/hydroxide nanosheets is observed especially in a wavelength range of 550–1000 nm. During discharging, the light absorption gradually decreases back to the original. In situ electrochemical XANES and EXAFS also indicate a reversible process of cobalt oxysulphide/hydroxide during charging and discharging. The oxidation state and the bond distance of Co–O and Co–S changes with chemical reaction lead to the distortion of crystal structure and the electrochromic property. The development on the electrochromic and charge storage property of cobalt oxysulphide/hydroxide nanosheets with high stability in alkaline solution might be used for many beneficial applications, for instance smart windows and/or smart energy storage devices in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Thailand Research Fund and Vidyasirimedhi Institute of Science and Technology (RSA6180031) as well as Energy Policy and Planning Office (EPPO), Ministry of Energy, Thailand. S. K. appreciated the Postdoctoral Fellowship from Vidyasirimedhi Institute of Science and Technology (VISTEC). Supports from the Frontier Research Centre at VISTEC and the Synchrotron Light Research Institute (Public Organization) BL5.2 SUT-Nanotec-SLRI XAS beamline in Thailand for XANES are also acknowledged.Notes and references
- P. Yang, P. Sun and W. Mai, Mater. Today, 2016, 19, 394–402 CrossRef CAS.
- E. L. Runnerstrom, A. Llordés, S. D. Lounis and D. J. Milliron, Chem. Commun., 2014, 50, 10555–10572 RSC.
- A. Nelson, K. E. Fritz, S. Honrao, R. G. Hennig, R. D. Robinson and J. Suntivich, J. Mater. Chem. A, 2016, 4, 2842–2848 RSC.
- J. Li, G. Liu, J. Fu, G. Jiang, D. Luo, F. M. Hassan, J. Zhang, Y.-P. Deng, P. Xu, L. Ricardez-Sandoval and Z. Chen, J. Catal., 2018, 367, 43–52 CrossRef CAS.
- U. K. Sultana, T. He, A. Du and A. P. O'Mullane, RSC Adv., 2017, 7, 54995–55004 RSC.
- X. Kuang, B. Kang, Z. Wang, L. Gao, C. Guo, J. Y. Lee, X. Sun and Q. Wei, Chem.–Eur. J., 2018, 24, 17288–17292 CrossRef CAS PubMed.
- M. A. R. Anjum, M. S. Okyay, M. Kim, M. H. Lee, N. Park and J. S. Lee, Nano Energy, 2018, 53, 286–295 CrossRef CAS.
- X. Ma, W. Zhang, Y. Deng, C. Zhong, W. Hu and X. Han, Nanoscale, 2018, 10, 4816–4824 RSC.
- Q. Liu and J. Zhang, CrystEngComm, 2013, 15, 5087–5092 RSC.
- L. Wang, D. Mitoraj, S. Turner, O. V. Khavryuchenko, T. Jacob, R. K. Hocking and R. Beranek, ACS Catal., 2017, 7, 4759–4767 CrossRef CAS.
- E. S. Wiedner and R. M. Bullock, J. Am. Chem. Soc., 2016, 138, 8309–8318 CrossRef CAS PubMed.
- C. L. Farrow, D. K. Bediako, Y. Surendranath, D. G. Nocera and S. J. L. Billinge, J. Am. Chem. Soc., 2013, 135, 6403–6406 CrossRef CAS PubMed.
- Y. Xie, F. Dong, S. Heinbuch, J. J. Rocca and E. R. Bernstein, Phys. Chem. Chem. Phys., 2010, 12, 947–959 RSC.
- Y. Chen, J. Zhou, P. Maguire, R. O'Connell, W. Schmitt, Y. Li, Z. Yan, Y. Zhang and H. Zhang, Sci. Rep., 2016, 6, 20704 CrossRef CAS PubMed.
- C. M. Hull, J. A. Koza and J. A. Switzer, J. Mater. Res., 2016, 31, 3324–3331 CrossRef CAS.
- X. Shen, S. Dai, C. Zhang, S. Zhang, S. M. Sharkey, G. W. Graham, X. Pan and Z. Peng, Chem. Mater., 2017, 29, 4572–4579 CrossRef CAS.
- J. P. Cheng, L. Liu, J. Zhang, F. Liu and X. B. Zhang, J. Electroanal. Chem., 2014, 722–723, 23–31 CrossRef CAS.
- D. Song, Y. Wang, Q. Wang, Y. Wang, L. Jiao and H. Yuan, J. Power Sources, 2010, 195, 7115–7119 CrossRef CAS.
- J. Yang, Z. Yang, L. H. Li, Q. Cai, H. Nie, M. Ge, X. a. Chen, Y. Chen and S. Huang, Nanoscale, 2017, 9, 6886–6894 RSC.
- P. F. Liu, S. Yang, L. R. Zheng, B. Zhang and H. G. Yang, J. Mater. Chem. A, 2016, 4, 9578–9584 RSC.
- J. Zhu, L. Xiang, D. Xi, Y. Zhou and J. Yang, Bull. Mater. Sci., 2018, 41, 54 CrossRef.
- Q. Wang, L. Jiao, H. Du, W. Peng, Y. Han, D. Song, Y. Si, Y. Wang and H. Yuan, J. Mater. Chem., 2011, 21, 327–329 RSC.
- W. Yuan, S. Wang, Y. Ma, Y. Qiu, Y. An and L. Cheng, ACS Energy Lett., 2020, 5, 692–700 CrossRef CAS.
- T. Magno de Lima Alves, B. F. Amorim, M. A. Morales Torres, C. G. Bezerra, S. Nóbrega de Medeiros, P. L. Gastelois, L. E. Fernandez Outon and W. Augusto de Almeida Macedo, RSC Adv., 2017, 7, 22187–22196 RSC.
- K. Zhu, C. Jin, Z. Klencsár, S. A. Ganeshraja and J. Wang, Catalysts, 2017, 7, 138 CrossRef.
- H. Li, W. Yuan, Q. Wang, X. Cui, J. Jiang, S. Chen, L. Song and X. Guo, ACS Sustainable Chem. Eng., 2019, 7, 17325–17334 CrossRef CAS.
- P. W. Faguy, N. Markovic, R. R. Adzic, C. A. Fierro and E. B. Yeager, J. Electroanal. Chem. Interfacial Electrochem., 1990, 289, 245–262 CrossRef CAS.
- Y. Li, L. Hu, W. Zheng, X. Peng, M. Liu, P. K. Chu and L. Y. S. Lee, Nano Energy, 2018, 52, 360–368 CrossRef CAS.
- R. Zou, K. Xu, T. Wang, G. He, Q. Liu, X. Liu, Z. Zhang and J. Hu, J. Mater. Chem. A, 2013, 1, 8560–8566 RSC.
- J. Wang, S. Fan, Y. Luan, J. Tang, Z. Jin, M. Yang and Y. Lu, RSC Adv., 2015, 5, 2405–2410 RSC.
- Y. E. Roginskaya, O. V. Morozova, E. N. Lubnin, Y. E. Ulitina, G. V. Lopukhova and S. Trasatti, Langmuir, 1997, 13, 4621–4627 CrossRef CAS.
- A. Shahraei, M. Kuebler, I. Martinaiou, K. A. Creutz, W. D. Z. Wallace, M. A. Nowroozi, S. Paul, N. Weidler, R. W. Stark, O. Clemens and U. I. Kramm, J. Mater. Chem. A, 2018, 6, 22310–22319 RSC.
- W. Du, Z. Wang, Z. Zhu, S. Hu, X. Zhu, Y. Shi, H. Pang and X. Qian, J. Mater. Chem. A, 2014, 2, 9613–9619 RSC.
- J. Shi, X. Li, G. He, L. Zhang and M. Li, J. Mater. Chem. A, 2015, 3, 20619–20626 RSC.
- J.-Y. Lin and J.-H. Liao, J. Electrochem. Soc., 2011, 159, D65–D71 CrossRef.
- A. Galtayries, C. Cousi, S. Zanna and P. Marcus, Surf. Interface Anal., 2004, 36, 997–1000 CrossRef CAS.
- K. M. Abraham and S. M. Chaudhri, J. Electrochem. Soc., 1986, 133, 1307–1311 CrossRef CAS.
- N. Alidoust, M. Lessio and E. A. Carter, J. Appl. Phys., 2016, 119, 025102 CrossRef.
- A. Luque, A. Martí and C. Stanley, Nat. Photonics, 2012, 6, 146 CrossRef CAS.
- A. Luque and A. Martí, Phys. Rev. Lett., 1997, 78, 5014–5017 CrossRef CAS.
- A. Martí, E. Antolín, C. R. Stanley, C. D. Farmer, N. López, P. Díaz, E. Cánovas, P. G. Linares and A. Luque, Phys. Rev. Lett., 2006, 97, 247701 CrossRef PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu and Y. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- S. Kim, D. A. Tryk, M. R. Antonio, R. Carr and D. Scherson, J. Phys. Chem., 1994, 98, 10269–10276 CrossRef CAS.
- W. Yuan, L. Cheng, Y. An, S. Lv, H. Wu, X. Fan, Y. Zhang, X. Guo and J. Tang, Adv. Sci., 2018, 5, 1700870 CrossRef PubMed.
- Y. He, C. Zhu, K. Chen, J. Wang, H. Qin, J. Liu, S. Yan, K. Yang and A. Li, J. Power Sources, 2017, 339, 13–19 CrossRef CAS.
- J. Wang, J. Liu, B. Zhang, F. Cheng, Y. Ruan, X. Ji, K. Xu, C. Chen, L. Miao and J. Jiang, Nano Energy, 2018, 53, 144–151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01714k |
| This journal is © The Royal Society of Chemistry 2020 |