
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA redox-sensitive core-crosslinked nanosystem combined with ultrasound for enhanced deep penetration of nanodiamonds into tumors†
Meixuan Lia,
Qianyan Lia,
Wei Houa,
Jingni Zhanga,
Hemin Yea,
Huanan Li*a,
Deping Zeng *ab and
Jin Bai*ab
*ab and
Jin Bai*ab
aState Key Laboratory of Ultrasound in Medicine and Engineering, College of Biomedical Engineering, Chongqing Medical University, Chongqing 400016, China. E-mail: 102733@cqmu.edu.cn; zengdp@cqmu.edu.cn; sajinbai@cqmu.edu.cn
bChongqing Key Laboratory of Biomedical Engineering, Chongqing Medical University, Chongqing 400016, China
First published on 17th April 2020
Abstract
Nanodiamonds (NDs) as drug delivery vehicles are of great significance in anticancer therapy through enhancing drug retention. However, the major barrier to clinical application of NDs is insufficient tumor penetration owing to their strong aggregation and low passive penetration efficiency. Herein, the core-crosslinked pullulan carrier, assembled using the visible light-induced diselenide (Se–Se) bond crosslinking method for encapsulating nanodiamonds-doxorubicin (NDX), is proposed to improve monodispersity. Furthermore, the core-crosslinked diselenide bond provides the nanosystem with redox-responsive capability and improved structural stability in a physiological environment, which prevents premature drug leakage and achieves tumor site-specific controlled release. What's more, ultrasound (US) is utilized to promote nanosystem intratumoral penetration via enlarged tumor vascular endothelium cell gaps. As expected, the nanosystem combined with ultrasound can enhance anti-tumor efficacy with deep penetration and excellent retention performance in a HepG2 xenograft mouse model. This study highlights the ability of the integrated therapeutic paradigm to overcome the limitation of nanodiamonds and the potential for further application in cancer therapy.
1. Introduction
Drug delivery technology is a critical issue in cancer therapy. Conventional small-molecule chemotherapeutic drugs are severely restricted from accessing the intratumoral environment due to heterogeneous vessels, high interstitial pressures, and other barrier effects, which results in relatively poor anti-tumor efficacy.1 Thus, nano-sized vehicles, including liposomes, polymeric micelles, dendrimers and nanoparticles, have been widely applied in delivering additional dosages and relying on the enhanced permeability and retention (EPR) effect to increase drug accumulation within the tumor.2 Although the EPR effect can improve delivery of nano-sized agents into the tumor compared to normal tissue, it is still inefficient and only a low concentration of nano-sized agents can accumulate intratumorally.3,4 Therefore, exploring a method to further enhance drug accumulation within the tumor may be pivotal to improving the therapeutic efficacy of nano-sized agents.Nanodiamonds (NDs), a new promising carbon nanomaterial with the property of preventing drug efflux, have attracted extensive attention as drug delivery vehicles to enhance drug retention in tumor cells.5,6 Moreover, NDs surfaces possess charge property that enables chemotherapeutic drugs to conjugate via physical absorption.7–9 The NDs-DOX complex has been shown to prevent drug efflux by overcoming ABC transporter-mediated multidrug resistance, thereby improving drug retention in tumor cells.6,10 However, NDs normally tend to form aggregates owing to their hydrophobic nature and high specific surface areas, which results in poor monodispersity, and limits their application in drug delivery.11–13
ND surface modification via polymer coating is a promising method for reducing the NDs' aggregation tendency and improving their monodispersity based on the shielding effect of the polymeric steric barrier, diminishing the surface interaction between NDs. ND surface modification using hydrophilic polyethylene glycol (PEG) or PEG derivatives exhibited excellent aqueous dispersion and markedly improved tumor therapeutic efficacy.14,15 Glycopolymer-coated NDs also reduced the tendency to aggregate, thus increasing NDs dispersion stability in aqueous solutions.16 In our previous work, hydrophilic pullulan polysaccharide nanocarriers were demonstrated to possess favorable properties of good stability and high drug loading contents.17–22 Therefore, we propose that the pullulan can be employed to form a hydrophilic shell to prevent the aggregation of NDs. However, achieving the optimal drug delivery effect still faces a challenge, as the NDs undergo early nanoparticle disintegration and premature drug release prior to reaching the target tumor site due to interaction between blood proteins and nanoparticles in the blood circulation.23
As such, the core crosslinking strategy was developed to minimize premature drug leakage during blood circulation and realize efficient drug delivery.24 Among the numerous available techniques, the reversible crosslinking method based on diselenide bond was determined to be the most effective. In addition to its crosslinked properties, diselenide bonds were confirmed to be responsive to the tumor redox microenvironment, which can be employed to achieve controlled tumor site-specific drug release. For instance, Zhai et al. reported three kinds of dual drug-loaded diselenide-rich polymers for pinpointed intracellular drug release.25 Given these advantages, diselenide bonds might endow nanoparticles with a robust and stable structure during systemic circulation, as well as the ability to efficiently and effectively perform drug delivery. Current nanoparticles based on EPR effect mainly accumulate around the tumor vessels due to the physiological barriers inside tumor tissue, leading to poor passive tumor penetration efficiency.26,27 As a drug delivery tool, ultrasound (US) is widely accessible, non-invasive, and can precisely target the tumor. Thus, delivery of nanoparticles to the tumor can be mediated by the combination of US, which enables deep tumor penetration by opening up tight junctions of tumor vascular endothelium cells and increasing vessel permeability.20,22
Herein, the rational design of a core–shell structure was reported to form a monodispersed and stable nanosystem for effective cancer drug delivery. The NDX core, prepared by the physical adsorption of DOX to NDs, facilitated enhanced drug retention and promoted delivery of the drug contents. The core-crosslinked pullulan shell, containing diselenide bonds, was responsive to the tumoral redox microenvironment and achieved site-specific drug release, while minimizing premature drug leakage under normal physiological conditions. US was simultaneously employed to increase blood vessel permeability and subsequently enhance intratumoral penetration of nanosystem. Therefore, this combined therapeutic design is expected to effectively deliver anticancer drugs to deep tumor sites and significantly inhibit tumor growth in vivo.
2. Results and discussion
2.1. Synthesis and characterization of NDX–CCS
NDX–CCS, a unique nanosystem structure comprised of a nanodiamonds-doxorubicin (NDX) core and pullulan-di-(4,1-hydroxybenzylene) diselenide (Pu-HBSe) shell, was rationally constructed in order to enhance drug retention, improve long-term stability, and facilitate site-specific drug release. Upon external ultrasound irradiation, NDX–CCS was readily transported within the blood vessel, deeply penetrated into the tumor vascular endothelium cells, and preferentially accumulated in the tumor regions (Scheme 1). This nano-sized NDX–CCS was fabricated by the self-assembly of Pu-HBSe conjugate and NDX, followed by visible light-induced metathesis and regeneration, among which, Pu-HBSe was synthesized by graft modification of HBSe containing rich Se–Se bond to carboxymethyl pullulan (CMP) backbone for excellent dispersibility of nanocarriers, stable blood circulation and minimal drug leakage, and then NDX was prepared through physical absorption hydrophobic DOX to the hydrophobic surfaces of NDs for overcoming drug efflux (Fig. 1a and Fig. S1†). The chemical structure of Pu-HBSe was determined using nuclear magnetic resonance (1H-NMR) and Fourier transform infrared (FTIR) spectroscopy by comparing the peaks of CMP, HBSe, and Pu-HBSe. The protons chemical shifts (ppm) of Pu-HBSe were indicated in the 1H-NMR spectrum (600 MHz, D2O, δ), corresponding to: 3.00–4.00 (4H, glucose C2, C3, C4, and C5), 4.70–5.60 (glucose, –OH), 4.82 (1H, s, 1-glueose a-1,6), 5.04 (1H, s, 2-glueose a-1,4), 7.17 (4H, d, a-H2), 7.44 (4H, d, b-H2). The 1H-NMR analysis showed that the Pu-HBSe resonance signal peaked at 3–4 ppm and 7–8 ppm, which corresponded to CMP and HBSe, respectively, and implies successful synthesis of the conjugate. In the FTIR spectra of Pu-HBSe, the characteristic absorption peak at 1600 cm−1 was attributed to the stretching vibration of phenyl ring of HBSe, indicating successful coupling of HBSe containing benzyl group to CMP. The absorption peaks at 1734 cm−1 could be clearly attributed to the carbonyl group, further indicating the successful incorporation of CMP onto HBSe (Fig. S2†).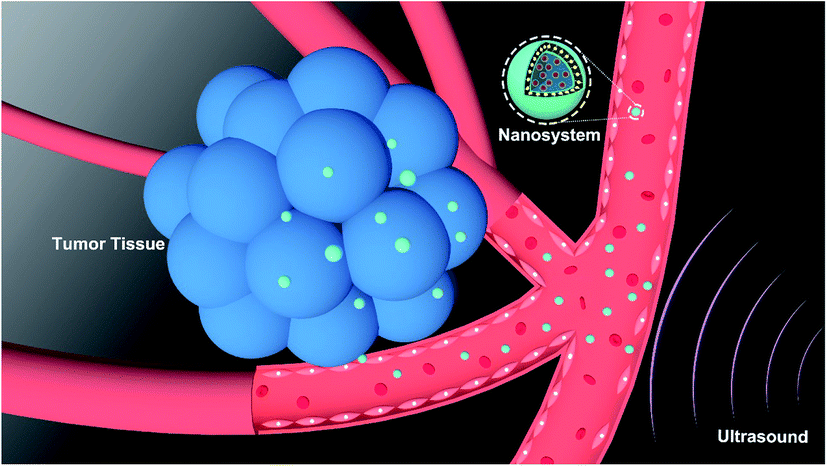 | ||
| Scheme 1 Schematic illustration of anti-tumor therapeutic process of NDX–CCS upon ultrasound irradiation. | ||
 | ||
| Fig. 1 Characterizations of NDX–CCS. (a) Schematic illustration of synthetic procedure of NDX–CCS. (b) TEM image of NDX–CCS. (c) Particle size and zeta potential analysis of (1) Pu-HBSe, (2) pristine ND, (3) NDX, (4) NDX–NCS, and (5) NDX–CCS. (d) The size stability of NDX, NDX–NCS and NDX–CCS at 37 °C in PBS with 10% FBS for 7 d. (e) In vitro cumulative DOX release from NDX–CCS. (f) Hemolysis test of NDX–CCS at different concentrations (0–200 μg mL−1) in PBS. The values of hydrodynamic size measured are based on intensity-mean. Data are presented as means ± SD, n = 3. | ||
As shown in Fig. 1b, the as-fabricated NDX–CCS displayed desirable monodispersity and uniform spherical core–shell morphology. Dynamic light scattering (DLS) measurement was performed to detect the hydrodynamic size and surface zeta potential of the different obtained nanosystems, and the values of hydrodynamic size measured were based on intensity-mean. Difference in functional groups on the NDs surface lead to differences in properties. The pristine NDs positive potential is indicative of the presence of hydrogenated surface of NDs surface.28,29 After the adsorption of DOX, NDX was detected by the slight shift of potential to become less positive. Based on the chemical structure of DOX, the formation of NDX complexes may be due to the π–π stacking interactions and polar interactions such as hydrogen bonding and electrostatic interactions.6 The NDX–NCS hydrodynamic diameters increased to ∼185 nm after NDX loading and decreased to ∼163 nm after crosslinking (Fig. 1c). We speculate that the Se–Se bond metathesis and regeneration under visible light may have resulted in a higher Pu-HBSe crosslinked density and hence the smaller NDX–CCS size.30 All of the obtained nanosystems exhibited only minor aggregation and narrow size distribution, with polydispersity indices (PDI) ranging from 0.07 to 0.15. The lower PDI of NDX–CCS (0.07) than that of NDX (0.13) indicates the enhanced monodispersity in solution, due to the hydrophilic Pu-HBSe coating on the NDX surface. Both properties are favorable for drug delivery systems, which should not be prone to excessive aggregation and precipitation. NDX–CCS exhibited a negative zeta potential of −34.1 mV after being coated with Pu-HBSe, which may be attributed to the CMP's residual carboxylic group. The negative surface charge would maintain the nanosystems stability and prolong blood circulation time, owing to low plasma protein adhesion and reduced clearance by the reticuloendothelial system (RES).31 The NDX–NCS and NDX–CCS in the PBS containing 10% FBS (Fig. 1d) displayed negligible diameter variation for 7 days, indicating long-term stability under this physiological condition. However, NDX was unstable in the same condition, and formed aggregation with the bulk of the nanoparticles already settled on the cuvette's bottom after storage for 7 d, attributed to the increased hydrophobicity or polar interaction, such as hydrogen bonding and electrostatic interactions.6
A key feature of any successful drug delivery system is its ability to release the drugs in a controlled biological setting. We expected that the redox-sensitive NDX–CCS would maintain stability during systemic circulation and minimize premature drug leakage, then achieve rapid and site-specific drug release at the tumor tissue or cells. To evaluate the redox sensitivity of NDX–CCS, an in vitro DOX release profile was measured in different concentrations of GSH and H2O2 at 37 °C. Only a small amount of DOX was released from the PBS (pH 7.4) without GSH and H2O2 (Fig. 1e) within a 48 h period. The GSH concentrations were selected to mimic the normal physiological (10 μM) and tumor intracellular (2 mM) conditions.32,33 In the presence of 2 mM and 10 μM GSH, 57% and 40.4% DOX was released from the NDX–CCS, respectively. 72.7% DOX leakage was observed in the presence of 100 μM H2O2 comparable to a tumor oxidative environment; and ∼54.1% DOX was released in the presence of 1 μM H2O2 comparable to a normal extracellular microenvironment.34,35 These results illustrate that NDX–CCS can inhibit premature drug diffusion and leakage from nanocarriers, suggesting excellent retention in normal tissues and effectively lessened systemic side effects.
As mentioned above, according to the DOX relative absorbance intensity in the UV-vis spectrum, we found that Pu-HBSe shells could serve as an excellent stabilizer and ensure DOX loading onto nanocarriers with 14.59% DOX loading efficiency and 29.79% encapsulation efficiency. More importantly, good nanomaterial biocompatibility can help prolong the circulation period and enhance the bioavailability in tumor regions.36 As such, hemolysis assay was performed to conduct a preliminary biocompatibility evaluation. The hemolysis rate was less than 5% and RBCs maintained their structural integrity at high concentrations (up to 200 μg mL−1) in the presence of NDX–CCS (Fig. 1f). These results indicate favorable nanosystem biocompatibility, which is beneficial for improving the bioavailability of tumor tissues.
2.2. Cellular drug uptake, distribution and retention in vitro
To assess the effectiveness of NDs as a drug-pumping-out inhibitor, intracellular DOX distribution and retention were measured via confocal laser scanning microscopy (CLSM) and flow cytometry (FCM) analysis. The results showed that free DOX, NDX, NDX–NCS, and NDX–CCS could all be internalized by HepG2 cells (Fig. 2b). After co-incubation for 4 h, there was a strong DOX fluorescence signal in the nuclei of HepG2 cells treated with free DOX. The red fluorescence from DOX was also clearly observed in cells treated with NDX, NDX–NCS, and NDX–CCS. However, most of the DOX fluorescence was observed in the perinuclei, indicating that the nanosystems were mainly located in the cytoplasm during the initial stage. After 4 h of co-incubation, the HepG2 cells were allowed to efflux drugs for 12 h, after which the DOX fluorescence signal was analyzed. There was significantly more intracellular DOX fluorescence in the HepG2 cells treated with the above three groups, compared to that in the free DOX-treated cells. This drug retention was also verified by FCM (Fig. 2c and d), demonstrating that NDs effectively enhance retention in tumor cells. One explanation for this phenomenon is that the nanodiamond-drug complex no longer meets the demand of ABC transporter substrates; thus the drug efflux induced by ABC transporter proteins is restrained and the drug remains within cells for longer periods.5,10 High DOX accumulation in the nuclei was observed with NDX–NCS and NDX–CCS, due to their favorable monodispersity and small particle size. Successful coating by Pu-HBSe creates a hydrophilic steric barrier on the surface of NDX, which could prohibit the interaction between the NDs rich chemistry surface and surrounding environment, leading to excellent stability. Compared to NDX–NCS and NDX–CCS, the uncoated NDX was unstable and tended to aggregate, which resulted in low cellular retention in tumor cells. In addition, DOX in NDX could be protonated in the tumor cellular slightly acidic environment, and might lead to increased hydrophilicity, followed by decreased transmembrane efficiency, which ultimately reduces the DOX concentration in the nuclei.37 It was noted that the punctate DOX fluorescence was observed in both the cytoplasm and nuclei of tumor cells treated with NDX, NDX–NCS, and NDX–CCS, suggesting the remarkable endosomal escape capability, accompanied by redox-sensitive diselenide bond cleavages and controlled drug release.25,38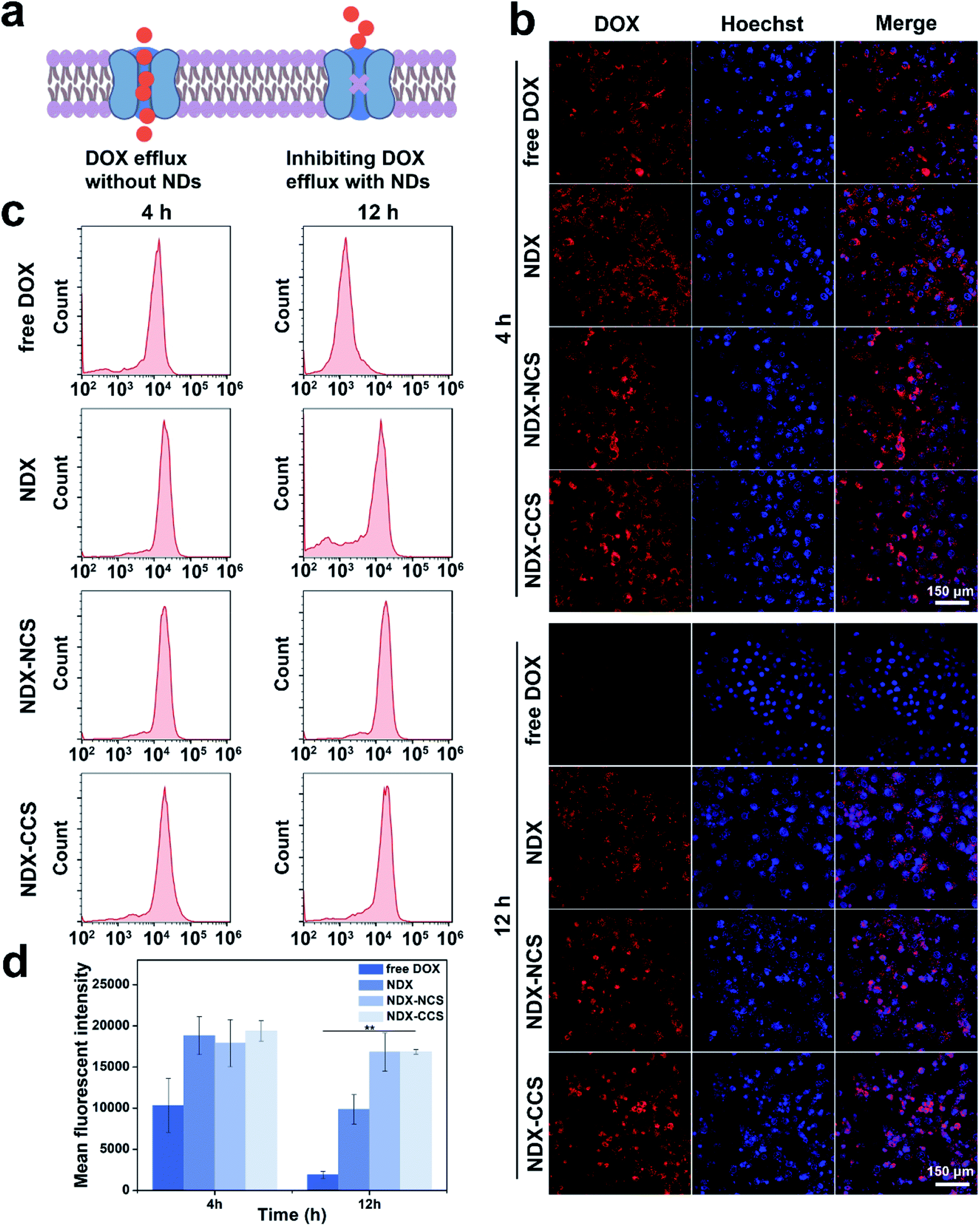 | ||
| Fig. 2 In vitro DOX uptake and retention analyses. (a) Illustration of the effect of NDs on inhibiting ABC transporter protein mediated drug efflux. (b) Representative CLSM images of HepG2 cells incubated with different formulations for 4 h before efflux and after 12 h efflux (blue fluorescence represented Hoechst and red represented DOX). (c) Flow cytometry analysis of DOX in HepG2 cells treated with different formulations for 4 h before efflux and after 12 h efflux (DOX = 10 mg L−1). (d) Mean fluorescence intensity of DOX in HepG2 cells from the flow cytometric result (**P < 0.01, n = 3). | ||
2.3. In vitro biosafety and cytotoxicity
The biosafety and biocompatibility of nanocarriers are of great significance for further in vivo applications. As such, they were evaluated by co-incubation with HUVEC and HepG2 cells using CCK-8 assay. The results (Fig. 3a) showed no obvious cytotoxicity for Pu-HBSe and NDs in the analyzed concentration range, even at high concentrations up to 100 mg L−1, indicating that the nanocarriers exhibit excellent biocompatibility. Consistent with the cellular uptake and retention study, NDX–CCS showed the highest cytotoxicity at a concentration range of 0.01–100 mg L−1 (Fig. 3b), while the cytotoxicity of other experimental groups was stronger than that of free DOX. The resulting cell viability suggests that the enhanced cellular uptake and retention of nanosystems could lead to increased cell growth inhibition. Furthermore, redox-sensitive potency of nanocarriers conserved the drug's potential activity and ensured simultaneous release within the tumor cells, which is the foundation of the superior anti-tumor efficacy in vivo. The calculated half maximal inhibitory concentrations (IC50) are listed in Table S1.† The IC50 value of DOX was 4.90 mg L−1 in the tumor cells co-cultured with free DOX, and decreased to 3.01 mg L−1 after absorption to the NDs. After being coated with Pu-HBSe, the DOX cytotoxicity further increased to an IC50 value of 0.86 mg L−1, which is ∼5.71-fold decrease from that of free DOX. Additionally, NDX–CCS showed a ∼2-fold lower IC50 value than that of NDX–NCS. A potential primary explanation is that the mild visible light-induced dynamic exchange of diselenide bonds in the nanosystem led to effective crosslinking. Compared with the non-crosslinked nanosystem (NDX–NCS), the physical stability of NDX–CCS was greatly enhanced, the drug leakage from the nanosystem in physiological conditions was effectively inhibited, and the blood circulation time was prolonged, leading to the highly effective tumor cellular accumulation.39–41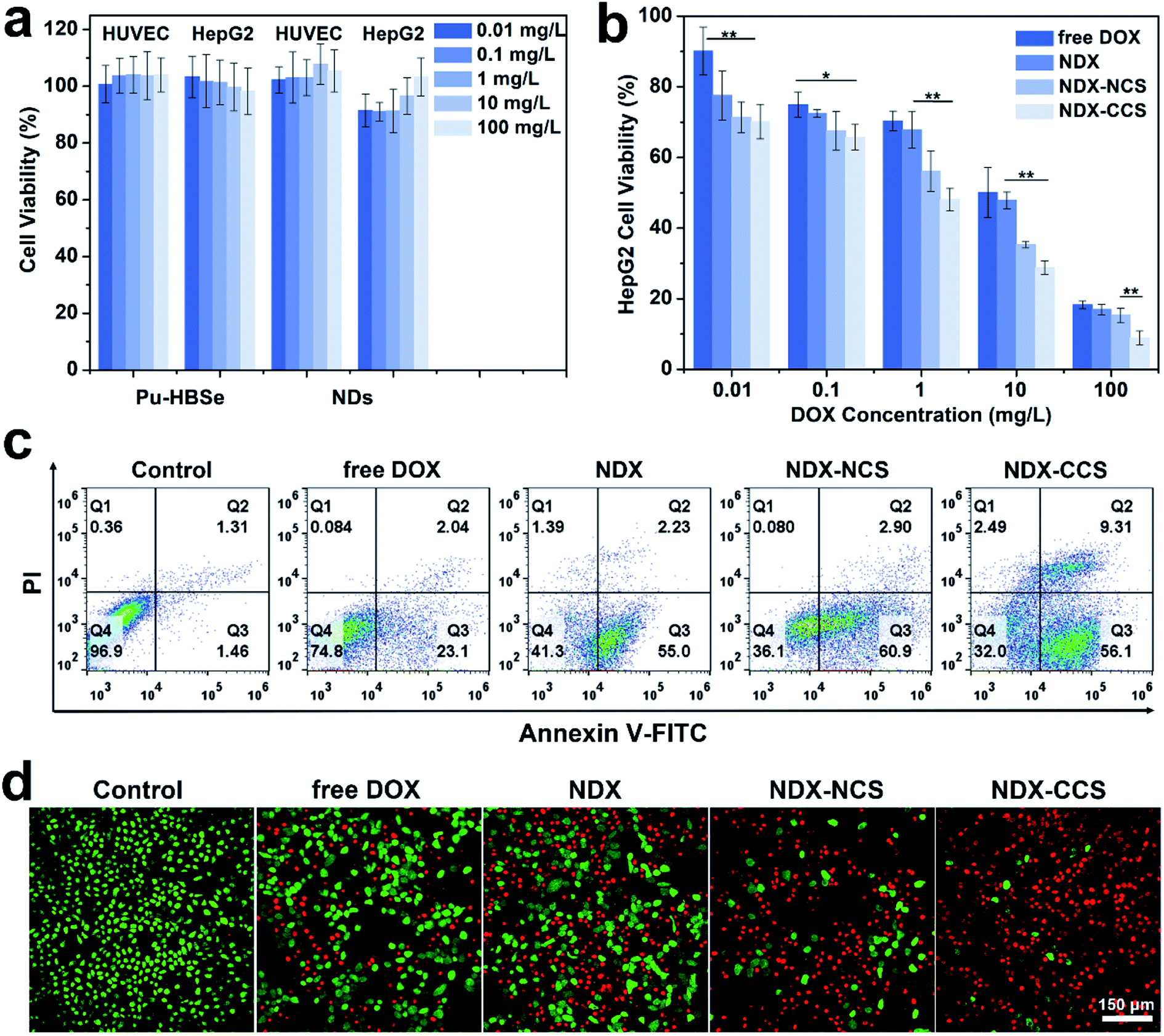 | ||
| Fig. 3 In vitro biosafety and cytotoxicity assay of NDX–CCS. (a) Cell cytotoxicity of nanocarriers for 24 h. (b) Cytotoxicity of HepG2 cells treated with different formulations with various concentrations (values are presented as means ± SD, *P < 0.05, **P < 0.01, n = 6). (c) Flow cytometry analysis of HepG2 cells apoptosis induced by different formulations for 24 h using the Annexin V-FITC/PI staining. (d) CLSM images of CAM and PI co-stained HepG2 cells after various treatments (DOX = 10 mg L−1). | ||
The flow cytometry-based Annexin V-FITC/PI apoptosis assays yielded results in accordance with those of the CCK-8 assay (Fig. 3c). The HepG2 cells co-incubated with NDX–CCS were confirmed to have the lowest viability of 32.0% and the highest total apoptosis ratio of 65.41% (56.1% and 9.31% for the early and late apoptosis, respectively). The enhanced cytotoxicity can be attributed to the efficient cellular accumulation of nanosystem and redox-sensitive release of DOX. It is worth noting that the tumor cell death caused by apoptosis is considered to be preferable to necrosis, because necrotic tissue can stimulate the host inflammatory response and possibly lead to treatment complications.42,43 The anti-tumor effect of nanosystems was further evaluated in vitro using Calcein-AM/PI to differentiate live (green fluorescence) and dead (red fluorescence) cells (Fig. 3d). Almost all the cells showed strong red fluorescence, indicating that extensive apoptosis and necrosis occurred under the NDX–CCS treatment.
2.4. In vivo biodistribution and fluorescence imaging
Evaluating the biodistribution of nanosystems in vivo is vital to determining the drug's bioavailability at the target lesion, as well as the potential toxicity to normal tissues or organs. Following intravenous (i.v.) administration of the nanosystems (DOX dose of 10 mg kg−1), fluorescence imaging was performed at predetermined time intervals. The free DOX fluorescence was widely distributed throughout the mouse body shortly after administration, and gradually weakened in the tumor region along with time (Fig. 4a and b). A time-dependent fluorescent signal was observed within the tumor region that peaked at 12 h and lasted until 24 h after injection of NDX–CCS, implying long-term stable circulation and excellent nanosystem site-specific release in the tumor. Ex vivo tumors and main organs collected at 24 h post-injection qualitatively and quantitatively confirmed the increased DOX accumulation in the tumor and lung in the NDX–CCS group compared with the free DOX group (Fig. 4c, d and f), and the tumors showed more accumulation of DOX than the other organs. One possible explanation is that the residual free DOX in the prepared NDX–CCS has been not completely removed, leading to accumulation in other organs. After administration, most DOX accumulated in the liver and kidneys, suggesting rapid hepatic clearance and renal excretion of this small molecule drug. Subsequently, the tumors were dissected into cryosections to observe the DOX distribution and intratumoral release via CLSM (Fig. 4e). At 24 h post-injection, strong DOX fluorescence signals of NDX–CCS group were evenly distributed throughout the tumor and localized in the nuclei, whereas there was less DOX fluorescence signal in the free DOX-treated group. This result further indicated that a core-crosslinked nanosystem can lead to massive accumulation and effective drug delivery in tumor cells by preventing premature drug leakage. | ||
| Fig. 4 In vivo drug biodistribution and penetration. (a) In vivo fluorescence images and (b) changes of fluorescence signal intensities of tumors in HepG2 tumor-bearing mice after i.v. injection of free DOX or NDX–CCS at predetermined time points. (c) Ex vivo fluorescence images and (d) corresponding fluorescence intensities of major organs and tumors dissected from mice 24 h post injection. (e) CLSM images of tumor cryosections at 24 h following i.v. injection. (f) Distribution profiles of total DOX in tissues at 24 h after a single i.v. injection of free DOX or NDX–CCS. (g) DOX content of tumor in the NDX–CCS + US-treated mice at different intervals. (h) DOX content of tumor at 24 h post injection in different formulations-treated mice. (i) Quantitative analysis of DOX concentrations in whole and central regions of tumors in mice after a single i.v. injection of free DOX or NDX–CCS for 24 h followed by different treatments. (j) Penetration indexes of free DOX, NDX–CCS and NDX–CCS + US (values are means ± SD, *P < 0.05, **P < 0.01, n = 6). (k) CLSM images of drug distribution in tumor cryosections. The equivalent dose of injected DOX was fixed at 10 mg kg−1. | ||
The dose of DOX accumulation at the tumor region in NDX–DOX + ultrasound group was quantitatively investigated via the tissue distribution method. As shown in Fig. 4g, at 12 h post-injection, the DOX accumulation reached 13.94% of the injected dose per gram of tissue (%ID g−1), which was almost equal to that at 24 h. Furthermore, quantitative data indicated that NDX–CCS was more prone to accumulate in the tumor under ultrasound irradiation (Fig. 4 h). NDX–CCS + ultrasound significantly increased DOX accumulation by ∼1.78-, 2.08-, 3.73-, and 8.31-fold compared to NDX–CCS, NDX–NCS, NDX, and free DOX, respectively. On one hand, these data demonstrate that prolonged blood circulation and improved physiological stability could benefit selective accumulation and long-term retention of the core-crosslinked nanosystems at the tumor site. On the other hand, they suggest that ultrasound might improve the tumor vessel permeability and drive nanosystems through the tumor vascular endothelial cells gap.22,44–46
2.5. Intratumoral drug penetration and pharmacokinetics
Ensuring the nanosystem deeply penetrates the tumor is crucial for highly efficient drug delivery. Thus, we anticipated that ultrasound would enhance nanosystem delivery to the tumor region. Mice bearing 600 mm3 HepG2 tumors were i.v. injected with free DOX, NDX–CCS, or NDX–CCS containing a 10 mg kg−1 dose of DOX before being exposed to ultrasound irradiation (1 MHz, 1 W cm−2, 5 min). Quantification of DOX accumulation in both the central region (100 mm3) and whole tumor was conducted at 24 h after a single i.v. administration (Fig. 4i). The small-molecule free DOX could permeate into deep tumor site and accumulate in the central tumor region. Nevertheless, the short half-life, fast clearance rate and insufficient cellular retention of free DOX led to drug efflux from tumor, decreasing the accumulation in the whole tumor. As a result, the penetration index of free DOX showed no difference with NDX–CCS + US, which was also verified by CLSM. Penetration index quantitatively confirmed a 1.39-fold difference in DOX accumulation between the NDX–CCS + ultrasound group and the NDX–CCS-only treatment group (Fig. 4j). Intratumoral penetration analysis of the tumor CLSM images showed prominent distribution of DOX throughout the tumor in the NDX–CCS + ultrasound group, as opposed to that in the NDX–CCS-only group with obvious peripheral contrast (Fig. 4k). Thus, these results demonstrate the superior tumor penetration ability of nanosystem upon ultrasound irradiation.Blood circulation is the first important bio-barrier encountered by drug delivery systems after i.v. injection, affecting the drug delivery efficiency, and therapeutic effect.47 The DOX in vivo pharmacokinetics was investigated in SD rats at different intervals after i.v. administration of free DOX, NDX–NCS, and NDX–CCS (DOX dose of 5 mg kg−1), respectively. Free DOX was rapidly cleared from blood circulation, as the DOX concentration in plasma dramatically decreased with a half-life (t1/2) of 1.7 h and an area under the curve (AUC0–24) of 26.2 μg mL−1 h−1 (Fig. S3 and Table S2†). In contrast, the t1/2 and AUC0–24 of NDX–NCS increased to 3.5 h and 33.9 μg mL−1 h−1, respectively. Furthermore, NDX–CCS remarkably slowed down the DOX clearance. The t1/2 and AUC0–24 of NDX–CCS were 5.7 h and 45.6 μg mL−1 h−1, respectively, implying that core-crosslinked nanosystems can improve the retention time in blood circulation by minimizing premature drug leakage.
2.6. In vivo anti-tumor activity and biosafety evaluation
After confirming the in vitro favorable efficacy of nanosystem and preferential intratumoral drug accumulation under ultrasound irradiation, we investigated the in vivo anti-tumor efficiency and biosafety of this strategy. Mice with an average tumor volume of ∼80 mm3 were intravenously injected with saline, free DOX, NDX, NDX–NCS, NDX–CCS, and NDX–CCS combined with ultrasound (DOX equivalent of 5 mg kg−1, ultrasound irradiation: 1 MHz, 1 W cm−2, 5 min) every three days (Fig. 5a). After performing the different therapies, the mice were photographed and tumor volumes were monitored during the therapeutic period (Fig. 5b and c). The saline group showed rapid tumor growth, considerably faster than all the other formulation-treated groups (Fig. 5d). The NDX and NDX–NCS-treated groups both exhibited more effective tumor inhibition as compared with the moderate tumor inhibition in the free DOX group. These results were in accordance with those of the drug biodistribution and pharmacokinetic experiments, suggesting the NDs have a positive effect on high drug retention and tumor inhibition. The tumor growth in the NDX–CCS group that was not exposed to ultrasound irradiation was partially inhibited with a ∼11.2-fold increase in the original tumor volume, probably due to the prolonged stable circulation time of crosslinked nanosystem and enhanced tumor accumulation. Upon coupling with ultrasound irradiation, the tumor growth in the NDX–CCS group was significantly inhibited with only a 2.4-fold increase in tumor volume, demonstrating the highest efficient anti-tumor effect and substantially enhanced therapeutic efficacy. The superior tumor inhibitory effect in vivo may be ascribed to ultrasound-induced preferential drug accumulation and enhanced deep penetration to the tumor central region.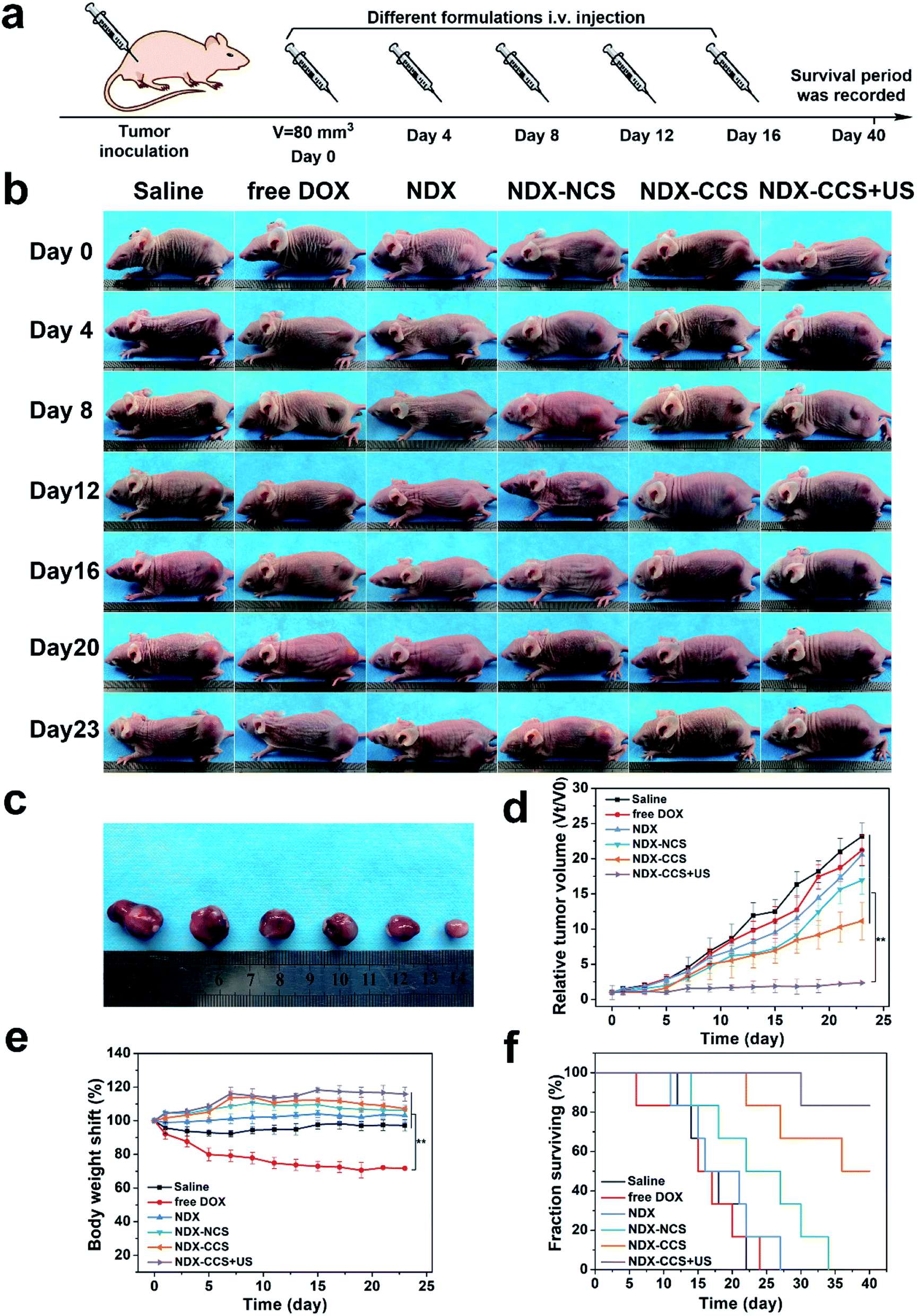 | ||
| Fig. 5 In vivo antitumor effect. (a) Schematic illustration of antitumor therapy. (b) Images of HepG2 tumor-bearing mice over 23 d period following various treatments (saline, free DOX, NDX, NDX–NCS, NDX–CCS and NDX–CCS + US, DOX dose of 5 mg kg−1). (c) Representative images of the HepG2 tumors after treatment with different formulations at day 23. (d) Tumor growth curves, (e) body weight changes and (f) survival rates of HepG2 tumor-bearing mice upon various treatments (value are means ± SD, n = 6, **P < 0.01). | ||
Hematoxylin–eosin (H&E), TdT-mediated dUPT Nick-End Labeling (TUNEL), and proliferating cell nuclear antigen (PCNA) staining on the tumor sections further confirmed the therapeutic efficacy in vivo. H&E staining showed most of the morphological changes (karyopyknosis, karyorrhexis, and karyolysis) in the tumor nuclei of the NDX–CCS + ultrasound group (Fig. 6a). Extensive brown nuclei (apoptotic nuclei) were observed using TUNEL assay and decreased proliferation-positive tumor cells were determined by PCNA assay (Fig. S4†). The above results showed that treatment with NDX–CCS + ultrasound was the most effective among all the treatment groups.
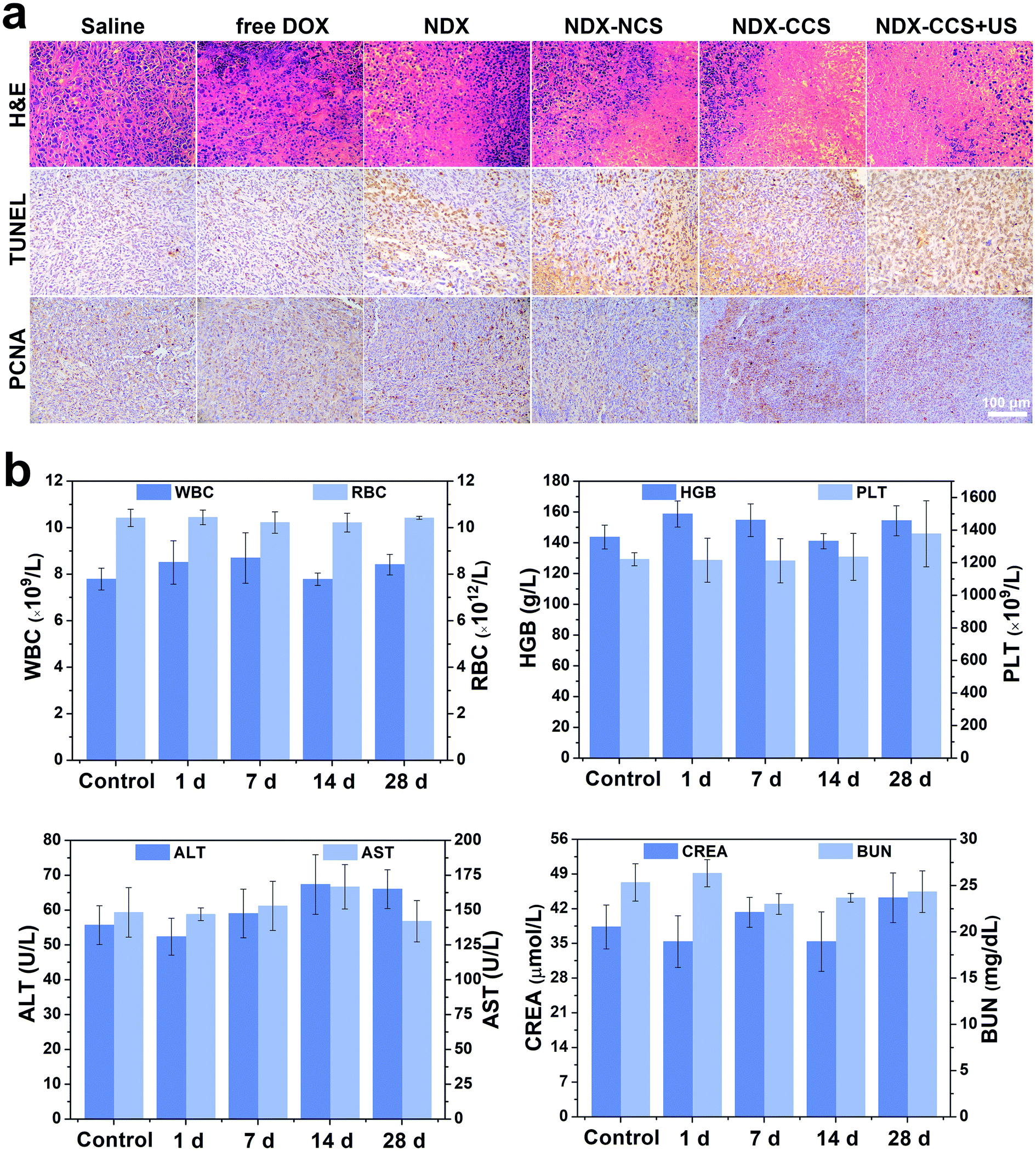 | ||
| Fig. 6 Biosafety evaluation in vivo. (a) H&E, TUNEL, and PCNA staining of tumor sections after various treatments. (b) Hematological assay including blood routine indexes, hepatic functions and renal functions in control group and experimental group at 1, 7, 14, and 28 d post i.v. injection of NDX–CCS (DOX dose of 5 mg kg−1). Value are means ± SD, n = 3. | ||
When developing nanosystems as therapeutic carriers, it is important that the nanosystem is safe for use. Thus, we studied the changes in the mice body weight and survival time to assess the biosafety of nanosystem in vivo. Results showed a significant weight decrease in the free DOX mice group, due to serious side effects associated with free DOX. A slight weight gain was observed in the other groups, implying nontoxicity of the nanocarriers (Fig. 5e). Furthermore, while no mice survived over 25 days in the saline and free DOX-treated control groups, 83.3% of the mice (5 out of 6) in the NDX–CCS + ultrasound group were still alive at day 40, significantly prolonging survival period for the tumor-bearing mice (Fig. 5f). H&E staining of the main organs (heart, liver, spleen, lung and kidney) was conducted on day 25 following the first therapy (Fig. S5†). Compared with the saline group, myocardial fiber denaturation, neutrophils infiltration, and glomerular pyknosis were observed in the free DOX-treated group, indicating that free DOX may induce acute cardiotoxicity and renal toxicity.48,49 Nevertheless, there were no changes in the physiological morphology of organs in other groups, which demonstrated the excellent biocompatibility and reduced systemic toxicity of the drug delivery vehicles. The blood routine examination and serum biochemical index reflecting the main organs' functions were measured and the results showed that there were no detectable changes between the NDX–CCS group at different intervals and the saline control group (Fig. 6b and S6†). These results strongly demonstrate the high biosafety of NDs and Pu-HBSe in vivo for anti-tumor therapy.
3. Conclusion
In summary, a core-crosslinked nanosystem (NDX–CCS) with good aqueous monodispersity and high stability has been successfully developed by coating a diselenide-rich pullulan shell onto NDX. The prepared NDX–CCS is able to overcome extracellular and intracellular physiological barriers that hinder drug delivery and retention in tumor cells, thus enhancing their accumulation at the specific tumor site. The introduced core-crosslinked diselenide bonds endowed NDX–CCS with redox-responsive capability, contributed to superior structure stability, and improved the efficacy of controlled drug delivery by minimizing premature drug leakage. Noticeably, the ultrasound-triggered tumor vessel permeability was increased to more readily drive the nanosystem deep into the tumor, thereby enabling the reinforced anti-tumor efficacy. Taken together, the combined therapeutic strategy developed here can perform high intracellular accumulation, site-specific drug release, and deep tumor penetration, showing great clinical potential for highly efficient tumor treatment.Ethical statement
All animal study followed procedures that were consistent with the Regulation for the Administration of Affairs Concerning Experimental Animal approved by the State Council of People's Republic of China and were approved by the Animal Ethics Committee of Chongqing Medical University.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the partial experimental facilities support from Institute of Ultrasound Imaging, Department of Ultrasound, The Second Affiliated Hospital of Chongqing Medical University.References
- K. Sano, T. Nakajima, P. L. Choyke and H. Kobayashi, ACS Nano, 2013, 7, 717–724 CrossRef CAS PubMed.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef PubMed.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2014, 4, 81–89 CrossRef CAS PubMed.
- L. Mei, Y. Liu, J. Rao, X. Tang, M. Li, Z. Zhang and Q. He, ACS Appl. Mater. Interfaces, 2018, 10, 17582–17593 CrossRef CAS PubMed.
- X. Wang, X. C. Low, W. Hou, L. N. Abdullah, T. B. Toh, M. Mohd Abdul Rashid, D. Ho and E. K. Chow, ACS Nano, 2014, 8, 12151–12166 CrossRef CAS.
- E. K. Chow, X. Q. Zhang, M. Chen, R. Lam, E. Robinson, H. Huang, D. Schaffer, E. Osawa, A. Goga and D. Ho, Sci. Transl. Med., 2011, 3, 21r–73r Search PubMed.
- M. Chipaux, K. J. van der Laan, S. R. Hemelaar, M. Hasani, T. Zheng and R. Schirhagl, Small, 2018, 14, 1704263 CrossRef PubMed.
- K. van der Laan, M. Hasani, T. Zheng and R. Schirhagl, Small, 2018, 14, 1703838 CrossRef PubMed.
- D. G. Lim, J. H. Jung, H. W. Ko, E. Kang and S. H. Jeong, ACS Appl. Mater. Interfaces, 2016, 8, 23558–23567 CrossRef CAS PubMed.
- D. G. Lim, R. E. Prim, K. H. Kim, E. Kang, K. Park and S. H. Jeong, Int. J. Pharm., 2016, 514, 41–51 CrossRef CAS PubMed.
- F. Neugart, A. Zappe, F. Jelezko, C. Tietz, J. P. Boudou, A. Krueger and J. Wrachtrup, Nano Lett., 2007, 7, 3588–3591 CrossRef CAS PubMed.
- O. A. Williams, J. Hees, C. Dieker, W. Jäger, L. Kirste and C. E. Nebel, ACS Nano, 2010, 4, 4824–4830 CrossRef CAS PubMed.
- Y. Liang, M. Ozawa and A. Krueger, ACS Nano, 2009, 3, 2288–2296 CrossRef CAS PubMed.
- J. Whitlow, S. Pacelli and A. Paul, J. Controlled Release, 2017, 261, 62–86 CrossRef CAS PubMed.
- D. Wang, Y. Tong, Y. Li, Z. Tian, R. Cao and B. Yang, Diamond Relat. Mater., 2013, 36, 26–34 CrossRef CAS.
- J. Zhao, H. Lai, H. Lu, C. Barner-Kowollik, M. H. Stenzel and P. Xiao, Biomacromolecules, 2016, 17, 2946–2955 CrossRef CAS PubMed.
- H. Li, Y. Cui, J. Liu, S. Bian, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2014, 2, 3500–3510 RSC.
- H. Li, Y. Cui, J. Sui, S. Bian, Y. Sun, J. Liang, Y. Fan and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 15855–15865 CrossRef CAS PubMed.
- H. Li, Y. Sun, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2015, 3, 8070–8078 RSC.
- H. Li, M. Ma, J. Zhang, W. Hou, H. Chen, D. Zeng and Z. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 20341–20349 CrossRef CAS PubMed.
- H. Li, Q. Li, W. Hou, J. Zhang, C. Yu, D. Zeng, G. Liu and F. Li, ACS Appl. Mater. Interfaces, 2019, 11, 43581–43587 CrossRef CAS PubMed.
- H. Li, D. Zeng, Z. Wang, L. Fang, F. Li and Z. Wang, Nanomedicine, 2018, 13, 981–996 CrossRef CAS PubMed.
- Y. Li, M. S. Budamagunta, J. Luo, W. Xiao, J. C. Voss and K. S. Lam, ACS Nano, 2012, 6, 9485–9495 CrossRef CAS PubMed.
- J. Zhao, C. Yan, Z. Chen, J. Liu, H. Song, W. Wang, J. Liu, N. Yang, Y. Zhao and L. Chen, J. Colloid Interface Sci., 2019, 540, 66–77 CrossRef CAS.
- S. Zhai, X. Hu, Y. Hu, B. Wu and D. Xing, Biomaterials, 2017, 121, 41–54 CrossRef CAS PubMed.
- R. Wang, Y. Han, B. Sun, Z. Zhao, Y. Opoku-Damoah, H. Cheng, H. Zhang, J. Zhou and Y. Ding, Small, 2018, 14, 1703110 CrossRef PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- H. Lai, M. H. Stenzel and P. Xiao, Int. Mater. Rev., 2020, 65, 189–225 CrossRef CAS.
- A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890–906 CrossRef CAS.
- Q. Bian, Y. Xiao, C. Zhou and M. Lang, J. Colloid Interface Sci., 2013, 392, 141–150 CrossRef CAS PubMed.
- Y. Li and X. Duan, Small, 2013, 9, 1521–1532 CrossRef PubMed.
- C. Sun, S. Ji, F. Li and H. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 12924–12929 CrossRef CAS PubMed.
- L. Yan, W. Wu, W. Zhao, R. Qi, D. Cui, Z. Xie, Y. Huang, T. Tong and X. Jing, Polym. Chem., 2012, 3, 2403 RSC.
- Y. Yuan, L. Zhao, C. Shen, Y. He, F. Yang, G. Zhang, M. Jia, R. Zeng, C. Li and R. Qiao, Mater. Sci. Eng. C, 2020, 106, 110159 CrossRef CAS PubMed.
- Q. Xu, C. He, C. Xiao and X. Chen, Macromol. Biosci., 2016, 16, 635–646 CrossRef CAS PubMed.
- M. Zhang, X. Wang, J. Zhu, M. Liu, C. Li, J. Feng and X. Zhang, Small, 2018, 14, 1800292 CrossRef PubMed.
- J. Xiao, X. Duan, Q. Yin, Z. Zhang, H. Yu and Y. Li, Biomaterials, 2013, 34, 9648–9656 CrossRef CAS PubMed.
- Y. Yuan, F. Wang, W. Tang, Z. Ding, L. Wang, L. Liang, Z. Zheng, H. Zhang and G. Liang, ACS Nano, 2016, 10, 7147–7153 CrossRef CAS PubMed.
- H. Zheng, L. Yin, X. Zhang, H. Zhang, R. Hu, Y. Yin, T. Qiu, X. Xiong and Q. Wang, J. Biomed. Nanotechnol., 2016, 12, 1641–1653 CrossRef CAS PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem. Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- V. G. Deepagan, S. Kwon, D. G. You, Van Q. Nguyen, W. Um, H. Ko, H. Lee, D. Jo, Y. M. Kang and J. H. Park, Biomaterials, 2016, 103, 56–66 CrossRef CAS PubMed.
- E. C. de Bruin and J. P. Medema, Canc. Treat Rev., 2008, 34, 737–749 CrossRef PubMed.
- H. Kiaris and A. Schally, Proc. Soc. Exp. Biol. Med., 1999, 221, 87–88 CrossRef CAS PubMed.
- M. Talelli, M. Barz, C. J. F. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10, 93–117 CrossRef CAS PubMed.
- E. Thomas, J. U. Menon, J. Owen, I. Skaripa-Koukelli, S. Wallington, M. Gray, C. Mannaris, V. Kersemans, D. Allen, P. Kinchesh, S. Smart, R. Carlisle and K. A. Vallis, Theranostics, 2019, 9, 5595–5609 CrossRef CAS PubMed.
- H. Sun, C. Chen, H. Cui and C. Cheng, Polym. Int., 2016, 65, 351–361 CrossRef CAS.
- Q. Sun, Z. Zhou, N. Qiu and Y. Shen, Adv. Mater., 2017, 29, 1606628 CrossRef PubMed.
- S. Deng, B. Kulle, M. Hosseini, G. Schlüter, G. Hasenfuss, L. Wojnowski and A. Schmidt, Eur. J. Heart Failure, 2007, 9, 986–994 CrossRef CAS PubMed.
- P. Dziegiel, E. Suder, P. Surowiak, Z. Jethon, J. Rabczynski, L. Januszewska, M. Sopel and M. Zabel, J. Pineal Res., 2002, 33, 95–100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01776k |
| This journal is © The Royal Society of Chemistry 2020 |