
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and investigation of sulfonated poly(p-phenylene)-based ionomers with precisely controlled ion exchange capacity for use as polymer electrolyte membranes†
Miru Yoshida-Hirahara ab,
Satoshi Takahashia,
Masahiro Yoshizawa-Fujitaa,
Yuko Takeokaa and
Masahiro Rikukawa*a
ab,
Satoshi Takahashia,
Masahiro Yoshizawa-Fujitaa,
Yuko Takeokaa and
Masahiro Rikukawa*a
aDepartment of Materials and Life Sciences, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: mlf19881231@163.com; Fax: +81 3 3238 4198; Tel: +81 3 3238 4250
bResearch and Development Bureau, Saitama University, Shimo-Okubo 255, Sakura-ku, Saitama-shi 338-8570, Japan
First published on 31st March 2020
Abstract
To achieve precise control of sulfonated polymer structures, a series of poly(p-phenylene)-based ionomers with well-controlled ion exchange capacities (IECs) were synthesised via a three-step technique: (1) preceding sulfonation of the monomer with a protecting group, (2) nickel(0) catalysed coupling polymerisation, and (3) cleavage of the protecting group of the polymers. 2,2-Dimethylpropyl-4-[4-(2,5-dichlorobenzoyl)phenoxy]benzenesulfonate (NS-DPBP) was synthesised as the preceding sulfonated monomer by treatment with chlorosulfuric acid and neopentyl alcohol. NS-DPBP was readily soluble in various organic solvents and stable during the nickel(0) catalysed coupling reaction. Sulfonated poly(4-phenoxybenzoyl-1,4-phenylene) (S-PPBP) homopolymer and seven types of random copolymers (S-PPBP-co-PPBP) with different IECs were obtained by varying the stoichiometry of NS-DPBP. The IECs and weight average molecular weights (Mws) of ionomers were in the range of 0.41–2.84 meq. g−1 and 143![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–465000 g mol−1, respectively. The water uptake, proton conductivities, and water diffusion properties of ionomers exhibited a strong IEC dependence. Upon increasing the IEC of S-PPBP-co-PPBPs from 0.86 to 2.40 meq. g−1, the conductivities increased from 6.9 × 10−6 S cm−1 to 1.8 × 10−1 S cm−1 at 90% RH. S-PPBP and S-PPBP-co-PPBP (4:1) with IEC values >2.40 meq. g−1 exhibited fast water diffusion (1.6 × 10−11 to 8.0 × 10−10 m2 s−1), and were comparable to commercial perfluorosulfuric acid polymers. When fully hydrated, the maximum power density and the limiting current density of membrane electrode assemblies (MEAs) prepared with S-PPBP-co-PPBP (4:1) were 712 mW cm−2 and 1840 mA cm−2, respectively.
000–465000 g mol−1, respectively. The water uptake, proton conductivities, and water diffusion properties of ionomers exhibited a strong IEC dependence. Upon increasing the IEC of S-PPBP-co-PPBPs from 0.86 to 2.40 meq. g−1, the conductivities increased from 6.9 × 10−6 S cm−1 to 1.8 × 10−1 S cm−1 at 90% RH. S-PPBP and S-PPBP-co-PPBP (4:1) with IEC values >2.40 meq. g−1 exhibited fast water diffusion (1.6 × 10−11 to 8.0 × 10−10 m2 s−1), and were comparable to commercial perfluorosulfuric acid polymers. When fully hydrated, the maximum power density and the limiting current density of membrane electrode assemblies (MEAs) prepared with S-PPBP-co-PPBP (4:1) were 712 mW cm−2 and 1840 mA cm−2, respectively.
Introduction
Polymer electrolyte fuel cells (PEFCs), which transform chemical energy directly into electricity, have been intensively developed as hydrogen energy technologies with high efficiency for use in automotive, and both stationary and portable electronic applications.1,2 Perfluorosulfonic acid polymers (PFSAs), particularly Nafion®, are typically used as polymer electrolyte membranes (PEMs) due to their high proton conductivity and excellent chemical stability. However, they have some drawbacks such as high manufacturing cost and limited operation temperatures below 90 °C, which results from a low glass transition temperature.3 Currently, sulfonated aromatic polymers have been studied as an alternative to PFSAs due to their low production cost, environmental compatibility, and high thermal stability.4–6Recently, sulfonated aromatic polymers containing pendant side chains between the polymer main chain and sulfonic acid group have received much attention. It is generally expected that the polymers with pendant sulfonic acid groups or side-chains are more stable against hydrolysis than those with sulfonic acid groups directly on the polymer backbones. Furthermore, the existence of flexible side chains promotes the nanophase separation of hydrophilic and hydrophobic domains, resulting in improved proton conductivity as well as dimensional stability of membranes.7–9 For example, Pang et al. synthesised sulfonated poly(arylene ether) copolymers containing pendant sulfonic acid groups with higher proton conductivity under fully hydrated conditions and lower swelling ratios than those of Nafion® membranes.10 Guiver et al. also reported that sulfonated poly(arylene ether sulfone) copolymers with pendant aliphatic sulfonic acid groups exhibit very low water uptake and dimensional changes at elevated temperature with unique nanophase separation architectures.11
In our previous study, we focused on poly(phenylene)-based polymer electrolytes and found that sulfonated poly(4-phenoxybenzoyl-1,4-phenylene) (S-PPBP), which contains sulfonic acid groups on the aromatic side chains, are attractive materials for PEFC applications.12,13 Generally, the rigid poly(p-phenylene) structure leads to high chemical and thermal stability as well as poor solubility and low film-forming ability due to the high bond dissociation energy between phenyl groups.7,14,15 Introduction of pendant sulfonic acid groups into the rigid poly(p-phenylene) backbone resulted in high thermal and hydrolysis stability, proton conductivity, and handling properties. S-PPBP could be synthesised via a nickel(0) catalysed coupling polymerisation technique, which was first demonstrated by Colon and Kelsey.16 The nickel(0) catalysed coupling reaction is a useful synthetic method for the formation of carbon–carbon aryl bonds. Excellent yields of biaryls can be prepared from aryl chlorides in a short time at mild temperature in the presence of a nickel salt, triphenylphosphine, and the reducing agent. In our work with the polymerisation technique, we optimised the synthetic conditions of the nickel(0) catalysed coupling polymerisation and successfully obtained high molecular weight PPBPs (Mw = 438000 g mol−1), which are the precursors of S-PPBP.17 Use of high molecular weight sulfonated aromatic polymers provides durable and reliable membranes for PEFC applications.
Our next target was the precise control of sulfonated polymer structures. There are two synthetic methods to obtain sulfonated polymers. One is the so-called post-sulfonation of preformed polymers. Various sulfonated polymer materials have been synthesised via post-sulfonation methods due to their simplicity and wide utility in materials selectivity, including use of commercially available polymers.18,19 While post-sulfonation is a simple and easy process, some serious side-reactions, such as cross-linking and chain scission reactions, might occur during the reaction. The other synthetic method is the so-called preceding sulfonation of precursor monomers. The direct polymerisation of preceding sulfonated monomers avoids the serious drawbacks of the former post-sulfonation process while also controlling the extent of sulfonation and position of the sulfonic acid group.2,20 Because of these advantages, the preceding sulfonation method has received much attention for the polymerisation of block copolymers and graft copolymers with well-defined morphologies. On the other hand, tedious synthesis of the monomer may be required because direct use of monomers with sulfonic acid groups for polymerisation is difficult; several research groups have reported the direct polymerisation of sodium or potassium sulfonate-type monomers via nucleophilic substitution reactions or nickel(0) catalysed coupling reactions.21–23 While it is relatively easy to prepare sulfonate-type monomers, their low solubility in organic solvents results in relatively low molecular weight polymers which may cause deterioration of film forming property and mechanical strength of polymer membranes. It is therefore necessary to design and synthesise novel sulfonated monomers that are more suitable for polymerisation processes. Recently, Goto et al. reported that the neopentyl (i.e., 2,2-dimethylpropyl) group has optimal properties as a protecting group of sulfonic acids for polymerisation.24 However, the synthetic procedures and characterisations of these polymers have not been reported. There are a few studies using sulfonic ester monomers for PEM applications with high molecular weights and ion exchange capacities (IECs).
Here, we synthesised a series of S-PPBP homopolymer and S-PPBP-PPBP random copolymers (S-PPBP-co-PPBPs) with well-controlled IECs and sulfonic acid group position by polymerisation of preceding sulfonated monomers with neopentylsulfonate esters. The thermal properties, mechanical properties, oxidative stability, water uptake, proton conductivity, water diffusion, gas permeability, and fuel cell performance of S-PPBP and S-PPBP-co-PPBPs were investigated. Comparison of S-PPBP-co-PPBPs with different IECs allowed us to understand the relationship between the IEC and copolymer properties.
Experimental
General information
Thionyl chloride and pyridine were purchased from FUJIFILM Wako Pure Chemical Co. and used as received unless otherwise noted. 2,2-Dimethyl-1-propanol (neopentyl alcohol), diethylamine hydrobromide ((C2H5)2NH·HBr), and chlorosulfuric acid were purchased from Sigma-Aldrich Co. LLC., Tokyo Chemical Industry Co., Ltd. and Kanto Chemical Co., Ltd., respectively, and used as received. All solvents were purchased from FUJIFILM Wako Pure Chemical Co. or Kanto Chemical Co., Ltd. and purified as appropriate where necessary. Zinc powder (Zn, 325 mesh; Merck Ltd.) was washed three times with 1.0 mol dm−3 HCl in diethyl ether and dried in vacuo at 100 °C for 6 h and at r.t. for 12 h. Bis(triphenylphosphine)nickel(II) dichloride (NiCl2(PPh3)2; Kanto Chemical) was dried in vacuo at 100 °C before use. Triphenylphosphine (PPh3; FUJIFILM Wako) was recrystallised from ethanol and dried in vacuo at 50 °C for 12 h. Sodium iodide (NaI; FUJIFILM Wako) was recrystallised from distilled water and dried in vacuo at 70 °C for 24 h. N-Methylpyrrolidone (NMP; Kanto Chemical) used for the polymerisation was purified by distillation under reduced pressure (1 mmHg, 54 °C), degassed by bubbling N2, then dehydrated over 200 °C-dried molecular sieves (MS4A; FUJIFILM Wako) before use. 2,5-Dichloro-4′-phenoxybenzophenone (DPBP) was synthesised according to the literature.171H NMR (500 MHz) and 13C NMR (125 MHz) measurements were performed on a ECA-500 spectrometer (JEOL Ltd.). Chloroform-d1 (CDCl3; FUJIFILM Wako), dichloromethane-d2 (CD2Cl2; Sigma-Aldrich), or dimethyl sulfoxide-d6 (DMSO-d6; Kanto Chemical) was used as the solvent, and tetramethylsilane (TMS; FUJIFILM Wako) was used as an internal standard for 1H and 13C NMR spectra. FT-IR spectra were recorded on a Nicolet 6700 spectrometer (Thermo Fisher Scientific Inc.) from 4000 to 650 cm−1. Solid samples were measured using KBr or attenuated total reflection (ATR) methods. High-resolution mass spectrometry was performed with a JMS-SX102A (JEOL) spectrometer in electron impact mode. Elemental analysis was performed using a PE2400-II instrument (PerkinElmer, Inc.) at 975 °C. Gel permeation chromatography (GPC) measurements were carried out using a LC-10AD chromatosystem (Shimadzu Co.) equipped with a Shodex LF-804 column (Showa Denko K.K.). GPC measurements were carried out at 50 °C using N,N-dimethylformamide (DMF; FUJIFILM Wako) containing 0.1% lithium bromide (LiBr; Wako) as an eluent at a flow rate of 1.0 mL min−1. Poly(ethylene oxide) standards (Sigma-Aldrich) were used as the GPC standards.
Synthesis of 4-[4-(2,5-dichlorobenzoyl)phenoxy]benzenesulfonylchloride (SC-DPBP)
DPBP (20.00 g, 58.28 mmol) was placed in a three-neck round-bottom flask filled with nitrogen. Chlorosulfuric acid (33.95 g, 291.4 mmol) was added to the flask, and the resulting mixture was stirred at r.t. for 3 h. The solution was then poured into ice water and extracted with CH2Cl2. The organic layer was washed with water and dried over MgSO4, then concentrated using a rotary evaporator. After filtration, the solid product was washed with chilled hexane and dried in vacuo at 40 °C for 12 h. SC-DPBP was obtained as a white solid in 85% yield (21.75 g, 49.24 mmol).1H NMR (500 MHz, CDCl3): δ 8.05 (d, J = 8.6 Hz, 2H), 7.89 (d, J = 8.6 Hz, 2H), 7.42–7.43 (m, 2H), 7.38 (t, J = 1.2 Hz, 1H), 7.21 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.6 Hz, 2H) ppm. EI-MS: m/z = 442. IR (KBr): 3093, 3074, 1664, 1573, 1502, 1487, 1462, 1414, 1377, 1309, 1287, 1255, 1184, 1167, 1153, 1093, 1086, 1053, 1012, 955, 904, 877, 833, 821, 807, 779, 771, 713, 686, 666 cm−1. Elemental analysis: calcd for (CHNS)n C, 51.66%; H, 2.51%; S, 7.26%. Found C, 51.71%; H, 2.42%; S, 7.20%.
Synthesis of 2,2-dimethylpropyl-4-[4-(2,5-dichlorobenzoyl)phenoxy]benzenesulfonate (NS-DPBP)
SC-DPBP (21.59 g, 48.89 mmol) and pyridine (15.0 mL) were placed in a three-neck round-bottom flask filled with nitrogen, and a mechanical stirring shaft was attached to the flask. A solution of neopentyl alcohol (5.696 g, 48.89 mmol) in pyridine (5.0 mL) was added to the flask, and the resulting mixture was stirred at r.t. for 3 h. The resulting solution was extracted with toluene. The organic layer was washed with HCl (aq), Na2CO3 (aq), and water, and then dried over MgSO4 and concentrated using a rotary evaporator. After filtration, the solid product was purified by recrystallisation from methanol at least three times and then dried in vacuo at 40 °C for 12 h. NS-DPBP was obtained as a white crystalline solid in 83% yield (17.94 g, 40.61 mmol).1H NMR (500 MHz, CDCl3): δ 7.91 (d, J = 8.6 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 7.45–7.44 (m, 2H), 7.38 (t, J = 1.2 Hz, 1H), 7.21 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.6 Hz, 2H), 3.69 (s, 2H), 0.90 (s, 9H) ppm. 13C NMR (125 MHz, CDCl3): δ 192.4, 160.9, 160.7, 140.2, 133.4, 133.0, 132.5, 131.8, 131.7, 131.6, 130.8, 129.8, 129.2, 119.8, 119.5, 80.2, 31.9, 26.1 ppm. EI-MS: m/z = 492. IR (KBr): 3095, 3071, 3040, 2976, 2958, 2868, 1666, 1585, 1491, 1463, 1421, 1387, 1360, 1285, 1244, 1182, 1165, 1093, 1051, 1013, 955, 935, 878, 839, 776, 764, 691, 674 cm−1. Elemental analysis: calcd for (CHNS)n C, 58.42%; H, 4.49%; S, 6.50%. Found C, 58.36%; H, 4.47%; S, 6.41%.
Synthesis of S-PPBP-co-PPBP
All reactants, catalyst reagents, and solvents were handled in a glove box under an argon atmosphere. NS-DPBP, DPBP, PPh3 (1.039 g, 3.962 mmol), NaI (0.203 g, 1.36 mmol), Zn (0.890 g, 13.6 mmol), and NiCl2(PPh3)2 (0.216 g, 0.330 mmol) were placed in a previously dried three-neck round-bottom flask. A mechanical stirring shaft and an addition funnel containing NMP (12.7 mL) was attached to the flask, and the flask was heated to 65 °C under a flow of argon. After NMP was added to the flask dropwise, the resulting mixture was stirred at 65 °C for 24 h with continuous and vigorous stirring. The resulting mixture was quenched with 550 mL of HCl/acetone (1:10, v/v). The crude product was then purified by reprecipitation from chloroform solution into HCl/methanol (1:10, v/v) at least twice in order to remove the residual catalyst, and then washed using Soxhlet extraction with methanol for three days and dried in vacuo at 50 °C for 12 h. NS-PPBP-co-PPBP was obtained as a white solid in 59–90% yield. Detailed polymerization conditions including the amounts of monomers are described in the ESI.†
The neopentyl-protecting group of NS-PPBP-co-PPBP was cleaved by acidolysis with (C2H5)2NH·HBr. NS-PPBP-co-PPBP was placed in a three-neck round-bottom flask filled with nitrogen, and a mechanical stirring shaft was attached to the flask. NMP was added to the flask, and the mixture was stirred at 80 °C. After NS-PPBP-co-PPBP completely dissolved in NMP, a solution of (C2H5)2NH·HBr in NMP was added to the flask, and the resulting mixture was stirred at 120 °C for 24 h. After cooling to room temperature, the resulting mixture was poured into an excess of methanol containing 10% HCl, and the solution was stirred for 24 h. The precipitate was immersed in 1 mol dm−3 HCl (aq) for 48 h to exchange the diethylammonium ions with protons. After conversion to the acid form, the crude product was purified twice by reprecipitation from NMP into methanol and then dried in vacuo at 80 °C for 12 h. S-PPBP-co-PPBP was obtained as a red-brown precipitate in 72–95% yield. Detailed synthetic conditions and characterisations are described in the ESI.†
Preparation of polymer membranes
Polymer membranes of S-PPBP and S-PPBP-co-PPBP were prepared by solution casting onto a flat glass plate with 5 wt% DMSO solution. Each polymer solution was cast on a previously washed glass substrate (10 × 10 cm), heated at 80 °C for 12 h at ambient pressure, and then dried in vacuo at 80 °C for 3 h. The membranes were immersed in deionised water to allow them to be peeled from the substrates, and then the detached membranes were immersed in 1 mol dm−3 HCl (aq) at 80 °C for 2 h, followed by treatment with deionised water at 80 °C for 3 h, and finally dried in vacuo at 80 °C for 12 h.Measurements
The experimental IEC values were determined using elemental analysis and back titration as described below. The dried membranes were immersed in 1.0 mol dm−3 HCl (aq) at 80 °C for 2 h followed by treatment with deionized water at 80 °C for 3 h. The IEC was then estimated by titration with 10 mmol dm−3 NaOH (aq). Thermogravimetric analyses (TGA) were performed with a TG-DTA7200 instrument (Seiko Instruments Inc.) at a heating rate of 10 °C min−1 from 25 to 500 °C under a flow of nitrogen. The oxidative stability was investigated using Fenton's reagents containing 3% hydrogen peroxide (H2O2, Wako) and 1.00 mmol dm−3 iron(II) chloride (aq) (FeCl2, Fe2+ = 4.0 ppm). The membrane samples were dried in vacuo and weighed before performing the Fenton's test. After immersing the membranes in the Fenton's reagent at 80 °C for 2 h, the residual weight of the membranes was measured; the oxidative stability was correlated with this residual weight of the membrane.25Mechanical tensile tests were performed with a Tensilon RTG-1210 instrument (A&D Company, Ltd.) equipped with a chamber in which the temperature and the humidity were controlled with a flow of humidified air. The tensile strength and ultimate elongation of membrane samples, which had areas of 10 mm × 20 mm, were measured under ambient conditions (r.t., non-humidified) and conditions of elevated temperature and humidity (80 °C, 90% RH).
Weight-based water uptake (WUw) was measured using a MSB-AD-V-FC isothermal adsorption measurement system (MicrotracBEL Corp.) equipped with a temperature and humidity controlled chamber. The measurement was performed at 80 °C with varying humidity from 25 to 85% RH. The value of water uptake was calculated using the following equation:
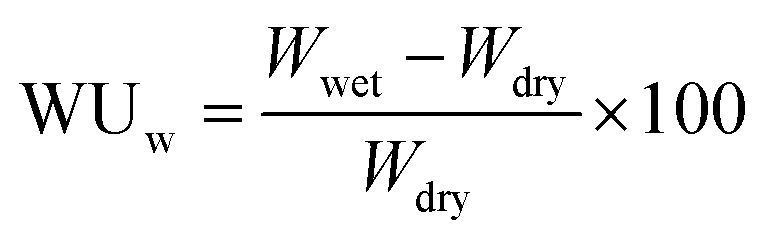
where 18.01 is the molecular weight of water.
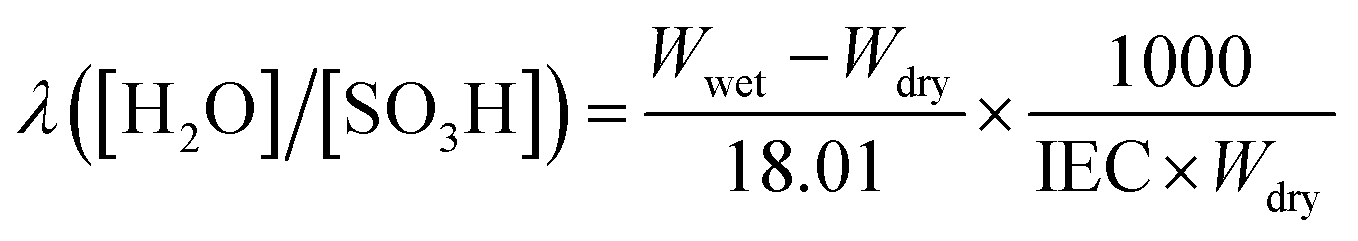
The in-plane proton conductivity σ (S cm−1) of the polymer membranes were measured using a SI1260 electrochemical impedance analyser (Solartron Group Ltd.). The samples were clamped between Pt electrodes and placed on a four-probe conductivity cell. The measurement was performed at 80 °C with varying humidity from 30 to 90% RH, and σ was calculated using the following equation:
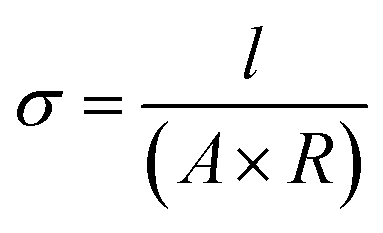
The pulsed-field-gradient nuclear magnetic resonance (PFG-NMR) measurements were carried out on an ECA-500 spectrometer (JEOL) with an 11.7 T superconducting magnet. To determine the diffusion coefficients of water, the sample membranes were equilibrated in a humidity–temperature chamber (ESPEC Corp.) at a specified RH and 30 °C for 24 h, and then inserted in a 5 mm ϕ NMR glass tube and tightly sealed with a Teflon rod, such that the water volume would not vary after sample conditioning. The bipolar pulse longitudinal eddy current delay (BPPLED) pulse sequence with half-sine shaped gradient pulses was employed and measurements were obtained by observing the NMR signal intensity I as a function of spin dephasing (gradient strength g). The self-diffusion coefficient was determined by fitting the data to the following equation:
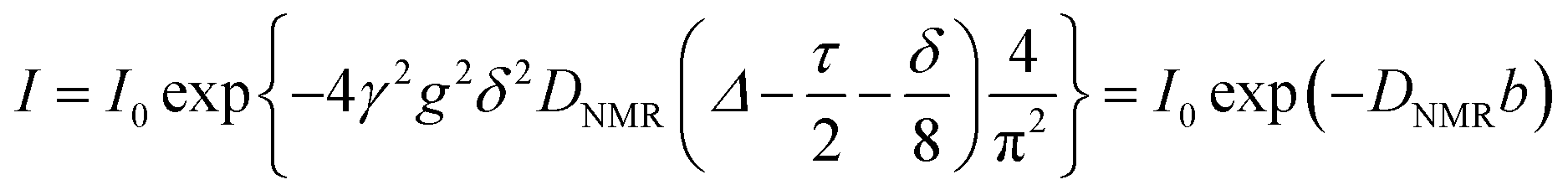
Hydrogen (H2) and oxygen (O2) permeability through membrane samples was measured with a GTR-30XFST gas permeation measurement apparatus (GTR Tec Corp.) equipped with a G2700T gas chromatograph (Yanaco) containing a Porapak-Q column and a thermal conductivity detector. Argon and helium were used as carrier gases for the measurement of H2 and O2, respectively. A membrane with an area of 35 mm × 35 mm, was set in a cell with a gas inlet and outlet on both sides of the membrane. The cell temperature was controlled by placing it in an oven. Dry or humidified test gas (H2 or O2) was supplied at a flow rate of 30 mL min−1. Before measurements, the membranes were equilibrated with the gases at the given temperature and humidity. The gas permeation rate, r (cm3(STP) cm−2 s−1), and the gas permeation coefficient, P (barrer; 1 barrer = 1 × 10−10 cm3(STP) cm cm−2 s−1 cmHg−1), were calculated according to the following equations:
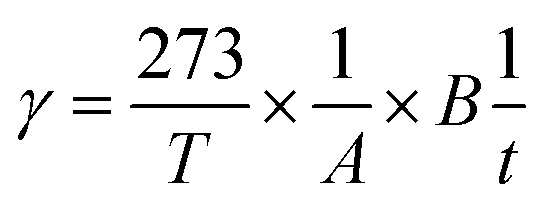
where T (K) is the absolute temperature of the cell, A (cm2) is the permeation area, B (cm3) is the volume of test gas permeated through the membrane, t (s) is the sampling time, l (cm) is the thickness of the membrane, and Pw (cmHg) is the water vapor pressure.26
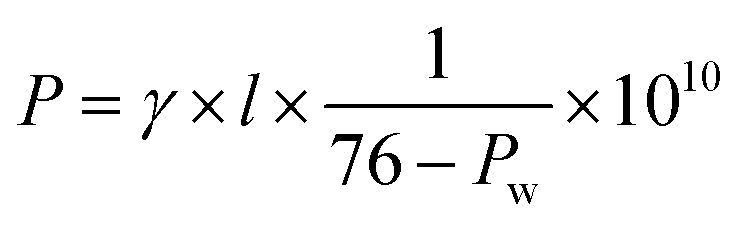
Membrane electrode assemblies (MEAs) were constructed according to the following protocol. A catalyst ink was prepared by mixing Pt on Vulcan XC-72 (amount of Pt = 47 wt%), purified water, ethanol, and 5 wt% Nafion® dispersion. The mixed catalyst ink was coated on SIGRACET® GDL 35BC gas diffusion layers (SGL Carbon Japan Co, Ltd.). The electrodes were then pressed onto membrane samples (35 mm × 35 mm) at 6 MPa and 125 °C for 10 min. The active area was 5 cm2, and the Pt catalyst loading was 1.0 mg cm−2. The MEA was assembled in a single cell (ElectroChem, Inc.) between bipolar plates made of graphite with serpentine flow channels. A single fuel cell test fed by H2/air was conducted using a 890B fuel cell test system (Scribner Associates, Inc.) at a cell temperature of 80 °C under humidified conditions. Dry or humidified H2 and air were continuously supplied at a flow rate of 500 and 1000 mL min−1, respectively, and the back pressure was maintained at 0.1 MPaG.
Results and discussion
Synthesis of sulfonated monomer
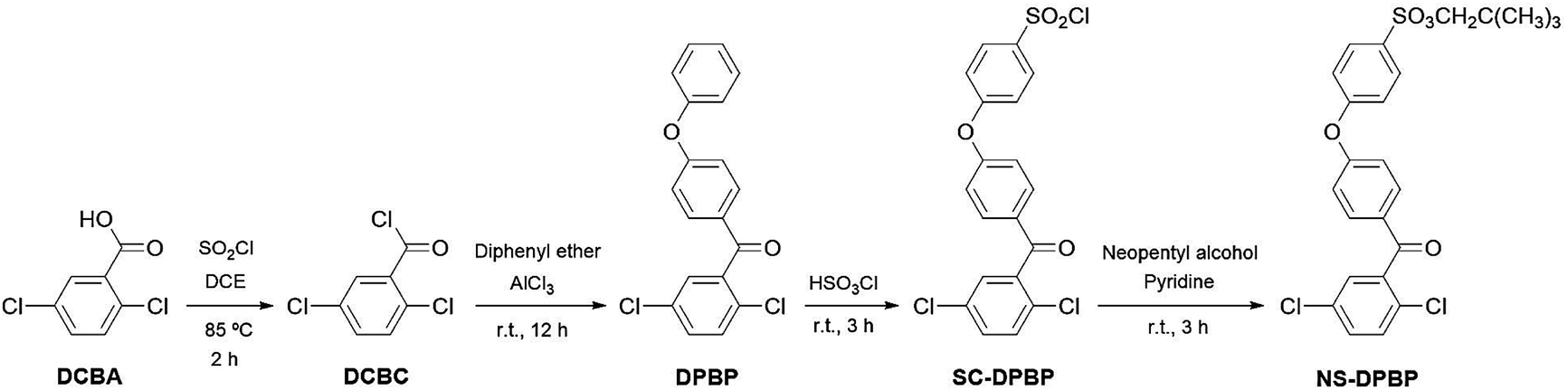
We synthesised S-PPBP and S-PPBP-co-PPBPs via the following three steps: (1) preceding sulfonation of monomers using a protecting group, (2) polymerisation or copolymerisation of NS-DPBP with a nickel(0) catalysed coupling reaction, and (3) cleavage of protecting groups from the polymer. Neopentyl groups were used as the protecting group of sulfonic acids due to both the high polymerisation stability and ease of deprotection. Neopentylsulfonate esters are more stable than their isopropyl counterparts.24,27The synthetic route to prepare the monomer is outlined in Scheme 1. In our synthesis of the sulfonated monomer, we started with the preparation of DPBP in 74% yield by Friedel–Crafts acylation of 2,5-dichlorobenzoyl chloride and diphenylether according to a literature procedure.17 Chlorosulfonation of DPBP with chlorosulfuric acid gave sulfonyl chloride DPBP (SC-DPBP) in 85% yield. SC-DPBP was then treated with neopentyl alcohol in pyridine to provide neopentylsulfonate ester DPBP (NS-DPBP) in 83% yield after purification from at least two recrystallisations from methanol.
 | ||
| Scheme 1 Synthesis of NS-DPBP. | ||
The identity of NS-DPBP was confirmed by EI-MS, FT-IR spectroscopy, 1H and 13C NMR spectroscopy, and elemental analysis. The FT-IR spectrum (KBr pellet) showed absorption bands at 2958, 1421, and 1360 cm−1 corresponding to the C–H stretching vibration, CH3 asymmetric vibration, and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, respectively (Fig. S1†). As shown in Fig. 1a, the 1H NMR signals were assigned, with two singlet peaks at 0.90 ppm (i) and 3.69 ppm (h) attributed to the aliphatic protons of the neopentylsulfonate ester group, and six peaks downfield (7.13–7.91 ppm) were assigned as aromatic protons. 1H NMR and 13C NMR spectra confirmed that one neopentylsulfonate ester group was successfully introduced to the para-position of the terminal phenoxy group. NS-DPBP was readily soluble in various organic solvents (e.g., CHCl3, NMP, DMF, and DMSO). This high solubility in organic solvents derives from the presence of the neopentyl group, which is suitable for our polymerisation strategy.
O stretching vibration, respectively (Fig. S1†). As shown in Fig. 1a, the 1H NMR signals were assigned, with two singlet peaks at 0.90 ppm (i) and 3.69 ppm (h) attributed to the aliphatic protons of the neopentylsulfonate ester group, and six peaks downfield (7.13–7.91 ppm) were assigned as aromatic protons. 1H NMR and 13C NMR spectra confirmed that one neopentylsulfonate ester group was successfully introduced to the para-position of the terminal phenoxy group. NS-DPBP was readily soluble in various organic solvents (e.g., CHCl3, NMP, DMF, and DMSO). This high solubility in organic solvents derives from the presence of the neopentyl group, which is suitable for our polymerisation strategy.
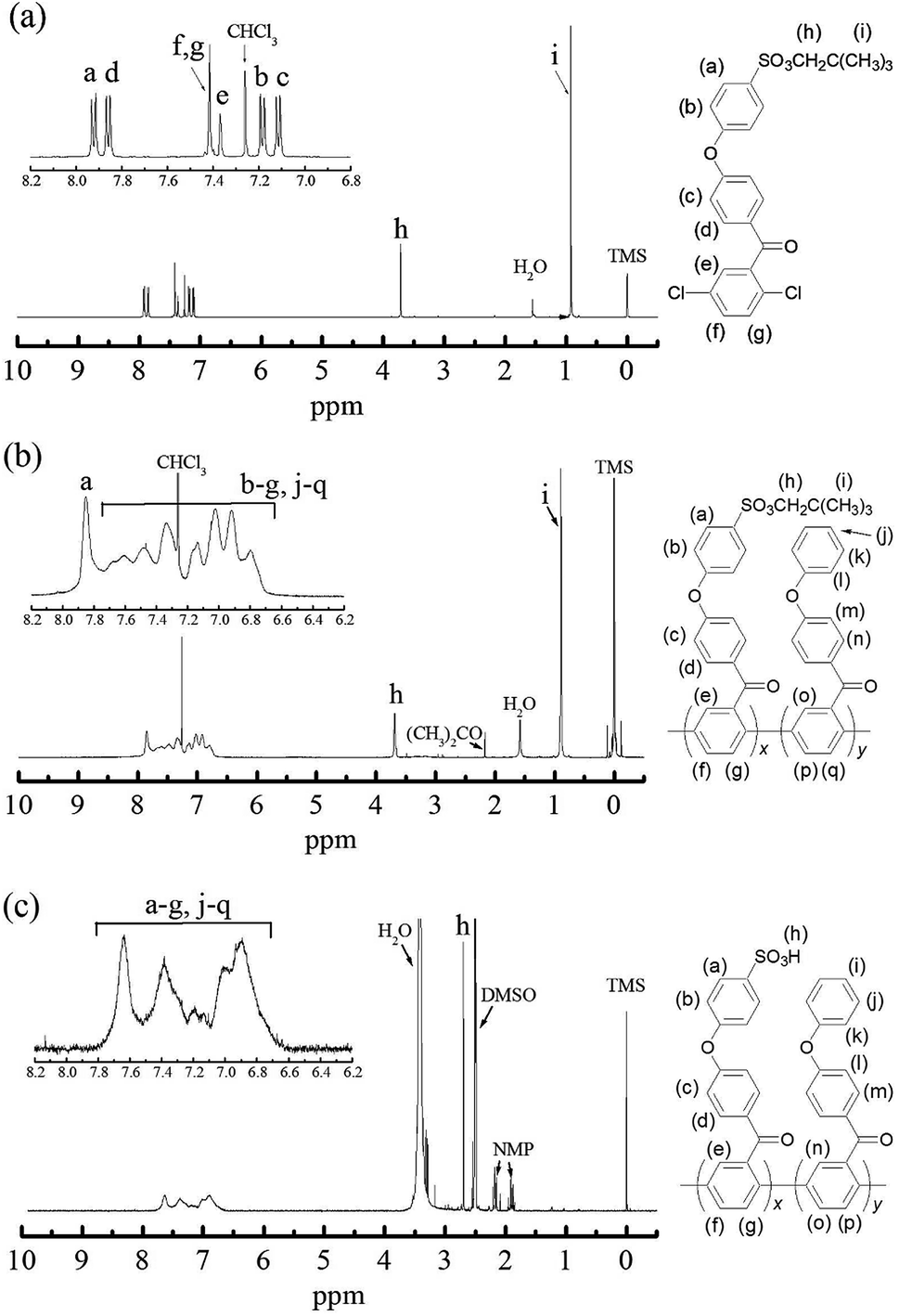 | ||
| Fig. 1 1H NMR spectra of (a) NS-DPBP in CDCl3, (b) NS-PPBP-co-PPBP (1:1) in CDCl3, and (c) S-PPBP-co-PPBP (1:1) in DMSO-d6. | ||
Nickel(0) catalysed coupling polymerisation
As shown in Scheme 2, S-PPBP homopolymer and seven types of random copolymers, S-PPBP-co-PPBP x:y with different molar ratios, were synthesised from NS-DPBP and DPBP via a nickel(0) catalysed coupling polymerisation. Herein, the (x:y) means the theoretical molar ratio of hydrophilic S-PPBP and hydrophobic PPBP units calculated from the feed ratio of NS-DPBP and DPBP.
 | ||
| Scheme 2 Synthesis of S-PPBP-co-PPBP. | ||
According to a previous report, a catalyst solution was prepared from Ni(PPh3)2Cl2, PPh3, NaI, and activated Zn in purified NMP under an argon atmosphere and stirred for 5 min at 40 °C.17 After formation of a deep-red color of the catalyst in solution, which indicated the reduction of nickel(II) to nickel(0) by activated zinc, a monomer solution of NS-DPBP and DPBP in NMP was added dropwise to the catalyst mixture with vigorous stirring at 65 °C for 24 h. A longer reaction time can be employed to prepare less dispersed copolymers.17 NS-PPBP and NS-PPBP-co-PPBPs were synthesised by varying the stoichiometry of NS-DPBP and DPBP with yields of 59–90%.
The identities of NS-PPBP and NS-PPBP-co-PPBPs were confirmed by FT-IR and 1H NMR spectroscopies, and with elemental analysis. The FT-IR spectra of NS-PPBP and NS-PPBP-co-PPBPs confirmed polymerisation or copolymerisation by disappearance of the absorption bands at 1093 cm−1 corresponding to the C–Cl stretching. The chemical structures of polymers were characterised via 1H NMR spectroscopy (Fig. 1b and S1†). The chemical shifts at 3.69 ppm (h) and 0.90 ppm (i) corresponding to the neopentylsulfonate ester group indicate that no cleavage of the neopentyl group occurs during the nickel(0) catalysed coupling reaction.
Preparation of the sulfonated polymer
Conversion of the neopentylsulfonate ester into a sulfonic acid group was realised by acidolysis with an NMP solution of diethylamine hydrobromide at 120 °C for 24 h. Due to adsorption of a large quantity of diethylammonium ions, the deprotected copolymers were subsequently immersed in a 1 mol dm−3 HCl (aq) for 48 h to obtain the acid form. All polymers were purified by reprecipitation in NMP/methanol.The identities of S-PPBP and S-PPBP-co-PPBPs were confirmed from FT-IR and 1H NMR spectroscopies, GPC, and elemental analysis. In the FT-IR spectra of S-PPBP and S-PPBP-co-PPBPs, absorption bands assigned to the SO stretching vibration at 1120, 1026, and 1003 cm−1 were observed (Fig. S2†). The intensity of absorption bands corresponding to the SO stretching vibration increased with increasing theoretical molar ratio of S-PPBP segments. Comparison of the spectra before and after the deprotection reaction indicates that the absorption bands at 2960 and 2868 cm−1, which were assigned to the C–H stretching of the neopentylsulfonate ester groups of NS-PPBP and NS-PPBP-co-PPBPs, disappear for S-PPBP and S-PPBP-co-PPBPs. These results indicate that deprotection proceeded successfully, as expected. The Mws of S-PPBP and S-PPBP-co-PPBPs determined by GPC with DMF as the eluent were in the range of 143000–465000 g mol−1. Slightly wide polydispersity of polymers is presumably due to the reduced reactivity of reduction reaction of Ni species. Since the polyphenylene-based polymer has low solubility, a viscosity of the reaction solution increased rapidly at the late stage of Ni(0) coupling polymerisation, which might cause the inhibition of Ni reduction, especially in high molecular weight products. The molecular weight did not depend on the molar ratio of S-PPBP and PPBP. All the samples of S-PPBP and S-PPBP-co-PPBPs were soluble in only polar aprotic solvents (e.g., NMP, DMF, and DMSO).
Polymer membranes of all polymers were prepared by solution casting onto a glass plate with DMSO. The thicknesses of prepared membranes were ∼50 μm. The experimental IEC values of polymer membranes were determined by back titration and elemental analysis, as shown in Fig. 2. All of the IEC data obtained are in good agreement with the theoretical IEC values calculated from the feed molar ratio of NS-DPBP and DPBP. These results indicate that we can successfully control the IEC by varying the feed ratio of monomers. PPBP homopolymer (Mw = 146000), Nafion®112, and Nafion®115 were used as controls. The properties of all polymer products are summarised in Table 1.
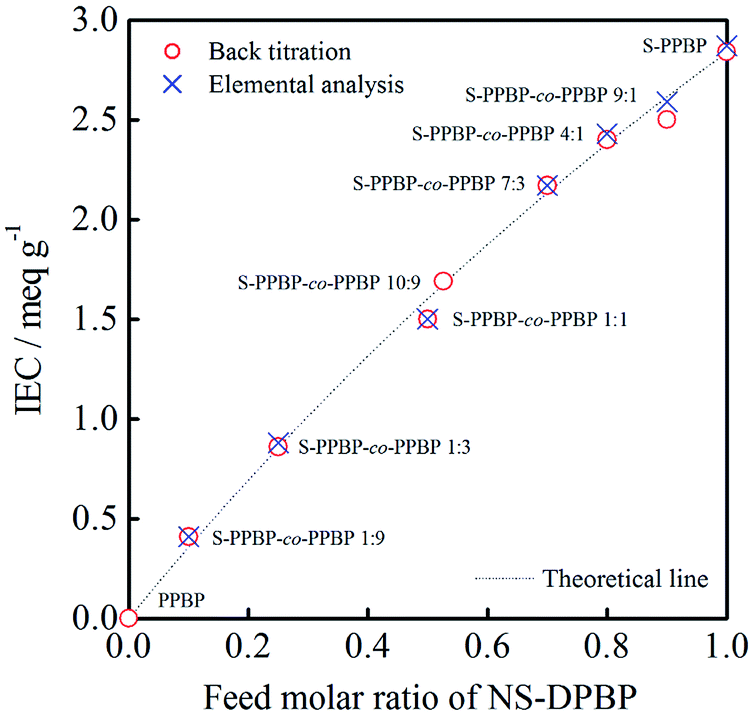 | ||
| Fig. 2 Relationship between IEC of S-PPBP-co-PPBP and feed molar ratio of NS-DPBP. | ||
| Sample | Mwa | Mw/Mna | IEC/meq. g−1 | |
|---|---|---|---|---|
| Exp.b | Theory | |||
| a Determined by GPC (eluent: DMF).b Determined by back titration. | ||||
| S-PPBP | 359000 |
3.04 | 2.84 | 2.84 |
| S-PPBP-co-PPBP 9:1 |
243000 |
3.20 | 2.50 | 2.61 |
| S-PPBP-co-PPBP 4:1 |
346000 |
4.94 | 2.40 | 2.38 |
| S-PPBP-co-PPBP 7:3 |
465000 |
4.84 | 2.17 | 2.13 |
| S-PPBP-co-PPBP 10:9 |
143000 |
3.00 | 1.69 | 1.68 |
| S-PPBP-co-PPBP 1:1 |
216000 |
3.32 | 1.50 | 1.60 |
| S-PPBP-co-PPBP 1:3 |
250000 |
2.69 | 0.86 | 0.86 |
| S-PPBP-co-PPBP 1:9 |
326000 |
3.36 | 0.41 | 0.36 |
| PPBP | 146000 |
2.53 | — | — |
Chemical, dimensional, and thermal stability
The oxidative stability of S-PPBP and S-PPBP-co-PPBPs was investigated with the Fenton degradation test, which is a standard method to gauge relative oxidative stability and simulate accelerated fuel cell operating conditions. S-PPBP and S-PPBP-co-PPBP membranes were immersed in Fenton's reagent (3% H2O2 containing 4 ppm Fe2+) at 80 °C for 2 h, and the residual weight of the membranes were measured, as summarised in Table 2. Generally, a high IEC leads to low oxidative stability because oxidative attack by a radical species, which is classified as an electrophilic addition, mainly occurs in hydrophilic domains and results in membrane degradation.28,29 As expected, the oxidative stability of membranes increased with decreasing IEC values. After immersion in the Fenton's reagent for 2 h, the membranes of S-PPBP and S-PPBP-co-PPBP 9:1 were completely dissolved in the Fenton's reagent. The residual weights of S-PPBP-co-PPBP 7:3, 1:1, 1:3, and 1:9 were approximately 24, 58, 78, and 95 wt%, respectively, comparable to reported random copolymers with pendant sulfoalkyl groups.11
| Sample (IEC) | Residual weighta/wt% | Swelling ratiob/% |
|---|---|---|
| a Residual weight of membranes after Fenton's tests. Each membrane was immersed in 3% H2O2 with 4 ppm FeCl2 aq. at 80 °C for 2 h.b Swelling ratios of membranes in the through-plane direction. Each membrane was immersed in water at 80 °C for 2 h. | ||
| S-PPBP (2.84 meq. g−1) | 0 | 90 |
| S-PPBP-co-PPBP 9:1 (2.50 meq. g−1) |
0 | — |
| S-PPBP-co-PPBP 7:3 (2.17 meq. g−1) |
24 | 38 |
| S-PPBP-co-PPBP 1:1 (1.50 meq. g−1) |
58 | 26 |
| S-PPBP-co-PPBP 1:3 (0.86 meq. g−1) |
78 | <1 |
| S-PPBP-co-PPBP 1:9 (0.41 meq. g−1) |
95 | — |
The dimensional stability of PEMs is an important technical problem that leads to serious physical degradation of MEAs under fuel cell operation. Huge swelling causes both loss of desired mechanical properties and interfacial detachment of the membranes and electrodes, which results in drastically reduced fuel cell performance. The dimensional stabilities of S-PPBP and S-PPBP-co-PPBPs were determined by measuring the swelling ratios in the through-plane direction. The polymer membranes were immersed in water at 80 °C for 2 h, and the changes in membrane thickness were measured (Table 2). Swelling ratios increased sharply with increasing IEC for all samples. Upon increasing the IEC of the membranes from 0.86 to 2.84 meq. g−1, the swelling ratio increased from 0 to 90%. While S-PPBP increased by 90% in the through-plane direction due to the high IEC, S-PPBP-co-PPBP (7:3) with relatively high IEC (2.13 meq. g−1) exhibited a much lower swelling ratio of 38%. Furthermore, S-PPBP-co-PPBP (1:3) demonstrated a negligible change that was comparable to the dry state. In a previous study, high molecular weight S-PPBPs exhibited less swelling than those of low molecular weight due to strong intermolecular interactions, which results in a tightly packed structure.17 These results indicate that controlling both the IEC and molecular weight are important for achieving desirable dimensional stability. Our synthetic strategy is effective for obtaining polyphenylene-based PEMs with precisely controlled IECs and high molecular weight.
The thermal properties of PPBP, NS-PPBP, S-PPBP, and S-PPBP-co-PPBP (10:9) were measured with TGA at a heating rate of 10 °C min−1 under a nitrogen atmosphere from 50 to 500 °C (Fig. 3). NS-PPBP exhibits two weight loss transitions. The first weight loss occurs from 170 to 180 °C due to degradation of the neopentyl group, and the second weight loss from 200 to 500 °C is associated with the degradation of the sulfonic acid group. The TG-MS spectrum of NS-PPBP measured at 177 °C shows the peaks of the neopentyl and isobutane groups, which reveals that the first weight loss of NS-PPBP is attributed to the degradation of the neopentyl group. Unlike PPBP and NS-PPBP, S-PPBP and S-PPBP-co-PPBP (10:9) exhibited three weight loss transitions. The first weight loss occurs below 150 °C and can be attributed to the release of free water molecules adsorbed by the sulfonic acid groups. The second continuous weight loss observed above 220 °C is due to degradation of the sulfonic acid groups. The third continuous weight loss above 400 °C is assigned to degradation of the polymer backbone. These results indicate that S-PPBP and S-PPBP-co-PPBP have good thermal stabilities for fuel cell operation.
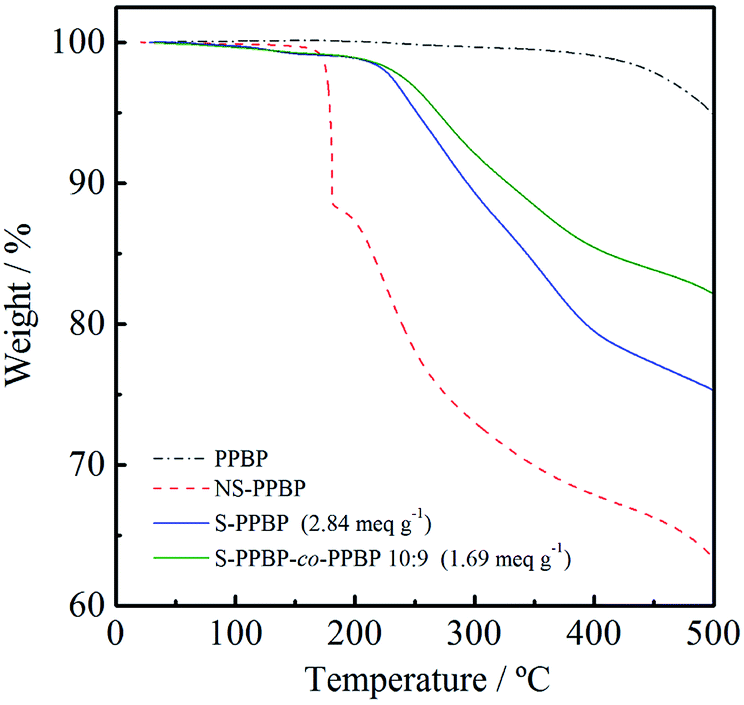 | ||
| Fig. 3 TGA curves of NS-PPBP, NS-PPBP-co-PPBP, and PPBP. | ||
Mechanical properties
It is important for polymer electrolyte membranes to achieve high mechanical properties under both dry and humid conditions that are suitable for MEA manufacturing conditions and PEFC operation. The tensile strength and ultimate elongation of PPBP, S-PPBP, and S-PPBP-co-PPBP membranes were determined with mechanical tensile tests under ambient atmospheric conditions (r.t., non-humidified) and elevated temperature and humidity (80 °C, 90% RH), as listed in Table 3. The polymer films of S-PPBP and S-PPBP-co-PPBPs under ambient conditions had tensile strengths ranging from 33.1 to 64.9 MPa, with elongation at break from 2.2 to 14.0%. S-PPBP tends to exhibit lower tensile strength and higher ultimate elongation than that of S-PPBP-co-PPBPs with low IECs. This is presumably due to the introduction of strong polar sulfonic acid groups in the polymer chain, which lowers the molecular interaction between the polymer backbones. Elongations of S-PPBP-co-PPBPs of <10% were attributed to the rigid poly(p-phenylene)-based backbone structure. A distinct relationship between mechanical properties and IECs under ambient (non-humidified) conditions was not observed. It is therefore possible that the molecular weight and backbone structure of the polymer chains have a greater influence on the mechanical properties in our copolymers.17 In the hydrated state (80 °C, 90% RH), S-PPBP and S-PPBP-co-PPBPs had tensile strengths between 6.2 and 50.5 MPa with elongations in the range of 3.7 to 48.9%. S-PPBP and S-PPBP-co-PPBP (4:1) showed much lower tensile strengths in the hydrated state. High levels of adsorbed water in the membranes results in excessive dimensional swelling and leads to poor mechanical properties.30 S-PPBP in the hydrated state exhibited much higher flexibility and lower tensile strength than those of S-PPBP-co-PPBPs because water molecules adsorbed on the membrane surface act to reduce the ion–ion interactions of the polymer chains.31 S-PPBP-co-PPBP membranes 10:9 and 1:3 maintained high tensile strength under humid conditions, which is attributed to both the low IEC and low water swelling. All S-PPBP and S-PPBP-co-PPBP membranes provide moderate mechanical integrities for their potential use as PEMs.
| Sample | Tensile strengtha/MPa | Elongationb/% | ||
|---|---|---|---|---|
| Ambient condition | 80 °C, 90% RH | Ambient condition | 80 °C, 90% RH | |
| a Tensile strength determined from mechanical tensile tests under ambient conditions (r.t., non-humidified) and elevated temperature and humidity (80 °C, 90% RH).b Ultimate elongation determined by mechanical tensile tests under ambient conditions (r.t., non-humidified) and elevated temperature and humidity (80 °C, 90% RH). | ||||
| S-PPBP | 47 | 27.0 | 13 | 49 |
| S-PPBP-co-PPBP 4:1 |
33 | 6.20 | 6.8 | 9.1 |
| S-PPBP-co-PPBP 10:9 |
65 | 50.5 | 2.2 | 3.7 |
| S-PPBP-co-PPBP 1:3 |
52 | 48.6 | 3.3 | 4.5 |
| PPBP | 23 | — | 2.2 | — |
Water uptake
Water uptake of sulfonated polymers has a profound effect on proton conductivity because water acts as a carrier for proton transportation through the polymer membrane. The water sorption properties of membranes expressed as water uptake and the number of water molecules per sulfonic acid group λ([H2O]/[SO3H]) were measured in the range of 30–85% RH at 80 °C (Fig. 4). The water uptake was 12–48 wt% for S-PPBP and 4–36 wt% for S-PPBP-co-PPBPs. Water uptake increases with increasing hydrophilicity of the polymer membranes. As expected, the water uptake increased with IEC values for all S-PPBP and S-PPBP-co-PPBP membranes.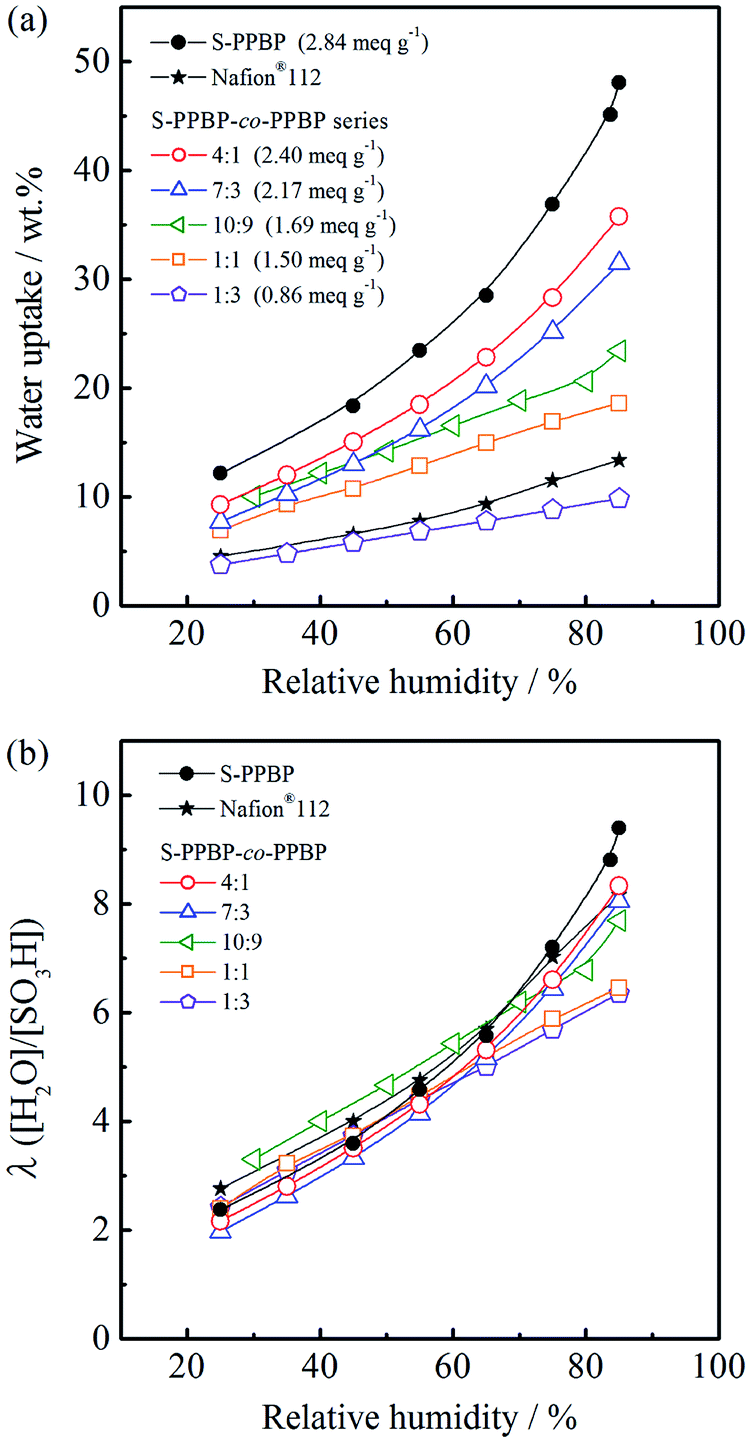 | ||
| Fig. 4 (a) Water uptake and (b) the number of adsorbed water molecules per sulfonic acid group (λ) for S-PPBP and S-PPBP-co-PPBP as a function of relative humidity at 80 °C. | ||
While S-PPBP exhibited the highest water uptake among those of S-PPBP-co-PPBPs and Nafion®112, there are few differences in their hydration numbers (λ) under low humidity (<60% RH). The λ values of S-PPBP-co-PPBP membranes (10:9, 1:1, and 1:3) with IEC values <1.69 meq. g−1 increased linearly with humidity. On the other hand, the λ values of S-PPBP, S-PPBP-co-PPBP (4:1 and 7:3), and Nafion®112 gently increase as a function of humidity below 60% RH, then sharply increase above 60% RH. This suggests that the observed changes of S-PPBP and S-PPBP-co-PPBPs with high IEC are influenced by the morphology and different states of water. It is known that there are at least three states of water associated with water residing in the hydrophilic phases of polymers, namely non-freezing bound water, freezable loosely bound water, and free water.32 The higher λ value of S-PPBP indicates the presence of more free water, which presumably reduces the extent of ionic interactions in the hydrophilic channels and increases the mobility of the protonic species.33 S-PPBP-co-PPBP (4:1 and 7:3) exhibit water sorption characteristics that are very similar to S-PPBP and Nafion®112, suggesting that the distribution of water types is similar among these polymers.
Proton conductivity
The proton conductivity of S-PPBP and S-PPBP-co-PPBP membranes in the in-plane direction was measured between 30 and 90% RH at 80 °C (Fig. 5). Conductivities increased linearly with increasing relative humidity for all samples. S-PPBP and S-PPBP-co-PPBP (4:1) showed high conductivities in the range of 2.1 × 10−3 to 2.2 × 10−1 S cm−1, which were comparable to or higher than those of Nafion®112. Generally, sulfonated aromatic polymers show lower conductivity than those of Nafion® membranes, especially under conditions of low humidity due to loss of interconnected water channels. The proton conductivity of polymer membranes is strongly related to the IEC values, water uptake, and microstructure. Compared to conventional sulfonated aromatic polymers based on sulfonated poly(arylene ether) backbone structures, S-PPBP exhibited high conductivities due to the existence of pendant side chains between the polymer main chain and the sulfonic acid groups, which enhances the mobility of the sulfonic acid groups and forms continuous ionic networks. This feature may be useful for increasing the water retention capacity under conditions of low humidity.
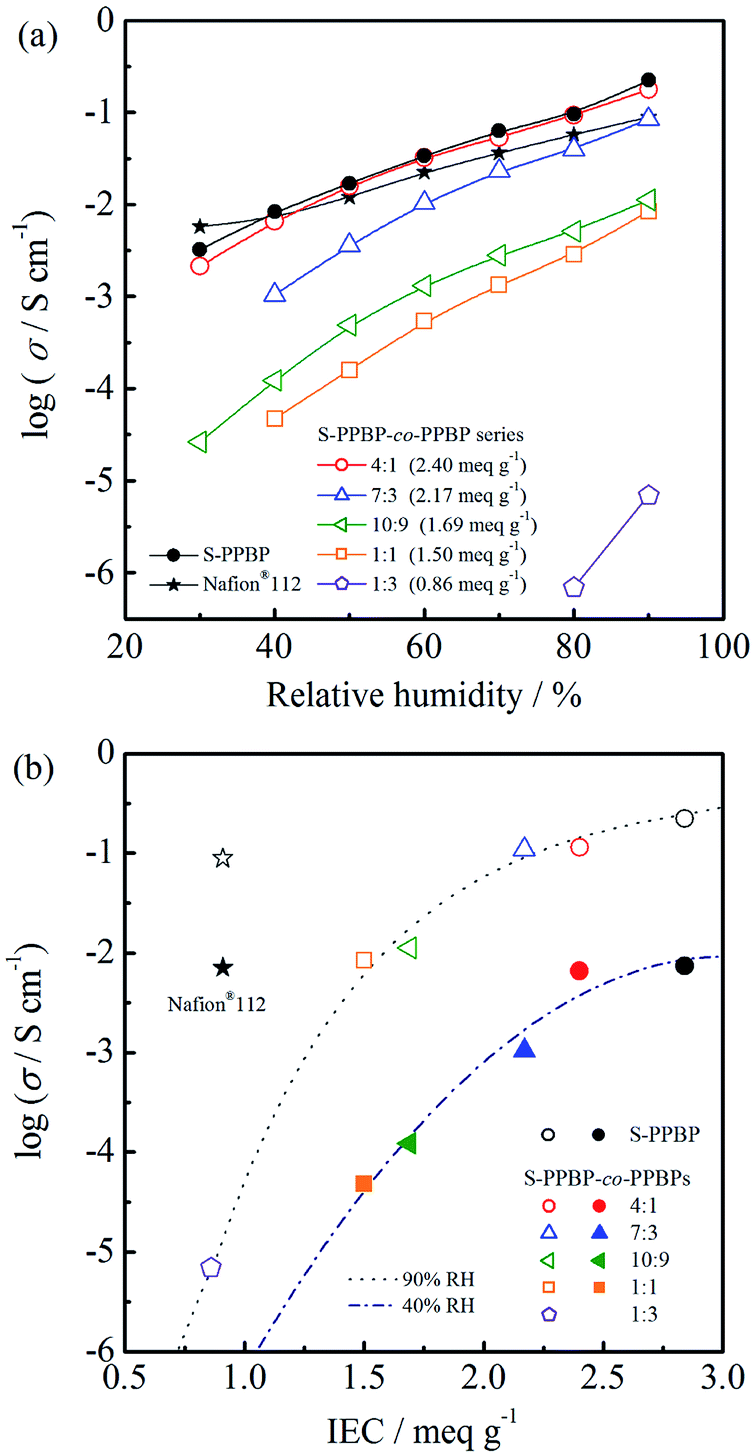 | ||
| Fig. 5 (a) Proton conductivity of S-PPBP and S-PPBP-co-PPBP as a function of relative humidity at 80 °C. (b) Relationship between proton conductivities and IEC of S-PPBP and S-PPBP-co-PPBP at 80 °C and either 90% RH or 40% RH. Symbols with white and coloured backgrounds correspond to 90% RH and 40% RH, respectively. | ||
The conductivities of S-PPBP-co-PPBP membranes increase with increasing IEC values. Upon increasing the IEC of S-PPBP-co-PPBPs from 0.86 to 2.40 meq. g−1, the conductivities increased from 6.9 × 10−6 S cm−1 to 1.8 × 10−1 S cm−1 at 90% RH and 80 °C. The S-PPBP-co-PPBP membranes with IEC values <1.69 meq. g−1 exhibited low conductivities. The conductivity changed by three orders of magnitude when the IEC increased from 0.86 to 1.50 meq. g−1. These results suggest that S-PPBP-co-PPBPs with low IEC form less connected and randomly distributed ionic channels, which results in low proton conductivity. When the ionic clusters become continuous and form interconnecting channels, the proton transport is facilitated, and proton conductivity is enhanced. It is expected that the conductivities of membranes with low IEC can be further improved by forming well-defined morphologies. We propose that block copolymerisation of S-PPBP and other hydrophobic segments is one promising strategy to achieve this aim. Further investigations including the synthesis of block copolymers of S-PPBP are ongoing in our laboratory.
Water diffusion properties
Pulsed-field-gradient (PFG) NMR techniques are widely used to study molecular diffusion of fluids and to probe the structures of restricted geometries on porous media.34–38 In order to investigate the water diffusion properties, the self-diffusion coefficients of water (DNMR) for S-PPBP and S-PPBP-co-PPBP membranes were measured using PFG-NMR under 30–95% RH at 30 °C.39 The NMR stimulated echo signal decay (I/I0) was measured as a function of b (=16γ2g2δ2(Δ − τ/2 − δ/8)/π2), and the values of DNMR were obtained from the slopes of the fitted lines (Fig. S4†). The DNMR for all samples as a function of the relative humidity are shown in Fig. 6b.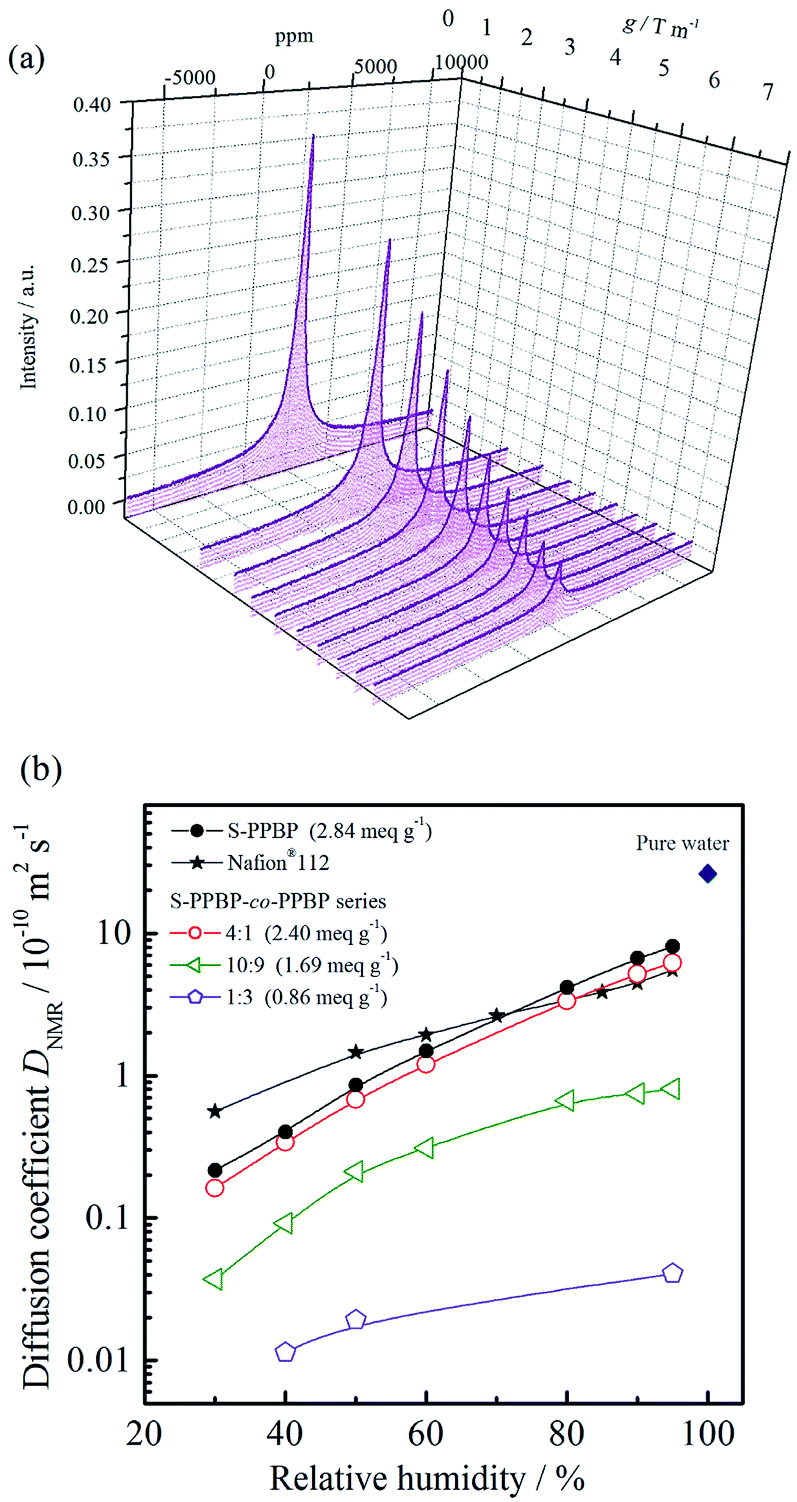 | ||
| Fig. 6 (a) PFG-NMR spectra of S-PPBP at 30 °C and 30% RH. (b) Diffusion coefficients of water (DNMR) for S-PPBP and S-PPBP-co-PPBP as a function of relative humidity at 30 °C. | ||
The DNMR for all the samples increased with increasing relative humidity, strongly suggesting that an increase in water content gives rise to faster diffusion in PEMs. Except for Nafion®112, the DNMR increased with IEC values as well. This trend is in good agreement with the proton conductivity in Fig. 5. S-PPBP and S-PPBP-co-PPBP (4:1) showed high DNMR values in the range of 1.6 × 10−11 to 8.0 × 10−10 m2 s−1. Under the same conditions, Nafion®112 exhibited DNMR values from 5.6 × 10−11 to 5.5 × 10−10 m2 s−1. While there are a few reports discussing the relationship between polymer structures and water diffusion properties of sulfonated aromatic polymers, some research groups have reported that the mobility of water molecules in the PFSA membranes depends on the channel structure of hydrophilic domains.40–42 As previous reports suggested, the high DNMR values of Nafion®112 reflect strongly phase separated structures, which water primarily transports within hydrophilic networks. Interestingly, S-PPBP and S-PPBP-co-PPBP (4:1) demonstrate higher water diffusion properties comparable to Nafion®112 above 70% RH. These results indicate that a continuous channel morphology without any morphological barriers exists for S-PPBP and S-PPBP-co-PPBP with high IEC values under conditions of high humidity. The high water diffusion properties of S-PPBP-co-PPBP (4:1), which is comprised of 20% hydrophobic component, suggests that the hydrophobic segments may not disturb the well-connected ionic channels when the amount of hydrophobic monomer introduced in the random copolymerised structure is low.
The DNMR of S-PPBP and S-PPBP-co-PPBPs showed a more pronounced dependence on humidity than Nafion®112. Below 70% RH, S-PPBP and all copolymers exhibited lower DNMR than Nafion®112. A decrease in the DNMR of the polymers reflects less connectivity and more isolated dead-end channels and pockets within the solvated hydrophilic domain. S-PPBP-co-PPBP (10:9 and 1:3) exhibited much slower water diffusion than the other membranes. Copolymers with IEC values <1.69 meq. g−1 also showed a drastic decrease in proton conductivity. These results indicate decreased connectivity within the solvated hydrophilic domain, and thus a decrease in both the water and proton mobility occurred in proportion to the random copolymerisation rate of hydrophobic monomers. As we mentioned above, controlling well-defined morphology is necessary for further improvement of desirable properties. Kreuer et al. also reported that the retardation of water diffusion in PEMs was caused by increased internal molecular friction within the solvated hydrophilic domain due to the confinement effect in addition to the effect of reduced connectivity of hydrophilic domain, especially under conditions of low water content.40
Gas permeation properties
Hydrogen and oxygen permeation properties of S-PPBP and S-PPBP-co-PPBP membranes were measured between 0 and 95% RH at 80 °C. The humidity dependence of the gas permeability coefficient, P (barrer; 1 barrer = 1 × 10−10 cm3(STP) cm cm−2 s−1 cmHg−1), for these samples are shown in Fig. 7. The hydrogen permeability coefficient (PH2) for S-PPBP and all S-PPBP-co-PPBP membranes was approximately one order of magnitude greater than the oxygen permeability coefficient (PO2) mainly due to a decrease in the kinetic diameters of the gases. All S-PPBP and S-PPBP-co-PPBPs possessed lower PH2 and PO2 than those of Nafion®115. For these membranes, PH2 decreased with increasing IEC values under dry conditions. PH2 of S-PPBP under dry conditions was 3.6 barrer, which was approximately three and five times lower than that of S-PPBP-co-PPBP (1:1; 13 barrer) and PPBP (20 barrer), respectively. The gas permeability of the sulfonated polymers under dry conditions mainly arise from the free volume in the hydrophobic domains.43,44 When dry, the S-PPBP domains are less permeable to gases than the PPBP domains due to their high cohesive energy density.45 We could only obtain PO2 values for S-PPBP-co-PPBP (1:1) under dry conditions; the amount of O2 that permeated across the membranes of the other samples was too low to be detected with gas chromatography.
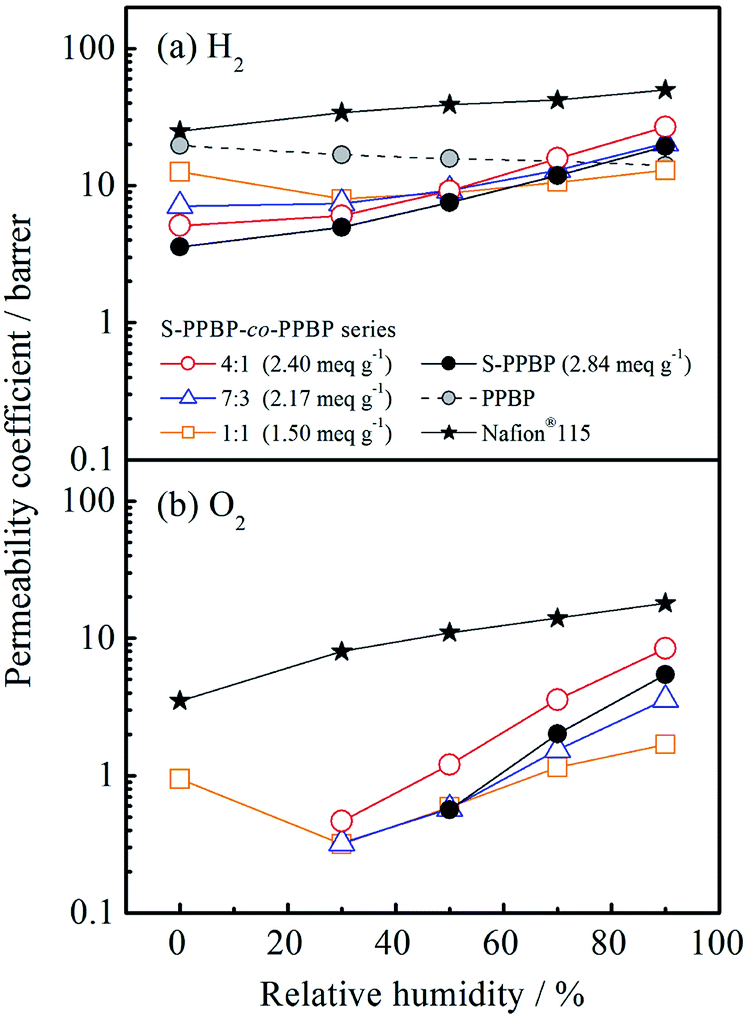 | ||
| Fig. 7 (a) Hydrogen permeability and (b) oxygen permeability of S-PPBP, S-PPBP-co-PPBP, and PPBP as a function of relative humidity at 80 °C. | ||
Upon hydration, the humidity dependence of PH2 of S-PPBP and S-PPBP-co-PPBPs first presented concave forms with increasing humidity, and then increased linearly at >30% RH. The first behaviour might be caused by water molecules filling the free volume as a result of stabilised intermolecular interactions.46 When sulfonated membranes contact water vapor, water molecules are adsorbed by sulfonic acid groups and gas molecules can only diffuse through the pathways formed from unoccupied spaces that are sufficiently large in size. The second linear behavior under conditions of >30% RH might be caused by the plasticising phenomenon induced by water sorption.45 When sulfonated membranes adsorb more water molecules, they begin to develop well-connected water clusters in hydrophilic domains, and these sorbed water molecules act to enhance the mobility of polymer chains, which results in increased gas permeation. Our results are in good agreement with these water sorption properties. Futhermore, S-PPBP and S-PPBP-co-PPBP (4:1) with high IEC showed high PH2 and PO2.
Fuel cell operation
Single-cell tests of S-PPBP and S-PPBP-co-PPBP membranes were carried out with a cell temperature of 80 °C and two different relative humidity conditions (100% RH and 42% RH) for both electrodes. The fuel cell polarisation curves of the membranes are shown in Fig. 8. The open circuit voltages (OCVs) of S-PPBP, S-PPBP-co-PPBP (4:1), and S-PPBP-co-PPBP (7:3) were 1.00, 1.00, and 0.97 V at 100% RH and 0.97, 0.94, and 0.92 V at 42% RH, respectively. Generally, a high OCV value indicates that the hydrogen gas permeability from the anode to the cathode through the membrane is very low. The trend of the OCV is in good agreement with the hydrogen permeation results of S-PPBP and S-PPBP-co-PPBPs, as discussed above.
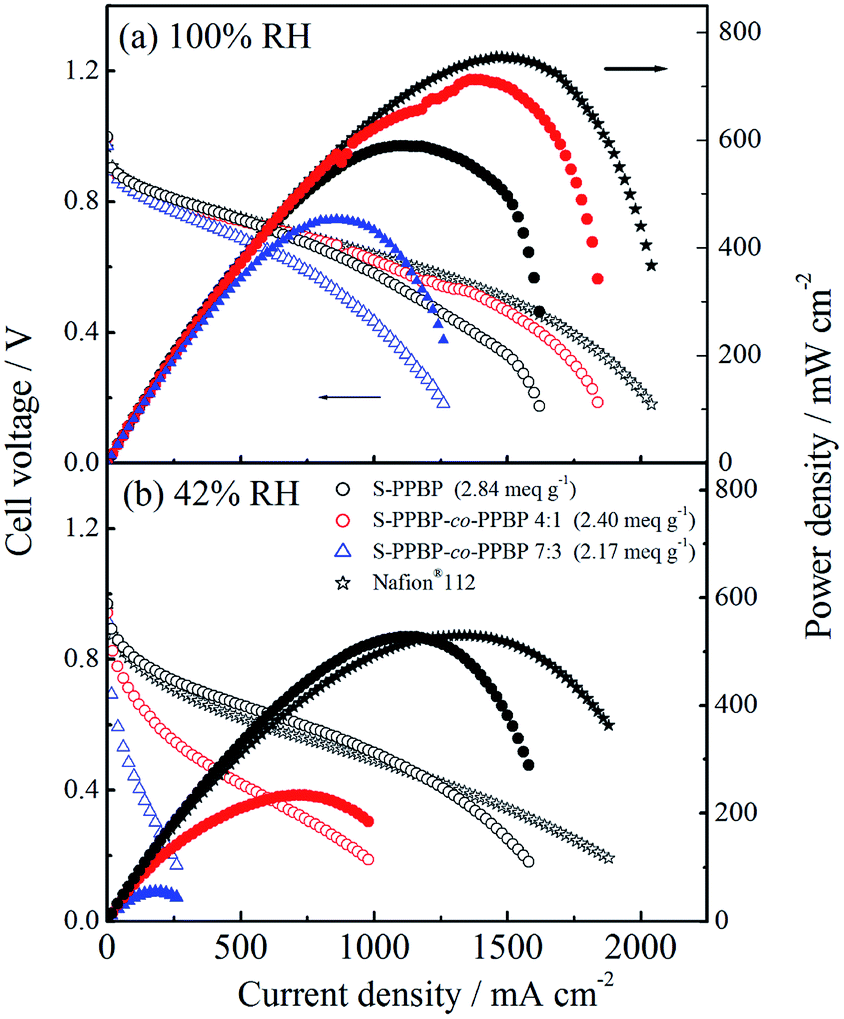 | ||
| Fig. 8 Cell voltage and power density as a function of current density for a 5 cm2 H2 (500 mL min−1, 0.1 MPaG)/air (1000 mL min−1, 0.1 MPaG) PEFC based on S-PPBP and S-PPBP-co-PPBP membranes at 80 °C at (a) 100% RH and (b) 42% RH. | ||
Under fully hydrated conditions, the maximum power density and the limiting current density of S-PPBP-co-PPBP (4:1) were 712 mW cm−2 and 1840 mA cm−2, respectively, which were higher than those of S-PPBP (589 mW cm−2 and 1620 mA cm−2) and S-PPBP-co-PPBP (7:3; 453 mW cm−2 and 1260 mA cm−2). S-PPBP and S-PPBP-co-PPBP with high IEC exhibit very promising fuel cell performance comparable to Nafion® membranes under fully hydrated conditions. At 42% RH, S-PPBP exhibited only a 10.5% reduction in power density (527 mW cm−2) while S-PPBP-co-PPBP (4:1 and 7:3) displayed reductions of 67.3% (233 mW cm−2) and 88.0% (54 mW cm−2) in power density, respectively. The high fuel cell performance of S-PPBP and Nafion® membranes under less humid conditions mainly depends on the well-connected proton channels which facilitate proton transfer. S-PPBP-co-PPBP (7:3) exhibited a drastic decrease in fuel cell performance with decreasing humidity due to the random structure. Controlling polymer morphology is important to improve both proton conductivity and fuel cell performance under conditions of low humidity.
Conclusions
We synthesised eight types of poly(p-phenylene)-based ionomers of S-PPBP and S-PPBP-co-PPBP via a nickel(0) coupling polymerisation of preceding sulfonated monomers protected with neopentylsulfonate esters. A series of random copolymers with well-controlled IECs of 0.41 to 2.50 meq. g−1 were successfully prepared by varying the stoichiometry of the sulfonated monomer. Well-controlled IECs and substantially high molecular weight of the obtained ionomers indicate that our preceding sulfonation process is suitable for PEM preparation.The relationship between the IEC and the fundamental membrane properties were investigated. The water uptake, dimensional stabilities, proton conductivities, and water diffusion properties of S-PPBP and S-PPBP-co-PPBPs clearly increased with increasing IEC values. S-PPBP and S-PPBP-co-PPBP (4:1) with IEC values >2.40 meq. g−1 showed high conductivities and water diffusion properties, which were comparable to Nafion®112. These results indicate that a continuous channel morphology exists in S-PPBP and S-PPBP-co-PPBP with high IEC values. The mechanical and gas permeation properties exhibit different trends when the membranes are under dry or humid conditions. While the mechanical properties under moderate conditions did not show a distinct dependence on IEC, the mechanical strength under higher humidity tends to decrease with increasing IEC values due to the excessive dimensional swelling caused by adsorbed water. S-PPBP, which possesses the highest IEC, exhibited much higher flexibility than the other copolymers. The gas permeation properties exhibited two effects due to the free volume of the polymer and the adsorption of water molecules. In the dry state, PH2 decreased with increasing IEC because gas permeability mainly contributes to the free volume in the hydrophobic domain. On the other hand, PH2 and PO2 increased greatly for samples with higher IEC under the humidification conditions of >30% RH. These distinctive properties of hydrated membranes are presumably due to the plasticising phenomenon induced by water adsorption. These results indicate that controlling IECs, in other words controlling water sorption properties, is significant in order to obtain PEMs with desirable properties.
Our synthetic procedure aimed to synthesise random copolymers with well-controlled IECs. A significant decrease in proton conductivity and water diffusion were exhibited below 1.69 meq. g−1. These results indicate that the decreasing connectivity within the solvated hydrophilic domain occurred in proportion to decreasing IEC due to the randomly distributed structure. It is expected that the PEM properties can be further improved by forming well-defined morphologies. Block copolymerisation of S-PPBP and other hydrophobic segments following the synthetic strategy presented here is one promising approach that we are currently exploring in our laboratory to develop membranes for use in PEFCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the New Energy and Industrial Technology Development Organisation (NEDO) and the Ministry of Education, Culture, Sports, Science and Technology, Japan, through a Grant-in-Aid for Scientific Research (17H03119).Notes and references
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587 CrossRef CAS PubMed.
- C. H. Park, C. H. Lee, M. D. Guiver and Y. M. Lee, Prog. Polym. Sci., 2011, 36, 1443 CrossRef CAS.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Int. J. Hydrogen Energy, 2010, 35, 9349 CrossRef CAS.
- D. W. Shin, S. Y. Lee, C. H. Lee, K.-S. Lee, C. H. Park, J. E. McGrath, M. Zhang, R. B. Moore, M. D. Lingwood, L. A. Madsen, Y. T. Kim, I. Hwang and Y. M. Lee, Macromolecules, 2013, 46, 7797 CrossRef CAS.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175 CrossRef CAS.
- E. A. Weiber, S. Takamuku and P. Jannasch, Macromolecules, 2013, 46, 3476 CrossRef CAS.
- S. G. Jo, T.-H. Kim, S. J. Yoon, S.-G. Oh, M. S. Cha, H. Y. Shin, J. M. Ahn, J. Y. Lee and Y. T. Hong, J. Membr. Sci., 2016, 510, 326 CrossRef CAS.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780 CrossRef CAS PubMed.
- J. Pang, X. Jin, Y. Wang, S. Feng, K. Shen and G. Wang, J. Membr. Sci., 2015, 492, 67 CrossRef CAS.
- J. Pang, H. Zhang, X. Li and Z. Jiang, Macromolecules, 2007, 40, 9435 CrossRef CAS.
- C. Wang, N. Li, D. W. Shin, S. Y. Lee, N. R. Kang, Y. M. Lee and M. D. Guiver, Macromolecules, 2011, 44, 7296 CrossRef CAS.
- T. Kobayashi, M. Rikukawa, K. Sanui and N. Ogata, Solid State Ionics, 1998, 106, 219 CrossRef CAS.
- M. Rikukawa and K. Sanui, Prog. Polym. Sci., 2000, 25, 1463 CrossRef CAS.
- H. Wang, A. S. Badami, A. Roy and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 284 CrossRef CAS.
- D. D. Andjelkovic and V. V. Sheares, Macromolecules, 2007, 40, 7148 CrossRef CAS.
- I. Colon and D. R. Kelsey, J. Org. Chem., 1986, 51, 2627 CrossRef CAS.
- I. Tonozuka, M. Yoshida, K. Kaneko, Y. Takeoka and M. Rikukawa, Polymer, 2011, 52, 6020 CrossRef CAS.
- Y. Sakaguchi, A. Kaji, K. Kitamura, S. Takase, K. Omote, Y. Asako and K. Kimura, Polymer, 2012, 53, 4388 CrossRef CAS.
- C. Wang, D. W. Shin, S. Y. Lee, N. R. Kang, G. P. Robertson, Y. M. Lee and M. D. Guiver, J. Mater. Chem., 2012, 22, 25093 RSC.
- A. Ghosh and S. Banerjee, e-Polym., 2014, 14, 227 CAS.
- N. Li, D. W. Shin, D. S. Hwang, Y. M. Lee and M. D. Guiver, Macromolecules, 2010, 43, 9810 CrossRef CAS.
- H. Xie, D. Tao, X. Xiang, Y. Ou, X. Bai and L. Wang, J. Membr. Sci., 2015, 473, 226 CrossRef CAS.
- Q. He, T. Xu, H. Qian, J. Zheng, C. Shi, Y. Li and S. Zhang, J. Power Sources, 2015, 278, 590 CrossRef CAS.
- K. Goto, I. Rozhanskii, Y. Yamakawa, T. Otsuki and Y. Naito, Polym. J., 2009, 41, 95 CrossRef CAS.
- J. Park and D. Kim, J. Membr. Sci., 2014, 469, 238 CrossRef CAS.
- S. Seesukphronrarak, K. Ohira, K. Kidena, N. Takimoto, C. S. Kuroda and A. Ohira, Polymer, 2010, 51, 623 CrossRef CAS.
- J. C. Roberts, H. Gao, A. Gopalsamy, A. Kongsjahju and R. J. Patch, Tetrahedron Lett., 1997, 38, 355 CrossRef CAS.
- H. Xie, D. Wang, D. Tao and L. Wang, J. Power Sources, 2014, 262, 328 CrossRef CAS.
- K. Kang and D. Kim, J. Power Sources, 2015, 281, 146 CrossRef CAS.
- R. M. Chen and G. Li, New J. Chem., 2016, 40, 3755 RSC.
- L. Lin, Z. Chen, Z. Zhang, S. Feng, B. Liu, H. Zhang, J. Pang and Z. Jiang, Polymer, 2016, 96, 188 CrossRef CAS.
- X. Wu, G. He, S. Gu, Z. Hu and X. Yan, Chem. Eng. J., 2010, 156, 578 CrossRef CAS.
- A. Siu, J. Schmeisser and S. Holdcroft, J. Phys. Chem. B, 2006, 110, 6072 CrossRef CAS PubMed.
- X. Gong, A. Bandis, A. Tao, G. Meresi, Y. Wang, P. T. Inglefield, A. A. Jones and W.-Y. Wen, Polymer, 2001, 42, 6485 CrossRef CAS.
- J. Li, K. G. Wilmsmeyer and L. A. Madsen, Macromolecules, 2009, 42, 255 CrossRef CAS.
- J. K. Park, J. Li, G. M. Divoux, L. A. Madsen and R. B. Moore, Macromolecules, 2011, 44, 5701 CrossRef CAS.
- J. Hou, J. Li, D. Mountz, M. Hull and L. A. Madsen, J. Membr. Sci., 2013, 448, 292 CrossRef CAS.
- D. W. Shin, S. Y. Lee, C. H. Lee, K.-S. Lee, C. H. Park, J. E. McGrath, M. Zhang, R. B. Moore, M. D. Lingwood, L. A. Madsen, Y. T. Kim, I. Hwang and Y. M. Lee, Macromolecules, 2013, 46, 7797 CrossRef CAS.
- M. Yoshida, Y. Zhao, M. Yoshizawa-Fujita, A. Ohira, Y. Takeoka, S. Koizumi and M. Rikukawa, ECS Trans., 2012, 50, 1045 CrossRef.
- K.-D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637 CrossRef CAS PubMed.
- M. Saito, N. Arimura, K. Hayamizu and T. Okada, J. Phys. Chem. B, 2004, 108, 16064 CrossRef CAS.
- Q. Berrod, S. Lyonnard, A. Guillermo, J. Ollivier, B. Frick, A. Manseri, B. Améduri and G. Gébel, Macromolecules, 2015, 48, 6166 CrossRef CAS.
- F. Piroux, E. Espuche and R. Mercier, J. Membr. Sci., 2004, 232, 115 CrossRef CAS.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2684 CrossRef CAS.
- S. Simon, E. Espuche, F. Gouanvé, E. Chauveau, C. Marestin and R. Mercier, J. Phys. Chem. B, 2012, 116, 12750 CrossRef CAS PubMed.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2009, 42, 1873 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01816c |
| This journal is © The Royal Society of Chemistry 2020 |