
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFused tetracyclic tris[1,2,4]triazolo[1,3,5]triazine as a novel rigid electron acceptor for efficient thermally activated delayed fluorescence emitters†
Suraj Kumar Pathakab,
Yepeng Xiangc,
Manli Huangc,
Taian Huanga,
Xiaosong Cao a,
He Liu*a,
Guohua Xiec and
Chuluo Yang*ac
a,
He Liu*a,
Guohua Xiec and
Chuluo Yang*ac
aGuangdong Research Center for Interfacial Engineering of Functional Materials, Shenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, P. R. China. E-mail: liuhe001@szu.edu.cn; clyang@szu.edu.cn
bKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, P. R. China
cHubei Key Lab on Organic and Polymeric Optoelectronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, P. R. China. E-mail: clyang@whu.edu.cn
First published on 20th April 2020
Abstract
Tris[1,2,4]triazolo[1,3,5]triazine, a new acceptor based on a fused triazole and triazine moiety, is utilized to construct D3–A star-shaped tristriazolotriazine derivatives, named 3,7,11-tris(4-(10H-phenoxazin-10 yl)phenyl)tris([1,2,4]triazolo)[1,3,5]triazine (TTT-PXZ) and 3,7,11-tris(4-(9,9-dimethylacridin-10(9H)yl)phenyl)tris([1,2,4])triazolo[1,3,5]triazine (TTT-DMAC). Both TTT-PXZ and TTT-DMAC emitters feature TADF activities and AIEE properties. Consequently, solution processed OLEDs based on TTT-PXZ green emitters exhibited good performances, with an external quantum efficiency (EQE) of up to 6.2%.
Introduction
Metal free thermally activated delayed fluorescence (TADF) has attracted substantial attention from the scientific community due to its competence of utilizing both singlet and triplet excitons for light emission.1–7 The seminal work of Adachi et al.2 elucidating the up conversion of a triplet exciton into a singlet state via a reverse intersystem crossing (RISC) process by regulating the singlet–triplet splitting energy (ΔEST), provided new prospects in the field of TADF research, with a bright future in the area of organic light emitting diodes (OLEDs). To realize efficient TADF materials from the quantum theory perspective, the spatial separation of electron densities of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) in one single molecule is needed to acquire a near-zero ΔEST; and consequently to realize an efficient RISC process.1–3 In addition, sufficiently high photoluminescent quantum yield (PLQY) is also important to ensure maximumly transition of excitons to photons. The design principles of twisted donor–acceptor (D–A) molecular structure generally depend on bulky substituents or a spiro-junction to realize a small ΔEST and reinforce the rigidity of the molecular structure leading to the suppression of non-radiative decay.4 Therefore, in designing TADF molecular structure, carefully choosing donor and acceptor moieties as well as spacer between them are utmost important.Recently, a renewed interest for star-shaped emitters (D3–A) to be used in OLEDs arises.5–7 In general, star-shaped molecules are of many advantages over their linear analogues as they encompass both the character of small molecules and polymers with well-defined molecular structure, superior chemical purity as well as good solution processability.8–15 There are several examples of fluorene containing star-shaped molecules showing improved device characteristics in comparison to the linear molecules.8,11–13 Besides, star-shaped D3–A twisted molecules can reduce intermolecular interaction in the solid state, resulting enhanced solid-state emission with better film forming ability.14,15 The solution processability and film forming ability encourage these materials to be used in solution processed OLEDs, which is economically feasible for low cost and large-area displays.16–26 Therefore, it is highly fascinating to develop molecular design to increase the luminescence efficiency and stability of solution-processed OLEDs, specifically on star-shaped TADF emitters. Generally, planar star-shaped molecule has an inherent problem of aggregation-caused quenching (ACQ) due to its planar intermolecular π–π stacking which is a glaring problem in decreasing the luminescence efficiency in solid state.27,28 Fortunately, the twisted D–A design strategy not only helps in rendering TADF features but also supports the integration of aggregation-induced enhanced emission (AIEE) features in the star-shaped molecules.29 In addition, star-shaped emitters having multiple D–A transitional channels can sustain large transitional integral for a high radiative rate. Thus, via wisely choosing different cores and branches in the star-shaped molecules, the photophysical, optoelectronic and thermal properties can be easily tuned. However symmetrical star-shaped TADF emitters were scarcely reported.5,6,30
Among efficient TADF materials, diverse electron deficient cores based on nitrogen heterocycles including triazine,5,30 oxadiazole,7 triazole,7 pyridine,31 pyrimidine,32 quinoxaline,33 pyrazine33 and heptazine34 have been utilized as acceptors. However, electron-acceptor moieties with a strong electron-withdrawing ability are far from sufficient. Heptazine was the latest included in the nitrogen heterocyclic family having seven sp2 hybridized nitrogen atoms in the fused ring, used as an acceptor in orange-red TADF emitter with high EQEmax of 17.5%.26 Tris[1,2,4]triazolo[1,3,5]triazine (TTT) is another heterocycle based on fused triazole and triazine moiety with nine sp2 hybridized nitrogen atoms and a C3 symmetry5–7,30–33,35–40 which can provide an striking and remarkable alternative. It can be considered as a strong acceptor unit for the construction of donor acceptor (D3–A) TADF structure because of its rigid large electron deficient plane. Recently our group reported an emitter based on fused rigid acridine donor moiety with high PLQY and EQE,41 indicating fused structure can strengthen the rigidity of the emitters and ultimately increases the radiative luminescence efficiency. Based on aforementioned strategy, fused tetracyclic tris[1,2,4]triazolo[1,3,5]triazine core was used as an rigid acceptor in uncharted territory of donor–acceptor charge transfer emitter.
Considering all these aspects, we report a new kind of star-shaped D3–A type tris[1,2,4]triazolo[1,3,5]triazine based molecules, named 3,7,11-tris(4-(10H-phenoxazin-10-yl)phenyl)tris([1,2,4]triazolo)[1,3,5]triazine (TTT-PXZ) and 3,7,11-tris(4-(9,9-dimethylacridin-10(9H)-yl)phenyl)tris([1,2,4]triazolo)[1,3,5]triazine (TTT-DMAC) (Fig. 1), with fused TTT as acceptor and 10H-phenoxazine (PXZ) and 9,9-dimethyl-9,10-dihydroacridine (DMAC) as donors. Both TTT-PXZ and TTT-DMAC not only exhibited excellent TADF properties with small ΔEST and short delay lifetime, but also exhibited AIEE properties.42 To the best of our knowledge, this is the first report on star-shaped TTT derivatives with both TADF, AIEE features and used as emissive materials in OLEDs. Encouragingly, solution processed devices based on TTT-PXZ green emitter exhibited good performances, with an EQE of 6.2% in CzSi doped film.
 | ||
| Fig. 1 The optimized geometric structures and DFT simulation of ground and excited states of TTT-PXZ and TTT-DMAC. (a) Optimized geometric structures and distribution of HOMO and LUMO, (b) natural transition orbitals (NTOs) of the hole-particle contribution for these two compounds at S1 and T1 excited state. | ||
Experimental
Materials
The intermediates 4-(10H-phenoxazin-10-yl)benzonitrile (PXZ-BN) and 4-(9,9-dimethylacridin-10(9H)-yl)benzonitrile (DMAC-BN) were synthesized according to reported literature.43,44 Cyanuric chloride, PXZ and DMAC were purchased from commercial sources and used as received.Synthesis
Results and discussion
Synthesis and thermal properties
The synthetic route for the preparation of two TTT based star-shaped molecules is depicted in Scheme 1. Target compounds of TTT-PXZ and TTT-DMAC were synthesized by Huisgen original procedure, where threefold reaction of corresponding tetrazoles precursors of PXZ-TZ and DMAC-TZ with cyanuric chloride produced the tetracyclic tris[1,2,4]triazolo[1,3,5]triazine derivatives in high yields, respectively.39 Both target compounds can be synthesized in large amount which will benefit further commercialization. Their chemical structures were established by 1H NMR, 13C NMR spectroscopy and high-resolution mass spectrometry. TTT-PXZ possesses high thermal decomposition temperature (Td, corresponding to 5% weight loss) of 428 °C than 241 °C of TTT-DMAC (Fig. S9(a)† in ESI and Table 1). The differential scanning calorimetry (DSC) traces disclose that there is no glass transition temperatures found in second heating cycles for both the emitters [Fig. S11(b) and (c)†]. This outcome points out that both compounds would be thermally stable and durable in the OLEDs application. | ||
| Scheme 1 The synthetic routes for TTT-PXZ and TTT-DMAC. | ||
Electrochemical properties
Using cyclic voltammetry (CV), the electrochemical properties of the two emitters were investigated. The CV curves of both the emitters show oxidation process corresponding to the oxidation process of electron donor PXZ and DMAC unit, respectively. The HOMO levels of TTT-PXZ and TTT-DMAC were estimated to be −5.38 eV and −5.61 eV, respectively (Table 1, Fig. S14†), providing that stronger electron donating ability of PXZ drives shallower HOMO level compared to DMAC which results in red shifted emission of TTT-PXZ compared to TTT-DMAC. LUMO levels of TTT-PXZ and TTT-DMAC were determined from their HOMO levels and optical bandgaps and come around −2.65 and −2.66 eV, respectively. The favorable HOMO/LUMO levels of both emitters might give an idea of selecting suitable hole and electron injection materials as well as adjacent transporting layers in OLEDs.Theoretical simulations
We are curious to know how these two molecules TTT-PXZ and TTT-DMAC obtain their geometrically optimized structure as well as HOMO and LUMO distribution in their ground state. Density functional theory (DFT) based on B3LYP-D3(BJ)/def2-SVP (Fig. 1a) was carried out and provided valuable insights.47 In both molecules, HOMOs are mostly distributed in donor units (PXZ or DMAC) and partially on the linking phenyl bridge resulting from the electron-donating properties of nitrogen. LUMOs act a little different. LUMO of TTT-DMAC extents more on the phenyl bridge compared to TTT-PXZ which may cause large overlap between HOMO and LUMO. Thus, charge transfer (CT) state will be weakened in TTT-DMAC. Further, the dihedral angle between PXZ and the phenyl bridge in case of TTT-PXZ is 89.2°, which is much larger than that of TTT-DMAC (51.6°). Therefore, it is quite obvious that TTT-PXZ is more twisted than TTT-DMAC, resulting in more efficient frontier molecular orbital (FMO) separation in the case of TTT-PXZ. The above results indicate that TTT-PXZ may possess better TADF properties than TTT-DMAC.To further gather the information of the excited states, time-dependent density functional theory (TD-DFT) based on PBE0/def2-SVP was used to determine the distribution of natural transition orbitals (NTOs) as well as the values of ΔEST of TTT-PXZ and TTT-DMAC (Fig. 1b, Table 1). Particle NTOs for both compounds are located on TTT unit for both S1 and T1. While, hole NTOs are mainly spread on the respective donor moieties. Multiple distribution of hole NTO of T1 of TTT-PXZ is found. This multiple D–A interaction of TTT-PXZ between donor and acceptor would stabilize the CT state as well as large transitional integral for a high radiative rate.5 Moreover, both the molecules display small ΔEST of 0.01 eV and 0.04 eV for TTT-PXZ and TTT-DMAC, respectively, which results from the efficient FMO separation as shown in Fig. 1b and Table 1. Slightly large ΔEST value of TTT-DMAC also confirms weaker CT state formed compared to TTT-PXZ. Overall, we can conclude that both molecules may demonstrate good TADF characteristics.
Photophysical properties
To evaluate the optical properties, photophysical characterization of the target compounds were performed. The photophysical properties of the star-shaped molecules TTT-PXZ and TTT-DMAC in solution as well as 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole (CzSi) (5 wt%) doped film are depicted in Table 2, Fig. 2 and 3. Absorption spectra in dilute toluene demonstrate broad characteristic band at 394 and 358 nm for TTT-PXZ and TTT-DMAC, respectively, assigned to intramolecular charge transfer (ICT). The ICT band slightly red shifted for TTT-PXZ compared to TTT-DMAC due to stronger electron donating ability of PXZ over DMAC. The shoulder near 322 nm for TTT-PXZ belongs to n–π* transition. No absorption was detected around the same region for TTT-DMAC due to the absence of oxygen atom that possesses lone pair electrons of the corresponding donor unit. The absorption band peaked 290 nm originated from π–π* transition for both the emitters (Fig. 2a and Table S2†). Emission spectra of TTT-PXZ and TTT-DMAC in dilute toluene solution showed green emission at 522 nm and sky-blue emission at 468 nm with clear ICT character proved by solvatochromism experiment (Fig. S12, Tables S1 and S2†). Similar trends were observed for neat films for both emitters (Fig. 2b and Table S2†). When compared to the solution emission spectra, emission of TTT-PXZ in neat film blue-shifted 6 nm, whereas TTT-DMAC in neat films red-shifted 10 nm. The interesting aggregation induced blue-shifted emission (AIBSE) of TTT-PXZ was ascribed to lower reorganization energies in aggregates than in the solution state, the dearth of π-stacking due to the highly twisted structure, and weak intermolecular interactions in the solid state.45 On the contrary, the less twisted structure of TTT-DMAC than TTT-PXZ, promotes some extent of π-stacking and intermolecular interactions, resulting in aggregation induced red-shifted emission.46| Compound | λPLa (nm) | S1b (eV) | T1c (eV) | ΔESTd (eV) | τpe (ns) | τde (μs) | ΦPLf (%) | Φp/Φdg (%) | krh (107 s−1) | kISCh (107 s−1) | kRISCh (106 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in doped CzSi films (5 wt%) at room temperature.b Calculated from the onset of the fluorescence spectra of two emitters doped into CzSi films (5 wt%) at room temperature.c Calculated from the onset of the phosphorescence spectra of two emitters doped into CzSi films (5 wt%) at room temperature.d ΔEST = Sc1 − Td1.e The prompt and delayed fluorescence lifetimes of two emitters doped into CzSi films (5 wt%) at room temperature.f The total PLQYs of two emitters doped into CzSi films (5 wt%) under oxygen free condition at room temperature.g The prompt and delayed PLQY under oxygen free conditions in CzSi host film.h Rate constant of radiative, intersystem crossing and reverse intersystem crossing process in CzSi host film. | |||||||||||
| TTT-PXZ | 522 | 2.71 | 2.64 | 0.07 | 36.2 | 4.2 | 39.5 | 3.5/36.0 | 0.098 | 2.55 | 2.69 |
| TTT-DMAC | 468 | 2.94 | 2.74 | 0.20 | 49.5 | 4.6 | 21.4 | 1.9/19.5 | 0.038 | 1.83 | 2.44 |
 | ||
| Fig. 2 (a) UV-vis and fluorescence spectra of TTT-PXZ and TTT-DMAC in 10−5 M toluene solution, (b) in spin coating film. | ||
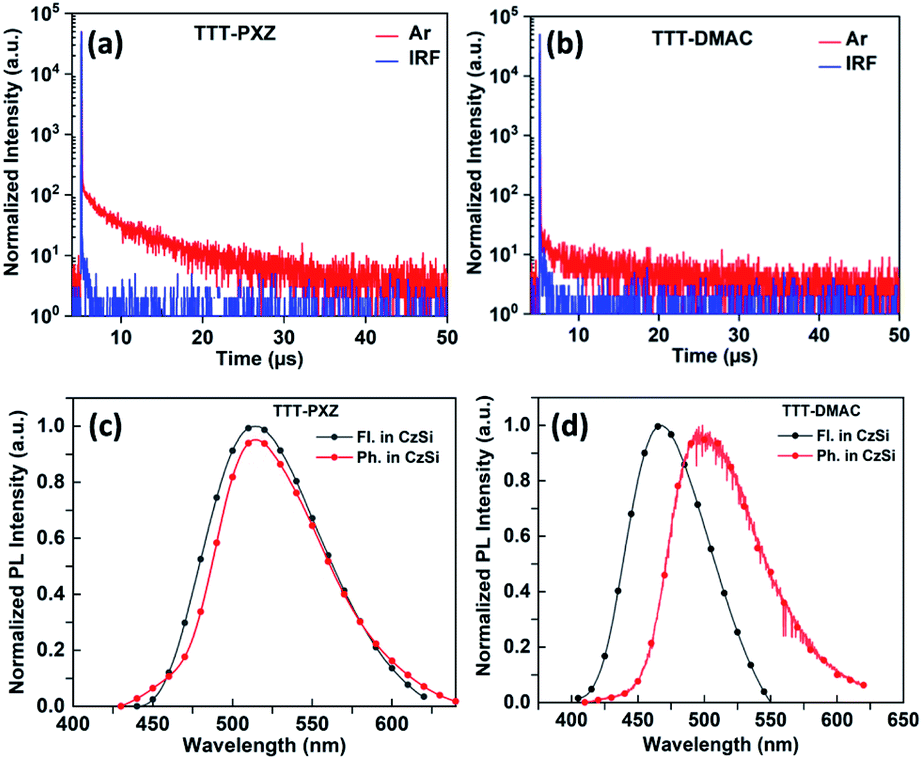 | ||
| Fig. 3 Transient photoluminescence decay curves of (a) TTT-PXZ, (b) TTT-DMAC in CzSi doping film (5 wt%) under oxygen free condition. Fluorescence spectra at 300 K and phosphorescence spectra at 77 K of (c) TTT-PXZ and (d) TTT-DMAC doped into CzSi films (5 wt%). | ||
Fluorescence and phosphorescence spectra were carried out for both the emitters with doping concentration of 5 wt% in CzSi film recorded at 300 K and 77 K. From the onset of fluorescence and phosphorescence spectra [Fig. 3(c) and (d)], S1/T1 were calculated to be 2.71/2.64 eV for TTT-PXZ and 2.94/2.74 eV for TTT-DMAC, respectively. Therefore, the respective ΔEST can be calculated to be 0.07 and 0.20 eV. The low ΔEST value of TTT-PXZ confers it on fast RISC process compared to TTT-DMAC which exhibited significantly larger ΔEST (Table 2). The total PLQYs (ФPL) of TTT-PXZ and TTT-DMAC doped into 5 wt% CzSi films were measured to be 40% and 21% under argon conditions, respectively. The more twisted D–A structure of TTT-PXZ with small ΔEST delivered high contribution of TADF.
To gain further insight into the photophysical kinetics of two emitters, transient photoluminescence (PL) decay profile was carried out in toluene (10−5 M) as well as in CzSi (5 wt%) doped films. Both TTT-PXZ and TTT-DMAC illustrate double exponential decay curve under free atmosphere accompanied by prompt and delayed fluorescence lifetimes of 36.2 ns/4.2 μs and 49.5 ns/4.6 μs, respectively (Fig. 3a and b). Via analyzing the life time decay curve of both the emitters in 5 wt% CzSi doped film, the kinetics parameters i.e., rate constants of prompt (kp), delayed (kd) fluorescence, radiative decay (kr), ISC (kISC) and RISC (kRISC) process can be calculated by using the equation as shown in ESI† and summarized in Table 2. The rate constant of radiative decay (kr) and RISC (kRISC) of TTT-PXZ are significantly higher than those of TTT-DMAC.
Both the emitters TTT-PXZ and TTT-DMAC were further tested for AIEE behavior in THF/water with various water fractions (fw) from 0 to 80% (Fig. S13†). The fluorescence intensity increases many fold when the water fraction fw reaches to 80%, signifying the AIEE behaviors of both the emitters (Fig. S13†).42 Obviously, this enhancement of emission intensity is most likely due to the formation of aggregates. Generally, the planar π-conjugated star-shaped molecule undergoes ACQ due to intermolecular π–π stacking which is detrimental for device performance. Fortunately, twisted D–A structural design not only favors TADF but also induces AIEE features by cutting down the intermolecular π–π stacking.
Electroluminescent properties
The detail photophysical studies encourage that both the emitters with doping concentration of 5 wt% in CzSi film can be used as emitters in OLEDs. To substantiate that, solution processed OLEDs with device configuration of ITO/PEDOT:PSS (35 nm)/CzSi: 5 wt% emitters (50 nm)/DPEPO (10 nm)/TmPyPB(50 nm)/Liq(1 nm)/Al were fabricated, where 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPB) were applied as electron transport materials, bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO) were used as electron transport material/exciton blocking layer and CzSi was served as the host matrix due to its wide band gap of 3.5 eV. The schematic diagram of the device design and the energy levels of the materials are depicted in Fig. 4. Table 3 summarizes the performance data of the devices. The current density–voltage–luminance (J–V–L) characteristics of fabricated OLEDs are shown in Fig. 4d. The device performance based on TTT-PXZ was observed with high luminance 1898 cd m−2, maximum current efficiency 19.2 cd A−1, power efficiency 5.2 lm W−1 and maximum external quantum efficiency 6.2% combined with slow efficiency roll-off 14.5% at high luminance of 1000 cd m−2. The device based on TTT-DMAC exhibited maximum luminance 346.6 cd m−2, maximum current efficiency 4.1 cd A−1, power efficiency 1.1 lm W−1 and maximum external quantum efficiency 1.9% as shown in Table 3. The improvement of device performance based on TTT-PXZ can be credited to its high PLQY, fast RISC process (Table 2). In addition, slow efficiency roll-off of device based on TTT-PXZ may be due to shorter lifetime of delayed component. Furthermore TTT-PXZ emits green emission with peak at 530 nm and Commission Internationale de I'Eclairage (CIE) coordinates of (0.32, 0.52), while TTT-DMAC based device emits in the sky blue region with peak at 480 nm and CIE coordinates of (0.19, 0.29) (Table 3). The utilization of electrical exciton (UE) for both the devices calculated from equation UE = EQE/(PLQY × ηout), where ηout is out-coupling efficiency (∼0.2–0.3), are 57–86% and 30–44% (without applying light out-coupling source), respectively. Both UEs are higher than 25% limitation due to the efficient TADF feature. | ||
| Fig. 4 (a) The energy level diagrams and (b) chemical structures of the materials used in the devices. (c) External quantum efficiency–current density curves; inset. (d) The EL spectra of devices. (e) Current density versus voltage versus luminance. | ||
| Emitter | V10a [V] | EQEb,c [%] | CEmaxb [cd A−1] | PEmaxb [lm W−1] | λmaxd [nm] | CIEe (x, y) |
|---|---|---|---|---|---|---|
| a Voltage in the luminance of 10 cd m2.b Maximum external quantum efficiency (EQE), maximum current efficiency (CE), maximum power efficiency (PE).c External quantum efficiency at 1000 cd m2.d Peak emission wavelength of the EL spectra.e The CIE coordinates. | ||||||
| TTT-PXZ | 8.7 | 6.2/5.3 | 19.2 | 5.2 | 530 | (0.32, 0.52) |
| TTT-DMAC | 10.6 | 1.9/— | 4.1 | 1.1 | 480 | (0.19, 0.29) |
In brief, the device performance based on TTT-PXZ was quite better than device based on TTT-DMAC which give assurance that by slight alteration of molecular structure in terms of choosing suitable rigid donor which not only increases the efficiency of exciton harvesting, but also provides a new prospect for the design of new kinds of efficient TADF emitters based on this unexplored fused tetracyclic rigid tris[1,2,4]triazolo[1,3,5]triazine core.
Conclusions
In conclusion, two new star-shaped D3–A emitters (TTT-PXZ and TTT-DMAC) have been designed and synthesized by connecting donor PXZ and DMAC in three symmetrical arms with tris[1,2,4]triazolo[1,3,5]triazine acceptor via phenyl bridges. Theoretical simulation revealed that TTT-PXZ take a highly twisted conformation as compared to TTT-DMAC, resulting in more efficient FMO separation and smaller ΔEST. Consequently, TTT-PXZ showed more DF component and delivered higher PLQY than TTT-DMAC. Encouragingly, economically feasible solution processed doping device based on TTT-PXZ green emitters exhibited decent performances, with an external quantum efficiencies (EQEs) of up to 6.2%. These results make it evident that the tris[1,2,4]triazolo[1,3,5]triazine moiety is a promising rigid acceptor for efficient TADF emitters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shenzhen Science and Technology Program (KQTD20170330110107046), the National Natural Science Foundation of China (No. 51803125 and 91833304), and Science and Technology Innovation Commission of Shenzhen (JCYJ20180507182244027). We thank the Instrumental Analysis Center of Shenzhen University for analytical support.References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- E. Ayataka, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu and J. Zhao, Chem. Soc. Rev., 2017, 46, 915 RSC.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766 CrossRef CAS.
- X. Cai, D. Chen, K. Gao, L. Gan, Q. Yin, Z. Qiao, Z. Chen, X. Jiang and S.-J. Su, Adv. Mater., 2018, 28, 1704927 Search PubMed.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599 RSC.
- F. Cherioux, L. Guyard and P. Audebert, Chem. Commun., 1998, 2225 RSC.
- J. Luo, Y. Zhou, Z.-Q. Niu, Q.-F. Zhou, Y. Ma and J. Pei, J. Am. Chem. Soc., 2007, 129, 11314 CrossRef CAS PubMed.
- W.-Y. Lai, R. Xia, Q.-Y. He, P. A. Levermore, W. Huang and D. D. C. Bradley, Adv. Mater., 2009, 21, 355 CrossRef CAS.
- C. Liu, Y. Li, Y. Zhang, C. Yang, H. Wu, J. Qin and Y. Cao, Chem.–Eur. J., 2012, 18, 6928 CrossRef CAS PubMed.
- C. Liu, Q. Fu, Y. Zou, C. Yang and J. Qin, Chem. Mater., 2014, 26, 3074 CrossRef CAS.
- L. Chen, P. Li, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23, 2986 CrossRef CAS PubMed.
- C. Coya, C. Ruiz, Á. L. Álvarez, S. Álvarez-García, E. M. García-Frutos, B. Gómez-Lor and A. d Andrés, Org. Electron., 2012, 13, 2138 CrossRef CAS.
- X. He, L. Chen, Y. Zhao, S. C. Ng, X. Wang, X. Sun and X. Hu, RSC Adv., 2015, 5, 15399 RSC.
- T. Huang, W. Jiang and L. Duan, J. Mater. Chem. C, 2018, 6, 5577 RSC.
- L. Duan, L. Hou, T.-W. Lee, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, J. Mater. Chem. C, 2010, 20, 6392 RSC.
- M. Shibata, Y. Sakai and D. Yokoyama, J. Mater. Chem. C, 2015, 3, 11178 RSC.
- M. Huang, Y. Li, K. Wu, J. Luo, G. Xie, L. Li and C. Yang, Dyes Pigm., 2018, 153, 92 CrossRef CAS.
- S. Gong, J. Luo, Z. Wang, Y. Li, T. Chen, G. Xie and C. Yang, Dyes Pigm., 2017, 139, 593 CrossRef CAS.
- Y. Li, T. Chen, M. Huang, Y. Gu, S. Gong, G. Xie and C. Yang, J. Mater. Chem. C, 2017, 5, 3480–3487 RSC.
- G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong and C. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef PubMed.
- Y. Li, G. Xie, S. Gong, K. Wu and C. Yang, Chem. Sci., 2016, 7, 5441 RSC.
- J. Luo, S. Gong, Y. Gu, T. Chen, T. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442 RSC.
- J. Luo, G. Xie, S. Gong, T. Chen and C. Yang, Chem. Commun., 2016, 52, 2292 RSC.
- Y. Zou, S. Gong, G. Xie and C. Yang, Adv. Opt. Mater., 2018, 6, 1800568 CrossRef.
- R. Cristiano, J. Eccher, I. H. Bechtold, C. N. Tironi, A. A. Vieira and F. Molin, Langmuir, 2012, 28, 11590 CrossRef CAS PubMed.
- Q. Niu, Y. Lu, H. Sun and X. Li, Spectrochim. Acta, Part A, 2013, 107, 377 CrossRef CAS PubMed.
- P. Das, A. Kumar, A. Chowdhury and P. S. Mukherjee, ACS Omega, 2018, 3, 13757 CrossRef CAS PubMed.
- Y. Wada, K. Shizu, S. Kubo, K. Suzuki, H. Tanaka, C. Adachi and H. Kaji, Appl. Phys. Lett., 2015, 107, 183303 CrossRef.
- J. Jayakumar, T.-L. Wu, M.-J. Huang, P.-Y. Huang, T.-Y. Chou, H.-W. Lin and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 21042 CrossRef CAS PubMed.
- P. Ganesan, R. Ranganathan, Y. Chi, X.-K. Liu, C.-S. Lee, S.-H. Liu, G.-H. Lee, T.-C. Lin, Y.-T. Chen and P.-T. Chou, Chem.–Eur. J., 2017, 23, 2858 CrossRef CAS PubMed.
- T. Huang, D. Liu, J. Jiang and W. Jiang, Chem.–Eur. J., 2019, 25, 10926 CrossRef CAS PubMed.
- J. Li, T. Nakagawa, J. MacDonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319 CrossRef CAS PubMed.
- S. Glang, T. Rieth, D. Borchmann, I. Fortunati, R. Signorini and H. Detert, Chem.–Eur. J., 2014, 15, 3116 CrossRef.
- H. Zhao, Y. Wang, Z. Liu and B. Dai, RSC Adv., 2014, 4, 13161 RSC.
- R. Cristiano, H. Gallardo, A. J. Bortoluzzi, I. H. Bechtold, C. E. M. Campos and R. L. Longo, Chem. Commun., 2008, 5134 RSC.
- V. A. Tartakovsky, A. E. Frumkin, A. M. Churakov and Y. A. Strelenko, Russ. Chem. Bull., 2005, 54, 719 CrossRef CAS.
- M. Sperner, N. Tober and H. Detert, Eur. J. Org. Chem., 2019, 4688 CrossRef CAS.
- A. G. Dal-Bo, G. G. L. Cisneros, R. Cercena, J. Mendes, L. M. d. Silveira, E. Zapp, K. G. Domiciano, R. d. C. Duarte, F. S. Rodembusch and T. E. A. Frizon, Dyes Pigm., 2016, 135, 49 CrossRef CAS.
- D. Chai, Y. Zou, Y. Xiang, X. Zeng, Z. Chen, S. Gong and C. Yang, Chem. Commun., 2019, 55, 15125 RSC.
- V. Anand, B. Sadhasivam and R. Dhamodharan, New J. Chem., 2018, 42, 18979–18990 RSC.
- D. Wei, F. Ni, Z. Wu, Z. Zhu, Y. Zou, K. Zheng, Z. Chen, D. Ma and C. Yang, J. Mater. Chem. C, 2018, 6, 11615 RSC.
- N. A. Drigo, L. G. Kudriashova, S. Weissenseel, A. Sperlich, A. J. Huckaba, Md. K. Nazeeruddin and V. Dyakonov, J. Phys. Chem. C, 2018, 122, 22796 CrossRef CAS.
- Q. Wu, T. Zhang, Q. Peng, D. Wang and Z. Shuai, Phys. Chem. Chem. Phys., 2014, 16, 5545 RSC.
- Z. Zhao, S. Chen, J. W. Y. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwoka and B. Z. Tang, Chem. Commun., 2010, 46, 2221 RSC.
- W. Zeng, S. Gong, C. Zhong and C. Yang, J. Phys. Chem. C, 2019, 123, 10081 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, TGA, DSC, CV, UV, fluorescence. See DOI: 10.1039/d0ra01925a |
| This journal is © The Royal Society of Chemistry 2020 |