
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Amino peptide scanning reveals inhibitors of Aβ42 aggregation†
Khalilia C. Tillett a and
Juan R. Del Valle*b
a and
Juan R. Del Valle*b
aDepartment of Chemistry, University of South Florida, Tampa, Florida 33620, USA
bDepartment of Chemistry & Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: jdelvalle@nd.edu
First published on 8th April 2020
Abstract
The aggregation of amyloids into toxic oligomers is believed to be a key pathogenic event in the onset of Alzheimer's disease. Peptidomimetic modulators capable of destabilizing the propagation of an extended network of β-sheet fibrils represent a potential intervention strategy. Modifications to amyloid-beta (Aβ) peptides derived from the core domain have afforded inhibitors capable of both antagonizing aggregation and reducing amyloid toxicity. Previous work from our laboratory has shown that peptide backbone amination stabilizes β-sheet-like conformations and precludes β-strand aggregation. Here, we report the synthesis of N-aminated hexapeptides capable of inhibiting the fibrillization of full-length Aβ42. A key feature of our design is N-amino substituents at alternating backbone amides within the aggregation-prone Aβ16–21 sequence. This strategy allows for maintenance of an intact hydrogen-bonding backbone edge as well as side chain moieties important for favorable hydrophobic interactions. An N-amino scan of Aβ16–21 resulted in the identification of peptidomimetics that block Aβ42 fibrilization in several biophysical assays.
Introduction
Amyloid protein aggregation has been linked to several neurodegenerative disorders, including Parkinson's and Alzheimer's Disease (AD).1 In AD, amyloid precursor protein (APP) is sequentially cleaved by α and β secretase to release pathogenic amyloid beta (Aβ). Aβ is prone to aggregation and forms toxic β-sheet fibrils that are the main component of neuritic plaques in AD patients.2 Of the two Aβ fragments formed during amyloidogenesis, the 42-residue variant (Aβ42) is more prone to aggregation and displays greater neurotoxicity in comparison to the 40-residue variant (Aβ40).3 Soluble oligomers of Aβ42 have been shown to impair hippocampal synaptic plasticity,4 induce tau hyperphosphorylation,5 activate microglial inflammation,6 and impair memory.7 Interestingly, studies have shown that high molecular weight oligomers are relatively inactive. However, when allowed to dissociate into low molecular weight oligomers (8–70 kDa) severe toxicity is observed. Additionally, soluble oligomers (but not monomers) cause synaptic death in a dose dependent manner.8Although Aβ42 is considered to be intrinsically disordered, its fibrils are comprised of parallel β-sheets that feature edge–edge hydrogen-bonding between Aβ42 monomers.9 This in-register association is driven in large part by a core hydrophobic sequence encompassing residues 16–21 (16KLVFFA21).10 Direct Aβ42 aggregation blockers that have entered clinical trials for the treatment of AD include homotaurine, scyllo-inositol, and sodium oligomannurarate.11 These and similar compounds face significant hurdles to advancement, as they are likely to interact in a non-specific manner with β-rich target proteins. In contrast, backbone modified peptides derived from aggregation-prone segments offer the prospect of sequence-specific inhibition of amyloidogenesis. Toward this end, several groups have reported Aβ16–21 peptidomimetics harboring D-amino acids, unnatural residues, or backbone amide substituents.12 In addition to optimizing affinity for Aβ42, such peptide modifications may serve to enhance proteolytic stability, conformational rigidity, and prospects for passage through the blood–brain barrier. Rationally designed peptidomimetics targeting Aβ42 fibrilization have shown efficacy in animal models of AD.12
Our laboratory has previously shown that peptide backbone N-amination stabilizes β-sheet-like conformation through cooperative non-covalent interactions.13 Like N-methylation, the incorporation of N-amino substituents within a peptide strand precludes canonical β-sheet associations along one of the hydrogen-bonding edges. This characteristic is useful for the design peptide derivatives that not only resist self-aggregation but also block β stacking of a target protein. Although backbone-methylated peptides are capable of disrupting Aβ aggregation,14 we viewed N-amination as an alternative strategy for the design of more conformationally rigid β-breaker peptides. Unlike N-methylation or proline incorporation, backbone amination does not promote a significant population of the cis amide rotamer. Given that Aβ42 fibrils adopt a β-strand conformation with an all-trans amide geometry, backbone-substituted mimics with high trans amide propensity should exhibit enhanced affinity for the conformationally extended core sequence. The N-amino group is also able to engage in an intraresidue C6 H-bond, which in turn stabilizes ψ and ϕ torsions in the β-sheet region of Ramachandran space.13a We sought to exploit these features in the design of β-strand peptidomimetics capable of blocking amyloid aggregation (Fig. 1). Here, we report the first example of N-amino peptide scanning and the identification of novel backbone-modified inhibitors of Aβ42 fibrilization.
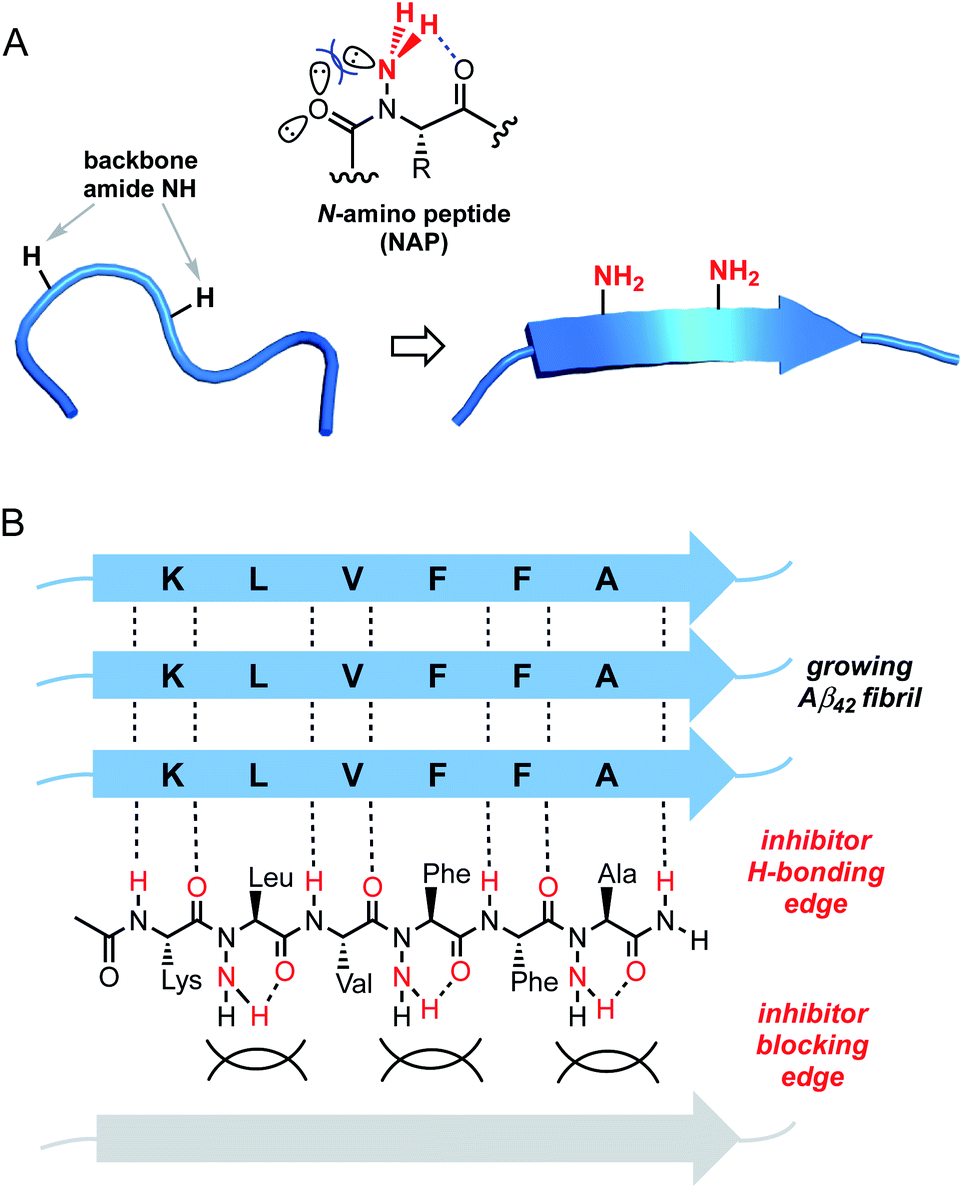 | ||
| Fig. 1 Design of N-amino peptide (NAP) Aβ16–21 mimics. (A) Conformational effects of peptide backbone N-amination. (B) Proposed model for inhibition of Aβ fibrilization by a representative NAP. | ||
Results
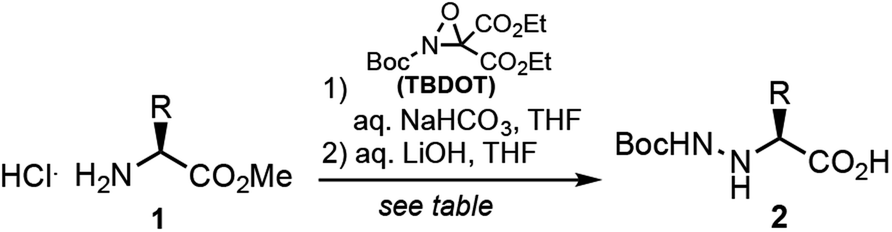
Peptide N-amino scanning of the aggregation-prone 16KLVFFA21 sequence required the synthesis of 5 distinct α-hydrazino acid derivatives starting from commercially available α-amino esters. We previously demonstrated that monomers suitable for solid-phase peptide synthesis (SPPS) can be readily accessed via electrophilic amination of α-amino esters.15 As shown in Table 1, the requisite building blocks were thus prepared by treatment of 1 with (2-t-butyl-3,3-diethyl)-oxaziridine-2,3,3-tricarboxylate (TBDOT)16 under aqueous conditions, followed by alkaline ester hydrolysis to give 2a–e in good yields.
|
|||
|---|---|---|---|
| Residue | R | Yield | |
| 2a | aLys(Boc) | (CH2)4NHBoc | 74 |
| 2b | aLeu | CH2CH(CH3)2 | 66 |
| 2c | aVal | CH(CH3)2 | 44 |
| 2d | aPhe | CH2Ph | 66 |
| 2e | aAla | CH3 | 61 |
Backbone-aminated analogues of Aβ16–21 were prepared on rink amide MHBA resin using Fmoc chemistry (Scheme 1). Condensation of the Boc-protected α-hydrazino acids onto the growing peptide chain was achieved by HCTU/NMM-mediated activation of the carboxylic acid in the presence of the unprotected Nα. The poorly nucleophilic Nα is only acylated upon exposure to pre-formed Fmoc-protected amino acid chloride derivatives.17 These were prepared by reaction of Fmoc-protected amino acids with thionyl chloride, or by activation with Ghosez' reagent in the case of the Fmoc-Lys(Boc)-OH residue (due to the acid-labile side chain protecting group). Following sequence elongation, the peptidomimetics were N-terminally acetylated, cleaved from the resin, and purified by RP-HPLC.
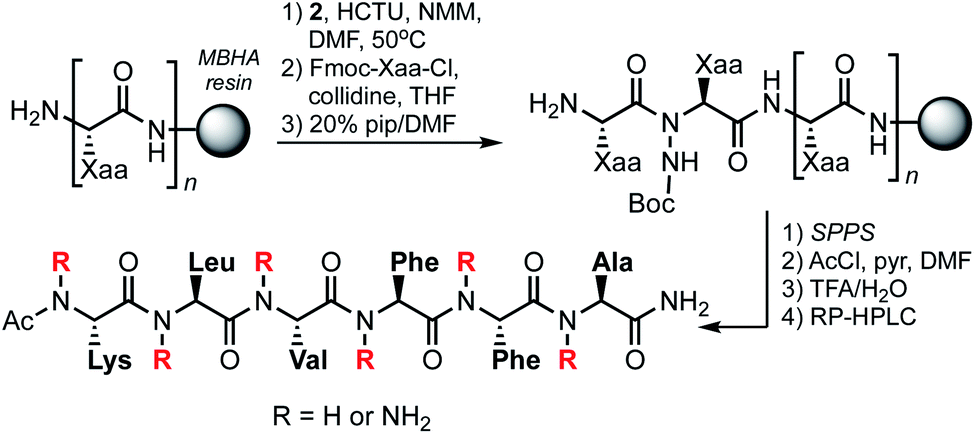 | ||
| Scheme 1 Solid-phase synthesis of NAP-based Aβ16–21 mimics. | ||
For the current study, we synthesized the 14 N-aminated Aβ16–21 variants shown in Table 2, along with the parent hexapeptide (3). This positional scanning library is comprised of all possible mono-aminated peptides, as well as di- and tri-aminated sequences that harbor backbone substituents on the same edge of the putative β-strand. We excluded analogues featuring alternate edge N-aminations since these compounds are presumably devoid of a fully intact hydrogen-bonding edge. The identity of purified peptidomimetics was confirmed by HRMS and their purities assessed by analytical HPLC and NMR. The 1H NMR spectra for 3–17 were recorded in DMSO-d6 and proton resonance assignments were made on the basis of TOCSY spectra.
| Peptide | Sequence | HRMS [M + H]obs+ |
|---|---|---|
| 3 | Ac-Lys-Leu-Val-Phe-Phe-Ala-NH2 | 779.4701 |
| 4 | Ac-aLys-Leu-Val-Phe-Phe-Ala-NH2 | 779.4697 |
| 5 | Ac-Lys-aLeu-Val-Phe-Phe-Ala-NH2 | 779.4714 |
| 6 | Ac-Lys-Leu-aVal-Phe-Phe-Ala-NH2 | 779.4711 |
| 7 | Ac-Lys-Leu-Val-aPhe-Phe-Ala-NH2 | 779.4680 |
| 8 | Ac-Lys-Leu-Val-Phe-aPhe-Ala-NH2 | 779.4687 |
| 9 | Ac-Lys-Leu-Val-Phe-Phe-aAla-NH2 | 779.4690 |
| 10 | Ac-aLys-Leu-aVal-Phe-Phe-Ala-NH2 | 794.4800 |
| 11 | Ac-aLys-Leu-Val-Phe-aPhe-Ala-NH2 | 794.4802 |
| 12 | Ac-Lys-aLeu-Val-aPhe-Phe-Ala-NH2 | 794.4811 |
| 13 | Ac-Lys-aLeu-Val-Phe-Phe-aAla-NH2 | 794.4802 |
| 14 | Ac-Lys-Leu-aVal-Phe-aPhe-Ala-NH2 | 794.4794 |
| 15 | Ac-Lys-Leu-Val-aPhe-Phe-aAla-NH2 | 794.4810 |
| 16 | Ac-aLys-Leu-Val-Phe-Phe-Ala-NH2 | 809.4920 |
| 17 | Ac-Lys-aLeu-Val-aPhe-Phe-aAla-NH2 | 809.4912 |
The aggregation of Aβ42 was monitored in the presence of 1 molar equivalent of NAPs 4–17 using thioflavin T (ThT), a dye that fluoresces upon binding to the perpendicular axis of amyloid fibrils (Fig. 2).18 Compounds 5, 14 and 15 were identified as the most effective inhibitors of Aβ42 aggregation in repeated experiments. Aβ42 fibrillization is typically characterized by a sigmoidal curve comprised of a lag phase, an elongation phase, and a plateau phase. Over a 12 h experiment, Aβ42 aggregation was inhibited by approximately 30–50% at an equimolar ratio for each inhibitor. Although the peak fluorescence values of all three inhibitors were comparable, the elongation/growth phase of 5 and 14 were slower than that of 15. In addition, 5 displayed increased lag times across all three runs (Fig. S1†). Although several other NAPs displayed some inhibitory effects on Aβ42 aggregation kinetics, observed variations in repeated experiments led us to focus on compounds 5, 14, and 15 for further study. Consistent with previous reports, the parent hexapeptide 3 exhibited accelerated aggregation when incubated in the presence of Aβ42 and ThT. Initial in vitro assay thus suggests that a single backbone amination, as in 5, is sufficient to convert a highly aggregation-prone peptide into an inhibitor of fibrilization.
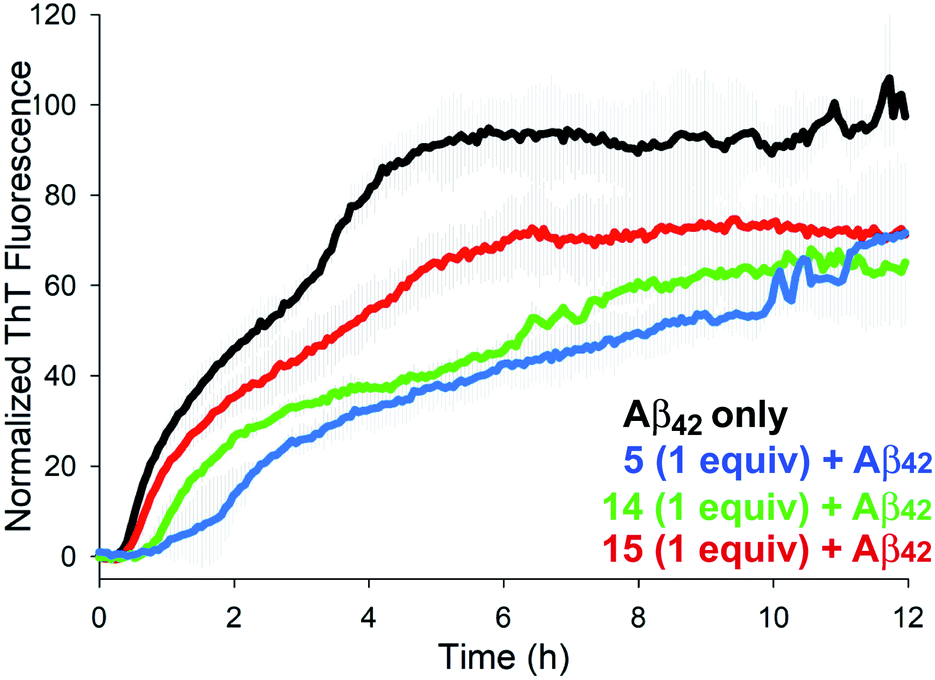 | ||
| Fig. 2 Effects of 1.0 equiv. of inhibitors 5, 14, and 15 on Aβ42 aggregation (40 μM) as monitored by ThT fluorescence. | ||
To gain further insight into the effects of NAPs 5, 14, and 15 on Aβ42 structure we carried out circular dichroism (CD) measurements over several time points (Fig. 3). Incubation of Aβ42 alone at 37 °C resulted in a transition from random coil to β-sheet structure over the course of 18 h, as evidenced by a pronounced minimum at 218 nm. Incubation of Aβ42 in the presence of an equimolar concentration of 15 for 18 h resulted in a significantly attenuated β-sheet signature by CD, although some β-sheet character was still evident. In contrast, analogues 5 and 14 showed a similar CD signature at 18 h indicative of a random coil conformation. These spectra were also similar to those observed for Aβ42 alone at 0 and 4 h timepoints. The Far UV-CD spectra of 40 μM 5, 14, and 15 revealed that each inhibitor showed irregular/random coil character in the absence of Aβ42 (Fig. S2†).
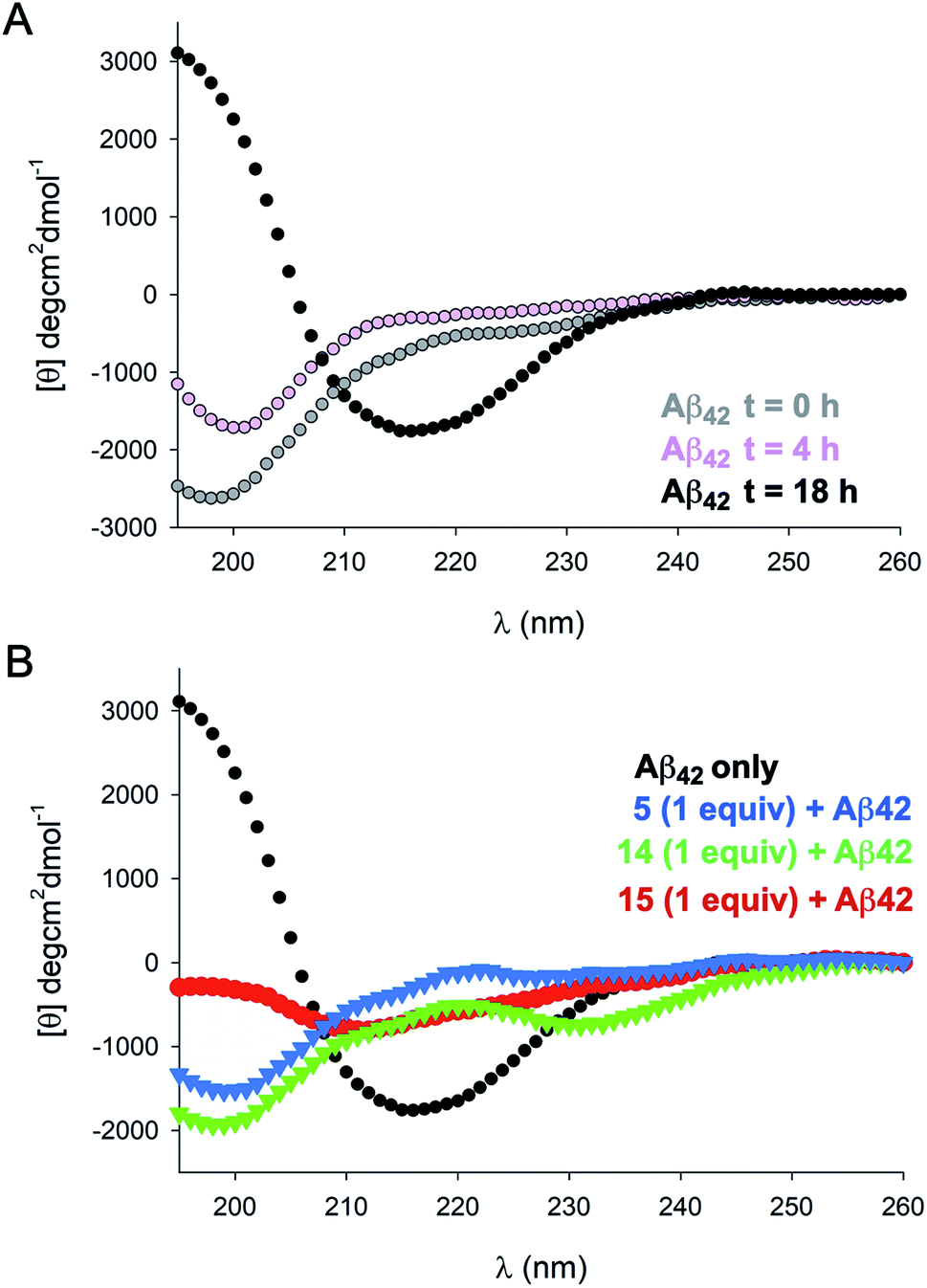 | ||
| Fig. 3 Circular dichroism spectra. (A) Time-dependent CD spectra of 40 μM Aβ42 recorded in phosphate buffer. (B) CD spectrum of Aβ42 at 18 h (black), and in the presence of an equimolar ratio of 5, 14, and 15. | ||
Protofibrils, fibrillar precursors to amyloid formation, are preceded by the appearance of small Aβ oligomers.19 Transmission electron microscopy (TEM) can be used to visualize the morphology of these fibril and pre-fibrillar species at high resolution. In order to assess the ability of 5, 14, and 15 to alter the dominant morphology of Aβ42, samples were taken directly from the ThT assays at the plateau phase (Fig. 4). Aβ42 incubated on its own formed a dense network of mature fibrils when incubated without inhibitor. Aβ42 incubated with 5 showed scattered, short, rod-like protofibrils. No evidence of mature fibril formation was visible. Aβ42 incubated with 14 showed little to no evidence of fibril formation, however, small seeding was visible. Aβ42 incubated with 15 showed sparse mature fibrils throughout the sample. These results are consistent with those from ThT assays and CD measurements showing that NAP 15 was not as effective as 5 or 14 in blocking the aggregation of Aβ42 or its transition to β structure.
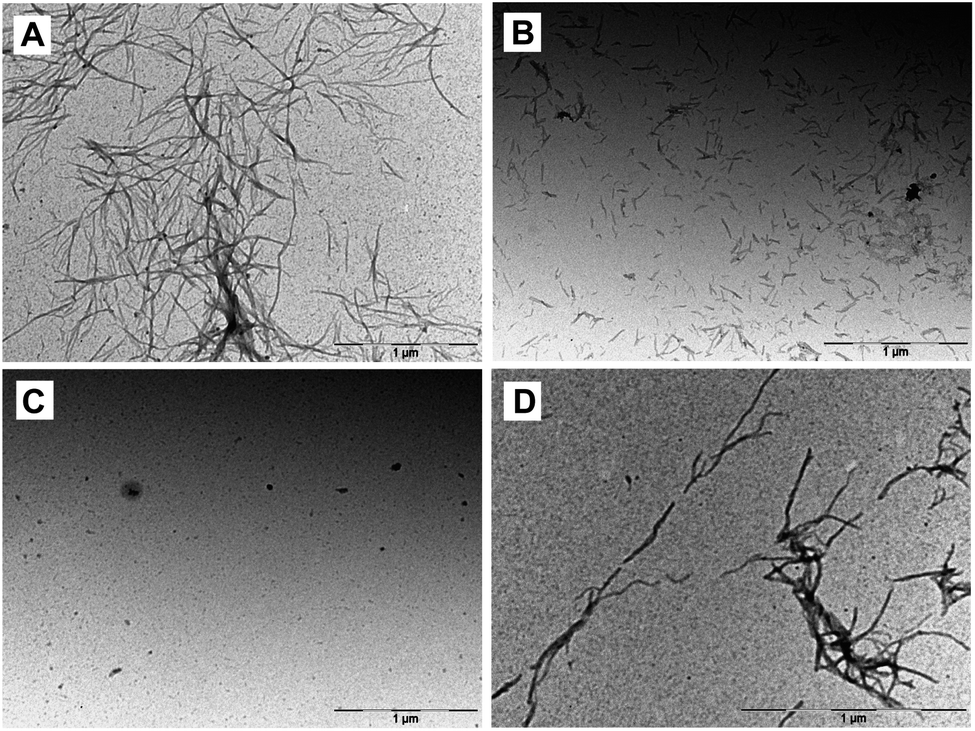 | ||
| Fig. 4 Representative transmission electron microscopy images at t = 6 h. (A) Aβ42 incubated alone, or in the presence of an equimolar amount of (B) 5, (C) 14, or (D) 15. | ||
Based on results from TEM experiments, we investigated the ability of our most effective inhibitor to delay fibrilization in a dose-dependent manner by ThT. As shown in Fig. 5, 14 had little effect on aggregation lag time at 0.5 equiv. concentration, but delayed the aggregation of Aβ42 by 80% and 220% at 1.0 equiv. and 4.0 equiv., respectively. Unlike 14, we did not observe a similar dose-dependency in the case of 5 or 15. Taken together, these data demonstrate that di-NAP 14 is effective in delaying Aβ42 aggregation in a dose-dependent manner, inhibiting the formation of mature fibrils, and in blocking the transition of Aβ42 from random coil to β structure in vitro.
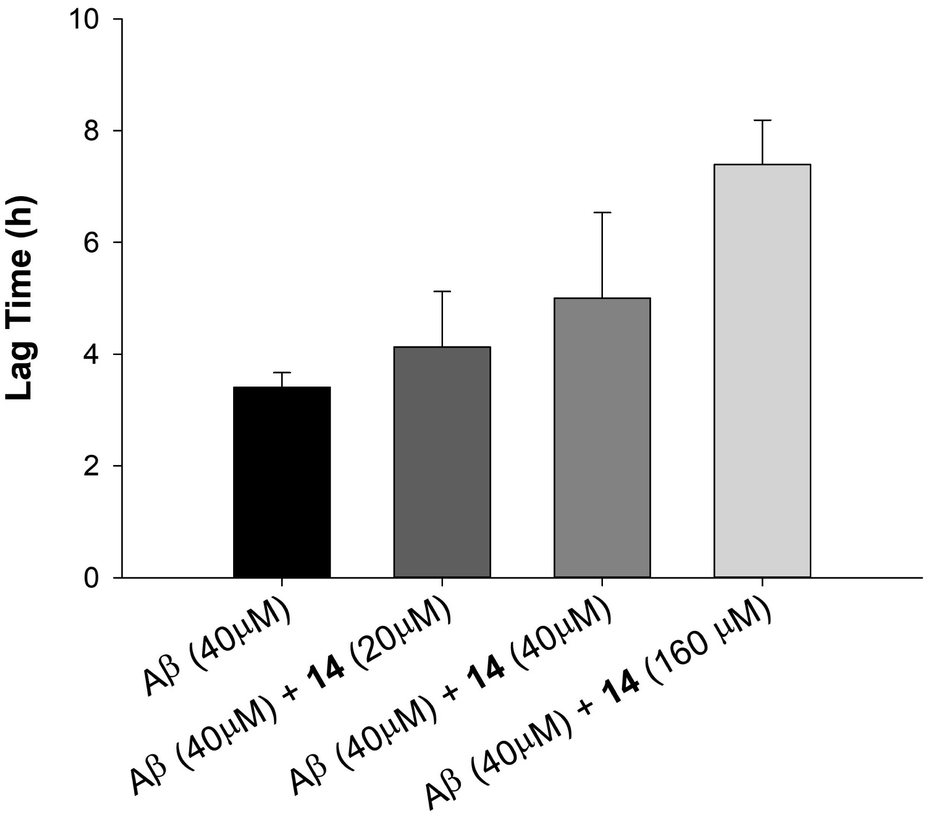 | ||
| Fig. 5 Effect of 0.5, 1.0 and 4.0 equiv. of 14 on 40 μM Aβ42 aggregation lagtime as monitored by ThT fluorescence assay. | ||
Conclusions
Here, we demonstrate the synthesis and evaluation of N-amino peptide inhibitors of Aβ42 aggregation that are readily accessible from α-hydrazino acid monomers. The inhibitory activity of selected NAPs was confirmed through ThT assay, CD, and TEM. Di-aminated derivative 14 emerged from this study as an effective inhibitor of Aβ42 aggregation in vitro. Interestingly, tri-aminated derivatives 16 and 17 were less effective by ThT assay, as were the majority of mono-aminated analogues. We previously observed that di-amination across a tripeptide strand is a particularly powerful approach for stabilizing β-sheet conformations within hairpin model systems.13 Peptidomimetics 14 and 15 likewise represent constrained β-strand mimics bearing two edge-blocking substituents in close proximity. This may explain their ability to engage Aβ42 and shield one hydrogen-bonding edge from further association with Aβ monomers. The current work adds backbone N-amination to the repertoire of peptidomimetic strategies for targeting β-sheet assembly. We anticipate that this approach can be readily applied to other disease-relevant protein interactions and will provide useful lead compounds for structure-based refinement.Experimental
Synthesis of compounds 3–17
Synthesis of all peptides/peptidomimetics was carried out on Fmoc-capped polystyrene rink amide MBHA resin (100–200 mesh). α-Hydrazino acid monomers were prepared from α-amino esters as previously described.15 Dry resin was washed with DMF three times and allowed to swell in DMF for 1 h prior to use. All reactions were carried out using gentle agitation. Fmoc deprotection steps were carried out by treating the resin with a solution of 20%piperidine/DMF (15 min × 2). Coupling of Fmoc-protected amino acids as well as (N′-Boc)-hydrazino acids was effected using 5 equiv. HCTU (0.5 M in DMF), 10 equiv. NMM (1.0 M in DMF), and 5 equiv. of the carboxylic acid in DMF at 50 °C (1 h). Coupling of residues N-terminal to the hydrazino acids was carried out with 30 equiv. collidine and 10 equiv. of pre-formed Fmoc amino acid chloride17 in THF at 45 °C (2 × 1 h). After each reaction the resin was washed with DMF three times. Peptides were cleaved from the resin by incubating with gentle stirring in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 TFA:H2O for 2 h. The cleavage mixture was filtered and the resin was rinsed with an additional portion of cleavage solution. The filtrate was concentrated to remove the bulk of the TFA and the remaining residue was treated with 8 mL of cold Et2O to induce precipitation. The mixture was centrifuged and the supernatant was removed. The remaining solid was washed 2 more times with Et2O and dried under vacuum. Peptides were analyzed and purified on C12 RP-HPLC columns (preparative: 4 μ, 90 Å, 250 × 21.2 mm; analytical: 4 μ, 90 Å, 150 × 4.6 mm) using linear gradients of MeCN/H2O (with 0.1% formic acid), then lyophilized to afford white powders. All peptides were characterized by LCMS (ESI), HRMS (ESI-TOF), 1H NMR, and TOCSY.
:5 TFA:H2O for 2 h. The cleavage mixture was filtered and the resin was rinsed with an additional portion of cleavage solution. The filtrate was concentrated to remove the bulk of the TFA and the remaining residue was treated with 8 mL of cold Et2O to induce precipitation. The mixture was centrifuged and the supernatant was removed. The remaining solid was washed 2 more times with Et2O and dried under vacuum. Peptides were analyzed and purified on C12 RP-HPLC columns (preparative: 4 μ, 90 Å, 250 × 21.2 mm; analytical: 4 μ, 90 Å, 150 × 4.6 mm) using linear gradients of MeCN/H2O (with 0.1% formic acid), then lyophilized to afford white powders. All peptides were characterized by LCMS (ESI), HRMS (ESI-TOF), 1H NMR, and TOCSY.
Thioflavin T fluorescence aggregation assay
ThT fluorescence assays were conducted in 96-well half area plates (black, flat bottom, non-binding surface) without shaking in a Biotek Synergy H1 microplate reader (444 nm excitation, 484 nm emission) at 37 °C for 12 h. Experiments were run in duplicate with 40 μM Aβ42, 40 μM ThT, and 40 μM of inhibitor in 10 mM PBS buffer (pH 7.4) with a total reaction volume of 100 μL.000 M−1 cm−1 at 412 nm] and diluted to final concentration using 10 mM PBS (pH 7.4).Transmission electron microscopy
Samples for TEM studies were taken from the ThT assay during the delayed lag phase. Each grid was prepared by aliquoting 10 μL of sample from the appropriate wells at the 6 h timepoint. The samples were pipetted onto formvar and carbon coated electron microscopy grids (Ted Pella, catalog no. 01754-F) and incubated for 3 min. Capillary action was used to remove the remaining liquid from the grids using filter paper. The grids were rinsed with Millipore water and stained with 1% (w/v) uranyl acetate solution. Samples were imaged using FEI M268D; Morgagni Series TEM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Kamlesh Makwana for assistance with data analysis and preparation of figures. This work was supported by a grant from the National Science Foundation (CHE1709927).Notes and references
- (a) F. Chiti and C. M. Dobson, Protein Misfolding, Functional Amyloid, and Human Disease, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed; (b) M. Bartolini and V. Andrisano, Strategies for the Inhibition of Protein Aggregation in Human Diseases, ChemBioChem, 2010, 11, 1018–1035 CrossRef CAS PubMed; (c) G.-f. Chen, T.-h. Xu, Y. Yan, Y.-r. Zhou, Y. Jiang, K. Melcher and H. E. Xu, Amyloid beta: structure, biology and structure-based therapeutic development, Acta Pharmacol. Sin., 2017, 38, 1205–1235 CrossRef CAS PubMed; (d) D. J. Selkoe and J. Hardy, The amyloid hypothesis of Alzheimer's disease at 25 years, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed; (e) G. S. Bloom, Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis, JAMA Neurology, 2014, 71, 505–508 CrossRef PubMed; (f) R. E. Tanzi and L. Bertram, Twenty Years of the Alzheimer's Disease Amyloid Hypothesis: A Genetic Perspective, Cell, 2005, 120, 545–555 CrossRef CAS PubMed.
- A. E. Roher, J. D. Lowenson, S. Clarke, C. Wolkow, R. Wang, R. J. Cotter, I. M. Reardon, H. A. Zürcher-Neely, R. L. Heinrikson and M. J. Ball, Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease, J. Biol. Chem., 1993, 268, 3072–3083 CAS.
- A. Lorenzo and B. A. Yankner, Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12243–12247 CrossRef CAS PubMed.
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan, C. A. Lemere, C. M. Regan, D. M. Walsh, B. L. Sabatini and D. J. Selkoe, Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory, Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed.
- Q. L. Ma, F. Yang, E. R. Rosario, O. J. Ubeda, W. Beech, D. J. Gant, P. P. Chen, B. Hudspeth, C. Chen, Y. Zhao, H. V. Vinters, S. A. Frautschy and G. M. Cole, Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin, J. Neurosci., 2009, 29, 9078–9089 CrossRef CAS PubMed.
- S. Mandrekar-Colucci and G. E. Landreth, Microglia and inflammation in Alzheimer's disease, CNS Neurol. Disord.: Drug Targets, 2010, 9, 156–167 CrossRef CAS PubMed.
- J. C. Dodart, K. R. Bales, K. S. Gannon, S. J. Greene, R. B. DeMattos, C. Mathis, C. A. DeLong, S. Wu, X. Wu, D. M. Holtzman and S. M. Paul, Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model, Nat. Neurosci., 2002, 5, 452–457 CrossRef CAS PubMed.
- T. Yang, S. Li, H. Xu, D. M. Walsh and D. J. Selkoe, Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate, J. Neurosci., 2017, 37, 152–163 CrossRef CAS PubMed.
- T. L. Benzinger, D. M. Gregory, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, R. E. Botto and S. C. Meredith, Propagating structure of Alzheimer's beta-amyloid(10-35) is parallel beta-sheet with residues in exact register, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13407–13412 CrossRef CAS PubMed.
- (a) C. Hilbich, B. Kisters-Woike, J. Reed, C. L. Masters and K. Beyreuther, Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer's disease βA4 peptides, J. Mol. Biol., 1992, 228, 460–473 CrossRef CAS PubMed; (b) N. S. de Groot, F. X. Aviles, J. Vendrell and S. Ventura, Mutagenesis of the central hydrophobic cluster in Aβ42 Alzheimer's peptide, FEBS J., 2006, 273, 658–668 CrossRef PubMed.
- F. Panza, M. Lozupone, G. Logroscino and B. P. Imbimbo, A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease, Nat. Rev. Neurol., 2019, 15, 73–88 CrossRef PubMed.
- D. Goyal, S. Shuaib, S. Mann and B. Goyal, Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer's Disease, ACS Comb. Sci., 2017, 19, 55–80 CrossRef CAS PubMed.
- (a) M. P. Sarnowski, C. W. Kang, Y. M. Elbatrawi, L. Wojtas and J. R. Del Valle, Peptide N-Amination Supports β-Sheet Conformations, Angew. Chem., Int. Ed., 2017, 56, 2083–2086 CrossRef CAS PubMed; (b) M. P. Sarnowski, K. P. Pedretty, N. Giddings, H. L. Woodcock and J. R. Del Valle, Synthesis and beta-sheet propensity of constrained N-amino peptides, Bioorg. Med. Chem., 2018, 26, 1162–1166 CrossRef CAS PubMed.
- (a) N. Kokkoni, K. Stott, H. Amijee, J. M. Mason and A. J. Doig, N-Methylated peptide inhibitors of beta-amyloid aggregation and toxicity. Optimization of the inhibitor structure, Biochemistry, 2006, 45, 9906–9918 CrossRef CAS PubMed; (b) D. J. Gordon, R. Tappe and S. C. Meredith, Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Abeta1-40 fibrillogenesis, J. Pept. Res., 2002, 60, 37–55 CrossRef CAS PubMed.
- C. W. Kang, M. P. Sarnowski, Y. M. Elbatrawi and J. R. Del Valle, Access to Enantiopure α-Hydrazino Acids for N-Amino Peptide Synthesis, J. Org. Chem., 2017, 82, 1833–1841 CrossRef CAS PubMed.
- A. Armstrong, L. H. Jones, J. D. Knight and R. D. Kelsey, Oxaziridine-Mediated Amination of Primary Amines: Scope and Application to a One-Pot Pyrazole Synthesis, Org. Lett., 2005, 7, 713–716 CrossRef CAS PubMed.
- (a) L. A. Carpino, B. J. Cohen, K. E. Stephens, S. Y. Sadat-Aalaee, J. H. Tien and D. C. Langridge, (Fluoren-9-ylmethoxy)carbonyl (Fmoc) amino acid chlorides. Synthesis, characterization, and application to the rapid synthesis of short peptide segments, J. Org. Chem., 1986, 51, 3732–3734 CrossRef CAS; (b) Y. M. Elbatrawi, C. W. Kang and J. R. Del Valle, Total Synthesis of L-156,373 and an oxoPiz Analogue via a Submonomer Approach, Org. Lett., 2018, 20, 2707–2710 CrossRef CAS PubMed.
- L. S. Wolfe, M. F. Calabrese, A. Nath, D. V. Blaho, A. D. Miranker and Y. Xiong, Protein-induced photophysical changes to the amyloid indicator dye thioflavin T, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16863–16868 CrossRef CAS PubMed.
- L. C. Serpell, Alzheimer's amyloid fibrils: structure and assembly, Biochim. Biophys. Acta, 2000, 1502, 16–30 CrossRef CAS.
- B. Bacsa, S. Bosze and C. O. Kappe, Direct solid-phase synthesis of the beta-amyloid (1-42) peptide using controlled microwave heating, J. Org. Chem., 2010, 75, 2103–2106 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02009e |
| This journal is © The Royal Society of Chemistry 2020 |