
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew diterpenoid quinones derived from Salvia miltiorrhiza and their cytotoxic and neuroprotective activities†
Zhao-Kun Yin,
Zi-Ming Feng,
Jian-Shuang Jiang,
Xu Zhang,
Pei-Cheng Zhang * and
Ya-Nan Yang*
* and
Ya-Nan Yang*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Peking Union Medical College, Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing 100050, China. E-mail: pczhang@imm.ac.cn; yyn@imm.ac.cn; Fax: +86 10 63017757; Tel: +86 10 63165231
First published on 8th April 2020
Abstract
One new tanshinone derivative, which possesses an unusual 6/6/5/6 fused-ring skeleton system (1), together with four new five-membered lactone benzohexa-membered ring compounds (2, 3, 4A and 4B), and three new carboxyl substituted 5,5-spiroketal compounds (5–7), were isolated from the dried rhizomes of Salvia miltiorrhiza. The structures of these compounds were determined by multiple spectral analyses (UV, IR, NMR, and HR-ESI-MS). In addition, the absolute configurations were established by X-ray diffraction experiments, calculated and experimental circular dichroism spectra. Evaluation of antitumor activity showed that 1 had strong cytotoxicity to tumor-repopulating cells (TRCs) with an IC50 value of 2.83 μM. In the evaluation of neuroprotective activity, 4A and 6 showed a strong improvement in the survival rates of SK-N-SH cell injury induced by oxygen glucose deprivation (OGD).
Introduction
Diterpenoid quinones are a kind of rosin diterpenes that are mainly composed of (11,12)-o-phenanthraquinone and (11,14)-p-phenanthraquinone. The root of Salvia miltiorrhiza Bunge (Labiatae) is known as a traditional Chinese medicine (TCM) which was found to be abundant in diterpenoid quinones.1–5 Long-term pharmacological studies have found that diterpenoid quinones have significant activities, especially in terms of antitumor and cardiovascular activities.4–9 In recent years, with continuous deep research on this kind of component from S. miltiorrhiza, new derivatizations of diterpenoid quinones have been discovered. For example, two diterpenoid quinones, which have a five-membered lactone benzohexa-membered ring structure,10 and five 5,5-spiroketals,12–14 have been isolated from S. miltiorrhiza Bunge (Labiatae), with these two components having neoteric basic skeleton forms of 6/6/5, 6/6/5/5 respectively. Although the amount of each compound was less, the discovery of these compounds with novel structures and significant activities reinvigorated our enthusiasm for the in-depth exploration of S. miltiorrhiza.In a study of the biologically active constituents in the ethyl acetate-soluble portion of S. miltiorrhiza root bark, which was acquired from an 80% EtOH extract, one new tanshinone derivative (1), which possessed an unusual ring-C compared with the common tanshinone skeleton, was obtained (Fig. 1). Furthermore, four new diterpenoid quinones (2, 3, 4A and 4B), which all contained a 6/6/5 skeleton, and three new 5,5-spiroketal compounds (5–7) that had the feature of a carboxylic acid-substituted helical lactone ring, were isolated. Based on the source route analysis, these three types of components were all derived from (11,12)-o-phenanthraquinone or (11,14)-p-phenanthraquinone. Evaluation of antitumor activity and neuroprotective activity results of these isolated products were also reported.
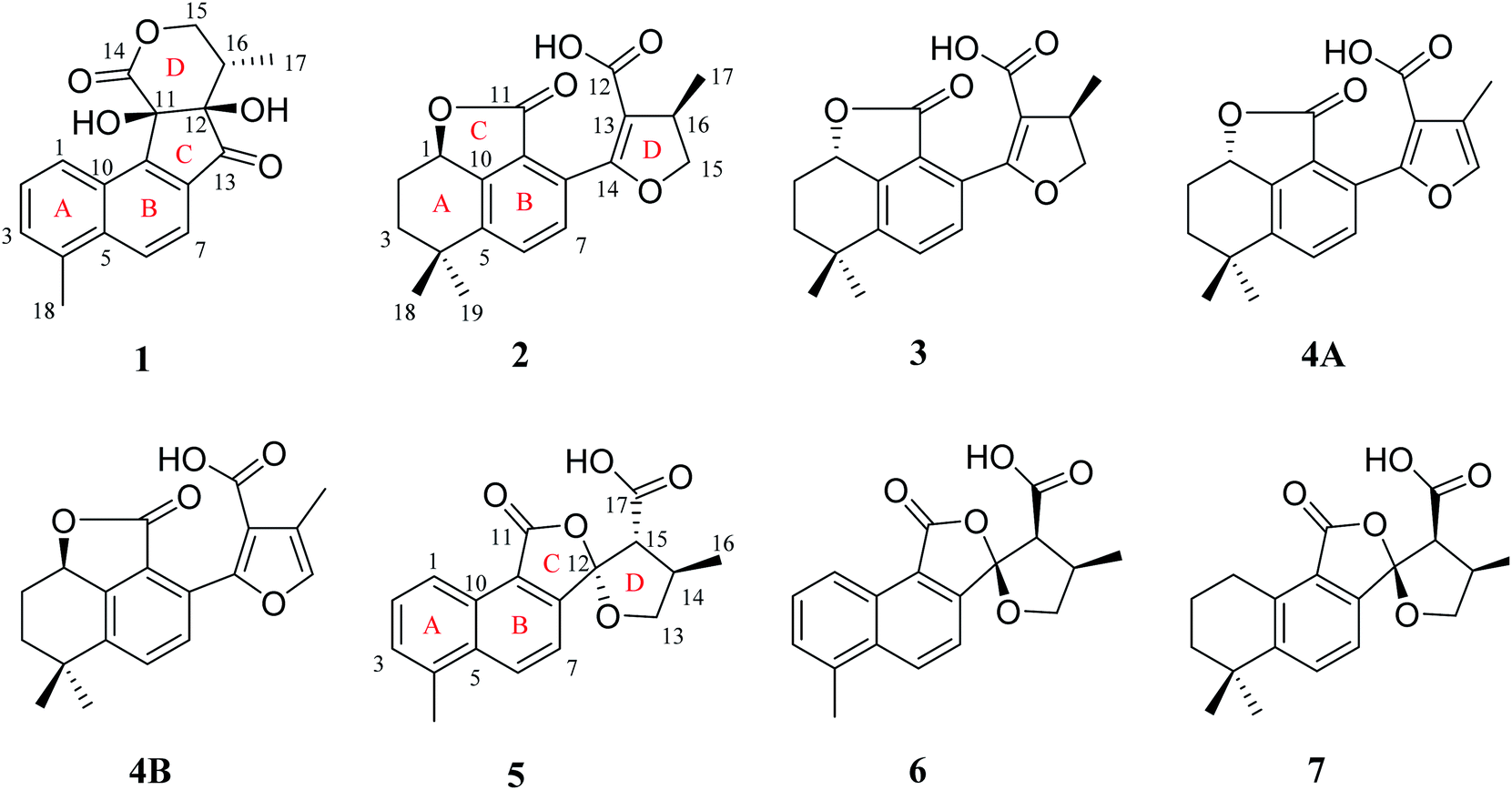 | ||
| Fig. 1 The structures of compounds 1–7. | ||
Results and discussion
Compound 1 was isolated as white amorphous powder. Its molecular formula was established as C18H16O5 by its negative HRESIMS ion at m/z 311.0924 [M − H]− (calcd for C18H15O5, 311.0925), which indicated 11 degrees of unsaturation. The IR spectrum showed the absorptions of hydroxy (3433 cm−1), carbonyl (1725 cm−1) and olefinic (1616 cm−1) groups. Its 1H NMR data (Table 1) were indicative of an AMX pattern for three aromatic protons at δH 8.32 (1H, d, J = 8.5 Hz, H-1), 7.65 (1H, dd, J = 6.5, 8.5 Hz, H-2), and 7.61 (1H, d, J = 6.5 Hz, H-3) and a pair of ortho-aromatic protons at δH 8.30 (1H, d, J = 8.5 Hz, H-6), 7.79 (1H, d, J = 8.5 Hz, H-7). This information, together with a methyl signal at δH 2.72 (3H, s, H-18), indicated that 1 contained a 4-methylnaphthalene unit, which was similar to that of tanshinone I.19 In addition, two oxygenated methylene protons at δH 3.95 (1H, dd, J = 3.5, 12.0 Hz, H-15a), 3.15 (1H, t, J = 12.0 Hz, H-15b), one methine proton at δH 2.28 (1H, m, H-16), and one methyl signal at δH 0.93 (3H, d, J = 7.5 Hz, H-17), were observed in the upfield region of the 1H NMR spectrum. The 13C NMR data (Table 1) and HSQC spectra revealed 18 carbon signals; apart from 11 carbons assigned to the 4-methylnaphthalene unit, the remaining seven carbons could be attributed to four quaternary carbons, one methine carbon, one methylene carbon and one methyl group. In the HMBC spectrum, the correlations from H-15 to C-12, C-16, and C-17, H-16 to C-12, C-13, C-15, and C-17, as well as OH-12 to C-12, C-13, and C-16 revealed the presence of a 2,4-dihydroxy-3-methyl butanone moiety in 1 (Fig. 2). Additionally, the characteristic HMBC correlations from H-15 to C-14, H-7 to C-13, OH-11 to C-11, C-9, and C-12 established the unusual five-membered ring (ring C) and six-membered ring (ring D) in 1, which consisted of C-8/C-9/C-11/C-12/C-13 and C-11/C-12/C-14/O/C-15/C-16. Then, the planar structure was elucidated as shown in Fig. 1 with a 6/6/5/6-membered ring skeleton.| No. | 1a | 2b | 3b | 4A/4Bb | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| a Data were measured in DMSO-d6.b Data were measured in CDCl3. | ||||||||
| 1 | 8.32, d (8.5) | 123.2 | 5.19, dd (5.5, 12.0) | 78.0 | 5.19, dd (5.5, 12.0) | 77.8 | 5.22, dd (5.5, 11.5) | 77.8 |
| 2 | 7.65, dd (6.5, 8.5) | 127.8 | 2.39, m, 1.62, m, 1.62, m | 26.3 | 2.39, m, 1.62, m | 26.3 | 2.40, m, 1.62, m | 26.3 |
| 3 | 7.61, d (6.5) | 130.5 | 1.92, m, 1.85, m | 38.0 | 1.92, m, 1.85, m | 38.2 | 1.92, m, 1.86, m | 37.1 |
| 4 | 133.5 | 34.9 | 34.9 | 34.9 | ||||
| 5 | 135.9 | 144.6 | 144.8 | 144.1 | ||||
| 6 | 8.30, d (8.5) | 128.4 | 7.55, d (8.0) | 131.1 | 7.55, d (8.0) | 131.5 | 7.65, d (8.0) | 131.2 |
| 7 | 7.79, d (8.5) | 118.3 | 7.47, d (8.0) | 130.0 | 7.47, d (8.0) | 130.0 | 7.52, d (8.0) | 130.3 |
| 8 | 129.2 | 126.9 | 126.8 | 126.5 | ||||
| 9 | 148.9 | 122.9 | 123.2 | 123.0 | ||||
| 10 | 136.4 | 148.0 | 148.0 | 148.3 | ||||
| 11 | 78.9 | 168.5 | 168.5 | 168.9 | ||||
| 12 | 83.4 | 170.1 | 170.1 | 169.0 | ||||
| 13 | 202.0 | 111.5 | 111.6 | 122.5 | ||||
| 14 | 173.0 | 163.3 | 163.0 | 154.0 | ||||
| 15 | 3.95, dd (3.5, 12.0), 3.15, t (12.0) | 68.2 | 4.72, t, (9.0), 4.23, dd (5.5, 9.0) | 78.9 | 4.70, t (9.0), 4.24, dd (5.5, 9.0) | 78.9 | 7.32, s | 140.6 |
| 16 | 2.28, m | 40.5 | 3.49, m | 37.2 | 3.40, m | 37.1 | 117.0 | |
| 17 | 0.93, d (7.5) | 11.3 | 1.34, d (6.5) | 19.6 | 1.38, d, (7.0) | 20.0 | 2.23, s | 10.1 |
| 18 | 2.72, s | 19.6 | 1.44, s | 31.8 | 1.44, s | 31.8 | 1.45, s | 31.8 |
| 19 | 1.19, s | 30.9 | 1.19, s | 31.0 | 1.22, s | 31.0 | ||
| OH-11 | 6.70, s | |||||||
| OH-12 | 6.34, s | |||||||
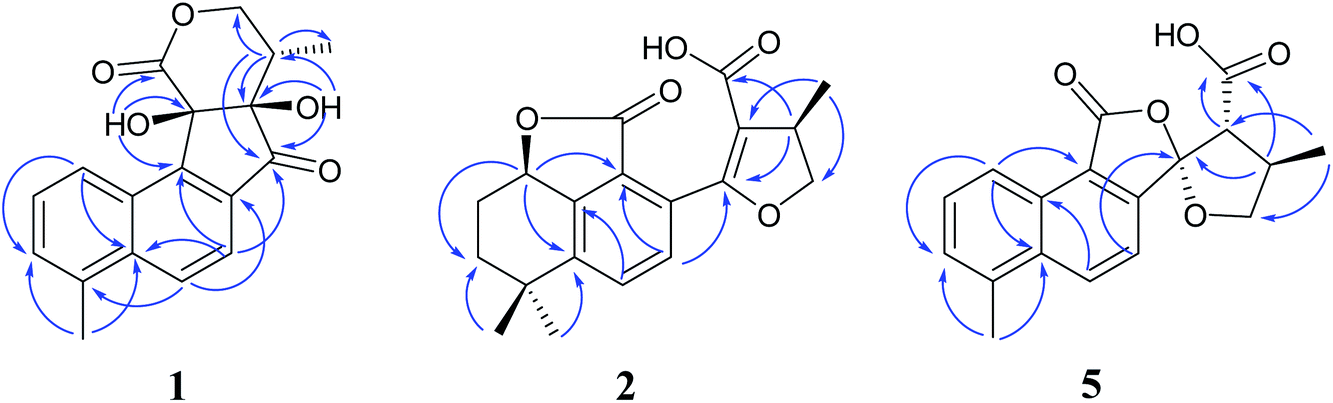 | ||
| Fig. 2 Key HMBC correlations of compounds 1, 2, 5. | ||
The absolute configurations of C-11, C-12, and C-16 were identified by the ROESY experiment and comparison of the experimental and calculated ECD spectra. In the ROESY experiment, the correlations from OH-11 to H-16 and OH-12 confirmed the cis-relationship between OH-11 and H-16, as well as the cis-relationship between OH-11 and OH-12 (Fig. 3). This result was further verified by a strong correlation from OH-12 to H-16, together with a weak correlation from OH-12 to H-17. From the above analysis, 1 had only one pair of enantiomers (1a: 11R,12R,16S and 1b: 11S,12S,16R). A systematic conformational analysis was performed for 1a using a molecular mechanics force field (MMFF94) calculation. The optimized conformation of 1a was further obtained using the time-dependent density functional theory (TDDFT) method at the B3LYP/6-311+G (d, p) level. The overall calculated ECD spectra of 1a was established based on the Boltzmann weighting of the lowest energy conformers. Finally, the calculated ECD spectrum of 1a was matched with the experimental result over the entire range of wavelengths (Fig. 4). Based on the above evidence, the structure of 1 was determined to be as shown in Fig. 1 and was named tanshin cyclopentanone A.
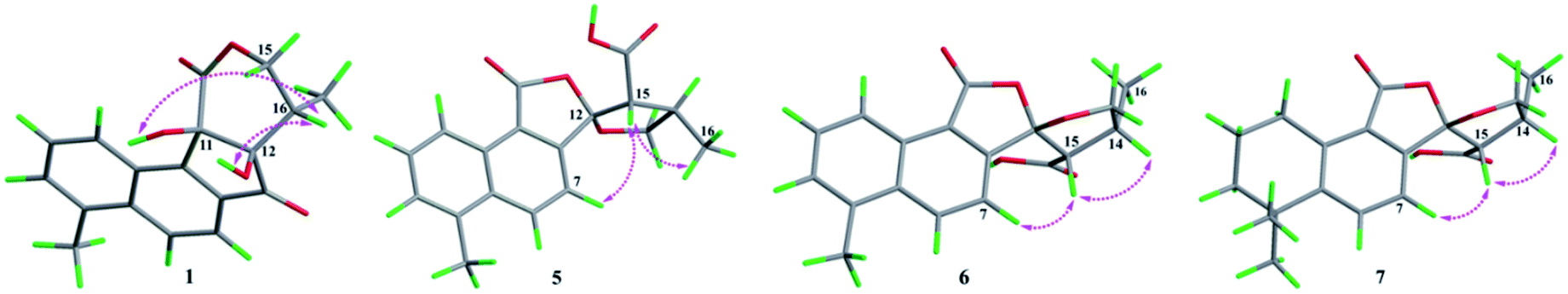 | ||
| Fig. 3 The ROESY correlations of compounds 1, 5–7. | ||
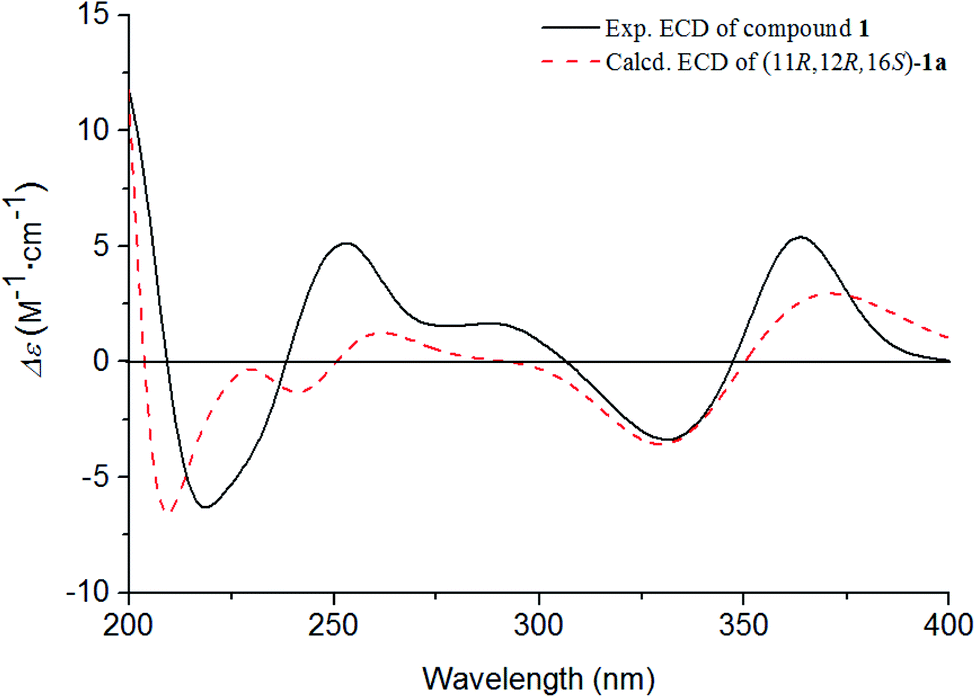 | ||
| Fig. 4 Experimental and calculated ECD spectra of 1. | ||
Compound 2, obtained as white massive crystal, was indicated to have the molecular formula of C19H20O5 according to the HRESIMS m/z 351.1198 [M + Na]+ (calcd for C19H20NaO5, 351.1203). The IR spectrum indicated that 2 contained carboxyl (2955, 1766 cm−1) and carbonyl (1666 cm−1) functional groups. Its 13C NMR data (Table 1) showed 19 carbon signals, including two carbonyl carbons, eight aromatic carbon signals and nine aliphatic carbon signals. In the 1H NMR data (Table 1), a group of aromatic hydrogen signals appeared in the downfield region at δH 7.55 (1H, d, J = 8.0 Hz, H-6), 7.47 (1H, d, J = 8.0 Hz, H-7). A set of –CH2CH2– characteristic signals were observed at δH 2.39 (1H, m, H-2a), 1.62 (2H, m, H-2b), 1.92 (1H, m, H-3a), 1.85 (1H, m, H-3b). In the upfield region, based on the HSQC spectrum, the characteristic signals of a methyl substituted dihydrofuran ring at δH 4.72 (1H, t, J = 9.0 Hz, H-15a), 4.23 (1H, dd, J = 5.5, 9.0 Hz, H-15b), 3.49 (1H, m, H-16), 1.34 (3H, d, J = 6.5 Hz, H-17) were observed. The 1D NMR information of 2 was almost identical to the 1R-hydroxy-anhydride of 16R-cryptotanshinone,15 which was obtained via biotransformation by Mucor rouxii. Moreover, the HMBC correlations found in 2 were also the same as those of the 1R-hydroxy-anhydride of 16R-cryptotanshinone (Fig. 2).
However, the single-crystal X-ray diffraction experiment (Cu Kα radiation) showed that 2 possessed a 6/6/5 skeleton structure rather than a 6/6/7/5 skeleton of 1R-hydroxy-anhydride of 16R-cryptotanshinone (Fig. 5). This result showed that it is difficult to distinguish 2 and 1R-hydroxy-anhydride of 16R-cryptotanshinone only by using the 2D NMR data. The absolute configurations of 2 were determined to be 1R,16R according to the X-ray diffraction analysis. This result was also confirmed by the calculated ECD data of (1R,16R)-2, which matched well with the experimental ECD data of 2 (Fig. S8, ESI†). Therefore, the structure of 2 was established and named salvianolactone acid A.
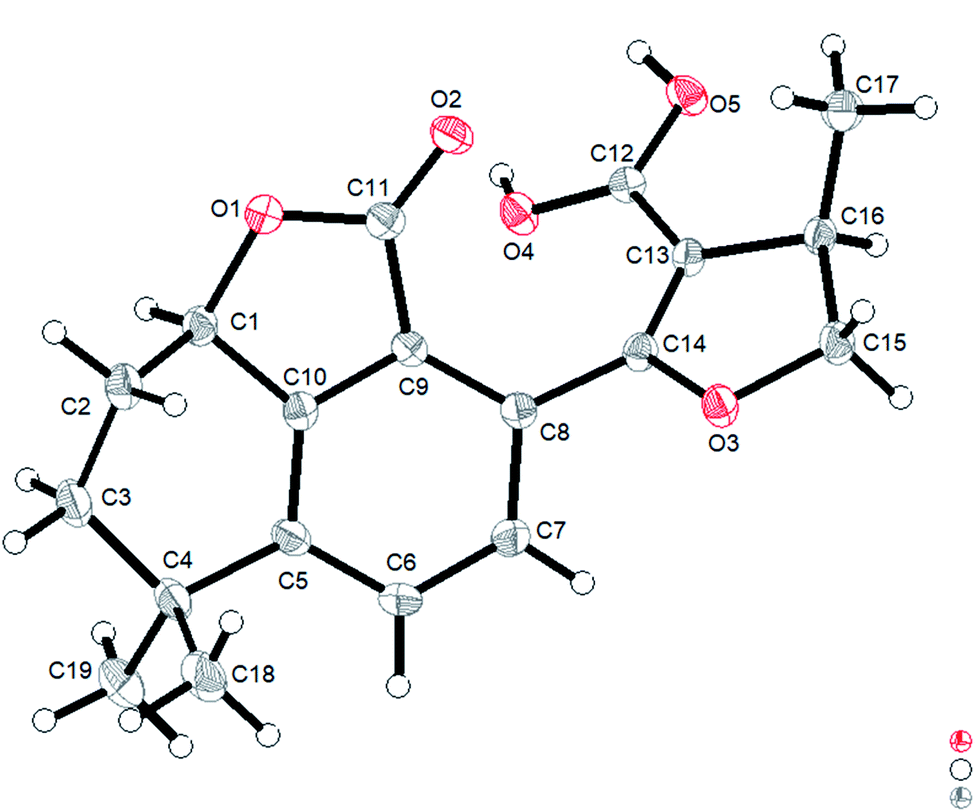 | ||
| Fig. 5 ORTEP diagram of 2. | ||
Compound 3 was isolated as white amorphous powder, and had the same molecular formula as 2 based on the HRESIMS m/z 351.1198 [M + Na]+ (calcd for C19H20NaO5, 351.1203). The UV spectrum and 1D, 2D NMR data of 3 were also similar to 2, and these features illustrated that 3 possessed the same planar construction with 2. However, the HPLC analysis and the nuances of the 1D NMR data between 2 and 3 indicated that 3 not an enantiomer but an epimer of 2. The absolute configurations of 3 might be 3a (1S,16R) or 3b (1R,16S). As a result of the experimental and calculated ECD spectra, the calculated ECD data of 3a matched well with the experimental ECD data of 3 (Fig. S9, ESI†). Thus, the structure of 3 was determined and named salvianolactone acid B.
Compounds 4A and 4B are a pair of enantiomers, which were obtained through chiral pre-HPLC. Their molecular formulas was determined to be C19H18O5 based on the HRESIMS m/z 325.1079 [M − H]− (calcd for C19H17O5, 325.1082). Analyzation of the 1D NMR data of 3 and 4A/4B revealed that the main difference between 3 and 4A/4B was ring D. The chemical shifts of C-15 (δC 140.6) and C-16 (δC 117.0) confirmed the furan ring moiety in 4A/4B, which was supported by the HMBC correlations of CH3-17 with C-13, C-15, C-16, H-7 with C-5, C-9, C-14, and H-15 with C-13, C-14, C-16.
The absolute configurations of this pair of enantiomers were established by experimental and calculated ECD. As a result, the (1S)-enantiomer matched well with the experimental ECD spectra of 4A, and the (1R)-enantiomer was in agreement with the experimental ECD spectra of 4B (Fig. S10, ESI†). Therefore, the structures of 4A and 4B were elucidated and named salvianolactone acid C and salvianolactone acid D, respectively.
Compound 5, a white amorphous powder, had the molecular formula of C18H16O5 as established by the HRESIMS ion at m/z 311.0927 [M − H]− (calcd for C18H15O5, 311.0925). The IR spectrum indicated that 5 contained carbonyl groups (1762 and 1726 cm−1). The 1H NMR data (Table 2) of 5 was showed to have the typical structure of the methyl substituted naphthalene ring and included an AMX pattern at δH 8.78 (1H, d, J = 8.5 Hz, H-1), 7.56 (1H, dd, J = 7.0, 8.5 Hz, H-2), 7.44 (1H, d, J = 7.0 Hz, H-3), a group of ortho-aryl hydrogen signals at δH 8.33 (1H, d, J = 8.5 Hz, H-6), 7.55 (1H, d, J = 8.5 Hz, H-7), and one methyl group at δH 2.71 (3H, s, H-18). The 13C NMR data (Table 2) displayed 18 carbon signals; in addition to the 11 carbon signals on the methyl substituted naphthalene ring unit, 5 contained two carbonyl groups (δC 168.2, 172.2), one oxygenated quaternary carbon group (δC 111.4), two methine groups (δC 34.6, 59.2), one methylene group (δC 76.1) and one methyl group (δC 17.1). These NMR data were similar to those of epi-danshenspiroketallactone A,12 except for the ethyl ester group in epi-danshenspiroketallactone A. The HMBC correlations (Fig. 2) of H-7 with C-5, C-9, C-12, CH3-16 with C-13, C-14, C-15, H-14 with C-17, and CH3-16 with C-17 were verified the planar structure of 5 as shown in Fig. 1.
| No. | 5 | 6 | 7 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 8.78, d (8.5) | 122.5 | 8.82, d (8.5) | 122.4 | 3.17, t (6.0) | 26.1 |
| 2 | 7.56, dd (7.0, 8.5) | 129.1 | 7.57, t (7.0, 8.5) | 129.1 | 1.82, m | 18.6 |
| 3 | 7.44, d (7.0) | 128.7 | 7.45, d (7.0) | 128.7 | 1.68, m | 38.4 |
| 4 | 129.3 | 129.3 | 34.6 | |||
| 5 | 133.7 | 133.7 | 149.2 | |||
| 6 | 8.33, d (8.5) | 132.3 | 8.32, d (8.5) | 132.1 | 7.62, d (8.0) | 133.2 |
| 7 | 7.55, d (8.5) | 118.0 | 7.48, d (8.5) | 117.7 | 7.18, d (8.0) | 118.9 |
| 8 | 146.2 | 146.4 | 144.4 | |||
| 9 | 122.2 | 122.9 | 124.5 | |||
| 10 | 135.3 | 135.2 | 137.9 | |||
| 11 | 168.2 | 168.5 | 169.0 | |||
| 12 | 111.4 | 109.8 | 109.8 | |||
| 13 | 4.51, t (8.0), 3.85, t (8.0) | 76.1 | 4.45, t (8.0), 4.07, dd (3.5, 8.5) | 76.6 | 4.36 t (7.5), 3.97, dd (3.5, 8.5) | 76.2 |
| 14 | 3.22, m | 34.6 | 3.04, m | 34.4 | 2.94, m | 34.3 |
| 15 | 3.20, overlap | 59.2 | 3.63, d (8.0) | 54.7 | 3.48, d (8.0) | 55.4 |
| 16 | 1.32, d (5.5) | 17.1 | 1.43, d (7.0) | 16.3 | 1.28, s | 16.1 |
| 17 | 172.2 | 171.6 | 172.3 | |||
| 18 | 2.71, s | 20.0 | 2.72, s | 20.0 | 1.30, s | 32.0 |
| 19 | 1.30, s | 31.9 | ||||
In the case of CDCl3 as a deuterated reagent, H-13 and H-16 overlapped. Therefore, DMSO-d6 was used as the deuterated reagent, and these two signals can be separated and appeared at δH 3.59 (1H, d, J = 11.0 Hz, H-13) and 2.95 (1H, m, H-16), respectively (Table S1, ESI†). In the NOE spectrum (Fig. S56, ESI†), irradiation of CH3-16 enhanced H-15. Furthermore, the ROESY correlations (Fig. 3) of H-7 with H-15, H-15 with CH3-16 indicated that the absolute configurations of 5 might be 5a (12S,14R,16R) or 5b (12R,14S,16S). The calculated ECD spectra of 5a and 5b showed that 5a agreed with the experimental spectrum of 5 (Fig. S11, ESI†); therefore, the structure of 5 was determined and named epi-danshenspiroketallactone B.
The planar structure of 6 was established as the same as 5 based on the 1D and 2D NMR data. The NOE spectrum (Fig. S67, ESI†) showed that irradiation with H-14 enhanced H-15. What's more, the ROESY experiment displayed that H-14 had correlation with H-15, and H-7 had correlation with H-15 (Fig. 3). Therefore, the absolute configurations of 6 might be 6a (12R,14R,15S) or 6b (12S,14S,15R). In the calculated ECD results, the spectrum of 6a agreed with the experimental spectrum of 6 (Fig. S12, ESI†), so the structure of 6 was established and named epi-danshenspiroketallactone C.
Compound 7 was isolated as white amorphous powder and had the molecular formula of C19H22O5 via the HRESIMS ion at m/z 329.1396 [M − H]− (calcd for C19H21O5, 329.1395). The 1H NMR data (Table 2) showed two aromatic protons, four methylene groups, two methine groups and three methyl groups. The 13C NMR spectrum (Table 2) of 7 displayed 19 carbon signals. Comparison of 7 with 6 showed that the main difference was in the structure of ring A. The chemical shifts of δH 3.17 (2H, t, J = 6.0 Hz, H-1), 1.82 (2H, m, H-2), 1.68 (2H, m, H-3) and δH 1.30 (6H, s, H-18,19) confirmed the dimethyl substituted six-membered ring of 7, which was also determined by the HMBC correlations of H-1 with C-2, C-3, C-5, C-9, C-10 and H-18/19 with C-3, C-4, and C-5.
In the NOE spectrum (Fig. S78, ESI†), irradiation of H-14 enhanced H-15. What's more, the ROESY correlations (Fig. 3) of H-15 with H-14, H-7 with H-15 illustrated that the absolute configurations of 7 might be either 7a (12R,14R,16S) or 7b (12S,14S,16R). Both 7a and 7b underwent ECD calculations, and 7a matched the experimental spectrum of 7 (Fig. S13, ESI†), so the structure of 7 was finally determined and named epi-danshenspiroketallactone D.
Structurally, 1 represents a new skeleton of tanshinone derivative with an unusual 6/6/5/6-membered ring skeleton. Its distinctive biogenetic route is proposed in Scheme 1. A literature survey indicated that the essential precursor neocryptotanshinone,16 which was isolated from the roots of S. miltiorrhiza previously, might be derived from ferruginol through a series of aromatization, oxidation, Diels–Alder reaction, rearrangement, hydrogenation and oxidation reactions. Subsequently, neocryptotanshinone formed XI through the oxidative cracking of ring C, hydrogenation, and cyclization. Finally, 1 was formed by aromatization and lactonization of X1. In particular, during the procedure of forming of 1, the key process is the construction of a cyclopentanone moiety, which is unique in the tanshinone derivative. According to the above biosynthetic pathway perspective, the absolute configuration of C-16 remained constant during the progression of ring cracking and recycling of neocryptotanshinone.17,18
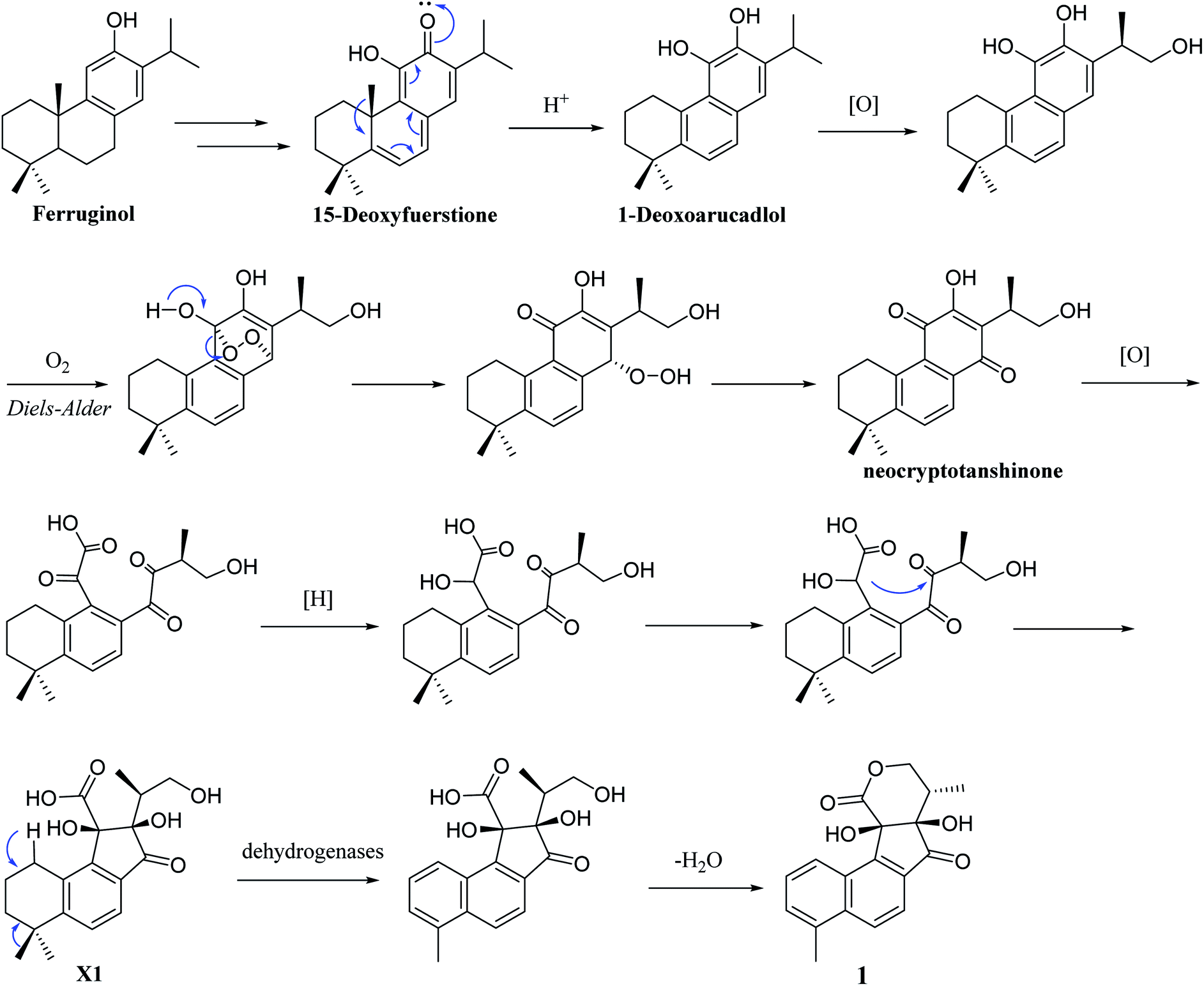 | ||
| Scheme 1 Plausible biogenetic pathway for 1. | ||
In addition, 2–7 contained two types of skeleton structures, which might all derivate from cryptotashinone (Scheme 2).11 During a series of oxidation, hydrogenation, and cracking rearrangement of ring C/D under active enzymatic steps, cryptotashinone could derive various products with multiple structures.10
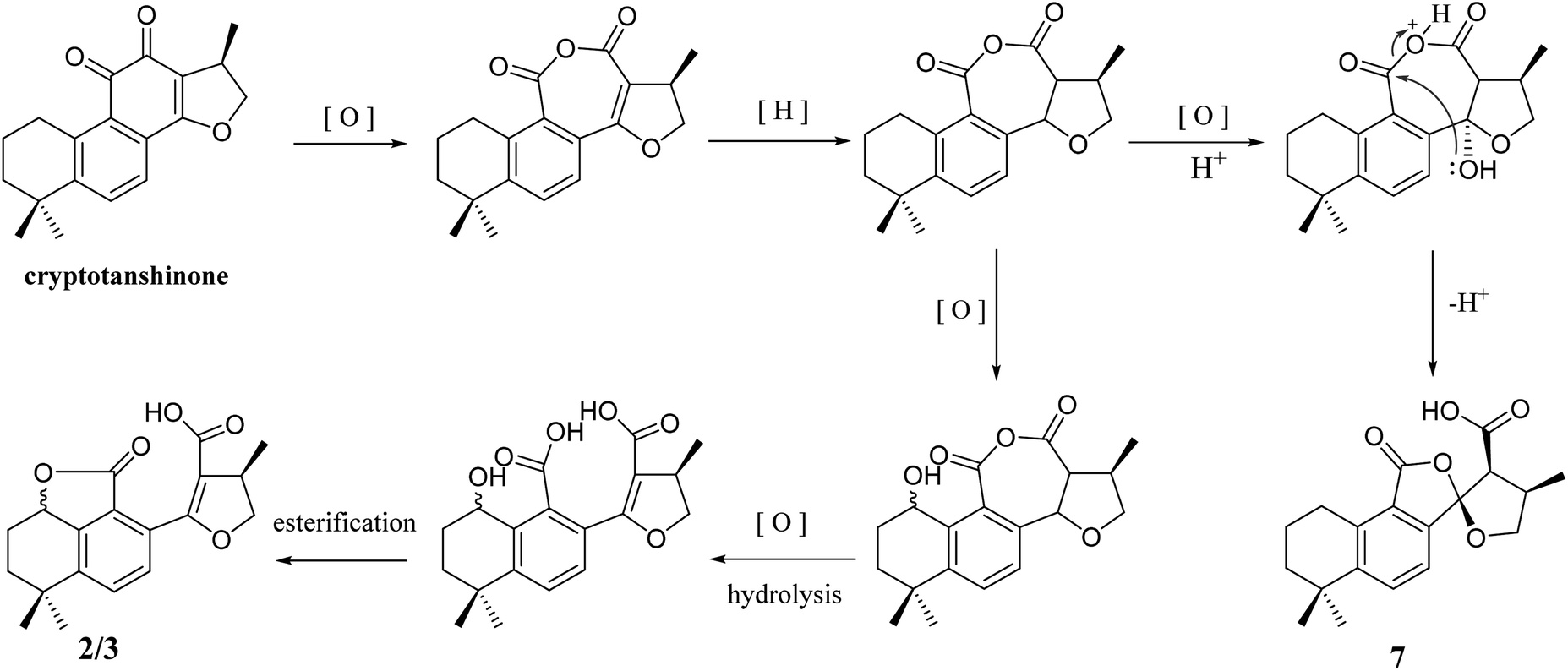 | ||
| Scheme 2 Plausible biogenetic pathway for 2/3 and 7. | ||
TRCs play an important role during the process of tumor migration and recurrence. Therefore, it is a research hotspot to explore an effective targeted agent to kill TRCs. An in vitro assay showed that 1 had strong cytotoxicity toward A375 TRCs (IC50 = 2.83 μM), which were generated from a 3D fibrin gel culture system.20 Delightedly, 1 exhibited no cytotoxicity to the nonstem-like A375 cancer cells at a concentration of 100 μM by the MTT method. This result implied that 1 might be a potent targeted antitumor agent with less adverse effects. In the evaluation of neuroprotective activities, 4A showed obvious activity to increase the survival rate (13.08%) of SK-N-SH cell injury induced by oxygen glucose deprivation (OGD) compared with the positive control drug PHPB (7.43%). And under the same activity screening model, compound 6 also showed a noteworthy improvement in the survival rate (10.48%) compared with PHPB.
Experimental
General experimental procedures
The optical rotations and ECD spectra were experimented by RUDOLPH automatic V polarimeter JASCO V650 and J-815 spectrometer (JASCO, Easton, MD, USA), respectively. The UV spectra was measured on JASCO V-650. The IR data were measured on Nicolet 5700 spectrometer (Thermo Scientific, FL, USA). The NMR spectra were recorded with Bruker 500 MHz (Bruker-Biospin, Billerica, MA, USA) and 600 MHz NMR spectrometers (Varian, Inc., Palo Alto, CA, USA). HRESIMS reports were obtained from Agilent 6520 HPLC-Q-TOF (Agilent Technologies, Waldbronn, Germany). Preparative HPLC was performed using a Shimadzu LC-10AT with an ODS-A column (250 mm × 20 mm, 5 μm; YMC Corp., Kyoto, Japan). The Agilent 1260 series system coupled with an Apollo C18 column (250 mm × 4.6 mm, 5 μm; Alltech Corp., KY, USA) were used for HPLC-DAD experiments. RP-18 (50 μm, YMC Corp., Kyoto, Japan), Sephadex LH-20 (Pharmacia Fine Chemicals, Uppsala, Sweden), and silica gel (200–300 mesh, Qingdao Ocean Chemical Plant) were used as chromatographic substrates. Chiral-phase separation was performed by the Chiralpak AD-RH and AD-H chiral column (250 mm × 10 mm, 5 μm; Daicel Corp., Tokyo, Japan). Analytical chiral-phase HPLC was performed by the Chiralpak AD-RH chiral column (250 mm × 4.6 mm, 5 μm; Daicel Corp., Tokyo, Japan).Fungal material
The dried rhizomes of Salvia miltiorrhiza were collected in Rizhao City (Shandong Province, China) in March 2017; the plant was authenticated by Lin Ma. A voucher specimen (herbarium no. ID-S-2944) has been deposited at the herbarium of the Department of Medicinal Plants, Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing, China.Extraction and isolation
The dried rhizomes of Salvia miltiorrhiza (70 kg) were smashed and extracted with 80% EtOH (3 × 100 L) at 85 °C for 2 h. The extract was concentrated under reduced pressure to obtained 23 kg of paste. Add water in the paste to make a suspension and extract four times with ethyl acetate. The ethyl acetate extract (2.2 kg) was subjected to silica gel (200–300 mesh) open column chromatography with a stepwise gradient of petroleum ether–acetone (100/0 to 0/100) gave twelve fractions (Fr.1–12). Fr.6 and Fr.7 (a total of about 100 g) were further subjected to column chromatography over silica gel and eluted with a gradient of PE–EtOAc to yield fractions y1–y30. Then, Fr.y22–Fr.y24 (27.6 g) was separated by silica gel column chromatography eluted with a gradient of PE–EtOAc mobile phase system and finally give six fractions A–F (2.38, 3.15, 5.87, 6.66, 6.89 and 1.80 g, respectively). Fraction E was further separated by Sephadex LH-20 (CH2Cl2–MeOH, gradient) and RP-HPLC (MeOH–H2O = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 for first time; MeCN–H2O = 50:50 for second time) to yield compound 2 (82.6 mg), compound 3 (27.3 mg), compound 4A/4B (21.5 mg). Fr.y25–Fr.y29 (16.8 g) was separated by silica gel column chromatography eluted with a gradient of dichloromethane–methanol mobile phase system to yield five fractions A1–E1 (4.85, 3.16, 2.65, 3.22 and 2.53 g, respectively). Fraction D1 were further separated by Sephadex LH-20 (CH2Cl2–MeOH, gradient) and RP-HPLC (MeOH–H2O = 70:30 for first time; MeCN–H2O = 50:50 for second time) to yield compound 1 (4.9 mg), compound 5 (10.2 mg), compound 6 (21.0 mg), compound 7 (7.0 mg). The flow rate of the RP-HPLC was 1 mL min−1, and the detection wavelength was 254 nm.
ε) 210 (2.10), 257 (2.51), 293 (1.69), 349 (1.22) nm; IR νmax 3433, 2958, 2929, 1725, 1616, 1593, 1469, 1383, 1246, 1125, 1022, 822, 779 cm−1; CD (MeOH) 219 (Δε −6.30), 253 (Δε +5.13), 331 (Δε −3.37), 364 (Δε +5.38) nm; 1H NMR (DMSO-d6, 500 MHz) and 13C NMR (DMSO-d6, 125 MHz) spectroscopic data, see Table 1; HR-ESI-MS m/z 311.0924 [M − H]− (calcd for C18H15O5, 311.0925).ε) 210 (2.39), 241 (2.01), 308 (1.61) nm; IR νmax 2955, 2649, 1766, 1666, 1493, 1338, 1247, 1072, 1007, 945, 844 cm−1; CD (MeOH) 215 (Δε +3.59), 234 (Δε −0.78), 260 (Δε +4.30), 322 (Δε −3.05); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 1; HR-ESI-MS m/z 351.1198 [M + Na]+ (calcd for C19H20NaO5, 351.1203).ε) 210 (2.36), 240 (2.00), 304 (1.58) nm; IR νmax 2961, 2871, 1767, 1664, 1497, 1440, 1072, 1045, 1005, 945, 843 cm−1; CD (MeOH) 231 (Δε +1.47), 257 (Δε −1.68), 296 (Δε +1.18) nm; 1H NMR (CDCl3, 500 MHz) spectroscopic data and 13C NMR (CDCl3, 125 MHz), see Table 1; HR-ESI-MS m/z 351.1198 [M + Na]+ (calcd for C19H20NaO5, 351.1203).ε) 207 (2.41), 241 (2.07), 295 (1.74), 329 (1.91) nm; IR νmax 2961, 2868, 1766, 1687, 1554, 1490, 1435, 1301, 1219, 1069, 1005, 970, 841 cm−1; CD (MeOH) 240 (Δε +2.16), 262 (Δε −6.09), 325 (Δε +3.01) nm (4A), CD (MeOH) 239 (Δε −3.89), 261 (Δε +4.50), 325 (Δε −2.32) nm (4B); 1H NMR (DMSO-d6, 500 MHz) spectroscopic data and 13C NMR (CDCl3, 125 MHz), see Table 1; HR-ESI-MS m/z 325.1079 [M − H]− (calcd for C19H17O5, 325.1082).ε) 211 (2.44), 244 (2.39), 313 (1.65) nm; IR νmax 3567, 2894, 1762, 1726, 1587, 1329, 1305, 1242, 1206, 1071, 985, 818 cm−1; CD (MeOH) 239 (Δε −7.52), 260 (Δε +4.13) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 2, 1H NMR (DMSO-d6, 500 MHz) spectroscopic data, see Table S1 in ESI;† HR-ESI-MS m/z 311.0927 [M − H]− (calcd for C18H15O5, 311.0925).ε) 211 (2.37), 244 (2.33), 313 (1.59) nm; IR νmax 3567, 3449, 2977, 1750, 1717, 1586, 1335, 1192, 1060, 974, 815 cm−1; CD (MeOH) 207 (Δε +1.90), 222 (Δε −0.94), 240 (Δε +3.86), 259 (Δε −2.75), 304 (Δε −0.93), 326 (Δε −0.76) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 2, 1H NMR (DMSO-d6, 500 MHz) spectroscopic data, see Table S1 in ESI;† HR-ESI-MS m/z 311.0927 [M − H]− (calcd for C18H15O5, 311.0925).ε) 208 (2.44), 241 (1.63), 291 (1.18) nm; IR νmax 3209, 2959, 1757, 1594, 1432, 1335, 1308, 1175, 1061, 927, 832 cm−1; CD (MeOH) 217 (Δε +1.50), 253 (Δε −1.25) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 2, 1H NMR (DMSO-d6, 500 MHz) spectroscopic data, see Table S1 in ESI;† HR-ESI-MS m/z 329.1396 [M − H]− (calcd for C19H21O5, 329.1395).
:30 for first time; MeCN–H2O = 50:50 for second time) to yield compound 2 (82.6 mg), compound 3 (27.3 mg), compound 4A/4B (21.5 mg). Fr.y25–Fr.y29 (16.8 g) was separated by silica gel column chromatography eluted with a gradient of dichloromethane–methanol mobile phase system to yield five fractions A1–E1 (4.85, 3.16, 2.65, 3.22 and 2.53 g, respectively). Fraction D1 were further separated by Sephadex LH-20 (CH2Cl2–MeOH, gradient) and RP-HPLC (MeOH–H2O = 70:30 for first time; MeCN–H2O = 50:50 for second time) to yield compound 1 (4.9 mg), compound 5 (10.2 mg), compound 6 (21.0 mg), compound 7 (7.0 mg). The flow rate of the RP-HPLC was 1 mL min−1, and the detection wavelength was 254 nm.
ε) 210 (2.10), 257 (2.51), 293 (1.69), 349 (1.22) nm; IR νmax 3433, 2958, 2929, 1725, 1616, 1593, 1469, 1383, 1246, 1125, 1022, 822, 779 cm−1; CD (MeOH) 219 (Δε −6.30), 253 (Δε +5.13), 331 (Δε −3.37), 364 (Δε +5.38) nm; 1H NMR (DMSO-d6, 500 MHz) and 13C NMR (DMSO-d6, 125 MHz) spectroscopic data, see Table 1; HR-ESI-MS m/z 311.0924 [M − H]− (calcd for C18H15O5, 311.0925).ε) 210 (2.39), 241 (2.01), 308 (1.61) nm; IR νmax 2955, 2649, 1766, 1666, 1493, 1338, 1247, 1072, 1007, 945, 844 cm−1; CD (MeOH) 215 (Δε +3.59), 234 (Δε −0.78), 260 (Δε +4.30), 322 (Δε −3.05); 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 1; HR-ESI-MS m/z 351.1198 [M + Na]+ (calcd for C19H20NaO5, 351.1203).ε) 210 (2.36), 240 (2.00), 304 (1.58) nm; IR νmax 2961, 2871, 1767, 1664, 1497, 1440, 1072, 1045, 1005, 945, 843 cm−1; CD (MeOH) 231 (Δε +1.47), 257 (Δε −1.68), 296 (Δε +1.18) nm; 1H NMR (CDCl3, 500 MHz) spectroscopic data and 13C NMR (CDCl3, 125 MHz), see Table 1; HR-ESI-MS m/z 351.1198 [M + Na]+ (calcd for C19H20NaO5, 351.1203).ε) 207 (2.41), 241 (2.07), 295 (1.74), 329 (1.91) nm; IR νmax 2961, 2868, 1766, 1687, 1554, 1490, 1435, 1301, 1219, 1069, 1005, 970, 841 cm−1; CD (MeOH) 240 (Δε +2.16), 262 (Δε −6.09), 325 (Δε +3.01) nm (4A), CD (MeOH) 239 (Δε −3.89), 261 (Δε +4.50), 325 (Δε −2.32) nm (4B); 1H NMR (DMSO-d6, 500 MHz) spectroscopic data and 13C NMR (CDCl3, 125 MHz), see Table 1; HR-ESI-MS m/z 325.1079 [M − H]− (calcd for C19H17O5, 325.1082).ε) 211 (2.44), 244 (2.39), 313 (1.65) nm; IR νmax 3567, 2894, 1762, 1726, 1587, 1329, 1305, 1242, 1206, 1071, 985, 818 cm−1; CD (MeOH) 239 (Δε −7.52), 260 (Δε +4.13) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 2, 1H NMR (DMSO-d6, 500 MHz) spectroscopic data, see Table S1 in ESI;† HR-ESI-MS m/z 311.0927 [M − H]− (calcd for C18H15O5, 311.0925).ε) 211 (2.37), 244 (2.33), 313 (1.59) nm; IR νmax 3567, 3449, 2977, 1750, 1717, 1586, 1335, 1192, 1060, 974, 815 cm−1; CD (MeOH) 207 (Δε +1.90), 222 (Δε −0.94), 240 (Δε +3.86), 259 (Δε −2.75), 304 (Δε −0.93), 326 (Δε −0.76) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 2, 1H NMR (DMSO-d6, 500 MHz) spectroscopic data, see Table S1 in ESI;† HR-ESI-MS m/z 311.0927 [M − H]− (calcd for C18H15O5, 311.0925).ε) 208 (2.44), 241 (1.63), 291 (1.18) nm; IR νmax 3209, 2959, 1757, 1594, 1432, 1335, 1308, 1175, 1061, 927, 832 cm−1; CD (MeOH) 217 (Δε +1.50), 253 (Δε −1.25) nm; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) spectroscopic data, see Table 2, 1H NMR (DMSO-d6, 500 MHz) spectroscopic data, see Table S1 in ESI;† HR-ESI-MS m/z 329.1396 [M − H]− (calcd for C19H21O5, 329.1395).X-ray crystallographic data for salvianolactone acid A
Salvianolactone acid A (2) was recrystallized from CH2Cl2 and MeOH (3:1) to give colorless block crystals. The X-ray crystallographic structure of 2 was obtained by anomalous scattering of Cu Kα radiation. Crystal data: C19H20O5, M = 328.35, hexagonal, a = 14.63788(16) Å, c = 13.6616(2) Å, U = 2535.06(7) Å3, T = 109.90(10), space group P64 (no. 172), Z = 6, μ(Cu Kα) = 0.767, 14929 reflections measured, 3057 unique (Rint = 0.0262) which were used in all calculations. The final wR(F2) was 0.0771 (all data). Flack parameter, x = 0.09(6). The complete data were deposited at the Cambridge Crystallographic Data Centre (CCDC 1975214).†
Antitumor activities of compounds 1–7
The details evaluation method of antitumor activities is same as involved in the literature.20Neuroprotective activities of compounds 1–7
The screening method of neuroprotective activities refer to the literature.21Conclusion
A tanshinone derivative (1) with an unusual 6/6/5/6 skeleton structure, four new diterpenoid quinones (2, 3, 4A and 4B), and three new 5,5-spiroketal compounds (5–7) were isolated from the roots of Salvia miltiorrhiza. All of the compounds were screened for their antitumor and neuroprotective activities. The results indicated that 1 had strong cytotoxicity to A375 TRCs (IC50 = 2.83 μM); 4A and 6 showed obvious neuroprotective activities based on the increased survival rate of SK-N-SH cell injury induced by oxygen glucose deprivation (OGD).Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This research was financially supported by Fundamental Research Funds for CAMS/PUMC (2018RC350011), The Drug Innovation Major Project (2018ZX09711001-008), and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (No. 2017-I2M-3-010).References
- M. H. Li, Q. Q. Li, C. H. Zhang, N. Zhang, Z. H. Cui, L. Q. Huang and P. G. Xiao, Acta Pharm. Sin. B, 2013, 3, 273–280 CrossRef.
- G. H. Du and J. T. Zhang, Her. Med., 2004, 23, 355–360 CAS.
- J. J. Wu, Q. L. Ming, X. Zhai, S. Q. Wang, B. Zhu, Q. L. Zhang, Y. B. Xu, S. S. Shi, S. C. Wang, Q. Y. Zhang, T. Han and L. P. Qin, Carbohydr. Polym., 2019, 223, 115125 CrossRef CAS PubMed.
- C. Y. Su, Q. L. Ming, K. Rahman, T. Han and L. P. Qin, Chin. J. Nat. Med., 2015, 13, 163–182 CAS.
- Y. B. Wu, Z. Y. Ni, Q. W. Shi, M. Dong, H. Kiyota, Y. C. Gu and B. Cong, Chem. Rev., 2012, 112, 5967–6026 CrossRef CAS PubMed.
- A. Watzke, S. J. O'Malley, R. G. Bergman and J. A. Ellman, J. Nat. Prod., 2006, 69, 1231–1233 CrossRef CAS PubMed.
- D. J. Cousins, in Medicinal, essential oil, culinary herb and pesticidal plants of the labiatae, ed. D. J. Cousins, CAB International, Wallingford, Oxford, U.K., 1994, part 2, pp. 244–284 Search PubMed.
- D. G. Kong, H. Oh, E. J. Sohn, T. Y. Hur, K. C. Lee, K. J. Kim, T. Y. Kim and H. S. Lee, Life Sci., 2004, 75, 1801–1816 CrossRef PubMed.
- B. Q. Wang, J. Med. Plants Res., 2010, 4, 2813–2820 CAS.
- L. Z. Li, X. Liang, X. Sun, X. L. Qi, J. Wang, Q. C. Zhao and S. J. Song, Org. Biomol. Chem., 2016, 14, 10050–10057 RSC.
- S. Y. Lee, C. D. Y. Choi and E. R. Woo, Arch. Pharmacal Res., 2005, 28, 909–913 CrossRef CAS PubMed.
- D. W. Zhang, X. Liu, D. Xie, R. D. Chen, X. Y. Tao, J. H. Zou and J. G. Dai, Chem. Pharm. Bull., 2013, 61, 576–580 CrossRef CAS PubMed.
- H. W. Luo, S. X. Chen, J. N. Lee and J. K. Snyder, Phytochemistry, 1988, 27, 290–292 CrossRef CAS.
- F. Asari, T. Kusumi, G. Z. Zheng, Y. Z. Cen and H. Kakisawa, Chem. Lett., 1990, 19, 1885–1888 CrossRef.
- W. N. He, Y. Li, Y. J. Qin, X. M. Tong, Z. J. Song, Y. Z. R. Wei, L. Li, H. Q. Dai, W. Z. Wang, H. W. Luo, X. Ye, L. X. Zhang and X. T. Liu, Appl. Microbiol. Biotechnol., 2017, 101, 6365–6374 CrossRef CAS PubMed.
- A. R. Lee, W. L. Wu, W. L. Chang, H. C. Lin and M. L. King, J. Nat. Prod., 1987, 50, 157–160 CrossRef CAS PubMed.
- Y. Tomita and Y. Ikeshiro, J. Chem. Soc., Chem. Commun., 1987, 520, 1311–1313 RSC.
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, Wiley, New York, 1994, pp. 413–442 Search PubMed.
- M. J. Don, C. C. Shen, W. J. Syu, Y. H. Ding and C. M. Sun, Phytochemistry, 2006, 67, 497–503 CrossRef CAS PubMed.
- Y. Y. Liu, X. Y. Liang, X. N. Yin, J. D. Lv, K. Tang, J. W. Ma, T. T. Ji, H. F. Zhang, W. Q. Dong, X. Jin, D. G. Chen, Y. C. Li, S. Y. Zhang, H. D. Q. Xie, B. Zhao, T. Zhao, J. Z. Lu, Z. W. Hu, X. T. Cao, F. X. F. Qin and B. Huang, Nat. Commun., 2017, 8, 15207 CrossRef PubMed.
- S. W. Huang, J. W. Qiao, X. Sun, P. Y. Gao, L. Z. Li, Q. B. Liu, B. Sun, D. L. Wu and S. J. Song, Funct. Foods, 2016, 24, 183–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1975214. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02022b |
| This journal is © The Royal Society of Chemistry 2020 |