
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding CO2 capture kinetics and energetics by ionic liquids with molecular dynamics simulation
Fan Yang†
a,
Xianjuan Wang†a,
Yang Liua,
Yanmei Yang b,
Mingwen Zhaoa,
Xiangdong Liu*a and
Weifeng Li*a
b,
Mingwen Zhaoa,
Xiangdong Liu*a and
Weifeng Li*a
aSchool of Physics and State Key Laboratory of Crystal Materials, Shandong University, Jinan, Shandong 250100, China. E-mail: xdliu@sdu.edu.cn; lwf@sdu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Institute of Molecular and Nano Science, Shandong Normal University, Jinan, 250014, China
First published on 6th April 2020
Abstract
Room temperature ionic liquids (ILs) are recognized to be potential media for CO2 capture, but the interaction nature is less documented which hinders the future development of ILs with high CO2 solvation capability. Here, through all atom molecular dynamics (MD) simulations, the solvation process of CO2 with four representative ILs, [EMIM][BF4], [BMIM][BF4], [EMIM]CL and [BMIM]CL was systematically studied. Our data clearly reflect the fact that hydrophobic components from both cations and anions dominate CO2 solvation because IL–CO2 attraction is mainly driven by the van der Waals attractions. Consequently, cations with longer alkyl chain (for instance, [BMIM]+ than [EMIM]+) and anions with higher hydrophobicity (for instance, [BF4]− than CL−) effectively enhance CO2 solvation. For all the ILs under study, addition of water into ILs abates CO2 solvation through regulating the anion–CO2 interactions. Moreover, it is worth mentioning that ILs with a hydrophobic anion ([BF4]− in this study) are more resistant to the existence of water to capture CO2 than ILs with a hydrophilic anion (Cl− in this study). Free energy decomposition analyses were conducted which support the above findings consistently. Generally, it is predicted that cations with long alkyl chain, anions with high hydrophobicity, and ILs with smaller surface tension are potentially effective CO2 capturing media. Our present study helps the deep understanding of the CO2 capturing mechanism by ILs and is expected to facilitate the future design and fabrication of a novel IL medium for gas capture and storage.
1. Introduction
Room temperature ionic liquids (ILs) are a new class of organic salts whose melting temperature falls below the conventional limit of 100 °C. Compared with conventional organic solvents, they possess many intriguing qualities, such as low vapor pressure, low toxicity, and high thermodynamic stability which have made these ionic compounds the solvents of choice for the so-called green chemistry.1,2ILs are regarded as potential candidates for CO2 storage, the dominant reason for the global warming process.3 It is remarkable that, compared with the conventional amine scrubbing approach,4 the organic and non-volatilizing nature of ILs can effectively decrease the energy cost for CO2 capture and avoid the concern of causing environmental pollution.5 For instance, Liu et al. have reported single-stage and multi-stage CO2 adsorption processes by ILs which could effectively decrease the electrical and thermal energy costs by 42.8% and 66.04%, respectively.6 Moreover, several ILs demonstrated CO2-selective capturing capability which can be utilized for the purpose of CO2 separation and purification from gas mixture.7 Numerous research groups have systematically studied the capturing capacity of various ILs for various gases, including CO2, CH4, H2 and so on, by both experimental and theoretical approaches.1,2,8–15 Ramdin et al. experimentally studied the CO2 solubility and diffusivity in numbers of ILs and proposed that ILs have great solubility capacity to CO2.1 Anthony et al. reported that 1-n-butyl-3-methlimidazolium hexafluorophosphates have strong dissolving ability for CO2.2 Bates et al. discovered that a new room temperature ionic liquid (TSIL) has a considerable capacity to capture CO2 effectively, compared to the commercial amine sequestering reagents.13 Kerle et al. further reported that CO2 solubility is dependent to the alkane chain length of ILs.12 Shi et al. discovered that the ionic liquid ([HMIM][Tf2N]) demonstrates strong interaction with CO2, by conducting Monte Carlo simulations, which is six times stronger than the interaction between ILs and H2.15
Albeit a large number of works that have been conducted experimentally, the effective CO2 capture and storage are still challenges. Previous experiments have evaluated the effects of introduction of water to ILs for CO2 capture. For instance, Bermejo et al. have systematically explored the effects of water in ILs on CO2 solubility and found that only small ratio of water in ILs is favorable to CO2 solvation. In contrast, high concentration of water reversely lowered the CO2 solubility.16 Unfortunately, the molecular mechanism of the influence of water on the performance of ILs is still unknown. This situation becomes to be the main obstacle to the design and development of high-performance ILs for the aim of effective CO2 capture and storage.
In this work, the solvation process of CO2 by several ILs is studied by atomic molecular dynamics (MD) simulations. Four experimentally widely-studied ILs and the corresponding hydrated states were adopted as the representative solvents. Several key characters of ILs, including the specific types of cations and anions, the length of the alkyl chain, etc., have been systemically assessed on CO2 solvation. We sincerely hope that our present study could help the better and deep understanding of CO2 capturing mechanism by ILs and facilitate the future design and fabrication of novel IL medium for gas capture and storage.
2. Model setup and simulation details
All the MD simulations were performed with the GROMACS 5.1.3 package17 with GPU acceleration. Four types of representative ILs which are commonly studied in the experiments have been considered: [BMIM][BF4], [EMIM][BF4], [BMIM]CL and [EMIM]CL. The force field parameters developed by Chaban et al. through quantum mechanics calculations were used for ILs.18 TIP3P water model19 and the EMP2 CO2 model20 were used. During the simulation, a leap-frog algorithm was used to integrate Newton's equations of motion, and the time step was set to 2 fs. All the covalent bonds were constrained by the LINCS algorithm.21 The cut-off for the short-range electrostatics and Lennard–Jones interactions were both set to 1.0 nm and the particle mesh Ewald (PME) algorithm22 was used to deal with the long-range electrostatic interactions. The simulation systems were firstly optimized by an energy minimization with the steepest descent algorithm to generate an appropriate starting configuration, and then equilibrated for 500 ps at NPT ensemble to reach the reference temperature of 300 K and the reference pressure of 1 bar, respectively. Then 100 ns trajectory was generated for data collection at the NVT ensemble.In addition to the normal MD simulations, we further studied the CO2 solvation energetics in ILs by calculating the solvation free energy (ΔG) using the Free Energy Perturbation (FEP) method.23,24 To study the effects of water contents in the ILs, we have considered a series of water molar concentrations in the range of 0–96.2%. The components in the simulation boxes were depicted in Fig. 1. In the FEP calculations, 21 values were used for the coupling parameter λ with a resolution of 0.05. The process was divided into two parts: the perturbation of electrostatic interaction and the perturbation of van der Waals (vdW) interaction.
 | ||
| Fig. 1 (a) Molecular structures of [BMIM]+, [EMIM]+ and [BF4]−; (b) snapshot of the simulated box. The CO2 molecule is placed at the box center. | ||
3. Results and discussion
3.1 CO2 solvation in pure water and ILs
As the main aim of the current work is to understand the CO2 storage capability of ILs through a mechanism view of the solvation free energetics, the benchmark study of the CO2 solvation free energy in pure water (abbreviated as ΔGwater) is desired. The current molecular models gave a theoretical prediction of ΔGwater = 1.80 kJ mol−1 at 300 K. Compared to previous studies (Table 1), this agrees well with the experimental value of 1.005 kJ mol−1.25 To verify the validity of our present models of CO2 and ILs, we have calculated the solvation free energy of CO2 in pure [BMIM][BF4] at the 323 K and obtained a value of −1.00 kJ mol−1. This is well consistent with experimental value of −1.005 kJ mol−1 reported by Cadena et al.26In the following, we explore the solvation characteristics of CO2 in pure ILs at room temperature (300 K). Using the same setups in the above calculations, the solvation free energy of CO2 in ILs (abbreviated as ΔGIL) are calculated which are: −1.69 kJ mol−1 in [EMIM][BF4], −2.09 kJ mol−1 in [BMIM][BF4], −0.87 kJ mol−1 in [EMIM]CL and −1.92 kJ mol−1 in [BMIM]CL, respectively. Compared with the value in pure water of 1.80 kJ mol−1, the negative values clearly indicate that CO2 molecule is better dissolved in ILs which is consistent with experimental reports.
Deduced from four ΔGIL, the trend of CO2 dissolving capacities of the four ILs is as follows: [BMIM][BF4] > [BMIM][CL] > [EMIM][BF4] > [EMIM][CL]. Amongst them, the [BMIM][BF4] has the highest dissolving capacity for CO2. When comparing the cations, it is found that the length of the alkyl chain of cations has a positive correlation to CO2 solubility when the anion is same: [BMIM][BF4] > [EMIM][BF4]. In detail, the ΔGIL in [BMIM][BF4] is −0.40 kJ mol−1 lower than that of [EMIM][BF4]. These results are consistent with the experimental reports by Camper29 and Anthony.30 For two Chloride ILs, the trend is same: [BMIM][CL] > [EMIM][CL]. The free energy difference is even larger, which is −1.05 kJ mol−1. This is well consistent with previous experimental observations by Yunus31 and Aki.32 In addition, the specific types of anions also play a minor role. The ΔGIL of two [EMIM]+ ILs differs by −0.82 kJ mol−1 when a [BF4]− is replaced by Cl−. While for the two [BMIM]+ ILs, the difference is smaller which is −0.17 kJ mol−1. This is mainly attributed to the effective volumes (in other words, the hydrophobicity) of the anions which will be presented below in detail. It is worth mention that Zhang et al. also have demonstrated that the increase in alkyl chain of cation and the fluoration of anions or cations are beneficial to capture CO2.33,34
In order to probe the dominating molecular components that play the key role in the solvation process of CO2 in pure ILs, we analyzed the radial distribution functions (RDFs) of the imidazole ring, alkyl chain and anion around the center of mass (COM) of the CO2 molecule. As depicted in Fig. 2a and b, the first interaction shell (beginning from 0.25–0.3 nm) around CO2 is uniformly from the anions for both [BF4]− and Cl−. For comparison, the RDF peaks representing cation begin at a larger distance, ∼0.42 nm from CO2. These results indicate the fact that anions form direction interactions with CO2. Especially that for the [BF4]−, it forms more intimate interactions, through the Fluorine atoms, with CO2 with a closer distance of ∼0.25 nm. The compact anion shell, to certain extent, prevents cations from interacting with CO2. These phenomena are consistent with experimental results by Anthony30 and Zhang.35 As monovalent anions, the [BF4]− clearly has a larger volume than Cl−. Thus, [BF4]− is relatively more hydrophobic than Cl−. Zhang et al. also reported that [HMIM][FEP] has higher CO2 solubility than [HMIM][TF2N] experimentally because of the longer fluoroalkyl chain of [FEP].35
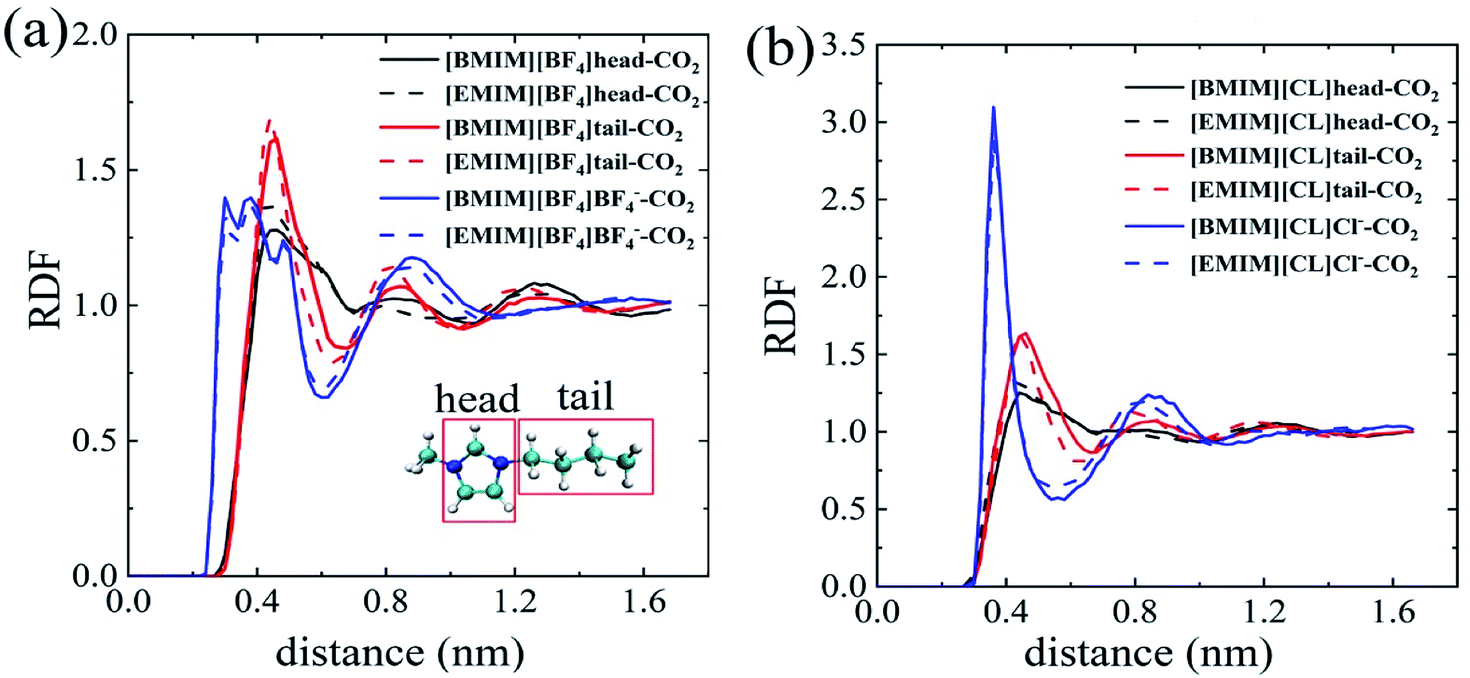 | ||
| Fig. 2 The radial distribution functions of ILs atoms around CO2 center of mass (COM). (a) [BMIM][BF4] solid line; [EMIM][BF4] dotted line; (b) [BMIM][CL] solid line; [EMIM][CL] dotted line. | ||
Further, we qualitatively compared different parts of cations in contacting with CO2. For brief, the imidazole ring of the cation is named as ‘head’ and the alkyl chain is named as ‘tail’ as depicted in the insert of Fig. 2a. For the four ILs, the heights of RDF peaks representing CO2–tail interactions are always higher than that of CO2–head interactions at the similar distance, regardless of the specific types of anions. Especially for the chloride ILs (Fig. 2b), there is almost no apparent peaks for the CO2–head interactions. This points to the fact that the alkyl chains of cations play the more important role in CO2 solvation and, more precisely, longer alkyl chain results in lower ΔGIL.
In combination with above anion analyses, it can be concluded that CO2 prefers to interact with hydrophobic components of cation and hydrophobic anions, although CO2 carries a dipole. Thus, it is a feasible way to search the suitable ILs with long alkyl chains or fluorinate anions for CO2 capture.
3.2 The effect of water in ILs on CO2 solvation
Previous experiments have demonstrated that the existence of water highly regulates CO2 capture. In detail, Bermejo et al. reported that small ratio of water in ILs is favorable to CO2 dissolving. In contrast, high concentration of water will reversely lower the CO2 solubility.16 Goodrich et al. also reported that the presence of water in ILs effectively reduced the CO2 storage capacity.36 Wang et al. reported that compared to pure triethylbutylammonium acetate ([N2224][Ac]), the introduction of H2O in [N2224][Ac] reduced the CO2 adsorption.37 However, the underline physical mechanism that accounts for the lowered performance of ILs is still poorly documented. To explain these experimental observations, we have considered the solvation of CO2 in a serious of hydrated ILs with molar ratios of water increased from 0% (pure ILs) to 96.2% (diluted ILs) and calculated the CO2 solvation free energies, ΔGh-IL.From Fig. 3a, it is very interesting to find that the tendencies of change of ΔGh-IL can be divided into two types according to anions. For Chloride ILs, the ΔGh-IL increases more quickly with the introduction of water. At water concentration of 83.3%, they approach to the value of ΔGwater (1.80 kJ mol−1). In contrast, ΔGh-IL in [EMIM][BF4] and [BMIM][BF4] increases in a much lower manner and reaches to a plateau of 0.8–1.0 kJ mol−1 when water content is as high as 96.2%.
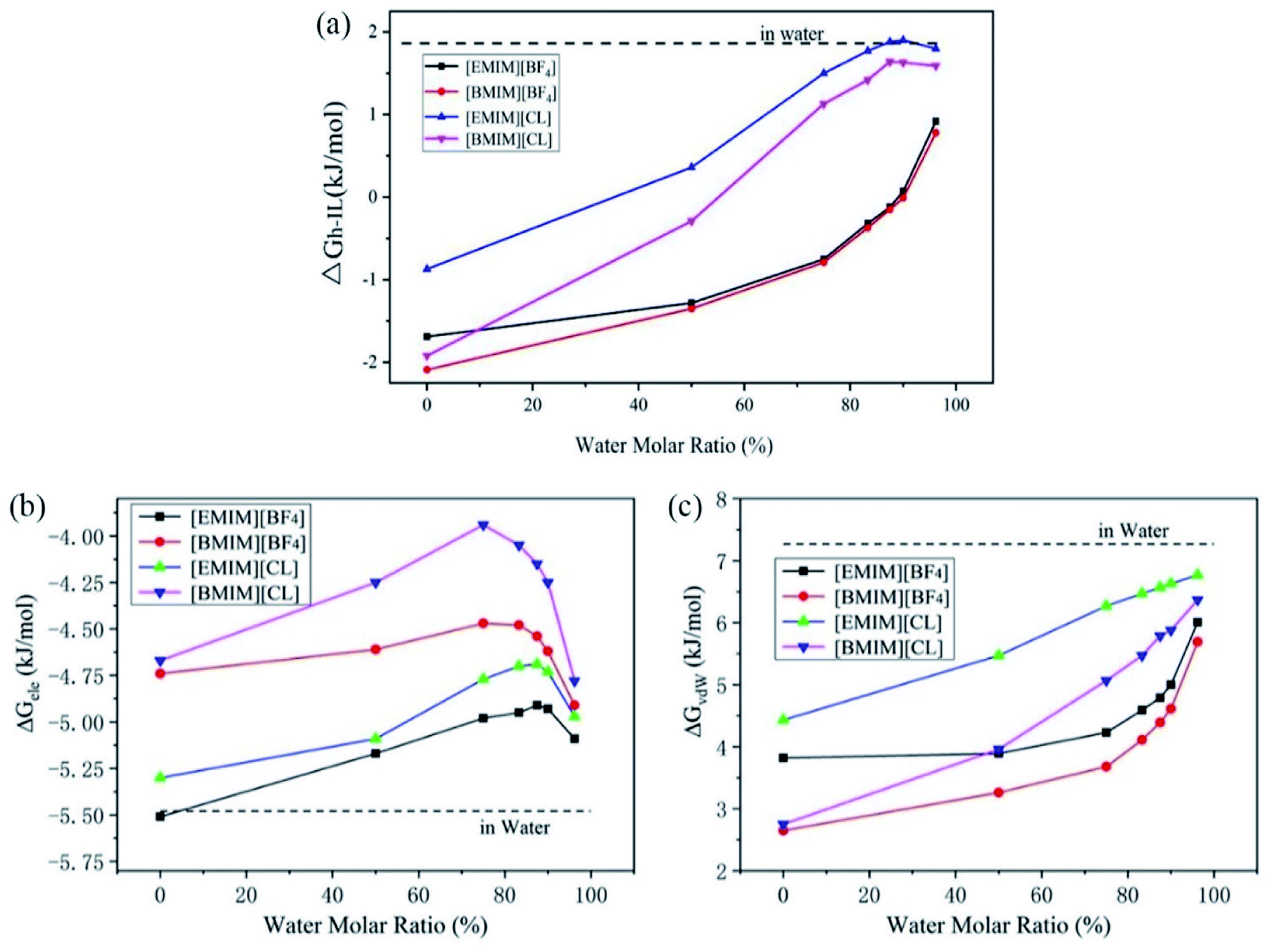 | ||
| Fig. 3 The change tendency of (a) total CO2 solvation free energy, (b) electronic component and (c) van der Waals component in hydrated ILs with different water molar ratio. The dashed lines represent the corresponding values in pure water. | ||
Then, the total ΔGh-IL is decomposed into the electrostatic (ΔGele) and vdW (ΔGvdW) contributions which is essential for a deep understanding of the physical interactions between CO2 and the solvents. As shown in Fig. 3b, the ΔGele term is always negative which reveals that energy–favorable interactions such as charge–charge and dipole–dipole attractions between CO2 and pure/hydrated ILs is the driving force for CO2 capture. In hydrated ILs, the ΔGele term firstly increases (weakened electrostatic interactions) at low water contents and the profiles reach to the maximum at a water content of ∼75% for two [BMIM]+ ILs, and ∼87.5% for two [EMIM]+ ILs. After that, further increasing water content reversely decreases the ΔGele (re-strengthened electrostatic interactions) which favors CO2 capture. This interesting transition is attributed to the formation of intimate interactions of CO2 with excess water molecules (more discussions are presented below from RDF analyses). It is worth mention that, the water fractions in ILs are relatively small in the experiments (which is typically lower than 30%). Thus, the current results support the previous observations because the experimental conditions correspond to the weakened ΔGele range in Fig. 3b.
For the van der Waals interactions, it represents the energy consumption for introducing a cavity inside the solution thus the ΔGvdW is positive. As shown in Fig. 3c, the ΔGvdW is monotonically increasing with the addition of water. Considering the nature of van der Waals interactions, this reflects that the surface tension of the hydrated ILs increases because more energy is required to introduce the cavity for CO2 molecule inside it. The ΔGvdW increase is considerably larger than ΔGele decrease. Thus, the overall solubility of CO2 in ILs generally decreases with addition of water. Moreover, as depicted in Fig. 3a, the CO2 solubility in ILs with [BF4]− anion is less regulated by water. From the decomposition analyses, this is mainly caused by the slower increase of ΔGvdW term in two [BF4]− ILs than that in Cl− ILs. Thus it can be seen that smaller surface tension and stronger electrostatic interaction with CO2 are two favorable factors to improve the CO2 solubility in ILs.
To probe the role of water in preventing ILs from adsorbing CO2, we analyzed spatial distributions of water, imidazole ring (head), alkyl chain (tail) and anions around the COM of CO2. For cations in IL with water ratio from 0–87.5%, the first peaks representing both head–CO2 and tail–CO2 bindings uniformly decreased in [EMIM][CL] (Fig. 4a) and [EMIM][BF4] (Fig. 4b). This reflects a fact that the cation–CO2 interactions were suppressed, resulting in ΔGele increase during this range which adopted the maximum value at water ratio of 87.5% as shown in Fig. 3b. Then, when further increasing water ratio from 87.5% to 96.2%, an interesting reverse trend was observed: these peaks reversely increased and were almost comparable to that in pure ILs, revealing that the interactions of cation–CO2 were almost recovered in diluted ILs. In addition to the formation of intimate binding of water to CO2 (dashed lines in Fig. 4c and d), ΔGele was found to decrease at this range (Fig. 3b). For anions, it is also interesting to find that two anions demonstrated distinct different behaviors in response to water. For Cl− (Fig. 4c), the first binding shell of Cl−–CO2 dramatically decreases with addition of water. This is attributed to the strong hydrophilic nature of Cl− which favors to bind to water instead of CO2. For [BF4]− (Fig. 4d), the first binding shell increases, but in a slow manner, when water content increases, indicating that water always enhances the binding between CO2 and [BF4]−. This is attributed to the large volume of [BF4]− which is classified to be hydrophobic anion. Thus, the ΔGh-IL of CO2 in hydrated ILs with Cl− anion increases more dramatically than [BF4]− as depicted in Fig. 3a.
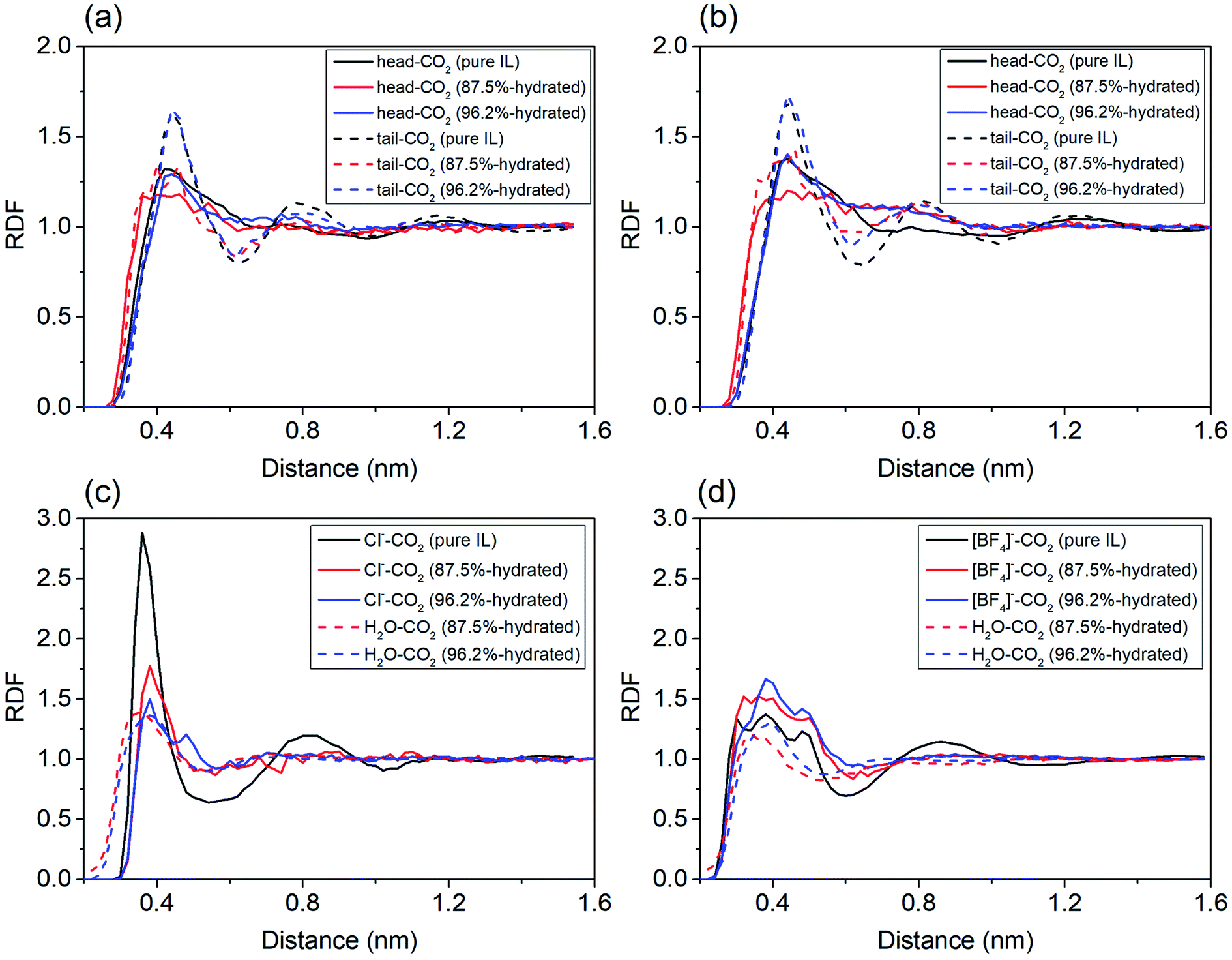 | ||
| Fig. 4 The radial distribution functions of solution components around CO2 in pure, 87.5% and 96.2% hydrated ILs. (a) and (c): [EMIM][CL]; (b)and (d) [EMIM][BF4]. | ||
By analyzing the RDFs, it is found that the dominate component to CO2 solubility of hydrated ILs is from the anion because the influence of water is closely connected to the hydrophilicity of anions. The current theoretical results predict that ILs with hydrophobic anions are more robust to capture CO2 when water exists.
3.3 Diffusions of ILs
One of the recognized features of ILs is the high viscosity that results in significantly lowered diffusion of the CO2 adsorbates. Bermejo et al. reported that small ratio of water in ILs is favorable to CO2 dissolving. In contrast, high concentration of water will reversely lower the CO2 solubility.16 While the introduction of water into ILs regulates the diffusion of CO2, less is known about the relationship between IL's viscosity and CO2 solubility. In the following, we examined the diffusion coefficients (D) of cation and anion, respectively, with respect to water contents using the Einstein relation.38As summarized in Fig. 5, it is found that the values of D of both anion and cation in hydrated ILs quickly increase with the increase of water content. For the four types of pure ILs under study, the D is considerably low, with an order of ∼0.6 ×10−11 m2 s−1. In diluted ILs, the D increases by around two orders. Moreover, comparing the four ILs, three aspects can be acquired: (1) the anions diffuse more quickly than cations, especially for Chloride ILs; (2) [EMIM]+ diffuses quicker than [BMIM]+ when anion is same; (3) Cl− diffuses quicker than [BF4]− when cation is same. These phenomena are consistent to the Stokes–Einstein theory which predicts that molecules with smaller size can diffuse faster. Generally, from these data, CO2 solvation in ILs is found to be negatively-correlated to the D of hydrated ILs. More specifically, hydrated ILs benefits CO2 diffusion, simultaneously it reduces CO2 storage capability.
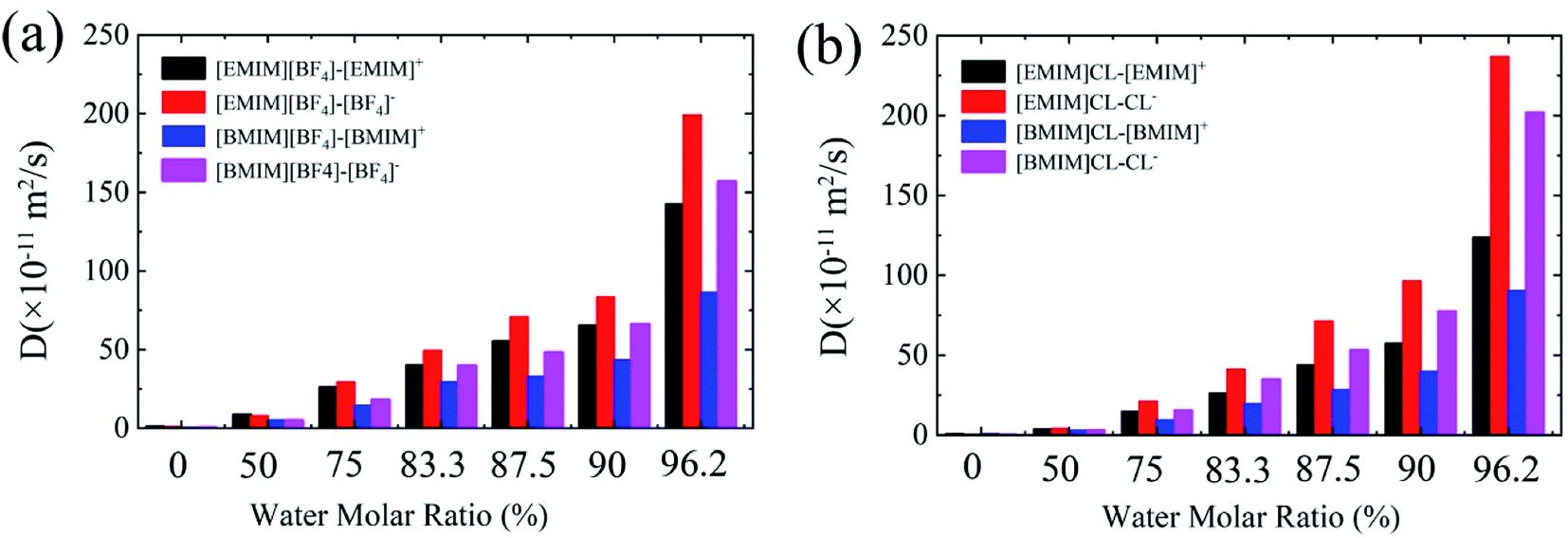 | ||
| Fig. 5 The diffusion coefficients of cations and anions in pure and hydrated ILs. | ||
4. Conclusions
To summarize, by MD simulations, the key factors of ILs that determine CO2 solvation were identified. For cations, the alkyl chain dominants CO2 capture because longer alkyl chain could effectively enhance CO2 solvation. While for anions, the hydrophobic species are proved to favor CO2 solvation. From free energy decomposition analyses, this is attributed to the interacting character between CO2 and the ILs which is mainly driven by the van der Waals attractions. The existence of water seriously abates CO2 solvation energy for all the ILs which is attributed to the weakened anion–CO2 interactions. More importantly, it is found that ILs with hydrophobic anions are more resistant to the existence of water to capture CO2 than ILs with hydrophilic anions. Through our analyses, a conclusion was made that ILs with long alkyl chain, hydrophobic anions, smaller surface tension and stronger electrostatic interaction with CO2 are indicative factors to improve the CO2 solubility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 11874238), the Basic Research Project of Natural Science Foundation of Shandong Province (Grant No. ZR2018MA034) and the Fundamental Research Funds of Shandong University.References
- M. Ramdin, T. W. de Loos and T. J. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef.
- J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315–7320 CrossRef.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef PubMed.
- H. Ahn, M. Luberti, Z. Liu and S. Brandani, Int. J. Greenhouse Gas Control, 2013, 16, 29–40 CrossRef.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross and J. P. Hallett, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- X. Liu, Y. Huang, Y. Zhao, R. Gani, X. Zhang and S. Zhang, Ind. Eng. Chem. Res., 2016, 55, 5931–5944 CrossRef.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668 RSC.
- S. Budhathoki, J. K. Shah and E. J. Maginn, Ind. Eng. Chem. Res., 2015, 54, 8821–8828 CrossRef.
- A. Yokozeki and M. B. Shiflett, Ind. Eng. Chem. Res., 2007, 46, 1605–1610 CrossRef.
- A. Yokozeki and M. B. Shiflett, Appl. Energy, 2007, 84, 351–361 CrossRef.
- D. Kerlé, R. Ludwig and D. Paschek, Z. Phys. Chem., 2013, 227, 167–176 CrossRef.
- D. Kerlé, R. Ludwig, A. Geiger and D. Paschek, J. Phys. Chem. B, 2009, 113, 12727–12735 CrossRef PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef PubMed.
- J. L. Anderson, J. K. Dixon and J. F. Brennecke, Acc. Chem. Res., 2007, 40, 1208–1216 CrossRef PubMed.
- W. Shi, D. C. Sorescu, D. R. Luebke, M. J. Keller and S. Wickramanayake, J. Phys. Chem. B, 2010, 114, 6531–6541 CrossRef PubMed.
- M. D. Bermejo, M. Montero, E. Saez, L. J. Florusse, A. J. Kotlewska, M. J. Cocero, F. van Rantwijk and C. J. Peters, J. Phys. Chem. B, 2008, 112, 13532–13541 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- V. V. Chaban, I. V. Voroshylova and O. N. Kalugin, Phys. Chem. Chem. Phys., 2011, 13, 7910–7920 RSC.
- M. F. Harrach and B. Drossel, J. Chem. Phys., 2014, 140, 174501 CrossRef PubMed.
- H. Liu, S. Dai and D.-e. Jiang, Nanoscale, 2013, 5, 9984–9987 RSC.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef.
- T. Darden, L. Perera, L. Li and L. Pedersen, Structure, 1999, 7, R55–R60 CrossRef PubMed.
- M. A. M. Latif, N. Micaêlo and M. B. A. Rahman, Chem. Phys. Lett., 2014, 615, 69–74 CrossRef.
- K. Jumbri, N. M. Micaelo and M. B. Abdul Rahman, Mol. Simul., 2017, 43, 19–27 CrossRef.
- J. Lide and H. P. R. Fredikse, CRC Handbook of Chemistry and Physics, CRC Press, 75th edn, 1995 Search PubMed.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef.
- N. Prasetyo and T. S. Hofer, J. Chem. Theory Comput., 2018, 14, 6472–6483 CrossRef.
- D. Jiao and S. B. Rempe, J. Chem. Phys., 2011, 134, 224506 CrossRef.
- D. Camper, C. Becker, C. Koval and R. Noble, Ind. Eng. Chem. Res., 2005, 44, 1928–1933 CrossRef.
- J. L. Anthony, J. L. Anderson, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2005, 109, 6366–6374 CrossRef.
- N. M. Yunus, M. A. Mutalib, Z. Man, M. A. Bustam and T. Murugesan, Chem. Eng. J., 2012, 189, 94–100 CrossRef.
- S. N. Aki, B. R. Mellein, E. M. Saurer and J. F. Brennecke, J. Phys. Chem. B, 2004, 108, 20355–20365 CrossRef.
- D. Almantariotis, T. Gefflaut, A. A. Padua, J.-Y. Coxam and M. Costa Gomes, J. Phys. Chem. B, 2010, 114, 3608–3617 CrossRef.
- X. Zhang, F. Huo, Z. Liu, W. Wang, W. Shi and E. J. Maginn, J. Phys. Chem. B, 2009, 113, 7591–7598 CrossRef.
- X. Zhang, Z. Liu and W. Wang, AIChE J., 2008, 54, 2717–2728 CrossRef.
- B. F. Goodrich, J. C. de la Fuente, B. E. Gurkan, Z. K. Lopez, E. A. Price, Y. Huang and J. F. Brennecke, J. Phys. Chem. B, 2011, 115, 9140–9150 CrossRef.
- G. Wang, W. Hou, F. Xiao, J. Geng, Y. Wu and Z. Zhang, J. Chem. Eng. Data, 2011, 56, 1125–1133 CrossRef.
- Z. Tang, L. Lu, Z. Dai, W. Xie, L. Shi and X. Lu, Langmuir, 2017, 33, 11658–11669 CrossRef.
Footnote |
| † These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |