
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDispersion-induced structural preference in the ultrafast dynamics of diphenyl ether†
Lian Wang ab,
Song Zhang*ab,
Ye Wangab and
Bing Zhang*ab
ab,
Song Zhang*ab,
Ye Wangab and
Bing Zhang*ab
aState Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Innovation Academy for Precision Measurement Science and Technology, Chinese Academy of Sciences, Wuhan 430071, China. E-mail: zhangsong@wipm.ac.cn; bzhang@wipm.ac.cn; Fax: +86-27-87198491; Tel: +86-27-87198491
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 11th May 2020
Abstract
Dispersion interactions are omnipresent in large aromatic systems and influence the dynamics as intermolecular forces. The structural preference induced by dispersion interactions is demonstrated to influence the excited state dynamics of diphenyl ether (DPE) using femtosecond time-resolved transient absorption (TA) associated with quantum chemical calculations. The experimental results in aprotic solvents show that the S1 state is populated upon irradiation at 267 nm with excess vibrational energy dissipating to solvent molecules in several picoseconds, and then decays via internal conversion (IC) within 50 ps as well as intersystem crossing (ISC) and fluorescence with a lifetime of nanoseconds. The polarity of the solvent disturbs the excited state energies and enhances the energy barriers of the ISC channel. Furthermore, the intermolecular dispersion interactions with protic solvents result in the OH–π isomer dominating in methanol and the OH–O isomer is slightly preferred in t-butanol in the ground state. The hydrogen bonded isomer measurements show an additional change from OH–O to OH–π geometry in the first 1 ps besides the relaxation processes in aprotic solvents. The time constants measured in the TA spectra suggest that the OH–O isomer facilitates IC. The results show that the OH–π isomer has a more rigid structure and a higher barrier for IC, making it harder to reach the geometric conical intersection through conformer rearrangement. This work enables us to have a good knowledge of how the structural preference induced by dispersion interactions affects excited state dynamics of the heteroaromatic compounds.
Introduction
Non-covalent interactions have been recognized as a very important factor in molecular recognition and aggregation processes,1–4 as well as the three-dimensional structure of proteins and DNA, which determines the function of these biomacromolecules.5,6 Undoubtedly, they determine deactivation pathways in light-driven processes. The chromophore of the green fluorescent protein can only emit fluorescence in the environments of noncovalent interactions with surrounding proteins, but not in the free condition without interactions.7–10 Hydrogen bonding and dispersion interactions, as important non-covalent interactions, act on the molecular systems individually or jointly and have attracted much attention.11–15 Hydrogen bonds have been proved to be of great significance in IC, photoinduced electron transfer, charge transfer, fluorescence quenching, etc.16–18 The formation of the hydrogen-bonded complex may be essential for the photoinduced reaction. It is reported that the intermolecular hydrogen-bonded aggregates of catechol elongate the lifetime for dissociation because of the excited state structural relaxation.19,20 However, dispersion interactions are ubiquitous in the chromophore-environment interactions but often neglected as inter- or intramolecular forces. In fact, dispersion forces sum up as the molecular size becoming larger and are comparable to the hydrogen bond in the large aromatic systems.21 The impact of the intramolecular dispersion interactions on the photochemical processes has been verified with theoretical calculations.22 Intramolecular dispersion interactions can shape the potential energy surface and the structural evolution of the photoexcited stilbene and its substituted derivatives, thus increasing the ratio of the photocyclization pathway. The effect of the solute–solvent dispersion interactions on the solute dynamics remains unclear. Two-type intermolecular hydrogen bonding OH–O and OH–π isomers are formed in the heteroaromatic compounds.23 And the effect of the structural preference for the OH–O and OH–π interactions on the dynamics of the heteroaromatic molecules requires further exploring.Because of the complexity of the intermolecular interactions in the large aromatic systems, the studies of dispersion interactions in small molecules have been concentrated using bottom-up approaches. Diphenyl ether (DPE) is a prototypical molecule for investigating the structural preference of hydrogen bound complexes since two hydrogen bond acceptor sites are available for alcohols, resulting in OH–π and OH–O isomers. The interaction energy of the OH–π and OH–O interactions can be partitioned into electrostatic, exchange, induction and dispersion.24,25 For DPE-alcohol, the primary contributions are electrostatics, but it happens that non-dispersion contributions largely cancel, leaving dispersion as the determining component.25–27 The preferred structure shows strong dependence on dispersion interactions in different alcohols. The structures of DPE in the ground and excited states in different alcohols have been inspected using the combination of FTIR, broadband microwave and IR/UV spectroscopy.25–27 In the ground state, the OH–π interaction in methanol is obviously preferred over classical OH–O hydrogen bonding, while the OH–O structure in tert-butyl alcohol (t-butanol) is slightly stabler than the OH–π isomer.26,27 Furthermore, a clear preference for the OH–π isomer in water is dominated and only the OH–O structure is observed in the heavier alcohol-adamantanol.28 It is concluded that OH–π structure is more stable for the smaller alcohols, which has a significant impact on all aspects of chemistry. And it is interesting that the OH–π isomer in methanol can still be maintained in the S1 state, whereas the initial preferred OH–O structure in t-butanol is converted to an OH–π isomer in the S1 state.26
In the present work, the effects of dispersion interactions on the photodeactivation dynamics of DPE are investigated employing a combination of femtosecond time-resolved transient absorption with quantum chemical calculations. Since there do not exist hydrogen bonds in aprotic environments, the relaxation processes of the S1 state are examined in different polarity solvents with the existence of dispersion interactions only. It is observed that the main ISC pathway is suppressed in the polar solvent possibly due to the increased barrier height for ISC. Moreover, we focus on the structural preference affects the excited state dynamics of the hydrogen bound complexes in protic environments. According to the previous reports,26,27 OH–π and OH–O isomers are formed in the protic methanol and t-butanol solvents, respectively. Due to the ππ* excited state character, the OH–π isomers trend to dominate in the S1 state where the π system enables a stronger dispersion interaction. The measurements of the TA spectra present a faster internal conversion in t-butanol and we speculated that the geometry rearrangement to reach the geometric conical intersection is hindered in the more rigid OH–π structure, which confirms the influence of dispersion interactions on the photodynamics.
Experimental
Diphenyl ether (DPE) was purchased from Sinopharm Chemical Reagent limited corporation (>99.5%) and used without further purification. The aprotic solvents cyclohexane, 1,4-dioxane and the protic solvents t-butanol and methanol were also obtained from Sinopharm Chemical Reagent limited corporation. All the solutions were prepared at concentrations of 2 mM at room temperature.The static electronic absorption spectra were measured on UV-vis spectrometer (INESA, L6) in a 1 mm quartz cell. Transient absorption measurements were carried out with an equipment described in detail before.29,30 Excitation was achieved with pulses at 267 nm delivered by the third harmonic generation of the output of a 1 kHz amplified Ti:sapphire laser system centered at 800 nm. The pulse energies focused on the sample were reduced to 2 mW. Probing was accomplished by a supercontinuum pulses generated in CaF2 with the range 350–700 nm. The pulse was split into two beam paths, where one was the probe beam and spatially overlapped with the pump pulse into the sample and the other was used as reference to eliminate the absorption by background. The polarization was set to the magic angle (54.7°) between the pump and probe pulses. All the samples were kept in a flow cell with 1 mm optical path length. Detection was carried out by the CCD camera (PI-MAX, 1024 × 256 pixel array) equipped with a spectrometer (Princeton, SpectraPro 2500i). The instrument response function (IRF) was measured to be about 250 fs.
Quantum chemical calculations were performed employing Gaussian 09 program.31 We conducted the density functional theory (DFT) calculations using B3LYP functional and a cc-pVDZ basis set with the D3 (BJ) empirical dispersion correction to optimize all the geometries of ground and excited states and obtain the vertical transition energies. The solvent effect was incorporated by using the polarized continuum formalism (PCM) model with the integral equation formalism.
Results and discussion
(A) Static absorption spectrum
The steady-state absorption spectra of DPE in aprotic solvents (cyclohexane and 1,4-dioxane) and protic solvents (t-butanol and methanol) are shown in Fig. 1, respectively. The moderate intensity band is located at between 250 and 280 nm, where three sharp peaks are exhibited and considered as vibrational structures. Besides, it has a strong absorption at the region above 250 nm. The origins of the S1 ← S0 transition were measured to be 4.45 eV in gas phase from REMPI spectra and 4.55 eV in heptane.32,33 Our calculated excitation energy of the S1 state is 4.65 eV in cyclohexane and 1,4-dioxane, and 4.67 eV in methanol and t-butanol, respectively, which is consistent with the calculated result of 4.64 eV in methanol by M. Gerhards.26 Upon pump at 267 nm, the S1 state is excited exclusively.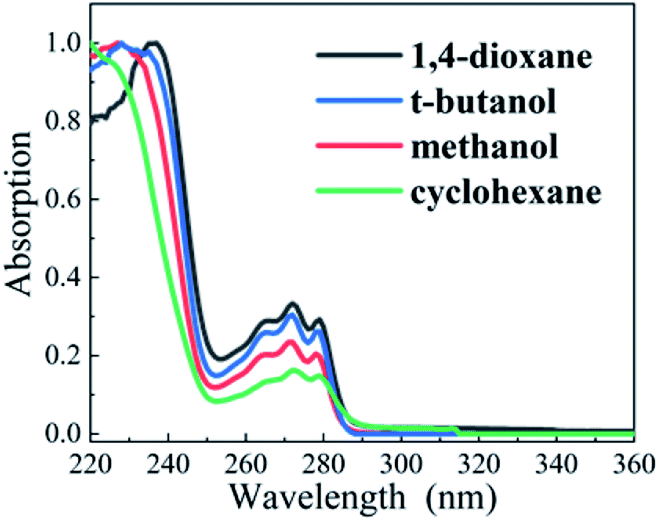 | ||
| Fig. 1 Normalized steady-state absorption spectra of DPE in cyclohexane (green), 1,4-dioxane (black), methanol (red), and t-butanol (blue). | ||
Compared with OH–π and OH–O structures in protic solvents, electron density transfers from the oxygen to the phenyl ring following photoexcitation and leads to the increasing electron density in phenyl ring, as shown in Fig. 2. It induces a stronger OH–π interaction while weakens the OH–O bond. Quantum chemical calculations are conducted to map out the potential curves of DPE in cyclohexane, 1,4-dioxane, methanol and t-butanol, respectively (see Fig. S1†). The calculated potential curves in all solvents are similar. In the S1 potential curve, it is obvious that the energy distribution shows an approximate parabola type depending on O–Ph bond distance in the region of 1.28–1.58 Å and approaches a maximum value at O–Ph bond distance of 1.68 Å. Subsequently, as the O–Ph bond continues to be lengthened, the vertical excitation energy decreases. The shapes of potential curves are in good agreement with the adiabatic potential hypersurfaces for the C–O bond dissociation of aromatic ethers calculated by Grimme et al.34 The energy barriers on the S1 vertical potential curves of DPE along the dissociation coordinate are estimated to be about 0.9 eV in the four solvents. However, the excitation energy of 267 nm (4.7 eV) is not enough to cross the barrier since the S1–S0 origin is about 4.44 eV in solutions.
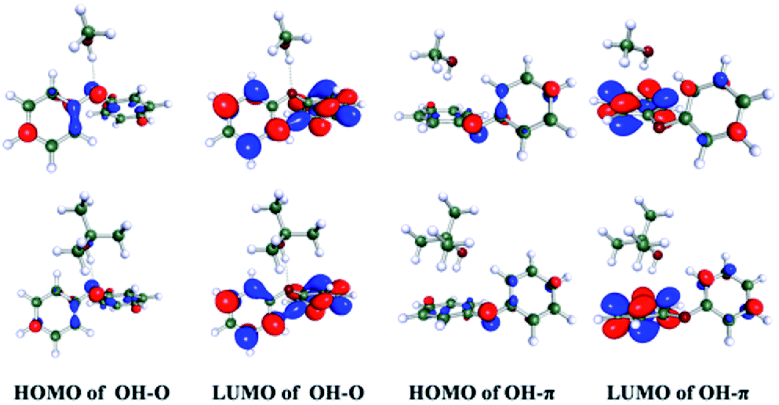 | ||
| Fig. 2 Molecular orbitals of OH–O and OH–π isomers of DPE in methanol (upper) and t-butanol (bottom), respectively. | ||
(B) The excited-state dynamics of DPE in aprotic solvents
In aprotic solvents without intermolecular hydrogen bond, the dispersion effect on the photodynamics of DPE in a nonpolar solvent (cyclohexane) and a polar solvent (1,4-dioxane) is explored. As shown in Fig. 3a and b, the measured TA spectra in cyclohexane and 1,4-dioxane display two absorption bands in the range of the whole measured spectral region. One is a broad band centered around 430 nm in cyclohexane and 415 nm in 1,4-dioxane, respectively, and the other band is in the range of above 580 nm. In both solvents, the two absorption bands have comparative intensity and decay towards the baseline with the increase of pump–probe delay, but not completely. Since DPE belongs to phenol derivatives, the characters of TA spectra measured in this work are akin to those of phenols. Two absorption bands are assigned to excited state absorption of the S1 state.35–40 It is noted that an additional absorption signal centered at 520 nm appears at tens of picoseconds in cyclohexane but absents in 1,4-dioxane. The triplet states (Tn) have been reported to be formed in the decay process of the S1 state and the quantum yield of ISC cannot be ignored in photoexcited phenols.41,42 In the TA spectra of guaiacol, there appears a rising absorption peak below ∼500 nm and it is assigned to the population of the triplet states.40 The yield of phosphorescence of DPE is measured to be 0.67 and stronger than that of fluorescence (0.14) in ethanol glass at 77 K, manifesting a very efficient ISC.43,44 Therefore, we attribute this growing signal centered at 520 nm to the absorption of the triplet states, suggesting that ISC is noticeable for DPE in cyclohexane. In addition, the vertical excitation energies of the S1 and Tn states were calculated (see Table S1†), and the result shows that the vertical excitation energies of T1–6 are lower than the S1 state.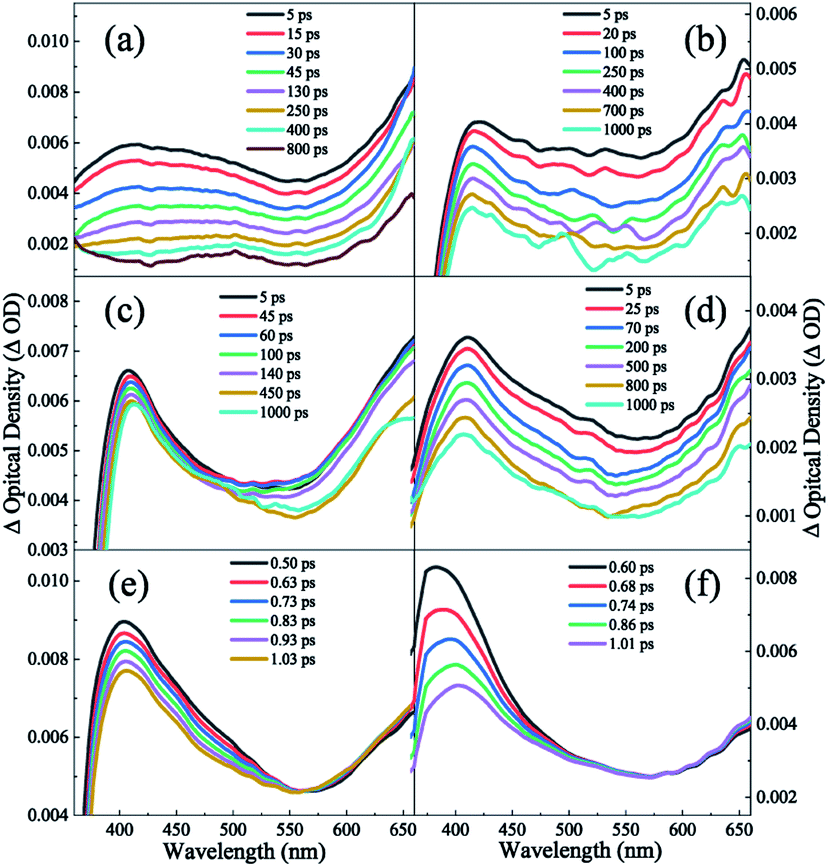 | ||
| Fig. 3 TA spectra of DPE in (a) cyclohexane (b) 1,4-dioxane, (c) methanol and (d) t-butanol solvents. The bottom shows the TA spectra in (e) methanol and (f) t-butanol collected in the first 1 ps. | ||
In order to obtain the dynamical information, the typical transient traces at 650 nm are extracted. The signals at this wavelength derive primarily from the absorption of the S1 state as the broad band below 600 nm overlaps substantially with the absorption of the triplet states. Three-exponential function kinetic model is employed to fit the traces and the fitting results are summarized in Table 1. Fig. 4a and b show the fitting curves of 650 nm in cyclohexane and 1,4-dioxane, respectively. A short lifetime τ1 = 1.8 ps in cyclohexane and 0.4 ps in 1,4-dioxane is determined. As mentioned above, the experimental S1–S0 origin was measured to be about 4.45 eV in the gas phase33 and 4.44 eV in cyclohexane and 1,4-dioxane from the UV-vis absorption spectra. Following photoexcitation at 267 nm (4.64 eV), the highly excited vibrational states are populated and the redundant vibrational energy dissipates to the solvents. Simultaneously, the similar time scales were also observed in aromatic ethers systems and assigned to the vibrational energy transfer process.19,20,45 The decay τ1 is attributed to the vibrational energy transfer process. According to the previous studies, phenols relax by photolysis via C–O bond cleavage and produce phenoxy radicals which have a signature absorption band at 400 nm.35–39 However, the dissociation pathway is not considered in DPE due to the absence of the absorption feature of the phenoxy radical in both TA spectra. Furthermore, it is noted that the lifetimes of the intersystem crossing and fluorescence pathways were measured on nanoseconds timescales.44 The second component τ2 with about 50 ps is significantly shorter than the lifetime of both ISC and fluorescence and assigned to IC. And the longest time constant τ3 = ns is attributed to depletion of the S1 state mediated through ISC and fluorescence.
| Polarity | τ1 (ps) | τ2 (ps) | τ3 | τ4 (ps) | |
|---|---|---|---|---|---|
| Cyclohexane | 0 | 1.8(4) | 59(6) | ns | — |
| 1,4-Dioxane | 4.8 | 0.4(2) | 56(5) | ns | — |
| t-Butanol | 3.9 | 1.4(3) | 26(4) | ns | 0.5(2) |
| Methanol | 6.6 | 1.0(2) | 63(5) | ns | 1.9(3) |
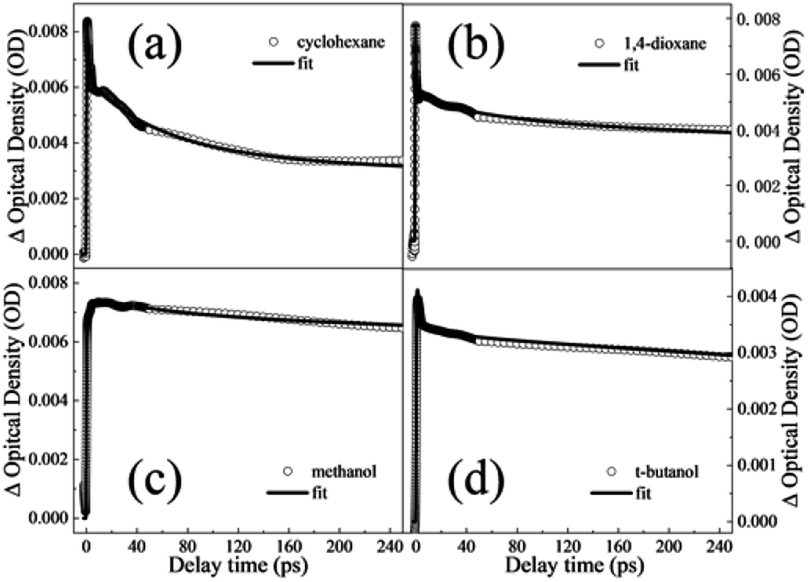 | ||
| Fig. 4 Time traces at 650 nm obtained from the TA spectra in (a) cyclohexane (b) 1,4-dioxane, (c) methanol and (d) t-butanol solvents for delay times up to 900 ps. Open circles are data; solid black lines are the overall least-squares fit curves. | ||
However, the band centered at 520 nm is not observed in 1,4-dioxane, which may be attributable to the low ISC quantum yield. It is inferred that the polarity of solvent modifies the excited state energies and leads to an enhanced barrier to ISC in 1,4-dioxane. Thus, the ISC channel is suppressed in 1,4-dioxane. Furthermore, the calculated results of the vertical excitation energies of the S1 and Tn states show that the energy gap between S1 and Tn states rises with the increasing polarity in aprotic solvents, which verifies the enhanced energy barrier of ISC in 1,4-dioxane. Thus, the ISC channel is suppressed in 1,4-dioxane. As a result, when dispersion interactions exist only, the increasing polarity of solvents leads to the suppression of ISC in DPE.
(C) The excited-state dynamics of DPE in protic solvents
Furthermore, the role of the structural preference for hydrogen bound complexes on the excited state dynamic is examined in protic solvents which could form OH–π and OH–O intermolecular hydrogen bound complexes with DPE. As shown in the Fig. 3c and d, the TA spectra measured in methanol and t-butanol resemble that in 1,4-dioxane. Two noticeable absorption bands are observed across the entire probe region: a peak centered around 410 nm and a broad band beyond 560 nm. We attribute these bands to the initially excited state absorption of the intermolecular hydrogen bound complex. It is worth noting that a rising trend in the band beyond 560 nm appears in the early 1 ps, shown in Fig. 3e and f, and increases with the decrease of the absorption centered at ∼410 nm. It implies a direct dynamical conversion between two states. In methanol and t-butanol, there exist OH–π and OH–O isomers in the ground state, respectively. Hinohara et al. pointed out that the S1 ← S0 transition of DPE primarily comes from ππ* contribution upon excitation.46 Bernhard et al. also indicated that the S1 ← S0 transition of DPE-methanol is seen to be primarily a ππ* transition, and the excitation in DPE-tert-butyl alcohol complex takes place as a ππ* transition with a small nπ* contribution.26,27 As mentioned above, in protic solvents, electron density transfers from the ether oxygen to the phenyl ring following photoexcitation and leads to the increasing electron density in phenyl ring, as shown in Fig. 2. It induces a stronger OH–π interaction while weakens the OH–O bond.26,27 The directly dynamical conversion process in first 1 ps is assigned to conformational rearrangement from OH–O to OH–π in the excited S1 states. As shown in Fig. 4c and d, the typical profiles of 650 nm in methanol and t-butanol were also extracted and fitted with four exponential functions. The fitting results provide the similar lifetimes with the values in 1,4-dioxane and are listed in Table 1. In term of dynamics in 1,4-dioxane, the components with the shorter lifetimes τ1 and τ2 are also assigned to the vibrational energy transfer and the IC process, respectively, and the longest decay time τ3 is ascribed to the ISC and fluorescence pathways. A new component τ4 is just observed in methanol and t-butanol and determined to be 1.9 and 0.5 ps in methanol and t-butanol, respectively. As mentioned earlier, the time component τ4 is attributed to the structural rearrangement of the hydrogen bound isomers due to the negative pre-amplifier and related to the rising trend in the bands beyond 560 nm.Dispersion interactions have a strong influence on the structural preference for the hydrogen bound isomers. In the ground state, the OH–π and OH–O isomers coexist actually in methanol and t-butanol.28 The OH–π structure dominates in methanol as the dispersion interaction with the phenyl ring is stronger than with the ether oxygen.26 On the contrary, a preference of the OH–O structure is observed in t-butanol. The OH–O structure has a stronger hydrogen bond and tert-butyl group is a better dispersion energy donor in t-butanol.27 In the S1 state, as the occurring of OH–O to OH–π conformational rearrangement, the OH–π isomer is the only structure observed in methanol and dominates in t-butanol. Since the OH–O isomers are stabilized by stronger dispersion interactions in larger alcohols both in the ground and S1 states, the OH–O isomers still exist in t-butanol but almost disappear in methanol. Besides, the structural preference for the hydrogen bound isomers induced by dispersion interactions affects the excited state dynamics. The time constant τ2 with 26 ps in t-butanol is shorter than 63 ps in methanol, suggesting a decreased barrier of IC in t-butanol. Furthermore, the motion of the phenyl in the OH–O structure is relatively freer than that in the OH–π structure. The latter one is more rigid due to the –OH groups binding to the phenyl. We deduced that the geometric relaxation to the conical intersection is easier for the OH–O isomer compared with the more rigid OH–π structure. Consequently, the barrier for IC is decreased in the OH–O structure and the IC process can be facilitated. Additionally, τ2 in methanol is comparable to the value in 1,4-dioxane. It suggests that the OH–π isomer has little impact on the photodynamics of DPE. Dispersion interactions enable the OH–O structure to become more stable in larger alcohols. Furthermore, this structural preference has a facilitation effect on IC process of DPE.
Conclusions
In conclusion, dispersion interactions not only play an important role in the three-dimensional structure of biomacromolecules, but also influence the dynamics of the chromophore in these systems. Utilizing bottom-up approaches, the effect of the structural preference induced by dispersion interactions on the photodeactivation mechanism for DPE is unravelled using a combination of femtosecond time-resolved transient absorption with quantum chemical calculations. In aprotic solvents without the existence of hydrogen bonds, the dispersion effect on the photodynamics was inspected. Upon excitation at 267 nm, the DPE molecule is excited to the S1 state and evolves through vibrational energy transfer in several picoseconds. Subsequently, the S1 state population decay to the ground state via IC within about 50 ps and ISC and fluorescence on ns time scale. However, the band centered at 520 nm is not observed in the polar solvent, which may exist an enhanced barrier to ISC. When only dispersion interactions are considered, the polarity of solvent can modify the excited state energies and lead to the suppression of the ISC channel in polar solvents. An energy scheme of the photoinduced mechanism of DPE is present in Fig. 5.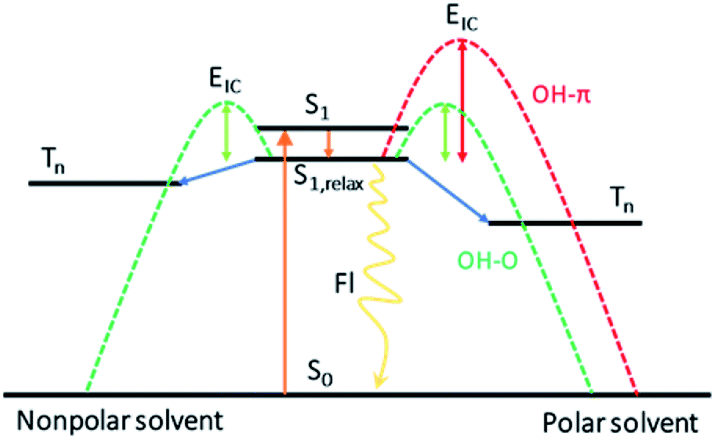 | ||
| Fig. 5 The scheme of the photodeactivation pathways for the S1 excitation in DPE. S0: the ground state, S1: the first excited singlet state, S1,relax: the relaxed S1 state, Tn: the triplet state folds, EIC: the barrier height for IC. | ||
Afterwards, the effect of the structural preference for hydrogen bound complexes on the excited state dynamic following excitation is explored. In the protic solvents-methanol and t-butanol, intermolecular OH–π and OH–O isomers form in the ground state. The structural preference induced by dispersion interactions leads to the domination of the OH–π isomer in methanol and a slight preference for the OH–O isomer in t-butanol.26,27 When initially excited to the S1 state, a geometric rearrangement from OH–O to OH–π occurs in the excited S1 states in about 1 ps. Besides the conformational rearrangement in protic solvents, the following dynamics relaxation on the S1 state is similar with that in 1,4-dioxane. The measured lifetime of IC in methanol is comparable to the value in 1,4-dioxane. It concludes that the OH–π isomer has little impact on the photodynamics of DPE since the OH–π isomer is the only structure in the S1 state in methanol. However, with the increasing side chains in t-butanol, the OH–O structure is stabilized caused by dispersion interactions. The time constant of IC in t-butanol is shorter than that in methanol, which implies a decreased barrier in t-butanol. We deduced that the ability to approach the geometric conical intersection for the freer structure in the OH–O isomer is increased and leads to a facilitation for IC. These results provide a dynamical insight into the impact of different preference for intermolecular hydrogen bound isomers induced by dispersion interactions on the photodeactivation mechanism of DPE. And it offers a foundation for future studies on how the structural changes induced by dispersion interactions affect and thus control the excited state dynamics in photoactivatable biomolecules or material molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2019YFA0307700), the National Natural Science Foundation of China (No. 11974381, 11674355, 21573279, 21773299, 21327804).Notes and references
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–167 CrossRef PubMed.
- J. M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89–112 CrossRef.
- J. Cerny and P. Hobza, Phys. Chem. Chem. Phys., 2007, 9, 5291–5303 RSC.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- N. H. Joh, A. Oberai, D. Yang, J. P. Whitelegge and J. U. Bowie, J. Am. Chem. Soc., 2009, 131, 10846–10847 CrossRef CAS PubMed.
- S. Hanlon, Biochem. Biophys. Res. Commun., 1966, 23, 861–867 CrossRef CAS PubMed.
- M. W. Forbes and R. A. Jockusch, J. Am. Chem. Soc., 2009, 131, 17038–17039 CrossRef CAS PubMed.
- H. Niwa, S. Inouye, T. Hirano, T. Matsuno, S. Kojima, M. Kubota, M. Ohashi and F. I. Tsuji, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13617–13622 CrossRef CAS PubMed.
- C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Nature, 2009, 462, 200–204 CrossRef CAS PubMed.
- T. Chatterjee, F. Lacombat, D. Yadav, M. Mandal, P. Plaza, A. Espagne and P. K. Mandal, J. Phys. Chem. B, 2016, 120, 9716–9722 CrossRef CAS PubMed.
- Y. Lalatonne, J. Richardi and M. P. Pileni, Nat. Mater., 2004, 3, 121–125 CrossRef CAS PubMed.
- J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef CAS PubMed.
- S. P. Koenig, N. G. Boddeti, M. L. Dunn and J. S. Bunch, Nat. Nanotechnol., 2011, 6, 543–546 CrossRef CAS PubMed.
- K. Autumn, M. Sitti, Y. A. Liang, A. M. Peattie, W. R. Hansen, S. Sponberg, T. W. Kenny, R. Israelachvili, J. N. Fearing and R. J. Full, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12252–12256 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- J. A. Berenbeim, S. Boldissar, S. Owens, M. R. Haggmark, G. Gate, F. M. Siouri, T. Cohen, M. F. Rode, C. S. Patterson and M. S. de Vries, Sci. Adv., 2019, 5, eaaw5227 CrossRef CAS PubMed.
- P. Song and F. C. Ma, Int. Rev. Phys. Chem., 2013, 32, 589–609 Search PubMed.
- M. A. P. Turner, R. J. Turner, M. D. Horbury, N. D. M. Hine and V. G. Stavros, J. Chem. Phys., 2019, 151, 084305 CrossRef CAS PubMed.
- C. Grieco, F. R. Kohl, Y. Zhang, S. Natarajan, L. Blancafort and B. Kohler, Photochem. Photobiol., 2019, 95, 163–175 CrossRef CAS PubMed.
- C. D. Zeinalipour-Yazdi and D. P. Pullman, J. Phys. Chem. B, 2006, 110, 24260–24265 CrossRef CAS PubMed.
- A. Fabrizio and C. Corminboeuf, J. Phys. Chem. Lett., 2018, 9, 464–470 CrossRef CAS PubMed.
- H. Sasaki, S. Daicho, Y. Yamada and Y. Nibu, J. Phys. Chem. A, 2013, 117, 3183–3189 CrossRef CAS PubMed.
- L. V. Slipchenko and M. S. Gordon, J. Phys. Chem. A, 2009, 113, 2092–2102 CrossRef CAS PubMed.
- C. Medcraft, S. Zinn, M. Schnell, A. Poblotzki, J. Altnoder, M. Heger, M. A. Suhm, D. Bernhard, A. Stamm, F. Dietrich and M. Gerhards, Phys. Chem. Chem. Phys., 2016, 18, 25975–25983 RSC.
- D. Bernhard, C. Holzer, F. Dietrich, A. Stamm, W. Klopper and M. Gerhards, ChemPhysChem, 2017, 18, 3634–3641 CrossRef CAS PubMed.
- D. Bernhard, F. Dietrich, M. Fatima, C. Perez, A. Poblotzki, G. Jansen, M. A. Suhm, M. Schnell and M. Gerhards, Phys. Chem. Chem. Phys., 2017, 19, 18076–18088 RSC.
- F. Dietrich, D. Bernhard, M. Fatima, C. Perez, M. Schnell and M. Gerhards, Angew. Chem., Int. Ed., 2018, 57, 9534–9537 CrossRef CAS PubMed.
- S. Zhang, S. Sun, M. Zhou, L. Wang and B. Zhang, Sci. Rep., 2017, 7, 43419 CrossRef CAS PubMed.
- K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z. Lu, S. Zhang and C. Yang, J. Am. Chem. Soc., 2018, 140, 8877–8886 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al., GAUSSIAN 09, Revision A02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. C. S. Paiva, P. G. Kistemaker and T. L. Weeding, Int. J. Mass Spectrom., 2002, 221, 107–115 CrossRef CAS.
- B. Uno, T. Kawakita, K. Kano, K. Ezumi and T. Kubota, Bull. Chem. Soc. Jpn., 1992, 65, 2697–2703 CrossRef CAS.
- S. Grimme, Chem. Phys., 1992, 163, 313–330 CrossRef CAS.
- M. D. Horbury, L. A. Baker, W. D. Quan, J. D. Young, M. Staniforth, S. E. Greenough and V. G. Stavros, J. Phys. Chem. A, 2015, 119, 11989–11996 CrossRef CAS PubMed.
- T. A. A. Oliver, Y. Y. Zhang, A. Roy, M. N. R. Ashfold and S. E. Bradforth, J. Phys. Chem. Lett., 2015, 6, 4159–4164 CrossRef CAS PubMed.
- Y. Y. Zhang, T. A. A. Oliver, M. N. R. Ashfold and S. E. Bradforth, Faraday Discuss., 2012, 157, 141–163 RSC.
- S. J. Harris, D. Murdock, M. P. Grubb, G. M. Greetham, I. P. Clark, M. Towrie and M. N. R. Ashfold, Chem. Sci., 2014, 5, 707–714 RSC.
- S. J. Harris, D. Murdock, Y. Y. Zhang, T. A. A. Oliver, M. P. Grubb, A. J. Orr-Ewing, G. M. Greetham, I. P. Clark, M. Towrie, S. E. Bradforth and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2013, 15, 6567–6582 RSC.
- S. E. Greenough, M. D. Horbury, J. O. F. Thompson, G. M. Roberts, T. N. V. Karsili, B. Marchetti, D. Townsend and V. G. Stavros, Phys. Chem. Chem. Phys., 2014, 16, 16187–16195 RSC.
- R. Hermann, G. R. Mahalaxmi, T. Jochum, S. Naumov and O. Brede, J. Phys. Chem. A, 2002, 106, 2379–2389 CrossRef CAS.
- D. V. Bent and E. Hayon, J. Am. Chem. Soc., 1975, 97, 2599–2606 CrossRef CAS PubMed.
- R. S. Becker, A. D. Jordan and J. Kolc, J. Chem. Phys., 1973, 59, 4024–4028 CrossRef CAS.
- S. K. Sarkar, A. Maiti and G. S. Kastha, Chem. Phys. Lett., 1984, 105, 355–358 CrossRef CAS.
- D. Murdock, S. J. Harris, T. N. V. Karsili, G. M. Greetham, I. P. Clark, M. Towrie, A. J. Orr-Ewing and M. N. R. Ashfold, J. Phys. Chem. Lett., 2012, 3, 3715–3720 CrossRef CAS PubMed.
- T. Hinohara, S. Cho and T. Morita, Bull. Chem. Soc. Jpn., 1971, 44, 629–637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02224a |
| This journal is © The Royal Society of Chemistry 2020 |