
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibitory effects of UDP-glucuronosyltransferase (UGT) typical ligands against E. coli beta-glucuronidase (GUS)
Ling Xiaoa,
Dehui Chib,
Guiju Shengb,
Wenjuan Lib,
Penghui Linc,
Sicheng Liangd,
Liangliang Zhu *b and
Peipei Dong*e
*b and
Peipei Dong*e
aSchool of Resources and Environment, Key Laboratory of Aqueous Environment Protection and Pollution Control of Yangtze River, Anqing Normal University, Anqing 246133, China
bDepartment of Food Science and Technology, School of Life Science and Research Center of Aquatic Organism Conservation and Water Ecosystem Restoration, Anqing Normal University, No. 1314, North Jixian Road, Anqing 246133, China. E-mail: zhuliangliang@aqnu.edu.cn
cCenter for Environmental and Systems Biochemistry, Markey Cancer Center, and Dept. of Toxicology & Cancer Biology, University of Kentucky, 789 S. Limestone St, Lexington, KY 40536, USA
dSchool of Pharmacy, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, China
eCollege of Integrative Medicine, Dalian Medical University, No. 9 West Section Lvshun South Road, Dalian 116044, China. E-mail: dongpeipei11@163.com
First published on 16th June 2020
Abstract
UDP-glucuronosyltransferases (UGTs) and β-glucuronidase (GUS) catalyze entirely distinct metabolism reactions. UGTs are responsible for the glucuronidation of a variety of drugs, endogenous and environmental chemicals, whereas GUS hydrolyzes glucuronides and liberates the parent substrates. Information on the overlap of ligand selectivity between UGT and GUS is essential for exploring the pharmacological or toxicological effects of the inhibitors of these two metabolic enzymes. This study is conducted to test whether UGTs and GUS share common ligands, by investigating the inhibitory effects towards E. coli GUS by a series of UGT typical substrates and inhibitors. Results showed that three typical ligands of UGTs, including two specific substrates (estradiol and trifluoperazine, E2 and TFP) and one selective inhibitor (magnolol, Mag), can inhibit the activity of GUS. Kinetic assays indicated that all the three UGT specific chemicals displayed competitive inhibition, with Ki values of 31.4 (E2), 56.9 (TFP), and 16.6 μM (Mag). Docking studies further revealed that the three chemicals can enter the active sites of GUS by forming contacts with residues Glu-413, Trp-549, Asp-163, Tyr-472, Arg-562, or bound water. Our study indicates that ligand selectivity overlaps between UGTs and GUS, and some chemicals can act as co-inhibitors of these two metabolic enzymes. The pharmacological or toxicological effects of those co-inhibitors require further investigations.
Introduction
UDP-glucuronosyltransferases (UGTs) and β-glucuronidase (GUS) catalyse entirely distinct reactions. UGTs are responsible for the conjugation of a variety of clinical drugs, endogenous hormones, food chemicals and environmental toxicants with the glucuronic acid moiety.1 After undergoing the glucuronidation metabolism, the parent chemicals are commonly inactivated and become more water soluble and are readily excreted.2 In contrast to UGTs, GUS catalyses the hydrolysis of glucuronides, liberating the parent endo- and xeno-biotics.3 As a result, the parent chemicals continue to exert their biological activities or even toxicities.4It is currently believed that the impaired glucuronidation activities can cause undesired effects resulting from the slow elimination of toxic chemicals (e.g. bilirubin and SN-38).5,6 To date, a massive number of chemicals have been reported to be potent in vitro inhibitors of UGT enzymes.7–9 However, only a limited number of these inhibitors are reported to result in pharmacokinetic drug–drug interactions via in vivo inhibition of UGTs.10 The mechanism for such in vitro–in vivo discrepancy was mainly attributed to those UGT substrates that are usually glucuronidated by different UGTs.10 However, the inconsistency may result from other aspects. One possible mechanism is that some UGT inhibitors may simultaneously inhibit the activity of β-glucuronidase (GUS). As a result, the decreased exposures from the glucuronidation metabolism are partly compensated.11 In the presence of one UGT inhibitor, glucuronide formation will be inhibited and the parent substrates will remain in relatively high levels.3,4 However, if the UGT inhibitor simultaneously inhibits GUS activity, glucuronide hydrolysis will also be decreased and there will be little replenishment for the parent substrates.11 Thus, the glucuronides will remain in similar levels regardless of the presence of the co-inhibitor.
Besides the undesired effects from the inhibition of UGTs, some positive effects may also be achieved via the inhibition of GUS.4 It is believed that the inhibition of GUS may help protect from gastrointestinal toxicities by hindering the liberation of parent toxins in the colon. One notable example is that the GUS inhibitor could decrease the irinotecan related digestive tract toxicity by inhibiting the hydrolysis of SN-38 glucuronides.12,13 A variety of natural and synthetic inhibitors are emerging as novel potential gastrointestinal protective agents.4 However, to our knowledge, in all these researches no consideration was given to the risk of the GUS inhibitors inhibiting UGTs. When the GUS inhibitors simultaneously inhibit the activity of UGTs, fewer toxic parent substrates are transformed into non-toxic glucuronides and the protective effects may be accordingly less obvious. It would be better to warrant little or no co-inhibition of UGT in the future GUS inhibitor discovery and development.
Since the reactions catalysed by UGTs and GUS are opposite to each other, it is generally thought that UGTs and GUS are unlikely to share common ligands. UGTs tend to bind lipophilic chemicals, while GUS binds relatively hydrophilic glucuronides.3,14 However, it was found that several lipophilic flavonoids (e.g. baicalein, quercetin and scutellarin) displayed potent inhibitory effects against GUS from E. coli.15 Interestingly, structures in these inhibitors do not contain the glucuronic acid moiety. These findings encouraged us to conduct this study to test whether the typical ligands of UGT, including specific substrates and selective inhibitors, can inhibit the activity of GUS. It is hoped that this study may provide evidences for ligand selectivity overlaps between UGTs and GUS.
Results
Screening of inhibitory effects
As displayed in Fig. 1, E2 (UGT1A1 probe substrate), TFP (UGT1A4 probe substrate) and Mag (UGT1A9 selective inhibitor) exerted potent inhibition towards GUS, with remaining activities at 37%, 43% and 26% of the control, respectively. For the other chemicals tested in this study, no significant inhibition was observed, where the remaining activities were at least higher than 70% of the control.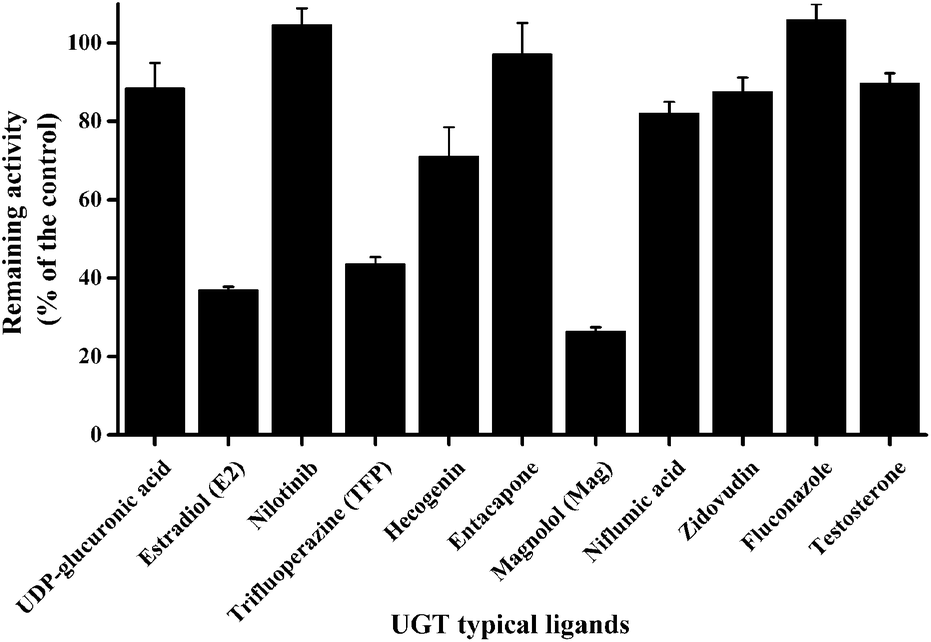 | ||
| Fig. 1 Inhibitory effects of a series of UGT probing ligands towards the activity of GUS in catalysing the hydrolysis of pNPG (200 μM). Data columns and error bars represent the mean and S.D. of triplicate determinations, respectively. | ||
Inhibition of GUS by E2
The structure of E2 is shown in Fig. 2A. Kinetic analysis indicated that E2 displayed competitive inhibition towards GUS mediated pNPG hydrolysis (Fig. 2B), with the Ki value determined to be 31.4 ± 2.0 μM. Docking studies further revealed that an H-bonding interaction existed between the E2-17-OH and carboxylate of Glu-413 in the active site. Two additional hydrogen bonds were also formed between the H2O in the crystal structure and E2.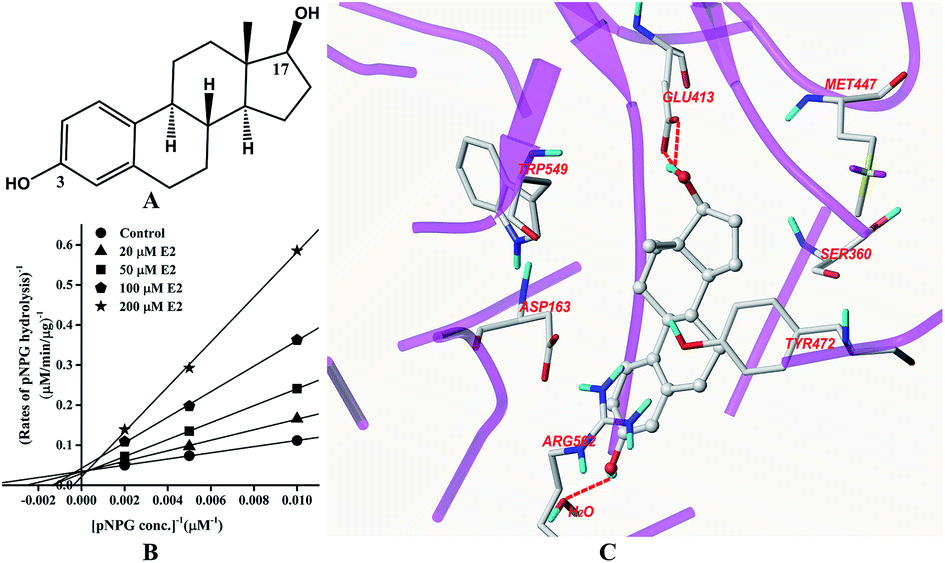 | ||
| Fig. 2 Molecular structure of E2 (A), Lineweaver–Burk plots for the inhibition of E2 on GUS (B), and the best ranked pose of E2 in the active-site cavity of E. coli GUS generated with docking (C). | ||
Inhibition of GUS by TFP
The structure of TFP is displayed in Fig. 3A. As shown in Fig. 3B, the inhibition of GUS by TFP follows the competitive model for the de-glucuronidation of pNPG (Fig. 3B). The Ki value was calculated to be 56.9 ± 2.1 μM. Docking analysis indicated that the fluorine atoms seem to be important for the inhibitor–protein interaction. The fluorine atoms formed an H-bonding interaction with the NH of Trp-549 and three extra H-bonds were formed with H2O.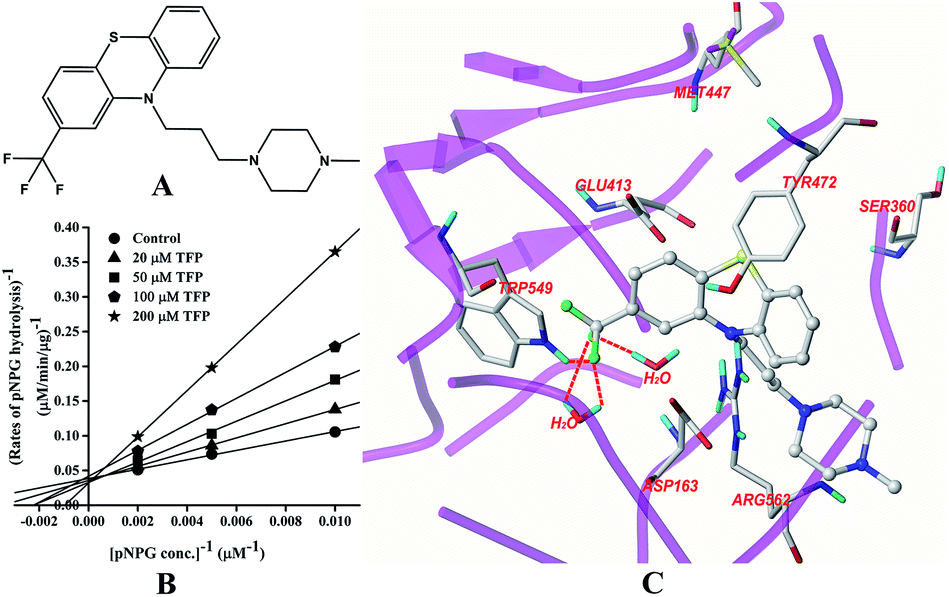 | ||
| Fig. 3 Molecular structure of TFP (A), Lineweaver–Burk plots for the inhibition of TFP on GUS (B), and the best ranked pose of TFP in the active-site cavity of E. coli GUS generated with docking (C). | ||
Inhibition of GUS by Mag
The structure of Mag is displayed in Fig. 4A. Fig. 4B indicated that Mag inhibited GUS competitively in catalysing the hydrolysis of pNPG, with the Ki value calculated to be 16.6 ± 0.6 μM. Docking results (Fig. 4C) indicated that four hydrogen bonds were formed with the residues Asp-163, Tyr-472 and Arg-562. π–π stacking interaction was additionally formed between the benzene rings of Mag and Tyr-472.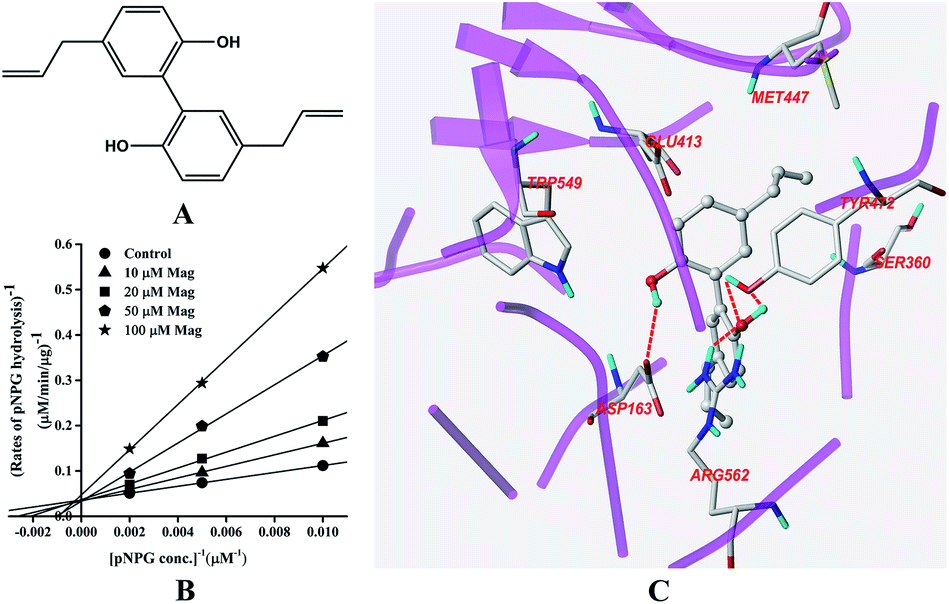 | ||
| Fig. 4 Molecular structure of Mag (A), Lineweaver–Burk plots for the inhibition of Mag on GUS (B), and the best ranked pose of Mag in the active-site cavity of E. coli GUS generated with docking (C). | ||
Discussion
After testing the inhibitory effects of a series of UGT probe ligands against GUS activity, it was clearly demonstrated that the ligand selectivity overlaps between UGTs and GUS. It is possible that some clinical drugs, environmental chemicals, and endogenous molecules may interfere in the glucuronidation pathway by hindering the hydrolysis of glucuronides. Among the chemicals tested in this study, nilotinib, hecogenin, Mag, niflumic acid and fluconazole were reported to be potent in vitro UGT inhibitors.16–20 In vitro–in vivo extrapolation suggested that nilotinib, Mag and fluconazole had potential to induce in vivo inhibition of UGTs.18–20 In vivo inhibition of UGTs has been confirmed for nilotinib and fluconazole.21,22 It is consistent with the current study that these two UGT inhibitors lack inhibition towards GUS (Fig. 1). For Mag, although one previous study demonstrated that it can potently inhibit the activity of UGT1A9,18 this study indicates that in vivo UGT inhibition will be neutralized due to the co-inhibition of GUS.E2, TFP and Mag were reported to be probe ligands for UGT1A1, 1A4 and 1A9, respectively.18,23 However, the results did not indicate that the overlapping selectivity with GUS is limited to these three isoforms. Actually, E2 undergoes glucuronidation at both 3-OH and 17-OH sites.24 Although E2-3-O-glucuronidation is commonly used as a probe reaction for UGT1A1 in human liver,23 it is additionally catalysed by other isoforms including UGT1A3, 1A8, 1A10, 2A1, and 2A2.24 For E2-17-O-glucuronidation, multiple UGTs including UGT1A3, 1A4, 1A8, 1A10, 2A1, 2A2, 2B7, and 2B17 are involved.24,25 Mag has been identified as a selective inhibitor of UGT1A9 in certain concentration ranges, whereas it also serves as a good substrate to several other different UGTs, in particular for UGT1A10 and 2B7.18,26 For TFP, available evidences indicated that it acts as a specific substrate for UGT1A4.23 However, there is still a lack of information about the inhibition selectivity for other UGTs. Therefore, ligand selectivity overlaps with GUS may occur for each individual UGT isoform.
In this study, all three compounds could be well docked into the active site cavity in a similar way to that adopted by the original ligand in crystallographic complexes. For E2, two H-bonding interactions were found between the 17-OH of E2 and the carboxylate of Glu-413 in the active sites. Besides, there is an additional hydrogen bond between the 3-OH of E2 and a H2O molecule in the crystal structure (Fig. 2C). As shown in Fig. 3C, the fluorine atoms are important for the inhibitor–protein interaction for TFP. F atoms formed an H-bonding interaction with the NH of Trp-549, as well as other three H-bonds with H2O molecules. For Mag, two phenolic hydroxyl groups are important for the inhibition activity. They formed four H-bonds with the residues Asp-163, Tyr-472 and Arg-562. In addition, π–π stacking interaction was formed between the benzene rings of Mag and Tyr-472. The smaller molecular volume was another merit for Mag to enter the active site cavity (Fig. 4C). From the above analysis, we can see that the polar functional groups including the hydroxyl, phenolic hydroxyl and fluoroalkane appear to be essential for the GUS inhibition.
In the active sites of GUS (E. coli), Glu-413 and Glu-504 are important catalytic residues for substrate recognition and hydrolysis.12 It was found that the competitive inhibitor D-saccharic acid 1,4-lactone can form contacts with Glu-413, Glu-504, Asp-163, Arg-562, Lys-568, Tyr-472, and His-330.12 Trp-549 was also proved important in binding a cyclic thioimidazole derivative.12 Rasmussen et al. (2011) demonstrated that the bound water could also interact with the potent competitive inhibitor uronic-noeurostegine.27 This study indicated that E2 and TFP could also interact with the bound water. Interactions with the catalytic residues and other important amino acids constituting the active site made these three ligands inhibitors of GUS. Without structural information, it is currently hard to describe the interaction pattern between UGTs and its probe ligands.
It is well known that the inhibition of GUS could help protect humans from irinotecan related digestive tract toxicity by blocking the hydrolysis of SN-38 glucuronides.4 E2 serves as an important endogenous chemical in humans, which is also widely used in hormone replacement treatment in clinics.28 It is found that the daily total fecal excretion of E2 can reach 393 μg.29 Given that the minimal colon cavity volume was reported to be only 180 mL,30 the normal physiological E2 concentration in the gut may reach approximately 7.8 μM, which is one quarter of the Ki value (31.4 μM) for the inhibition of GUS. Moreover, when E2 is used in the treatment of breast cancer, the daily oral dosage can reach up to 30 mg.31 Therefore, the inhibition of GUS may be achieved by administering E2. This deserves further investigations, in particular consideration of potential application of irinotecan in the treatment of breast cancer.31 TFP acts as an anti-psychotic medicine, which is mainly used to treat anxiety or psychotic disorders.32 Based on a study conducted in rats, 87% of the administered TFP will be excreted into the gut.33 The single oral dosage of TFP can reach 5 mg,34 which may result in the TFP level to be higher than 56.9 μM in the human gut. It is possible that after the administration of TFP, GUS activity in the gut is correspondingly decreased.
Mag containing Magnolia Officinalis has been widely used in the treatment of multiple common diseases such as gastrointestinal disorders and anxiety in China and Japan for a very long time.35 It was reported that a single dose of 5 g Sai-boku-to contained 2.1 mg Mag, and the urinary excretion was less than 10% of the total dosage.36 Most of the chemical may stay in the colon with the concentration higher than 16.6 μM. One thing that needs to be noted here is Mag may also undergo extensive glucuronidation in humans.26 This study did not test the inhibitory effects of Mag glucuronide towards GUS. As a good substrate for GUS, it is likely that Mag glucuronide can potentially inhibit the activity of GUS. Interestingly, after taking Mag, whether the chemical exists as a parent or a glucuronidated form, the inhibition of GUS is going to happen. As discussed previously, Magnolia Officinalis has been used in the treatment of gastrointestinal disorders.35 Based on this study, it seems that the inhibition of GUS may partly contribute to gastrointestinal protective effects, since Mag can prevent the release of toxic parent chemicals from glucuronides.
Materials and methods
Chemicals and enzymes
Estradiol (E2, purity > 98%), testosterone (purity ≥ 98%), entacapone (purity ≥ 98%), and nilotinib (purity ≥ 99%) were purchased from Aladdin Co., Ltd. (Shanghai, China). Trifluoperazine (TFP, purity ≥ 99%), fluconazole (purity ≥ 98%), and niflumic acid (purity ≥ 98%) were purchased from Macklin Co., Inc. (Shanghai, China). Magnolol (MAG, purity ≥ 98%) and hecogenin (purity ≥ 98%) were purchased from Pufei De Biotech Co., Ltd. (Chengdu, China). 4-Nitrophenol (pNP, purity > 99%), 4-nitrophenol-β-O-glucuronide (pNPG, purity > 98%), and 3′-azido-3′-deoxythymidine (purity ≥ 98%) were purchased from J&K Chemical Ltd. (Shanghai, China). Uridine diphosphate glucuronic acid (UDPGA, purity > 96%) and E. coli β-glucuronidase (GUS, Cat No. G8295, 25KU/2.1 mg) were purchased from Sigma-Aldrich (Shanghai, China).Incubation conditions
Hydrolyses of pNPG by GUS were carried out at 37 °C in 200 μL mixture of 50 mM phosphate buffer (pH = 6.8). The reactions were initiated by adding 20 μL pNPG solution. Preliminary experiments demonstrated that the linear ranges of GUS and time required are 0–2 U mL−1 and 0–40 min, respectively. In the inhibition assays, GUS consumption in the reaction mixture and reaction time were set at 0.5 U mL−1 and 20 min, respectively. The reaction was stopped by adding 100 μL ice-cold methanol. The samples were then centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 minutes to remove the protein, and 10 μL of the supernatants were analysed in the UPLC system.
000g for 20 minutes to remove the protein, and 10 μL of the supernatants were analysed in the UPLC system.
Inhibition screening
Inhibitory effects towards GUS were tested for various typical substrates and inhibitors of UGTs. pNPG (200 μM) was incubated with GUS (0.1 U) in the presence of UDPGA (4 mM), E2 (100 μM), nilotinib (100 μM), TFP (100 μM), hecogenin (100 μM), entacapone (100 μM), Mag (100 μM), niflumic acid (100 μM), zidovudine (1 mM), fluconazole (1 mM) or testosterone (100 μM). Information on chemicals probing UGTs is available in the literature. Specifically, UDPGA acts as the universal substrate for all UGT isoforms.37 E2, TFP, entacapone, zidovudine, and testosterone were specific substrates for UGT1A1, 1A4, 1A9, 2B7 and 2B17, respectively.16,23,38,39 Nilotinib, hecogenin, Mag/niflumic acid and fluconazole serve as the selective inhibitors of UGT1A1, UGT1A4, 1A9, and 2B7, respectively.17–19,23 The concentrations used here were set much higher (at least 5-folds) than the known affinity values to the individual UGT isoforms. All incubations were conducted at 37 °C for 20 min in triplicate.Kinetic assays
Inhibitory constants (Ki) were determined for E2, TFP, and Mag. Various concentrations of pNPG (100, 200, 500 μM), E2 (0–200 μM), TFP (0–200 μM) and Mag (0–100 μM) were employed in the kinetic assays. Either competitive inhibition kinetic model, non-competitive inhibition model, un-competitive inhibition model, or mixed inhibition model were fitted into the kinetic data.18 The Ki values were generated from the inhibition kinetic model displaying the highest R2 values by using Graphpad Prism 5.0. All incubations were conducted at 37 °C for 20 min in triplicate.Docking studies
Molecular docking studies were performed to probe the interaction mode of E2, TFP, and Mag with the active sites of GUS using Surflex-Dock of the SYBYL procedure (x-1.2). The structure of GUS in a complex with inhibitor (PDB code 3LPF) was used as the receptor.12 The B-chain of protein and hetero-atoms including the cofactors were removed from the original protein data file. The 3D structure of these three compounds were optimized by energy minimization using the default Tripos force field parameters. The Gasteiger–Hückel charges were calculated. The active pocket for inhibitor binding was generated around the crystallographic ligand in an automatic mode with the setup of float radius as zero. Compounds were docked into the active sites to observe the interaction mode.Analysis methods
PNPG hydrolysis samples were analysed on a Waters Acquity UPLC system equipped with an ultraviolet detector. A waters C18 analytical column (50.0 mm × 2.1 mm, I.D., 1.7 μm) was used and kept at 40 °C during the analyses. The mobile phase was composed of acetonitrile (A) and 0.1% formic acid in water (B) at a flow rate of 0.3 mL min−1, with a gradient: 0–4 min, 90% B–30% B; 4.01–8 min, 5% B; 8.01–12 min, balance to 90% B. To detect pNP and pNPG, the detector wavelength was set at 310 nm. pNP and pNPG were eluted at 3.6 and 2.4 min, respectively. Hydrolysis rates were quantified by the standard curve of pNP, which was linear at least within 0.1 to 20 μM. The intra- and inter-day RSD were both less than 2%.Conclusions
This study identified that ligand selectivity overlaps between UGTs and GUS deserve attention in the exploration of inhibitors of these two enzymes. UGT typical ligands E2, TFP and Mag could act as competitive inhibitors of GUS via contacts with the residues or bound water in the enzyme active site. The inhibitory potential of the three UGT ligands could induce the inhibition of gut hydrolysis of glucuronides, further displaying protective effects on the intestinal tract.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was supported by National Natural Science Foundation of China (81503151), Provisional Educational & Natural Science Foundation of Anhui (2018jyssf086, 1808085QC97 and KJ2018A0355), Dalian Youth science and technology star project (2017RQ046).References
- E. C. C. Mano, A. L. Scott and K. M. Honorio, Curr. Med. Chem., 2018, 25, 3247–3255 CrossRef PubMed.
- A. Rowland, J. O. Miners and P. I. Mackenzie, Int. J. Biochem. Cell Biol., 2013, 45, 1121–1132 CrossRef CAS PubMed.
- B. Sperker, J. T. Backman and H. K. Kroemer, Clin. Pharmacokinet., 1997, 33, 18–31 CrossRef CAS PubMed.
- P. Awolade, N. Cele, N. Kerru, L. Gummidi, E. Oluwakemi and P. Singh, Eur. J. Med. Chem., 2020, 187, 111921 CrossRef CAS PubMed.
- G. Ma, Y. Zhang, W. Chen, Z. Tang, X. Xin, P. Yang, X. Liu, W. Cai and M. Hu, Mol. Pharm., 2017, 14, 2952–2966 CrossRef CAS PubMed.
- M. Iwase, K. ichi Fujita, Y. Nishimura, N. Seba, Y. Masuo, H. Ishida, Y. Kato and Y. Kiuchi, Cancer Chemother. Pharmacol., 2019, 83, 993–998 CrossRef CAS PubMed.
- D. Liu, L. Zhang, L. xin Duan, J. jun Wu, M. Hu, Z. qiu Liu and C. yan Wang, Pharmacol. Res., 2019, 150, 104510 CrossRef CAS PubMed.
- K. M. Knights, A. Rowland and J. O. Miners, Br. J. Clin. Pharmacol., 2013, 76, 587–602 CAS.
- K. Grancharov, Z. Naydenova, S. Lozeva and E. Golovinsky, Pharmacol. Ther., 2001, 89, 171–186 CrossRef CAS PubMed.
- J. A. Williams, R. Hyland, B. C. Jones, D. A. Smith, S. Hurst, T. C. Goosen, V. Peterkin, J. R. Koup and S. E. Ball, Drug Metab. Dispos., 2004, 32, 1201–1208 CrossRef CAS PubMed.
- Z. Y. Zhong, B. Bin Sun, N. Shu, Q. S. Xie, X. G. Tang, Z. L. Ling, F. Wang, K. J. Zhao, P. Xu, M. Zhang, Y. Li, Y. Chen, L. Liu, L. Z. Xia and X. D. Liu, Acta Pharmacol. Sin., 2016, 37, 1002–1012 CrossRef CAS PubMed.
- B. D. Wallace, H. Wang, K. T. Lane, J. E. Scott, J. Orans, J. S. Koo, M. Venkatesh, C. Jobin, L. A. Yeh, S. Mani and M. R. Redinbo, Science, 2010, 330, 831–835 CrossRef CAS PubMed.
- T. Kodawara, T. Higashi, Y. Negoro, Y. Kamitani, T. Igarashi, K. Watanabe, H. Tsukamoto, R. Yano, M. Masada, H. Iwasaki and T. Nakamura, Basic Clin. Pharmacol. Toxicol., 2016, 118, 333–337 CrossRef CAS PubMed.
- X. Lv, J. Bin Zhang, J. Hou, T. Y. Dou, G. B. Ge, W. Z. Hu and L. Yang, Biotechnol. J., 2019, 14, e1800002 CrossRef PubMed.
- Z. M. Weng, P. Wang, G. B. Ge, Z. R. Dai, D. C. Wu, L. W. Zou, T. Y. Dou, T. Y. Zhang, L. Yang and J. Hou, Food Chem. Toxicol., 2017, 109, 975–983 CrossRef CAS PubMed.
- V. Uchaipichat, P. I. Mackenzie, D. J. Elliot and J. O. Miners, Drug Metab. Dispos., 2006, 34, 449–456 CrossRef CAS PubMed.
- J. O. Miners, K. Bowalgaha, D. J. Elliot, P. Baranczewski and K. M. Knights, Drug Metab. Dispos., 2011, 39, 644–652 CrossRef CAS PubMed.
- L. Zhu, G. Ge, Y. Liu, G. He, S. Liang, Z. Fang, P. Dong, Y. Cao and L. Yang, Xenobiotica, 2012, 42, 1001–1008 CrossRef CAS PubMed.
- L. Ai, L. Zhu, L. Yang, G. Ge, Y. Cao, Y. Liu, Z. Fang and Y. Zhang, Xenobiotica, 2014, 44, 320–325 CrossRef CAS PubMed.
- V. Uchaipichat, L. K. Winner, P. I. MacKenzie, D. J. Elliot, J. A. Williams and J. O. Miners, Br. J. Clin. Pharmacol., 2006, 61, 427–439 CrossRef CAS PubMed.
- R. J. Liang, Y. N. Shih, Y. L. Chen, W. Y. Liu, W. L. Yang, S. Y. Lee and H. J. Wang, Eur. J. Pharm. Sci., 2020, 141, 105093 CrossRef CAS PubMed.
- S. P. L. Chen, W. T. Poon, C. M. Mak, C. W. Lam, Y. L. Kwong, A. Y. W. Chan and S. Tam, Pathology, 2011, 43, 273–274 CrossRef CAS PubMed.
- J. O. Miners, K. M. Knights, J. B. Houston and P. I. Mackenzie, Biochem. Pharmacol., 2006, 71, 1531–1539 CrossRef CAS PubMed.
- K. Itäaho, P. I. Mackenzie, S. I. Ikushiro, J. O. Miners and M. Finel, Drug Metab. Dispos., 2008, 36, 2307–2315 CrossRef PubMed.
- L. Zhu, L. Xiao, Y. Xia, K. Zhou, H. Wang, M. Huang, G. Ge, Y. Wu, G. Wu and L. Yang, Toxicol. Appl. Pharmacol., 2015, 283, 109–116 CrossRef CAS PubMed.
- L. Zhu, G. Ge, H. Zhang, H. Liu, G. He, S. Liang, Y. Zhang, Z. Fang, P. Dong, M. Finel and L. Yang, Drug Metab. Dispos., 2012, 40, 529–538 CrossRef CAS PubMed.
- T. S. Rasmussen, H. Koldsø, S. Nakagawa, A. Kato, B. Schiøtt and H. H. Jensen, Org. Biomol. Chem., 2011, 9, 7807–7813 RSC.
- V. N. Luine, Horm. Behav., 2014, 66, 602–618 CrossRef CAS PubMed.
- A. C. Johnson and R. J. Williams, Environ. Sci. Technol., 2004, 38, 3649–3658 CrossRef CAS PubMed.
- S. E. Pritchard, L. Marciani, K. C. Garsed, C. L. Hoad, W. Thongborisute, E. Roberts, P. A. Gowland and R. C. Spiller, Neurogastroenterol. Motil., 2014, 26, 124–130 CrossRef CAS PubMed.
- M. J. Ellis, F. Gao, F. Dehdashti, D. B. Jeffe, P. K. Marcom, L. A. Carey, M. N. Dickler, P. Silverman, G. F. Fleming, A. Kommareddy, S. Jamalabadi-Majidi, R. Crowder and B. A. Siegel, J. Am. Med. Assoc., 2009, 302, 774–780 CrossRef CAS PubMed.
- M. Tardy, M. Dold, R. R. Engel and S. Leucht, Cochrane Database Syst. Rev., 2014, CD009396 Search PubMed.
- N. R. West and W. H. Vogel, Arch. Int. Pharmacodyn. Ther., 1975, 215, 318–335 CAS.
- K. K. Midha, E. D. Korchinski, R. M. H. Roscoe, E. M. Hawes, J. K. Cooper and G. McKay, J. Pharm. Sci., 1984, 73, 261–263 CrossRef CAS PubMed.
- Y. J. Lee, Y. M. Lee, C. K. Lee, J. K. Jung, S. B. Han and J. T. Hong, Pharmacol. Ther., 2011, 130, 157–176 CrossRef CAS PubMed.
- M. Homma, K. Oka, H. Kobayashi, T. Nhtsuma, S. Yamamoto, H. Itoh and N. Takahashi, J. Pharm. Pharmacol., 1993, 45, 839–841 CrossRef CAS PubMed.
- R. Meech and P. I. Mackenzie, Clin. Exp. Pharmacol. Physiol., 1997, 24, 907–915 CrossRef CAS PubMed.
- P. Lautala, B. T. Ethell, J. Taskinen and B. Burchell, Drug Metab. Dispos., 2000, 28, 1385–1389 CAS.
- H. Zhang, A. Basit, D. Busch, K. Yabut, D. K. Bhatt, M. Drozdzik, M. Ostrowski, A. Li, C. Collins, S. Oswald and B. Prasad, Biochem. Pharmacol., 2018, 156, 32–42 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |