
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical insights into a colorimetric azo-based probe to detect copper ions†
Juan Panga,
Li Shub,
Ming Lia and
Xiaohong Hu *a
*a
aCollege of Material Science and Engineering, Jinling Institute of Technology, Nanjing 211169, People's Republic of China. E-mail: hxh@jit.edu.cn; Tel: +86 18913805386
bDepartment of Chemical and Materials Engineering, Hefei University, Hefei 230601, People's Republic of China
First published on 17th June 2020
Abstract
In the present study, a colorimetric azobenzene-based probe (AZO 1) was reported that exhibits high selectivity toward Cu2+ and undergoes a red to yellow colour change upon its detection. Density functional theory (DFT) calculations were carried out to investigate the mechanism of the probe discoloration. The differences in the binding energies of complexes of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 stoichiometry indicated that a two-step complexation process takes place as the Cu2+ content increases. However, the calculated absorption spectra suggested that a significant colour change would only be observed for the 1:1 AZO 1:Cu2+ complex. A HOMO–LUMO electronic transition was a key factor for the blue shift of the absorption bands of the probe. Further studies indicated that solvent molecules participate in the complexation and that the presence of the o-methoxy group in AZO 1 led to formation of an octahedral complex because of the additional chelating site. A significant change in the conformation of AZO 1, namely the rotation of the N,N-di(carboxymethyl)amino group around the N–CAr bond by approximately 90°, resulted in a larger HOMO–LUMO energy gap, and the corresponding alteration of the intramolecular charge transfer (ICT) from the N,N-di(carboxymethyl)amino group to the phenyl ring led to the observed colour change.
:1 and 1:1 stoichiometry indicated that a two-step complexation process takes place as the Cu2+ content increases. However, the calculated absorption spectra suggested that a significant colour change would only be observed for the 1:1 AZO 1:Cu2+ complex. A HOMO–LUMO electronic transition was a key factor for the blue shift of the absorption bands of the probe. Further studies indicated that solvent molecules participate in the complexation and that the presence of the o-methoxy group in AZO 1 led to formation of an octahedral complex because of the additional chelating site. A significant change in the conformation of AZO 1, namely the rotation of the N,N-di(carboxymethyl)amino group around the N–CAr bond by approximately 90°, resulted in a larger HOMO–LUMO energy gap, and the corresponding alteration of the intramolecular charge transfer (ICT) from the N,N-di(carboxymethyl)amino group to the phenyl ring led to the observed colour change.
1. Introduction
Since the mid 1800s,1 azobenzene and its derivatives have been considered an important family of synthetic dyes. Numerous studies describe the synthetic methods, absorption spectra, and trans–cis isomerisation of these compounds, among other characteristics. The trans-form is generally stable and predominant.2,3 The absorption spectrum of π-conjugated trans-azobenzene has a strong and a weak band at approximately 320 and 440 nm, respectively.4 However, the absorption wavelength depends on various factors such as substituents,5–7 solvents,8 pH,9–11 and complexation to metal ions.12,13 In addition to their use as synthetic colouring agents in the dye industry, azo compounds are promising candidates for application in molecular switches,14–17 optical memory devices,18,19 and ion detection,20–24 among others.Ion detection, especially the selective determination of heavy transition metal ions such as Cu2+, is essential to the analysis of biological and environmental samples. Copper is an indispensable trace element for human health that plays an important role in the formation and functioning of blood cells, central nervous system, immune system, and internal organs. Although it is vital to human health in small amounts, it may be toxic in large quantities. With the continuous accumulation of copper in the body, severe health problems, such as gastrointestinal disturbance and liver or kidney damage, occur.25 Hence, the concentration of Cu2+ in drinking water must be lower than 1.3 ppm (20 mM) according to the US Environmental Protection Agency.26
Among many approaches to detect Cu2+, colorimetric methods27,28 are an attractive option because of their convenience, high speed, and low cost. However, colorimetric probes for the naked eye detection of Cu2+ reported to date are few, and include dynamic metal–organic framework receptors,29 amido-imine based receptors,30 and an azobenzene disperse dye-based colorimetric probe.31–33 A large shift in the UV-vis absorption band is an important criterion for an ideal visualization probe. In 2004, Gunnlaugsson et al.34 synthesized the dipotassium salts of {carboxymethyl-[2-methoxy-4-(4-nitro-phenylazo)-phenyl]-amino}-acetic acid and {carboxymethyl-[4-(4- nitro-phenylazo)-phenyl]-amino}-acetic acid (salts 1 and 2, respectively, Fig. 1). Although both salts are azobenzene-based, the absorption of salt 1 was highly affected by the presence of Cu2+ with a large blue shift of approximately 184 nm, whereas salt 2 lacking an o-methoxy group did not detect copper ions. These findings raise the issue of whether the additional chelation site provided by the o-methoxy group is solely responsible for the different responses of salts 1 and 2 to copper ions.
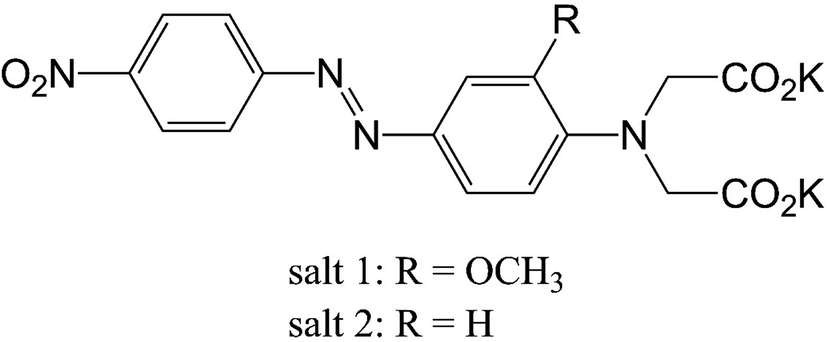 | ||
| Fig. 1 Chemical structure of the investigated salts. | ||
Currently, computational chemistry is widely accepted by scientists in different fields as a tool to provide explanations at the atomic or molecular levels. Herein, density functional theory (DFT) methods were used to explore the effect of the o-methoxy group in salt 1 and reveal the mechanism of Cu2+ detection by a colorimetric azo-based probe. The molecular models and essential details of the DFT calculations, described in the next section, allowed for calculations of various parameters, including molecular geometries, complexation process, interaction energies, spectra, and major molecular orbitals involved in the transitions. We expect that the present study will promote the development of ion detection techniques.
2. Theoretical studies
2.1. Molecular models
Considering that salts dissociate in aqueous solutions, {carboxymethyl-[2-methoxy-4-(4-nitro-phenylazo)-phenyl]-amino}-acetate (AZO 1) and {carboxymethyl-[4-(4-nitro-phenylazo)-phenyl]-amino}-acetate (AZO 2) were used in the modelling. The two most common spatial structural models for copper ions, the planar four-coordination and octahedral six-coordination models, were adopted. As complex structures can also be formed between copper ions and solvent molecules, the participation of chloride ions and water molecules in complexation was taken into account. Based on these considerations, distinct initial complexes of 2:1 and 1:1 stoichiometry were modelled for the AZO ligands and Cu2+.
2.2. Computational details
Geometry optimisation and frequency calculations of the molecular models were performed using B3LYP/6-31G(d,p), which enables a qualitative prediction of the absorption properties of azobenzenes.2,5 The LanL2DZ effective core potentials and a spin-unrestricted model for Cu2+ were employed. The absorption spectra were predicted by the time-dependent density functional theory (TDDFT) and 50 states were calculated. Solvent effects were considered within the polarizing continuum model (PCM) framework by including water in the geometry optimisation and excited state calculations. All quantum calculations were performed using Gaussian 16 programmes35 on a Linux system.3. Results and discussion
Geometry optimisation and frequency calculations for AZO 1 and AZO 2 in aqueous solutions were performed to locate and verify the minima. The electrostatic potential can clarify the molecule's reactivity since negative electrostatic potentials correspond to a high electron density and the ability to attract a proton and vice versa.36 The mapped electrostatic potential surfaces generated from the electron density cubes are illustrated in Fig. S1.† The colour changed from blue to red along with the value of the electrostatic potential which varied from 0.28 to −0.28. As expected, the increased electron density in the vicinity of the methoxy and carboxyl groups of AZO 1 represented an additional Cu2+ chelation site.3.1. Complexation of AZOs and Cu2+ in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 stoichiometry
:1 stoichiometry
When the copper ion concentration was relatively low, complexation of AZOs and Cu2+ occurred in 2:1 stoichiometry. The optimised structures are shown in Fig. 2, which shows (a) the complexation of AZO 1 and Cu2+, 1 + Cu2+ + 1, and (b) that of AZO 2 and Cu2+, 2 + Cu2+ + 2. For AZO 1, the oxygen and nitrogen atoms of the N,N-di(carboxymethyl)amino groups and the oxygen atoms of the o-methoxy groups participated in the chelation of Cu2+. However, chelation of Cu2+ by AZO 2, lacking o-methoxy groups, occurred only via the oxygen and nitrogen atoms of the N,N-di(carboxymethyl)amino groups, resulting in an approximately square-planar coordination geometry and a “Z” or “П” arrangement of the azo groups. The distance between Cu2+ and the chelation site, N–O–Cu angles, and binding energies are listed in Table S1.† The latter was calculated as the energy of complexation minus the energies of each molecule. A careful analysis of the relationship between structure and binding energy indicated that higher binding energy values corresponded to Cu2+ complexes closer to square planar or octahedral geometries (Fig. 2 and Table S1†).
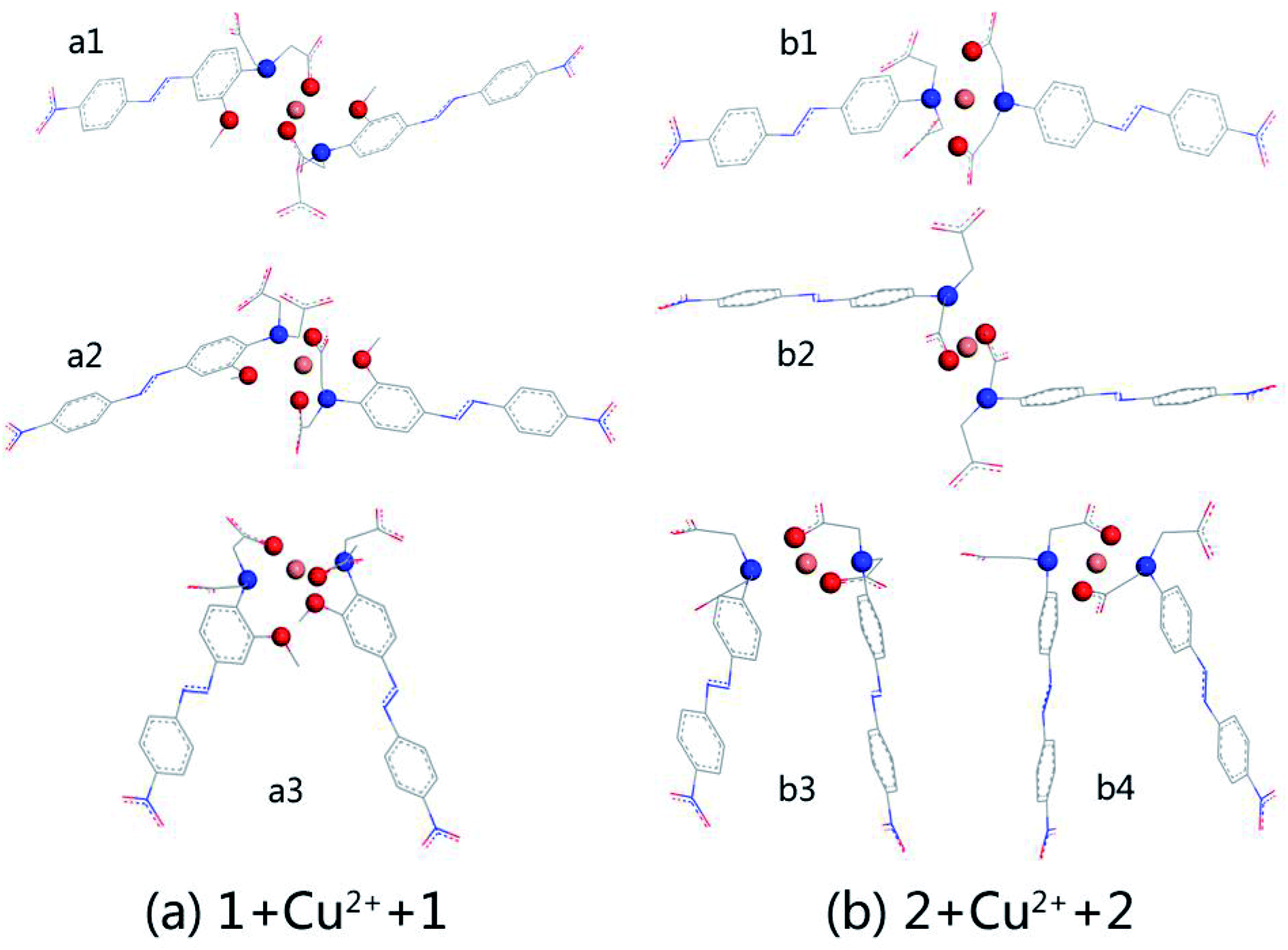 | ||
| Fig. 2 Optimized structures of 2:1 stoichiometry for AZOs and Cu2+ calculated at the levels of B3LYP/6-31G(d,p)//LanL2DZ: (a) complexation of AZO 1 and Cu2+; (b) complexation of AZO 2 and Cu2+. The chelation sites and Cu2+ are all highlighted by using balls of different colors. N is in blue, O is in red, and Cu2+ is in orange. All hydrogen atoms are not shown. | ||
We selected the complexation of a2 and b4 with binding energies of −223.606 and −213.894 kcal mol−1, respectively, for further study. Their UV-vis spectra are shown in Fig. 3 along with the corresponding main molecular orbitals, whereas the spectra of the other structures were depicted in Fig. S2.† The absorption peak of a2 in the range of 550–650 nm was noticeably weaker than those of the other structures (Fig. 3 and S2†). The involved transition orbitals indicate that these peaks originate from the intramolecular charge transfer (ICT) from the N,N-di(carboxymethyl)amino group to the phenyl rings, rendering this ICT dependent on the spatial arrangement of the groups. In a2, slight rotation of the N,N-di(carboxymethyl)amino group to form an octahedral Cu2+ complex weakened the ICT. In addition, the calculated absorption spectra suggest that no obvious colour changes accompanied the chelation of Cu2+ by AZO 1 or AZO 2 in solution when the stoichiometry was 2:1.
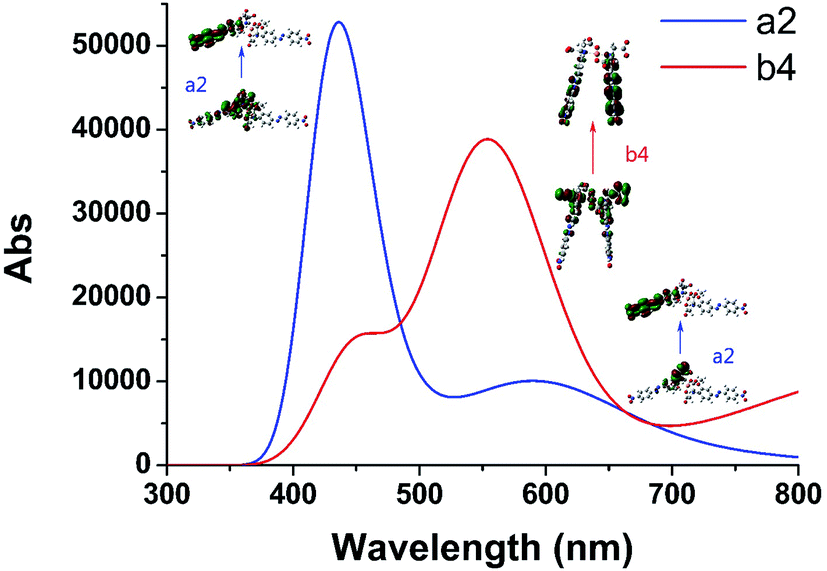 | ||
| Fig. 3 Calculated UV-vis spectra of complexation of 2:1 stoichiometry for AZOs and Cu2+ of a2 and b4 which have larger binding energies than other complexation. Some main molecular orbitals were presented next to the corresponding absorption peaks. | ||
3.2. Complexation of AZOs and Cu2+ in 1:1 stoichiometry
With the increase in the copper ion content, complexation of AZOs and Cu2+ in 1:1 stoichiometry takes place. Out of different initial models, optimised structures were selected for detailed studies taking into account the planar four-coordination or octahedral six-coordination of Cu2+. The structures of 1 + Cu2+ + 2Cl−, 1 + Cu2+ + 2H2O, 2 + Cu2+ + Cl−, and 2 + Cu2+ + H2O, together with the optimised AZO 1 and AZO 2 structures are displayed in Fig. 4, whereas other optimised complexation modes are presented in Fig. S3.† The chelation sites and Cu2+ are highlighted by differently coloured spheres (blue for N, red for O, green for Cl−, and orange for Cu2+). The artificially added light grey plate and arrow indicate the position of the atoms in Fig. 4, which shows the participation of the coplanar O1, O2, and N atoms in the complexation of Cu2+. Additionally, the o-methoxy group in AZO 1 forces the central copper ion to adopt the six-coordinated octahedral geometry. However, complexation of Cu2+ by AZO 2 is different than that by AZO 1, as the lack of the o-methoxy group results in a change in the spatial location of the copper ion from the side of the azobenzene plane to its front. Due to the repulsion of the azobenzene conjugated plane, stable octahedral structures are not obtained. Instead, the four-coordinated planar geometry is acquired for 2 + Cu2+ + Cl− and 2 + Cu2+ + H2O (Fig. 4(e) and (f), respectively).
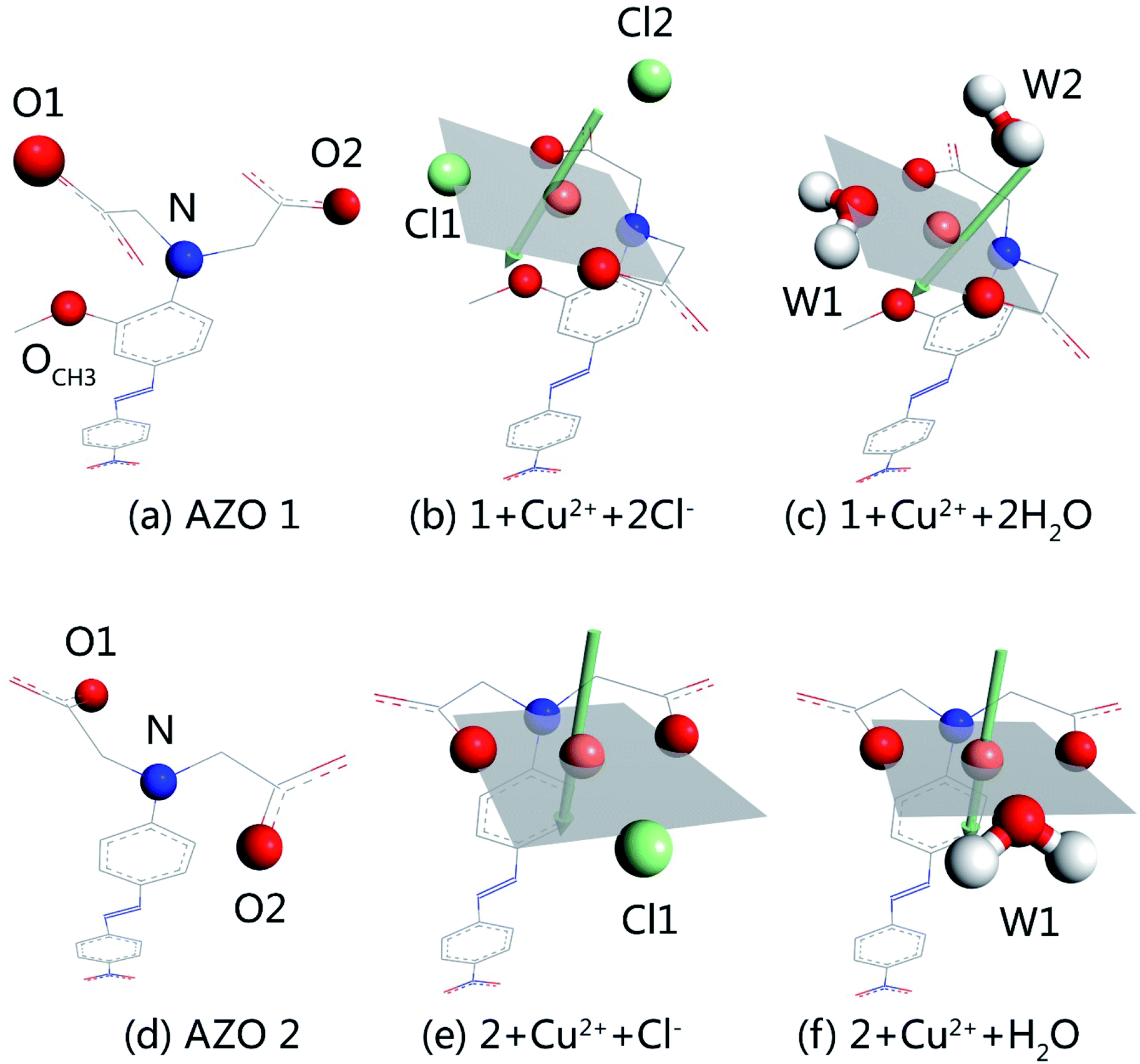 | ||
| Fig. 4 Optimized structures of AZO 1, AZO 2 and complexation of 1:1 stoichiometry for AZOs and Cu2+ calculated at the levels of B3LYP/6-31G(d,p)//LanL2DZ: (a) AZO 1, (b) 1 + Cu2+ + 2Cl−, (c) 1 + Cu2+ + 2H2O, (d) AZO 2, (e) 2 + Cu2+ + Cl− and (f) 2 + Cu2+ + H2O. A light gray plate and an arrow were added artificially only to help make clear the position of atoms. | ||
Table S2† shows the binding energies in the complexes, which consist of azobenzenes, Cu2+, and solvent particles (chlorine ions or water molecules) and have therefore two distinct binding energies. That between AZO and Cu2+ (ΔE1), calculated as ΔE1 = Ecomplex − EAZO − Ecopper ion + solvent particles, is −119.263, −160.862, −127.272, and −174.463 kcal mol−1 for 1 + Cu2+ + 2Cl−, 1 + Cu2+ + 2H2O, 2 + Cu2+ + Cl−, and 2 + Cu2+ + H2O respectively, whereas the energy between Cu2+ and solvent particles (ΔE2), calculated as ΔE2 = Ecomplex − EAZO + copper ion − Esolvent particles, is −32.925, −32.421, −27.833, and −23.001 kcal mol−1 for the same complexes. The total binding energy (ΔE) was calculated as the energy of complexation minus the energies of each molecule, and were −241.589, −248.296, −224.407, and −222.576 kcal mol−1 for 1 + Cu2+ + 2Cl−, 1 + Cu2+ + 2H2O, 2 + Cu2+ + Cl−, and 2 + Cu2+ + H2O, respectively. The values of ΔE1 reveal that the interaction between AZO and Cu2+ is enhanced by water molecules more significantly than by chlorine ions. Additionally, comparison of the binding energies of AZOs and Cu2+ complexes of 1:1 and 2:1 stoichiometry shows that the former are more stable structures. Hence, a two-step complexation process (AZOs and Cu2+ from 2:1 to 1:1 stoichiometry) should occur as a result of an increase in the copper ion content.
The vertical electronic excitation energies were calculated at the TD-B3LYP/6-31G(d,p)//LanL2DZ level. The absorption spectra were artificially broadened using the GaussSum 2.2.5 software. The simulated UV-vis spectra of AZO 1, 1 + Cu2+ + 2H2O, AZO 2, and 2 + Cu2+ + H2O, and the UV-vis spectra of 1 + Cu2+ + 2Cl− and 2 + Cu2+ + Cl− shown in Fig. 5 and S4,† respectively, reveal that the influence of the chlorine ions on the absorption spectra is similar to that of the water molecules. Since the latter can significantly increase the interaction between the AZO and copper ion, only the absorption spectra of Fig. 5 are analysed herein. Two distinct absorption bands of AZO 1 are calculated at 672 and 465 nm (the experimental values28 are 509 and 325 nm), and those of AZO 2 are at 644 and 432 nm, indicating a red shift of approximately 30 nm upon introduction of the o-methoxy group. However, the spectra of 1 + Cu2+ + 2H2O and 2 + Cu2+ + H2O are distinct. The former shows an extinct peak at 366 nm and a shoulder at 416 nm. The clear blue shift with regard to the spectrum of AZO 1 explains the colour change of the solution from red to yellow. However, the absorption spectrum of 2 + Cu2+ + H2O shows a weak band in the range of 500–800 nm. As the absorption intensity increases with the concentration and the solution contained more than one AZO 2, this absorption cannot be ignored in the real experiment. Since a 1:1 mixture of AZO 2 and Cu2+ absorbed in the green spectral region, no obvious colour change was observed during the experiment.
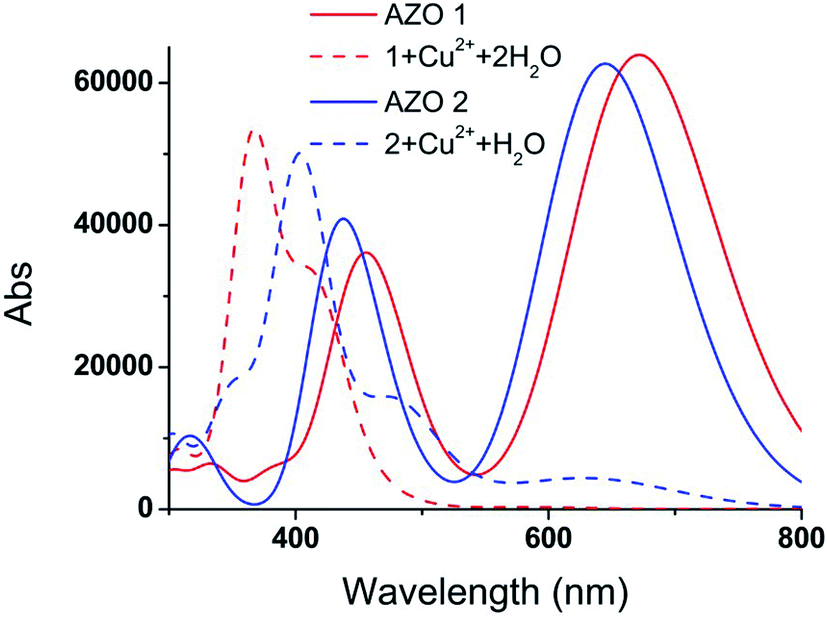 | ||
| Fig. 5 Calculated UV-vis spectra of AZO 1, 1 + Cu2+ + 2H2O, AZO 2 and 2 + Cu2+ + H2O. | ||
In order to explain the observed >250 nm blue shift of the AZO 1 absorption bands in contrast to the unchanged AZO 2 absorption in the range of 500–800 nm upon combining the AZOs with Cu2+ in 1:1 stoichiometry, the major molecular orbitals involved in the transitions were examined (Fig. 6). In the case of AZO 1, the red colour results from the first lowest excited state of 672 nm, which corresponds to the HOMO → LUMO transition (98%, Osc. = 0.8804). The data of 1 + Cu2+ + 2H2O show that the 11th to 25th excited states were relevant to the colour change, among which the 12th and 23rd excited states greatly contributed to the absorption. Specifically, the 12th excited state (416 nm, Osc. = 0.3762) was related to the ICT from the N,N-di(carboxymethyl)amino group to the phenyl rings (HOMO → LUMOα, 53%), while the 23rd excited state (366 nm, Osc. = 0.6931) mainly indicated the ICT from the phenyl rings to the nitro group (HOMO-4 → LUMOα, 46%).
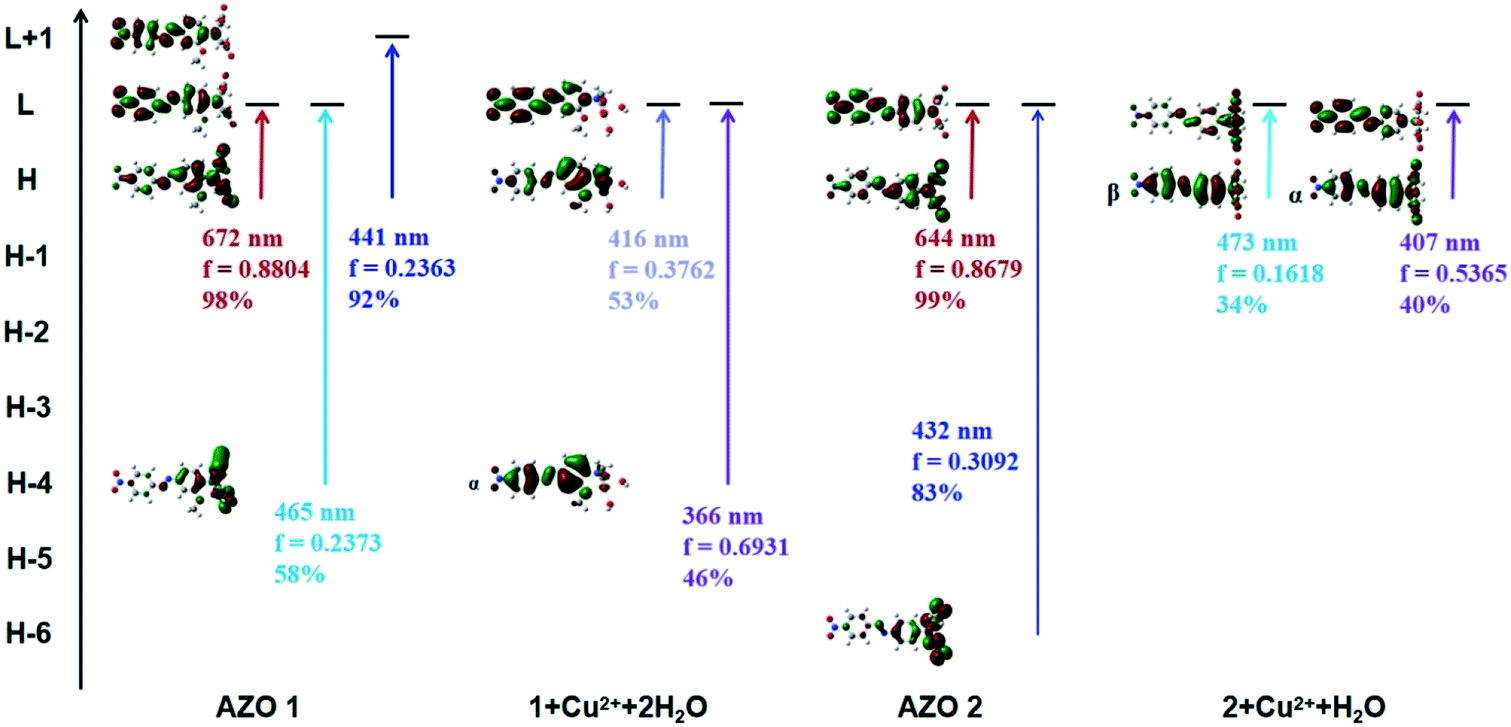 | ||
| Fig. 6 Some main molecular orbitals transitions of AZO 1, 1 + Cu2+ + 2H2O, AZO 2 and 2 + Cu2+ + H2O. | ||
Similarly, we found that the AZO 2 absorption peaks resulting from the HOMO → LUMO transition (644 nm, Osc. = 0.8679, ICT from the N,N-di(carboxymethyl)amino group to phenyl rings) accounted for the red colour. A weak peak at 656 nm in the spectrum of 2 + Cu2+ + H2O indicated a HOMO → LUMOβ transition (54%, ICT from the phenyl rings to the N,N-di(carboxymethyl)amino group). A shoulder at 473 nm was also observed due to the HOMO → LUMOβ transition (34%), whereas the strong peak at 407 nm results from a HOMO → LUMOα transition (40%, ICT from the N,N-di(carboxymethyl)amino group to the phenyl rings). Hence, we infer that the HOMO to LUMO electronic transition is a key factor for colour change (or lack thereof) upon combining AZO 1 and AZO 2 with Cu2+ in a 1:1 stoichiometry. The HOMO–LUMO energy gap was calculated as ΔEH–L = EHOMO − ELUMO. The ΔEH–L of AZO 1 and AZO 2 are 1.9 and 2.01 eV, respectively, whereas the ΔEH–L of 1 + Cu2+ + 2H2O are 3.37 and 3.34 eV for the α and β orbitals, respectively, and the corresponding values for 2 + Cu2+ + H2O are 3.32 and 2.87 eV (α and β orbitals, respectively). Clearly, the AZO 1 and AZO 2 strong bands and the 2 + Cu2+ + H2O weak bands in the range of 500–800 nm were due to the small ΔEH–L (<3 eV). Hence, the presence of the o-methoxy group in AZO 1 led to formation of an octahedral copper complex. As a result, rotation of the N,N-di(carboxymethyl)amino group by approximately 90° around the N–CAr bond, which in turn led to a larger HOMO–LUMO gap of approximately 3.34 or 3.37 eV related to the ICT from the N,N-di(carboxymethyl)amino group to the phenyl ring.
4. Conclusion
To investigate the mechanism of Cu2+ detection by AZO 1 in aqueous solution, DFT calculations at the B3LYP/6-31G(d,p)//LanL2DZ level were performed. For comparison, the related AZO 2 molecule lacking an o-methoxy group on the phenyl ring was also studied. The mapped electrostatic potential surfaces suggested the o-methoxy group in AZO 1 is an additional chelation site for Cu2+. Complexation of AZOs and Cu2+ in a 2:1 stoichiometry would occur at low Cu2+ concentrations without colour change, according to the corresponding calculated absorption spectra. With the increase in the Cu2+ concentration, complexation of AZOs and Cu2+ in a 1:1 stoichiometry would occur. The geometry of the optimised structures revealed that the formation of an octahedral complex requires rotation of the N,N-di(carboxymethyl)amino group by approximately 90° around the N–CAr bond in AZO 1. In contrast, AZO 2 lacking a o-methoxy group led to a four-coordinated, planar Cu2+ complex in which the N,N-di(carboxymethyl)amino group did not rotate. Further studies revealed that electronic transitions from HOMO to LUMO were the key factors in the colour changes of the solutions, thus enabling the detection of Cu2+. The change in the conformation of AZO 1 in the 1:1 complex is associated with a larger HOMO–LUMO gap (ΔEH–L was 3.34 and 3.37 eV for α and β orbitals, respectively). Hence, the ICT from the N,N-di(carboxymethyl)amino group to the phenyl ring with a larger ΔEH–L results in >250 nm blue shift of the absorption bands, which corresponds to the change in the colour of the solution from red to yellow.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper was financially supported by National Natural Science Foundation of China (21702082) and Natural Science Foundation of Jiangsu Province (BK20171113).References
- V. Shibaev, A. Bobrovsky and N. Boiko, Prog. Polym. Sci., 2003, 38, 729–836 CrossRef.
- Y. Ye, J. Pang, X. Zhou and J. Huang, Comput. Theor. Chem., 2016, 1076, 17–22 CrossRef CAS.
- C. J. Barrett, J. Mamiya, K. G. Yager and T. Ikeda, Soft Matter, 2007, 3, 1249–1261 RSC.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- J. Pang, Z. Tian and J. Ma, Chem. Phys. Lett., 2014, 613, 110–114 CrossRef CAS.
- O. Sadovski, A. A. Beharry, F. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484–1486 CrossRef CAS PubMed.
- Z. Yan, S. Guang, H. Xu and X. Liu, Dyes and Pigments, 2013, 99, 720–726 CrossRef CAS.
- D. L. Isac, A. Airinei, M. Homocianu, N. Fifere, C. Cojocaru and C. Hulubei, J. Photochem. Photobiol., A, 2020, 390, 112300 CrossRef CAS.
- A. D. W. Kennedy, I. Sandler, J. Andreasson, J. Ho and J. E. Beves, Chem.–Eur. J., 2020, 26, 1103–1110 CrossRef CAS PubMed.
- W. Yuan, C. Wang, S. Lei, J. Chen, S. Lei and Z. Li, Polym. Chem., 2018, 9, 3098–3107 RSC.
- N. J. Dunn, W. H. Humphries, A. R. Offenbacher, T. L. King and J. A. Gray, J. Phys. Chem. A, 2009, 113, 13144–13151 CrossRef CAS PubMed.
- T. Muraoka, K. Kinbara and T. Aida, Nature, 2006, 440, 512–515 CrossRef CAS PubMed.
- J. Pang, Y. Ye, Z. Tian, X. Pang and C. Wu, Comput. Theor. Chem., 2015, 1066, 28–33 CrossRef CAS.
- W. R. Browne and B. L. Feringa, Annu. Rev. Phys. Chem., 2009, 60, 407–428 CrossRef CAS PubMed.
- J. M. Mativetsky, G. Pace, M. Elbing, M. A. Rampi, M. Mayor and P. Samori, J. Am. Chem. Soc., 2008, 130, 9192–9193 CrossRef CAS PubMed.
- J. Pang, Z. Gao, H. Tan, X. Mao, J. Xu, J. Kong and X. Hu, Front. Chem., 2019, 7, 620 CrossRef CAS PubMed.
- J. Pang, Z. Gao, L. Zhang, H. Wang and X. Hu, Front. Chem., 2018, 6, 217 CrossRef PubMed.
- A. Sobolewska, J. Zawada and S. Bartkiewicz, Langmuir, 2014, 30, 17–21 CrossRef CAS PubMed.
- M. kamenjicki, I. K. Lednev, A. Mikhonin, R. kesavamoorthy and S. A. Asher, Adv. Funct. Mater., 2003, 13, 774–780 CrossRef CAS.
- H. Hosseinjani-Pirdehi, N. O. Mahmoodi, M. P. Nadamani and A. Taheri, J. Photochem. Photobiol., A, 2020, 391, 112365 CrossRef CAS.
- Y. Liu, L. Huang, S. Mahmud and H. Liu, J. Cluster Sci., 2020, 31, 549–560 CrossRef CAS.
- X. Cheng, Y. Zhou, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2133–2138 CrossRef CAS PubMed.
- L. Hu, L. Nie, G. Xu, H. Shi, X. Xu, X. Zhang and Z. Yan, RSC Adv., 2014, 4, 19370–19374 RSC.
- Z. Yan, L. Hu, L. Nie and H. Lv, Spectrochim. Acta, Part A, 2011, 79, 661–665 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, X. Kang, M. Ge, H. Feng, J. Han, D. Wang and D. Zhao, J. Photochem. Photobiol., A, 2018, 356, 652–660 CrossRef CAS.
- Z. Liu, C. Zhang, X. Wang, W. He and Z. Guo, Org. Lett., 2012, 14, 4378–4381 CrossRef CAS PubMed.
- G. Zhao, F. Song, G. Wei, R. Wu, Z. Yan, F. Zhang, S. Guang and H. Xu, Sens. Actuators, B, 2019, 286, 163–172 CrossRef CAS.
- Q. Zhao, H. Yuan, X. Xu, L. Hu, P. Gong and Z. Yan, Dyes and Pigments, 2019, 165, 217–222 CrossRef CAS.
- C. Qiao, X. Qu, Q. Yang, Q. Wei, G. Xie, S. Chen and D. Yang, Green Chem., 2016, 18, 951–956 RSC.
- A. Bhattacharyya, S. Ghosh and N. Guchhait, RSC Adv., 2016, 6, 28194–28199 RSC.
- E. Hrishikesan, C. Saravanan and P. Kannan, Ind. Eng. Chem. Res., 2011, 50, 8225–8229 CrossRef CAS.
- H. Ren, P. Wu, F. Li, L. Jin and D. Lou, Inorg. Chim. Acta, 2019, 487, 234–239 CrossRef CAS.
- L. Hu, L. Yin, F. Wang, D. Yu, C. Wang, M. Hui, L. Chu, X. Zhu and Z. Yan, Spectrochim. Acta, Part A, 2019, 220, 117130 CrossRef CAS PubMed.
- T. Gunnlaugsson, J. P. Leonard and N. S. Murray, Org. Lett., 2004, 6, 1557–1560 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. B. Foresman, and Æ. Frisch, Exploring chemistry with electronic structure methods, Gaussian Inc., Wallingford CT, 3rd edn, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02468f |
| This journal is © The Royal Society of Chemistry 2020 |