
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalytical and preparative separation and isolation of functionalized fullerenes by conventional HPLC stationary phases: method development and column screening†
Merve Ergun Dönmez and
Helena Grennberg*
and
Helena Grennberg*
Uppsala University, Department of Chemistry – BMC, Box 576, 75123 Uppsala, Sweden. E-mail: helena.grennberg@kemi.uu.se
First published on 20th May 2020
Abstract
Isolation and purification of functionalized fullerenes from often complex reaction mixtures is challenging due to the hydrophobic nature and low solubility in regular organic solvents. We have developed an HPLC method that efficiently, employing regular reversed phase stationary phases, separates not only C60 from C70 in a model mixture, but also C60 monoadducts from polyadducts and unreacted C60 from fulleropyrrolidine and hydroarylation example reaction mixtures. Six HPLC columns with regular reversed phase stationary phases were evaluated using varying proportions of acetonitrile in toluene as eluent; with C18 and C12 stationary phases with high surface area (450–400 m2 g−1) being the most efficient regarding separation efficiency and analysis time for all mixtures. The analytical method is effectively transferrable to a preparative scale to isolate the monoaddition products from complex fullerene reaction mixtures.
1. Introduction
With more than three decades of fullerene chemistry, modification reactions have been developed which allow addition of a wide range of functional groups, thus transforming the all-carbon molecules into organic compounds with interesting properties, with applications ranging from organic photovoltaics to biomedicine.1–15 Due to permanent curvature, the π-bonds of fullerenes are not conjugated. In contrast to planar aromatic compounds where a new substituent will affect the reactivity even at remote π-conjugated positions, fullerenes react at multiple positions leading to complex hard-to-separate mixtures of mono-, bis- and more highly functionalized fullerenes.16–19 Due to poor solubility in most solvents, purification of product mixtures with isolation of the desired components and assessment of purity are major challenges.20–23Already in 1990, Kroto et al.24 identified liquid chromatography (LC) as a purification method that could provide C60 and C70 as clean fractions from their complex synthesis soot mixtures. More recently, chromatographic separations on silica and alumina have been reported, however employing solvents with poor environmental profile.25–27 Alternatives, in particular the fullerene-specific stationary phases, opened for LC methods that employ less harmful solvents for both preparative and analytical HPLC applications.28–30 However, a more generally applicable method for analysis and separation of complex fullerene mixtures using regular HPLC columns and standard solvents has been lacking. Herein, we report a method that give efficient analytical and preparative separation using standard stationary phases and toluene–acetonitrile mobile phases for model mixtures of C60 and C70 as well as for two example reaction mixtures: pyrrolidination31 and hydroarylation,32 (Scheme 1) reactions that result in different proportions of unreacted C60, monoadducts and higher adducts.
 | ||
| Scheme 1 General representation of pyrrolidination and hydroarylation of C60 by literature conditions, resulting in the two reaction product mixtures of the present study. | ||
2. Materials and methods
2.1. Reagents and materials
All chemicals, including C60 and C70 were purchased from Sigma Aldrich and used as received. Acetonitrile and toluene was purchased from VWR Chemicals as HPLC grade. The columns used are listed in Table 1. The analytical columns were used with a Gemini C18 pre-guard cartridge (2.0 × 4.0 mm, Phenomenex, USA), the preparative column was used with ACE 5 C18 (10.0 × 10.0, Advanced Chromatography Technologies Ltd, UK) pre-guard cartridge.| Column code | Brand name | Inner dimensions length × radius | Particle size | Surface area (m2 g−1) | Carbon load (%) | Column packing | End-capping |
|---|---|---|---|---|---|---|---|
| a From GL Sciences, Japan.b From Phenomenex, USA.c From Advanced Chromatography Technologies Ltd, UK. | |||||||
| Col 1a | Inertsil ODS 4 | 250 × 4.6 mm | 5.0 μm | 450 | 11 | C18 | TMS |
| Col 2b | Synergi Hydro-RP | 100 × 4.6 mm | 4.0 μm | 400 | 19 | C18 | Hydrophilic |
| Col 3b | Synergi Max-RP | 150 × 4.6 mm | 4.0 μm | 400 | 17 | C12 | TMS |
| Col 4b | Gemini NX | 100 × 3.0 mm | 3.0 μm | 375 | 14 | C18 | TMS |
| Col 5b | Kinetex Biphenyl | 100 × 4.6 mm | 2.6 μm | 200 | 11 | C12 (biphenyl) | TMS |
| Col 6b | Kinetex Phenyl-Hexyl | 150 × 4.6 mm | 5.0 μm | 200 | 11 | C12 (phenyl-hexyl) | TMS |
| Col 7c | ACE 5 | 150 × 21.0 mm | 5.0 μm | 300 | 15.5 | C18 | TMS |
In order to describe the quality of the peaks and the efficiency of the separation for all analytical columns, retention times (Rt), total analysis time (run time), capacity factors (k′) and resolution (Rs) were evaluated and summarized in Tables 2 and 3. The capacity factor (k′) is a measure that gives the molecules retention compared to the un-retained component or solvent, typically preferred to be higher than 0.8. Resolution (Rs) is a measure to describe the separation between two adjacent peaks, typically preferred to be higher than 2.0.
| Rt (C60) | Rt (2) | Run time | k′ (2) | Rsa | |
|---|---|---|---|---|---|
| a Calculated according to the closest peak.b 55% (v/v) acetonitrile in toluene was used as mobile phase. NS: not separated. The values in bold/italic are outside the ideal range. | |||||
| Col 1 | 9.9 | 8.3 | 13 | 1.1 | 2.2 |
| Col 2 | 5.7 | 4.8 | 9 | 1.3 | 2.4 |
| Col 3 | 5.7 | 5.0 | 9 | 0.9 | 1.7 |
| Col 4 | 4.2 | 3.6 | 7 | 0.7 | NS |
| Col 4b | 6.5b | 5.2b | 10b | 1.3b | 1.6b |
| Col 5 | 3.5 | 2.9 | 7 | 0.2 | NA |
| Col 6 | 4.0 | 3.7 | 7 | 0.3 | NA |
2.2. Instrumentation
All analyses were carried out using a Gilson HPLC system consisting of a Gilson 331 Pump, Gilson 156 Dual UV/VIS detector, Gilson 215 Nebula Liquid Handler and fraction collector, Gilson 819 Injection Module and Gilson 506C System Interface, using Gilson Unipoint Software (version 5.11, USA). The HPLC separations were performed using isocratic flow rate and the columns were kept at room temperature (between 20 °C and 23 °C). Detection was carried out at 285 nm and 350 nm simultaneously. In order to monitor the mobile phase effects, four different mobile phases; toluene, 45% (v/v) acetonitrile in toluene, 50% (v/v) acetonitrile in toluene and 55% (v/v) acetonitrile in toluene were used. The flow rate was adjusted between 0.4–1 mL min−1 based on the backpressure of the columns. Injection volumes were between 30–50 μL for analytical separation, and 4000–5000 μL for preparative separation. Preparative separations were conducted by applying the corresponding analytical methodology but increasing the injection volume and employing two flow rates; starting at 13 mL min−1 and, after the elution of monoadduct, increased to 16 mL min−1 for faster elution of unreacted C60. All the injections were made within 30 minutes of the sample preparation.NMR spectra of reaction mixtures and isolated monoadducts were recorded on a Varian 500 MHz and Agilent 400 MHz NMR Spectrometers. Mass spectrometry of reaction mixtures and isolated monoadducts was carried out using a Bruker Autoflex 2 MALDI-TOF spectrometer in positive and negative ion mode and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as matrix. Matrix solution was prepared by dissolving 10 mg of DCTB in 1 mL of chloroform. The analytes to be measured were then dissolved in chloroform and mixed with a small amount of matrix solution and loaded on the target plate.
2.3. Sample preparation for HPLC analyses
2.3.1.1. C60 solution. 15 mg of C60 was weighed to a 10 mL volumetric flask and dissolved using toluene by sonication and marked up to its volume by toluene. Then 4 mL of this stock solution was pipetted into a 10 mL volumetric flask and diluted to its volume by 50% (v/v) toluene in acetonitrile or toluene depending on the mobile phase. The solution is then mixed by shaking thoroughly and filtered through 0.45 μm PVDC filter into an HPLC vial.
2.3.1.2. C70 solution. 7 mg of C70 was weighed to a 5 mL volumetric flask and dissolved using toluene by sonication and marked up to its volume by toluene. Then 2 mL of this stock solution was pipetted into a 10 mL volumetric flask and diluted to its volume by 50% (v/v) toluene in acetonitrile or toluene depending on the mobile phase. The solution is then mixed by shaking thoroughly and filtered through 0.45 μm PVDC filter into an HPLC vial.
2.3.1.3. C60 + C70 mixture solution. 2 mL of C60 solution and 2 mL of C70 solution were pipetted into a 10 mL volumetric flask and diluted to its volume using 50% (v/v) toluene in acetonitrile. The solution is then mixed by shaking thoroughly and filtered through 0.45 μm PVDC filter into an HPLC vial.
For analytical HPLC sample preparation, approximately 15 mg of RPM1 was weighed to a 10 mL volumetric flask and dissolved using toluene by sonication and marked up to its volume by toluene. Then 4 mL of this solution was pipetted into a 10 mL volumetric flask and diluted to its volume by 50% (v/v) toluene in acetonitrile or toluene depending on the mobile phase. The solution is then mixed by shaking thoroughly and filtered through 0.45 μm PVDC filter into an HPLC vial.
For preparative HPLC sample preparation, approximately 12 mg of RPM1 was weighed to a 10 mL volumetric flask and dissolved using 6 mL of toluene by sonication. Then the solution is diluted to its volume with acetonitrile, mixed by shaking thoroughly and sonicated for 10 minutes. The solution is then centrifuged for 5 minutes at 3000 rpm.
For analytical HPLC sample preparation, approximately 15 mg of RPM2 was weighed to a 10 mL volumetric flask and dissolved using toluene by sonication and marked up to its volume by toluene. Then 4 mL of this solution was pipetted into a 10 mL volumetric flask and diluted to its volume by 50% (v/v) toluene in acetonitrile or toluene depending on the mobile phase. The solution is then mixed by shaking thoroughly and filtered through 0.45 μm PVDC filter into an HPLC vial.
For preparative HPLC sample preparation, approximately 12 mg of RPM2 was weighed to a 10 mL volumetric flask and dissolved using 6 mL of toluene by sonication. Then the solution is diluted to its volume with acetonitrile, mixed by shaking thoroughly and sonicated for 10 minutes. The solution is then centrifuged for 5 minutes at 3000 rpm.
3. Results and discussion
3.1. Fullerene separation by different mobile phases and columns
To understand the response of different column packings to fullerenes, C60 + C70 mixture solution was injected to all columns using toluene as a mobile phase. None of the columns showed any retention of the fullerenes as the molecular interactions of fullerenes with the stationary phase are much weaker than the solubilizing interactions of fullerenes with toluene. C60 and C70 were eluted together without any separation. Adding acetonitrile, a very convenient solvent for HPLC analyses20 in which fullerenes are very poorly soluble as co-eluent decreased the solubilizing strength of the eluent and resulted in improved separation.The mobile phase compositions were scanned by injecting C60 + C70 mixture solution using Col 1 (Fig. 1). At 25% (v/v) acetonitrile in toluene as mobile phase composition, C70 peak started to get separated from C60 peak and with 35% (v/v) acetonitrile in toluene, the peaks resolved completely with a run time of 10 minutes. The chromatogram shows that this mobile phase composition holds great potential for separating higher fullerene compounds which will elute later than C60 and C70 (ref. 33 and 34) within a reasonably short run time. A gradient method can be developed to elute higher fullerenes by gradually decreasing the amount of acetonitrile in the eluent or using an additional co-eluent in which fullerenes are more soluble such as chlorobenzene or carbon disulfide. On the other hand, since our focus was on separation of functionalized fullerenes which are expected to be eluted faster than unreacted fullerenes35–39 and on developing a method with better solvents, higher retention rather than lower is required. This was obtained by increasing the proportion of acetonitrile to 45% (v/v) in toluene or higher.
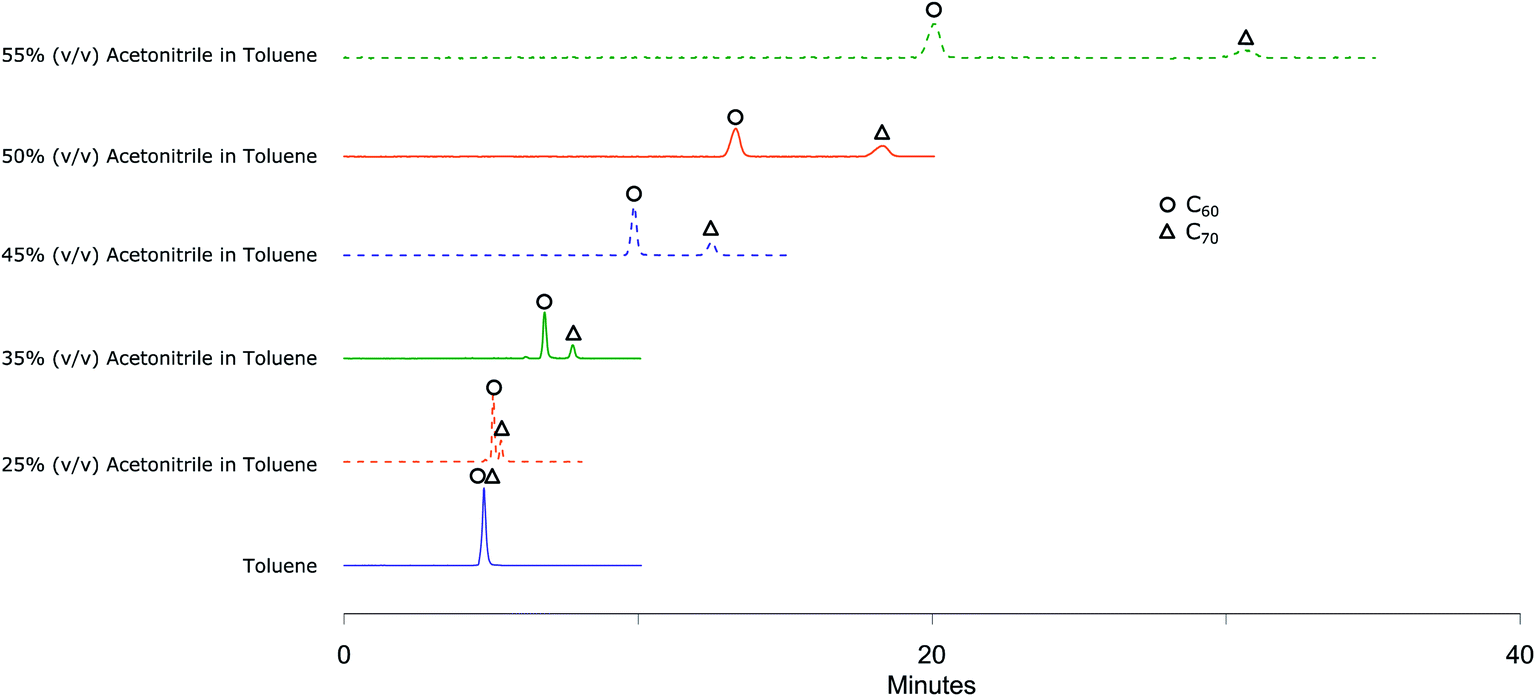 | ||
| Fig. 1 C60 + C70 mixture chromatograms obtained by eluting with 1 mL min−1 from Col 1 at 350 nm UV detection showing no separation in toluene, moderate separation in 25% (v/v) acetonitrile in toluene, very good separation in all other mobile phase compositions. | ||
3.2. Analysis of reaction product mixture 1 (RPM1)
Fulleropyrrolidination (Prato reaction)31 is one of the most well-studied reactions for functionalizing fullerenes. The reaction usually results in near complete conversion of the starting material to a mixture of different products including mono-, di- and poly-adducts. Several separation systems are reported in the literature but most are either relying on CS2 as chromatography solvent or are built around special stationary phases. Encouraged by the efficient separation of C60 and C70 in regular stationary phases we applied the method on fullerene fulleropyrrolidination reaction mixtures (RPM 1) prepared in house (Scheme 2). | ||
| Scheme 2 Pyrrolidination/1,3-dipolar cycloaddition reaction of fullerene C60 with 4-anisaldehyde and sarcosine. Before HPLC sample preparation, all reagents were washed away by extraction, having only unreacted C60, product 1, bis, tris and higher adducts in the RPM1. | ||
Solutions of RPM1 were injected to all six analytical columns using 45%, 50% and 55% (v/v) acetonitrile in toluene as eluent. According to the recorded chromatograms using 45% (v/v) acetonitrile in toluene as mobile phase (Fig. 2), Col 1, Col 2 and Col 3 showed very good resolution between 1 and unreacted C60 as well as the poly-addition products, with a short run time of 10 minutes for Col 2 and Col 3. Col 4 showed moderate separation and Col 5 and Col 6 were unable to separate the product with this mobile phase (Table 2). However, Resolution (Rs) for 1 is quite high also for Col 4 which indicates that effective separation may be achieved if a longer column is used. Additionally, by increasing the proportion of acetonitrile amount in the mobile phase to 55% (v/v), Col 4 also showed very good base separation of the corresponding peaks, whereas Col 5 and Col 6 still did give enough separation (ESI†). The chromatograms obtained using two of the best columns (Col 1 and Col 2) with 45%, 50% and 55% (v/v) acetonitrile in toluene as eluent are given in Fig. 3.
 | ||
| Fig. 2 RPM1 chromatograms obtained by eluting with 45% (v/v) acetonitrile in toluene at 285 nm UV detection where Col 1, Col 2 and Col 3 show very good separation in a short analysis time, Col 4 shows moderate separation and Col 5 and Col 6 does not separate RPM1 product 1 from bis, tris and polyadducts and almost no separation from unreacted C60. | ||
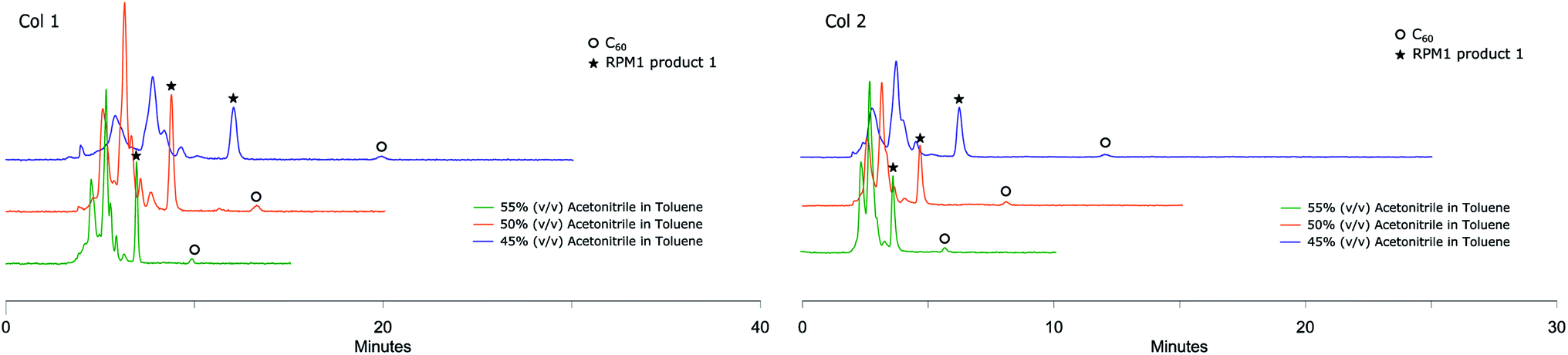 | ||
| Fig. 3 RPM1 chromatograms obtained by Col 1 and Col 2 eluting with different mobile phases, detection at 285 nm, both columns showing excellent separation of RPM1 product 1 from other components of the mixture. | ||
The analytical method conditions were transferred to preparative scale and 1 was successfully isolated with Col 7 using 55% (v/v) acetonitrile in toluene with 13 mL min−1 flow (Fig. 4). With this methodology, 4000 μL reaction mixture solution (4.8 mg of reaction crude) was injected in each run with excellent separation of 1 from the other components in the mixture. The collected fractions belonging to RPM1 product 1 were then injected to the analytical column Col 1, to prove that the separation had given pure fractions (Fig. 4). MALDI-MS analysis verifies that the isolated compound is RPM1 product 1 (ESI†). Our method, using standard column stationary phases, is thus not only very efficient for separation but also for isolation of components from this type of reaction mixtures.
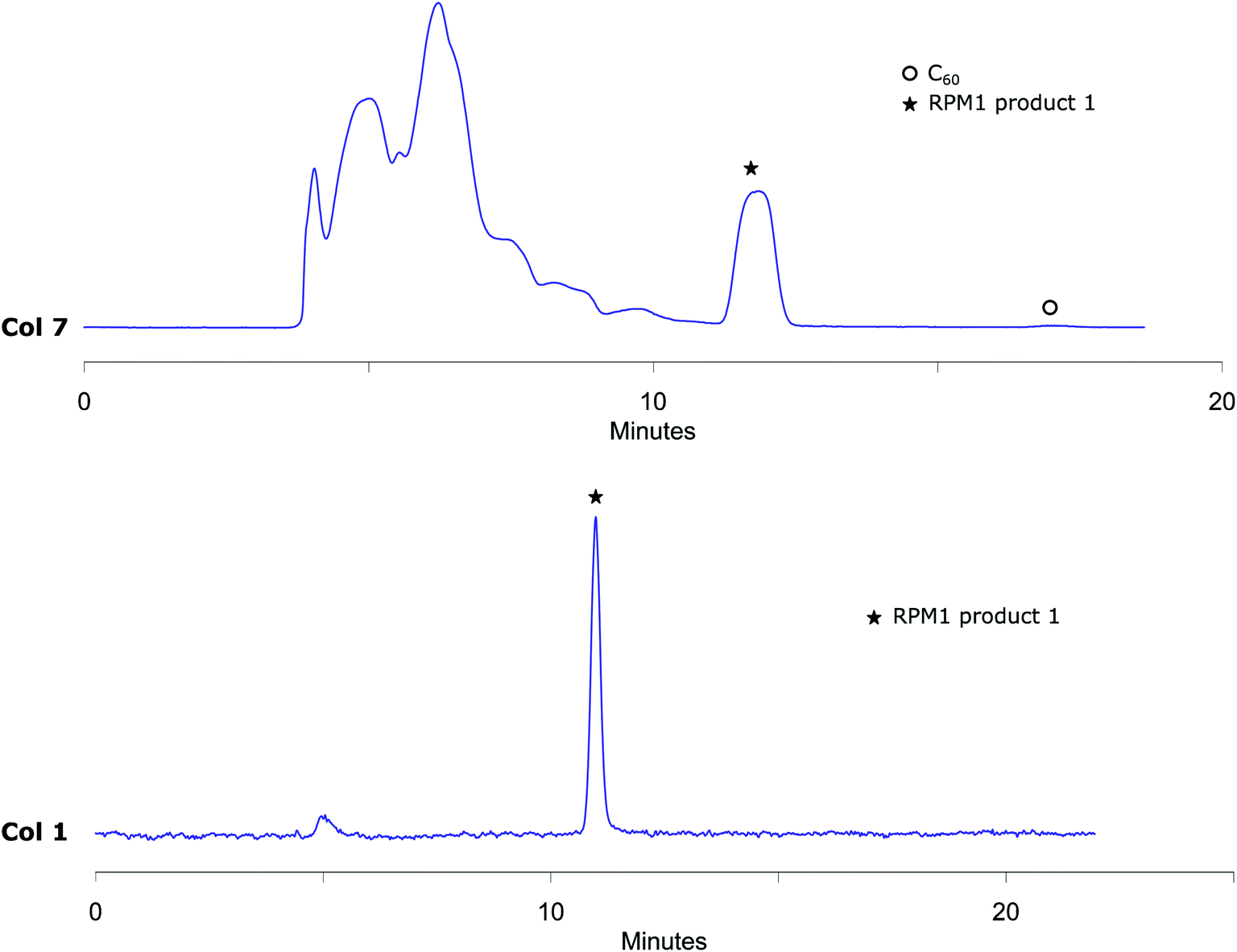 | ||
| Fig. 4 Upper trace: preparative RPM1 chromatogram obtained by Col 7 eluting with 55% (v/v) acetonitrile in toluene at 285 nm UV detection that shows very effective separation of RPM1 product 1 from higher adducts and unreacted C60. Lower trace: analytical chromatogram obtained by injecting collected RPM1 product 1 fraction from preparative RPM1 injection by Col 1 eluting with 55% (v/v) acetonitrile in toluene at 285 nm UV detection that shows RPM1 product 1 is well separated from other peaks and isolated pure. | ||
3.3. Analysis of reaction product mixture 2 (RPM2)
Metal-catalyzed hydroarylation of fullerenes32 has potential to become another highly applicable wide-scope functionalization reaction for fullerene modification. The reported procedures however all suffer from poor conversion of C60 or overreaction to polyadducts. These mixtures of compounds are more challenging to separate than systems such as RPM1 due to the chemical and structural similarities of the components and reported methods rely on special stationary phases. In order to challenge this, in-house prepared hydroarylation reaction mixtures (RPM2, Scheme 3) were injected to all analytical columns using 45%, 50% and 55% (v/v) acetonitrile in toluene as eluent.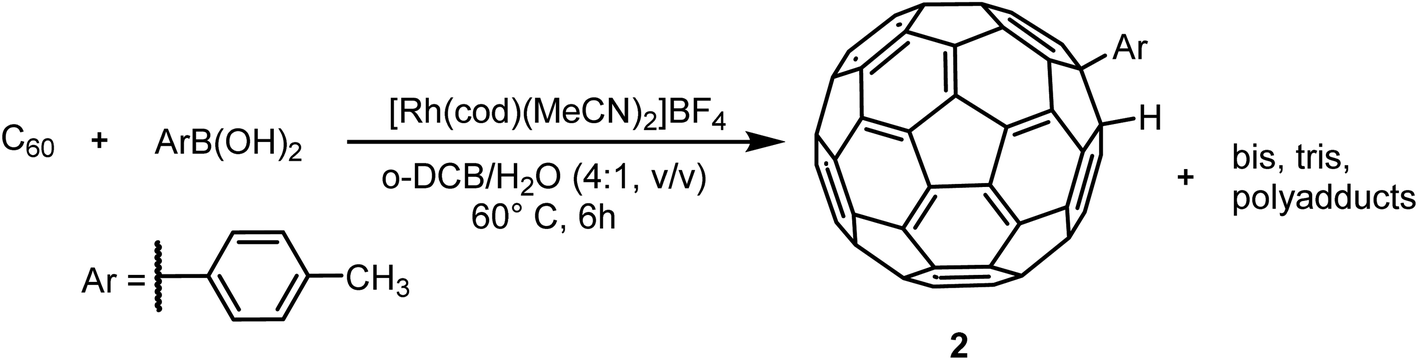 | ||
| Scheme 3 Hydroarylation reaction of C60 with p-tolylboronic acid using a Rh catalyst. Before HPLC sample preparation, all reagents were washed away by extraction, having only unreacted C60, 2 and higher adducts in the RPM2. | ||
The small amounts of poly-addition products were eluting faster than the mono-addition product, which in turn eluted faster than unreacted C60. Using 45% (v/v) acetonitrile in toluene as mobile phase (Fig. 5), Col 1 and Col 2 showed excellent resolution between 2 and both unreacted C60 and poly-addition products with a short run time of 10 minutes for Col 2 whereas Col 3, Col 4, Col 5 and Col 6 did not separate the product well enough (Table 3). However, Resolution (Rs) for 2 is quite high also for Col 3 which indicates that effective separation may be achieved if a longer column is used. In accord with the observation for RPM1, increasing the proportion of acetonitrile to 55% (v/v), Col 4 displayed very good base separation of the corresponding peaks whereas Col 5 and Col 6 still did not separate the components well enough (ESI†). The chromatograms obtained using two of the best columns (Col 1 and Col 2) with 45%, 50% and 55% (v/v) acetonitrile in toluene as eluent are given in Fig. 6.
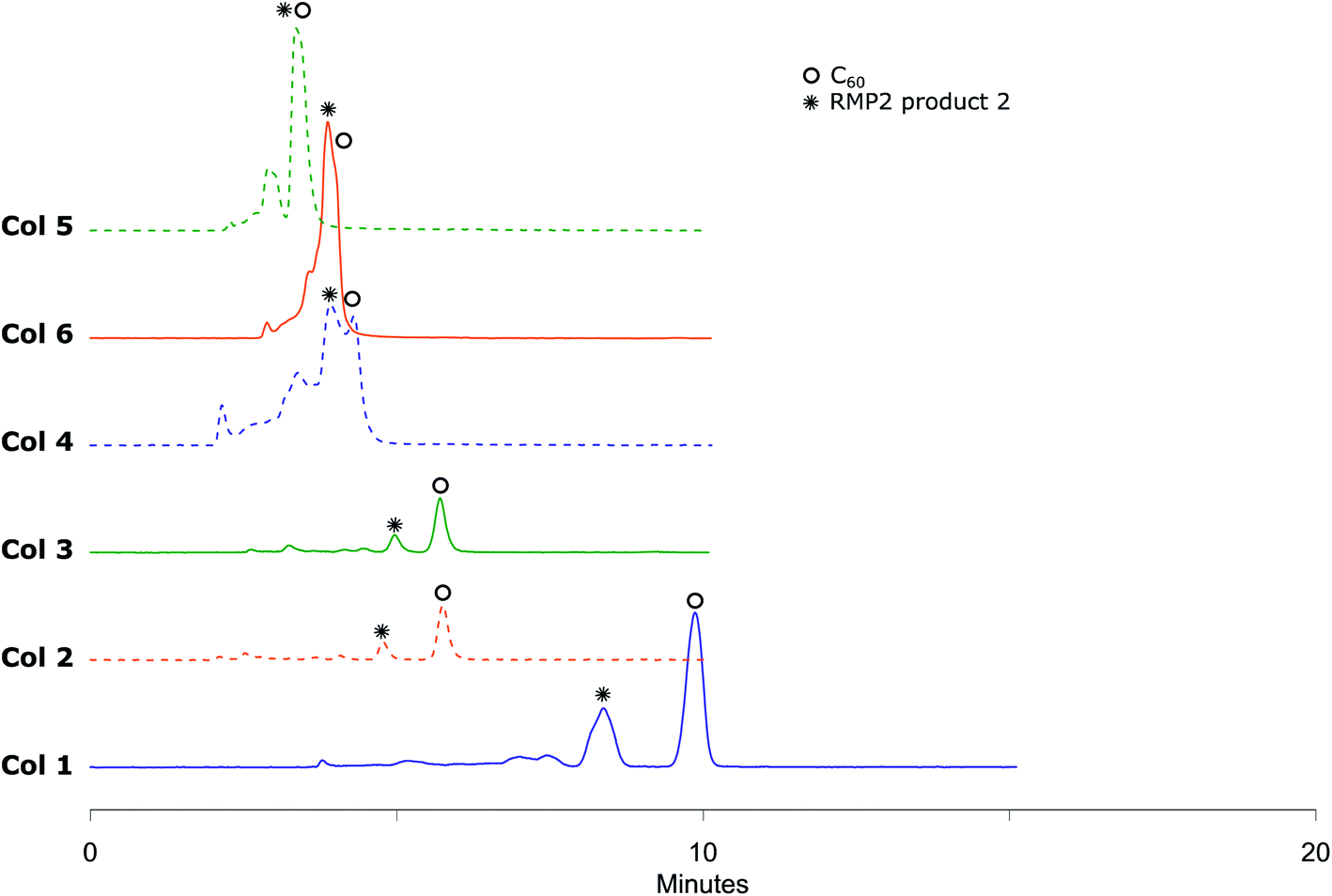 | ||
| Fig. 5 RPM2 chromatograms obtained by eluting with 45% (v/v) acetonitrile in toluene at 285 nm UV detection where Col 1, Col 2 and Col 3 show very good separation in short analysis time, Col 4, Col 5 and Col 6 does not separate RPM2 product 2, neither from bis, tris and polyadducts nor from unreacted C60. | ||
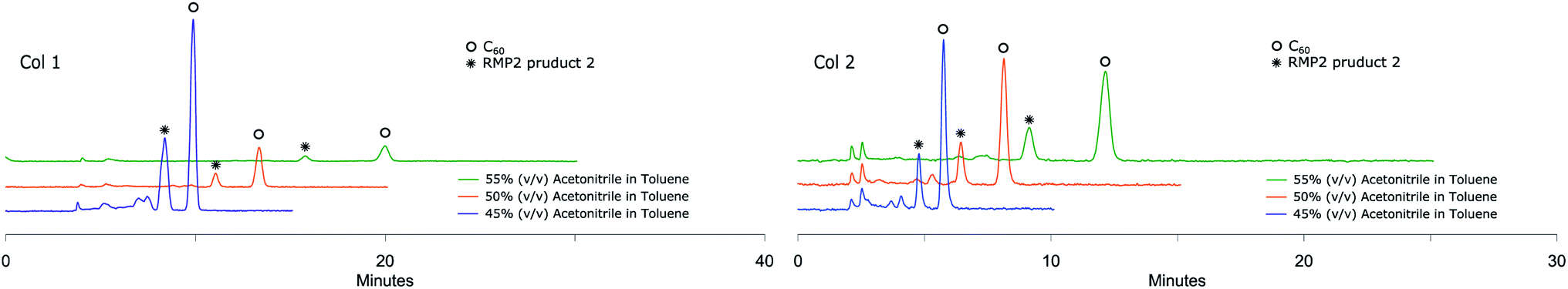 | ||
| Fig. 6 RPM2 chromatograms obtained by Col 1 and Col 2 eluting with different mobile phases at 285 nm UV detection both showing excellent separation of RPM2 product 2. | ||
The analytical method conditions were transferred to preparative scale and 2 was successfully isolated with Col 7 using 45% (v/v) acetonitrile in toluene with 10 mL min−1 flow. With this methodology, 4800 μL reaction mixture solution (5.8 mg of reaction crude) was injected in each run with excellent separation of 2 from the other components of the mixture. The collected fractions belonging to RPM2 product 2 were then injected to the analytical column Col 1 to prove that the separation had given pure fractions (Fig. 7). MALDI-MS analysis verifies that the isolated compound is RPM2 product 2 (ESI†). Our method, using standard column stationary phases, is thus not only very efficient for separation but also for isolation of components from this type of reaction mixtures as well.
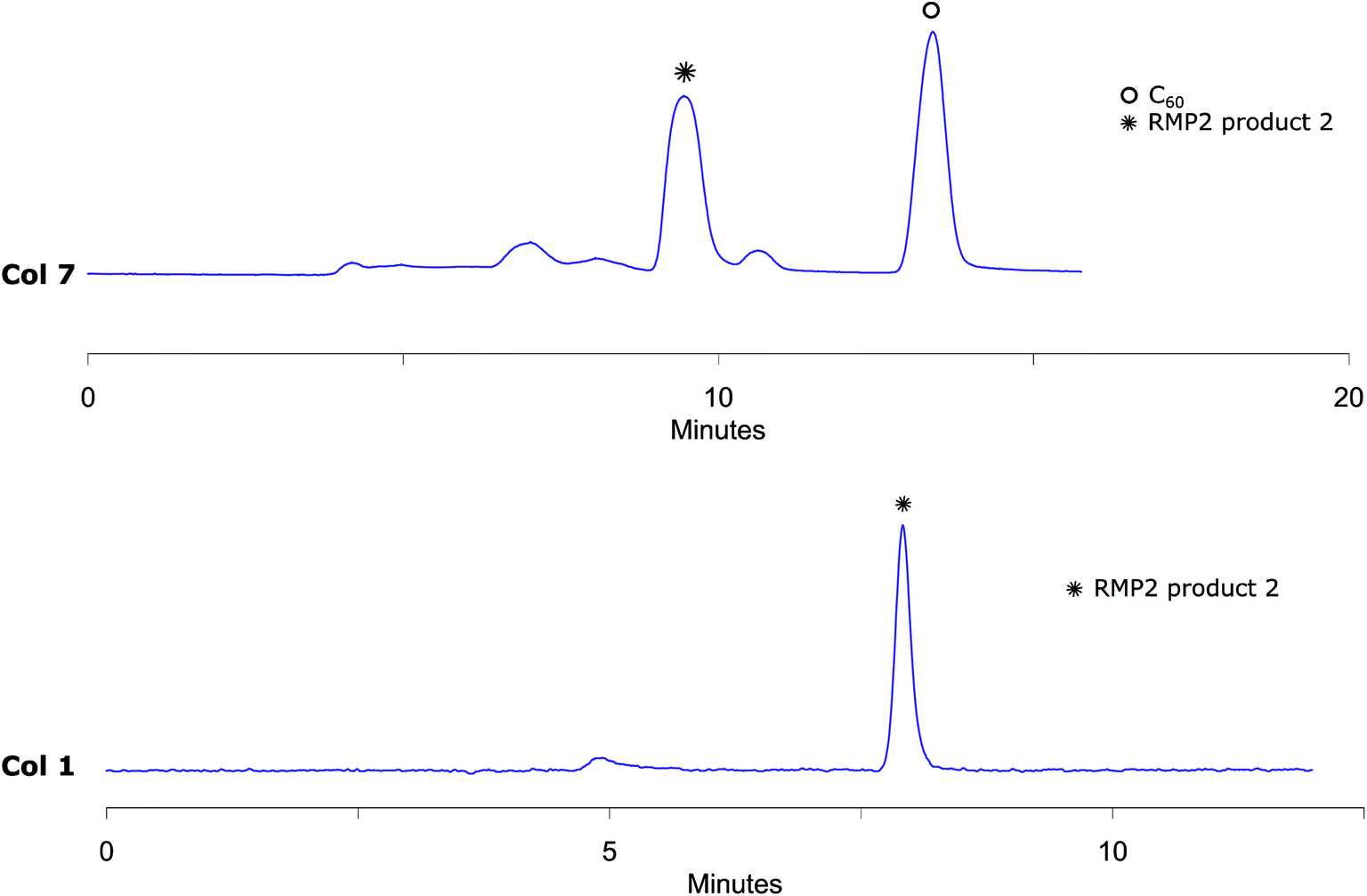 | ||
| Fig. 7 Upper trace: preparative RPM2 chromatogram obtained by Col 7 eluting with 45% (v/v) acetonitrile in toluene at 285 nm UV detection that shows very effective separation of RPM2 product 2 from higher adducts and unreacted C60. Lower trace: analytical chromatogram obtained by injecting collected RPM2 product 2 fraction from preparative RPM2 injection by Col 1 eluting with 45% (v/v) acetonitrile in toluene at 285 nm UV detection that shows RPM2 product 2 is well separated from other peaks and isolated pure. | ||
4. Conclusions
In this study, six analytical HPLC columns with different hydrophobic stationary phases and dimensions were tested for their separation power on a reference C60 + C70 mixture, a fulleropyrrolidination reaction mixture and a C60 hydroarylation reaction mixture, using different proportions of acetonitrile in toluene as mobile phase. From the C60 + C70 mixture analyses, it can be concluded that all columns can separate these molecules efficiently using 55% (v/v) acetonitrile in toluene, however separation power decreases as the stationary phase changes from straight-chained C18 to aromatic C12. Moreover, smaller particle size for the stationary phase in a longer column results in significantly better separation. The fulleropyrrolidination reaction mixtures were excellently separated on columns with C18 stationary phases with high surface area (450–400 m2 g−1). For the C60 hydroarylation reaction mixture, in addition to C18, also C12 stationary phase with high surface area was efficient using 45%, 50% and 55% (v/v) acetonitrile in toluene as eluent. The findings opens for simple and very fast analysis such as reaction conversion monitoring, product profile investigation or quantification of the components of reaction mixtures without the need for special column packings. Different column packings other than what is reported in this study can also be tested for separation by first injecting C60 and adjusting toluene/acetonitrile composition in the mobile phase to achieve suitable retention. Additionally, this separation method is easily applied to HPLC preparative scale for isolation of functionalized fullerene products from reaction mixtures yielding pure products in reasonably short time. The poor solubility of fullerenes in acetonitrile is the biggest challenge and diluted reaction mixture solutions (1.2 mg mL−1) were injected in large volumes to our 150 × 21.0 mm, 5.0 μm C18 column to overcome this issue which resulted isolating 2–4 mg of the product per injection. In conclusion, we have developed simple and fast HPLC methods that employ regular stationary phase columns and standard solvents; the method give efficient analytical as well as preparative separation of complex fullerene reaction mixtures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from O E Nycander Foundation and the Uppsala University KoF priority project on graphene.References
- D. M. Guldi and N. Martin, Fullerenes: From Synthesis to Optoelectronic Properties, Springer Netherlands, Dordrecht, 1st edn, 2002 Search PubMed.
- D. Chirvase, J. Parisi, J. C. Hummelen and V. Dyakonov, Nanotechnology, 2004, 15, 1317–1323 CrossRef CAS.
- M. Izquierdo, S. Osuna, S. Filippone, A. Martín-Domenech, M. Solà and N. Martín, J. Org. Chem., 2009, 74, 6253–6259 CrossRef CAS PubMed.
- H. S. Lin and Y. Matsuo, Chem. Commun., 2018, 54, 11244–11259 RSC.
- M. Yamada, R. Ochi, Y. Yamamoto, S. Okada and Y. Maeda, Org. Biomol. Chem., 2017, 15, 8499–8503 RSC.
- L. Sánchez, M. Á. Herranz and N. Martín, J. Mater. Chem., 2005, 15, 1409–1421 RSC.
- K. Itami, Chem. Rec., 2011, 11, 226–235 CrossRef CAS PubMed.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–303 CrossRef CAS.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- Y. Shirai, J. F. Morin, T. Sasaki, J. M. Guerrero and J. M. Tour, Chem. Soc. Rev., 2006, 35, 1043–1055 RSC.
- N. Tagmatarchis and H. Shinohara, Mini-Rev. Med. Chem., 2001, 1, 339–348 CAS.
- S. Jennepalli, S. G. Pyne and P. A. Keller, RSC Adv., 2014, 4, 46383–46398 RSC.
- C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- Y. N. Biglova and A. G. Mustafin, RSC Adv., 2019, 9, 22428–22498 RSC.
- F. Beuerle and A. Hirsch, Chem.–Eur. J., 2009, 15, 7434–7446 CrossRef CAS PubMed.
- J. L. Shi, X. F. Zhang, H. J. Wang, F. B. Li, X. X. Zhong, C. X. Liu, L. Liu, C. Y. Liu, H. M. Qin and Y. S. Huang, J. Org. Chem., 2016, 81, 7662–7674 CrossRef CAS PubMed.
- T. J. Kop, J. Đorđević, M. S. Bjelaković and D. R. Milić, Tetrahedron, 2017, 73, 7073–7078 CrossRef CAS.
- L. Isaacs, R. F. Haldimann and F. Diederich, Angew. Chem., Int. Ed. Engl., 1994, 33, 2339–2342 CrossRef.
- R. S. Ruoff, D. S. Tse, R. Malhotra and D. C. Lorents, J. Phys. Chem., 1993, 97, 3379–3383 CrossRef CAS.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229–231 CrossRef CAS.
- Y. Shoji, K. Tashiro and T. Aida, J. Am. Chem. Soc., 2004, 126, 6570–6571 CrossRef CAS PubMed.
- Y. D. Yang and H. Y. Gong, Chem. Commun., 2019, 55, 3701–3704 RSC.
- R. Taylor, J. P. Hare, A. K. Abdul-Sada and H. W. Kroto, J. Chem. Soc., Chem. Commun., 1990, 74, 551–558 Search PubMed.
- M. Santella, V. Mazzanti, M. Jevric, C. R. Parker, S. L. Broman, A. D. Bond and M. B. Nielsen, J. Org. Chem., 2012, 77, 8922–8932 CrossRef CAS PubMed.
- M. Zhang, H. J. Wang, F. B. Li, X. X. Zhong, Y. S. Huang, L. Liu, C. Y. Liu, A. M. Asiri and K. A. Alamry, J. Org. Chem., 2017, 82, 8617–8627 CrossRef CAS PubMed.
- Y. F. Li, D. Zhang, H. J. Wang, F. B. Li, L. Sun, L. Liu, C. Y. Liu, A. M. Asiri and K. A. Alamry, J. Org. Chem., 2019, 84, 2922–2932 CrossRef CAS PubMed.
- K. Kimata, K. Hosoya, T. Araki and N. Tanaka, J. Org. Chem., 1993, 58, 282–283 CrossRef CAS.
- T. Hirose, K. Moriuchi, K. Hosoya, T. Araki, N. Tanaka and K. Kimata, Anal. Chem., 1995, 67, 2556–2561 CrossRef.
- S. Selmani, M. Yue Shen and D. J. Schipper, RSC Adv., 2017, 7, 19026–19029 RSC.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- M. Nambo, R. Noyori and K. Itami, J. Am. Chem. Soc., 2007, 129, 8080–8081 CrossRef CAS PubMed.
- F. Diederich, R. Ettl, Y. Rubin, R. L. Whenren, R. Beck, M. Alvarez, S. Anz, D. Sensharma, F. Wudl, K. C. Khemani and A. Koch, Science, 1991, 252, 548–551 CrossRef CAS PubMed.
- F. Diederich, R. L. Whetren, C. Thilgen, R. Etrl, I. T. O. Chao and M. M. Alvarez, Science, 1991, 254, 1768–1771 CrossRef CAS PubMed.
- A. Carboni, E. Emke, J. R. Parsons, K. Kalbitz and P. de Voogt, Anal. Chim. Acta, 2014, 807, 159–165 CrossRef CAS PubMed.
- K. Komatsu, Y. Murata, N. Takimoto, S. Mori, N. Sugita and T. S. M. Wan, J. Org. Chem., 1994, 59, 6101–6102 CrossRef CAS.
- S. Mori, M. Nambo, L. C. Chi, J. Bouffard and K. Itami, Org. Lett., 2008, 10, 4609–4612 CrossRef CAS PubMed.
- A. Astefanei, O. Núñez and M. T. Galceran, J. Chromatogr. A, 2014, 1365, 61–71 CrossRef CAS PubMed.
- A. Kolkman, E. Emke, P. S. Bäuerlein, A. Carboni, D. T. Tran, T. L. Ter Laak, A. P. Van Wezel and P. De Voogt, Anal. Chem., 2013, 85, 5867–5874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02814b |
| This journal is © The Royal Society of Chemistry 2020 |