
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of M(III)-cyclam (M = Rh, Ru; cyclam = 1,4,8,11-tetraazacyclotetradecane) complexes as novel methanol resistant electrocatalysts for the oxygen reduction reaction
I. L. Vera-Estradaa,
J. Uribe-Godínezb and
O. Jiménez-Sandoval *a
*a
aCentro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Unidad Querétaro, Libramiento Norponiente 2000, Fracc. Real de Juriquilla, Querétaro, Qro. 76230, MEXICO. E-mail: ojimenez@cinvestav.mx; Tel: +52 442 2119900
bCentro Nacional de Metrología, Km 4.5 Carretera a Los Cués, El Marqués, Qro. 76246, MEXICO
First published on 12th June 2020
Abstract
Transition metal macrocyclic complexes have acquired relevance as electrocatalysts to perform oxygen electroreduction in acid media as an alternative to platinum. This work presents two macrocyclic complexes using cyclam as ligand, which has a much simpler molecular structure (smaller size, no π-electrons, etc.) than the well studied porphyrins and phthalocyanines as transition metal complexes. Such compounds are usually subjected to thermal treatments at relatively high temperatures (800–900 °C) which result in ligand decomposition, leaving the so-called MNx active sites. In contrast, the complexes reported in this work are efficient electrocatalysts for the oxygen reduction reaction (ORR) in their original molecular structure, with no thermal treatments of any kind applied. The electrocatalytic activity of the Rh(III)-cyclam and Ru(III)-cyclam complexes during the ORR in the absence and presence of methanol (2 mol L−1) was evaluated by voltammetry techniques. The kinetic parameters of the novel materials for the reaction were determined. The exchange current density (j0) values, directly related to the charge transfer velocity, are of the same order as or higher than those of platinum/Vulcan® nanoparticles. In addition, they are practically unaffected by methanol, therefore, becoming interesting candidates to be evaluated as cathodes in polymer electrolyte membrane and direct methanol fuel cells.
1. Introduction
Fuel cells are low emission and high efficiency energy conversion devices and are suitable for the present energy demand and the use of renewable sources. The commercial viability of fuel cells highly depends on using an efficient catalyst for the oxygen reduction reaction (ORR). Current commercial fuel cells use platinum due to its relatively good catalytic activity, however, it is an expensive material with important technical disadvantages like facile poisoning with numerous species, such as methanol and carbon monoxide, among others.1–4 Thus, the search for more affordable and efficient alternatives has led to the development of two main catalyst groups: Pt-based and Pt-free catalysts.The first group, Pt-based catalysts, are materials with a low platinum loading and are commonly combined with other metals as alloys or core–shell structures. Different works summarize recent advances on these materials1,5–7 and show some enhancements by combining Pt with metals like Ni, Co or Fe. The decrease in platinum loading is also achieved by tuning the predominant phases that are present on the surface5–7 or by using a catalyst support, which generally is a carbon-based material, although there have been also studies of non-carbonaceous supports such as conductive polymers or electroconductive oxides.6,8
Pt-free catalysts, on the other hand, have proved an optimal long-term alternative for their use in fuel cells due to their abundance and lower cost compared to Pt-based catalysts, however, their activity and stability do not generally reach the level of Pt-based materials. The efforts to develop Pt-free catalysts cover a broad variety of materials such as metal chalcogenides, nitrides, carbides, organometallic compounds, transition metal macrocyclic complexes, nitrogen–carbon supported materials, conductive polymer-based complexes, metal oxides, etc., several of which have been reviewed in recent years.1,3,6,9
Transition metal macrocyclic complexes have acquired special attention since 1964, when Jasinsky demonstrated that a cobalt phthalocyanine was active for ORR in basic media.10 This compound is similar to the porphyrins found in biological molecules such as chlorophyll and in the heme group; the latter is also present in enzymes such as cytochrome c oxidase, which is used to reduce O2 to water.11–13
The catalytic activity of the nitrogen-based macrocyclic complexes can be modified by changing the central atom or varying the structure of the macrocycle. Therefore, different metallophthalocyanines (MPC) along with compounds with other macrocyclic ligands like porphyrins or their derivatives were developed and evaluated in acid or alkaline media for the ORR.11,13,14 The most promising results were obtained with pyrolyzed Fe or Co complexes.11,14 Unfortunately, the heat treatments used to improve the activity and stability of the compounds affect the structure of the macrocycle and consequently the active site is not well defined. Even though the macrocycle is altered, an M–Nx core seems to remain,9,14,15 which allows to infer that it is possible to use smaller macrocycles to catalyze the ORR.
Furthermore, some transition metal macrocyclic compounds, like iron phthalocyanine16 or heat-treated Co–Fe tetraphenylporphyrin17 perform the oxygen reduction reaction even in the presence of methanol, an advantage over platinum for their application as cathode catalysts in direct methanol fuel cells (DMFC).
One of the simplest N4-macrocycles is 1,4,8,11-tetraazacyclotetradecane (cyclam). This macrocycle along with its derivatives are able to coordinate various transition metals.18–25 Some of these complexes have been used for catalyzing different reactions such as CO2 reduction and olefins epoxidation and are used for medical and radiopharmaceutical applications as well.26–30 However, there are few evidences of the use of cyclam complexes to catalyze the oxygen reduction reaction. Kalvelage et al.31 analyzed the activity of Co and Fe cyclam complexes and the results show that they have a poor activity and only after a heat treatment. The work of Claude et al.32 also shows that a cobalt cyclam compound does not exhibit catalytic activity before heat treatment.
This work presents two cyclam complexes, obtained by simple synthesis routes without any heat treatment that can perform the electroreduction of oxygen in an acid medium, in the absence and presence of methanol, allowing their possible use as cathodes in hydrogen PEMFC and DMFC.
2. Experimental
2.1 Synthesis
Rh-cyclam complex: 0.7 mmol of RhCl3·xH2O (Sigma-Aldrich) were dissolved in 25 mL of methanol (J. T. Baker) and then 0.7 mmol of 1,4,8,11-tetraazacyclotetradecane (Sigma-Aldrich) were added slowly. The solution was stirred for 30 min at room temperature, then it was filtered and the resulting red solid was washed with methanol and acetone and dried at room temperature.Ru-cyclam complex: 1.0 mmol of 1,4,8,11-tetraazacyclotetradecane (Sigma-Aldrich) was dissolved in 20 mL of methanol (J. T. Baker). This solution was added dropwise to a solution of 1.25 mmol of RuCl3·xH2O (Sigma-Aldrich) in 20 mL of methanol (J. T. Baker). The suspension obtained was stirred for 30 minutes at room temperature, then it was filtered and the blackish solid formed was washed with methanol and acetone and dried at room temperature.
2.2 Structural characterization
The cyclam compounds obtained were analyzed by diffuse reflectance FT-IR spectroscopy on a PerkinElmer-GX3 spectrometer, with a resolution of 4 cm−1. The samples were previously dissolved in FTIR grade KBr (Sigma-Aldrich).A qualitative analysis of the chemical composition of the products was made by Energy Dispersive Spectroscopy (EDS). The measurements were performed in a Philips XL30 ESEM microscope with a coupled EDS system.
Elemental combustion analysis was used to quantify carbon, nitrogen and hydrogen. The measurements were performed in a Thermo Scientific Flash 2000 elemental analyzer at a temperature of 950 °C.
The concentration of the metal in the complexes was estimated by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) in a Horiba Ultima 2 equipment. The sample was treated by acid digestion (HCl/H2O2, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a Multiwave PRO equipment, and Crescent Chemical standards were used for each metal (Rh, Ru).
:1) in a Multiwave PRO equipment, and Crescent Chemical standards were used for each metal (Rh, Ru).
The scanning electron micrographs were obtained on a JEOL JXA-8530F Electron Probe Microanalyzer (a thin gold film was evaporated on the samples before their analysis).
2.3 Electrochemical measurements
Ru-cyclam: a catalyst ink was prepared by ultrasonically mixing 1.5 mg of the catalyst and 1.5 mg of carbon powder (Vulcan® XC72R), previously triturated, and 30 μL of 5% wt. Nafion® solution (Sigma-Aldrich) for 20 minutes. The ink was deposited on the glassy carbon RDE (Area = 0.072 cm2) and dried in air at 300 rpm.
Linear sweep voltammetry (LSV) was used to study the oxygen reduction reaction. The electrolyte was saturated with oxygen (Infra UHP) (15 min) to determinate the open circuit voltage (EOC). Current–potential curves were obtained in the EOC to 0 V/NHE range at a 5 mV s−1 rate. The rotational speed was varied from 100 to 600 rpm.
Once the LSV was finished, the electrolytic solution was deoxygenated for 30 minutes (UHP N2 Infra) and methanol (J. T. Baker) was added to reach a 2.0 mol L−1 concentration. Then 3 new potential sweeps were done by CV as described before, and finally the LSV measurements were repeated under the above conditions.
3. Results and discussion
3.1 Structural characterization
Fig. 1 shows the FTIR spectra of the cyclam complexes synthesized, as well as that of the ligand as reference. Fig. 2 shows a representation of the structure of the cyclam ligand. A stretching O–H band is observed at 3538 and 3482 cm−1 for the rhodium and ruthenium compound, respectively. This band as well as the one located at ≈1610 cm−1 indicate the presence of water in both compounds.33 | ||
| Fig. 1 FTIR spectra of the metal–cyclam complexes and the cyclam ligand. | ||
 | ||
| Fig. 2 Representation of the cyclam ligand structure. | ||
The N–H stretching modes in the complexes are found at lower frequencies than those present in the spectrum of the cyclam ligand,34 as expected for the complexation effect, i.e. the stronger the M–N bond, the weaker is the N–H bond.35 The stretching C–H modes, which are observed as intense bands between 2650 and 3000 cm−1 in cyclam, are expected to be less intense in metal complexes.34 Therefore, a combined band in the 2750–3300 cm−1 range is observed in the compounds as a result of the νNH modes shift and the low intensity νCH modes.
The deformation N–H modes are also weakened for the M–N bond, therefore in the complexes the δNH band should appear at lower frequencies compared to the bands observed in the ligand (1518 cm−1). However, the δNH band could be masked by deformation C–H modes, which appear in the 1450–1300 cm−1 range. Thus, the band found in 1455 cm−1 for the Rh complex and in 1447 cm−1 for the Ru compound is attributed to the combination of δCH and δNH modes.
The stretching C–C and C–N modes are observed in the 1400–1000 cm−1 range. As expected, these modes should also be influenced by complexing. The two synthesized complexes present a combined band in the 1110–1010 cm−1 region corresponding to νCC, νCN and δNH modes.33–35 The remaining bands at about 517 cm−1 and 533 cm−1 for the rhodium and ruthenium complexes, respectively, could be assigned to ring deformations involving the CNC and CCC bonds that are far from the coordination sites.34
The chemical composition of the compounds was determinated using different techniques given the nature of the materials. EDS analyses were made to identify the elements present in the compounds. The results show the presence of carbon, nitrogen, hydrogen, oxygen, chlorine and the respective metal (rhodium or ruthenium) in each complex; however, due to the overlapping energies of some of these elements, it was not possible to establish the composition of the compounds by this technique. Therefore, elemental combustion analyses were used to identify the organic part of the compounds. The rhodium complex presents the follow composition: C = 17.59, N = 7.62, H = 4.59 (mass percentage) and the values for the ruthenium complex are: C = 16.8, N = 6.96, H = 3.93 (mass percentage). The concentration of the metal determined by ICP is 25.5% for the ruthenium complex and 26.7% for the rhodium complex.
It is observed that both complexes present a similar chemical composition. These results, together with the type of the synthesis processes, which does not involve heating, the ligand flexibility, as well as the oxidation state of the metals used, and the evidence of the presence of water in the complexes, allowed us to propose complexes of the type: [M2(cyclam)(H2O)4Cl4]Cl2 (M = Rh or Ru). In both compounds, the carbon concentration is nearly half the expected for the more common 1:1 macrocyclic complexes with a single metal atom inside the cavity of the ring, hence giving support to the proposal of binuclear complexes in our case.
Fig. 3 shows the SEM images for the two complexes synthesized. The rhodium compound exhibits an irregular morphology, like a cluster composed of nearly planar structures of different sizes. In contrast, the ruthenium compound shows a highly homogeneous porous morphology, which could imply an advantage for its catalytic activity.
 | ||
| Fig. 3 Scanning electron micrographs of the macrocyclic complexes synthesized. | ||
3.2 Electrochemical characterization
 | ||
| Fig. 4 Cyclic voltammograms of the macrocyclic complexes in the absence and presence of 2.0 mol L−1 methanol. Electrolyte: H2SO4 0.5 mol L−1. Sweep rate: 20 mV s−1. | ||
A most remarkable feature of both materials is that in the presence of methanol (2.0 mol L−1) the cyclic voltammograms remain practically unchanged, except for a small current decrease in the hydrogen adsorption/desorption and evolution zones of the rhodium complex voltammogram. The absence of the distinctive methanol oxidation peaks can be considered a sign of selectivity of both catalysts.
 | ||
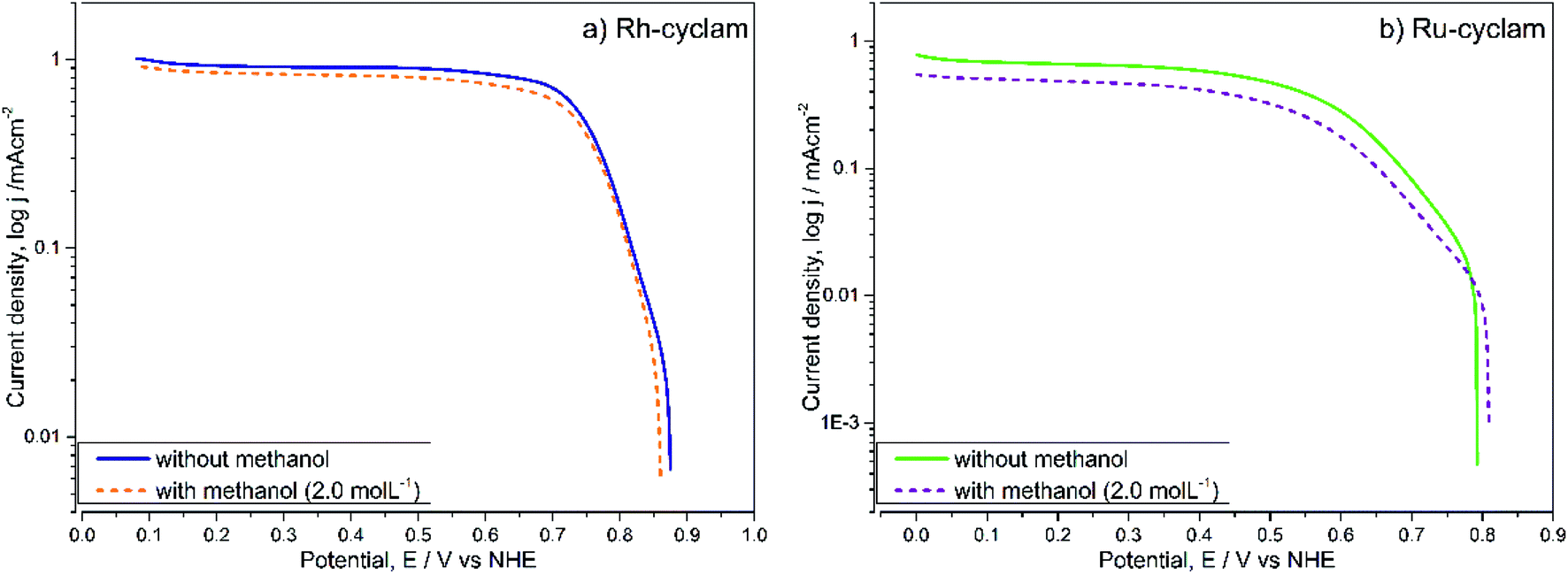
| Fig. 5 ORR current–potential curves of the: (a) Rh-cyclam and (b) Ru-cyclam complexes, as well of (c) Pt/Vulcan® (30%), in the absence and presence of methanol (2.0 mol L−1). Electrolyte: H2SO4 0.5 mol L−1. Sweep rate: 20 mV s−1. | ||
| Material | [Methanol] (mol L−1) | EOC (V/NHE) | B (mV dec−1) | α | j0 (mA cm−2) | Effective area (cm2) |
|---|---|---|---|---|---|---|
| Rh-cyclam | 0 | 0.877 | 82.72 | 0.725 | 8.92 × 10−7 | 0.121 |
| 2 | 0.862 | 76.46 | 0.785 | 3.58 × 10−7 | 0.136 | |
| Ru-cyclam | 0 | 0.793 | 142.33 | 0.422 | 1.530 × 10−5 | 0.106 |
| 2 | 0.809 | 155.29 | 0.386 | 1.944 × 10−5 | 0.112 | |
| Platinum/Vulcan® (30%)46 | 0 | 0.974 | 78.12 | 0.75 | 1.4 × 10−6 |
For each case, the current–potential curves show a very similar behavior when the activity is evaluated in the presence of 2 mol L−1 methanol, in contrast with the Pt/Vulcan® polarization curves (Fig. 5c), which in the presence of methanol do not show the three ORR characteristic zones. This is indicative that, unlike the latter catalytic material, both cyclam complexes are selective to oxygen reduction at relatively high methanol concentrations, which was later confirmed by the kinetic parameter values.
The total current observed in the polarization curves has two main contributions: kinetic current (ik) and diffusion current (id) and can be described by the Koutecky–Levich equation (eqn (1)).
 | (1) |
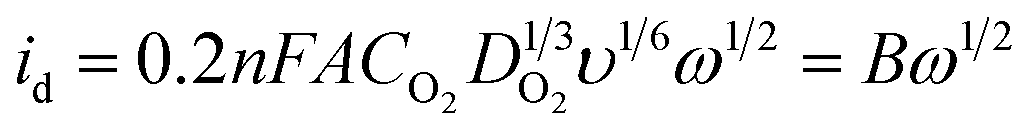 | (2) |
To obtain the total electrocatalytic activity, the diffusion current must be calculated. In the laminar regimen it is determined by the Levich equation (eqn (2)), where n is the number of electrons exchanged per mol of O2, F the Faraday constant, A the catalytic effective surface area, υ the electrolyte kinematic viscosity, DO2 the oxygen diffusion coefficient, CO2 the bulk oxygen concentration in the electrolyte and ω the rotation speed (rpm); the values used in this work were 0.01 cm2 s−1 for the kinematic viscosity, 1.4 × 10−5 cm2 s−1 for the oxygen diffusion coefficient and 1.1 × 10−6 mol cm−3 for the bulk oxygen concentration.39
The value of B from eqn (2) is obtained by graphing 1/i vs. 1/ω1/2. These graphs known as Koutecky–Levich plots are shown in Fig. 6. They present the theoretical (2 and 4 electron mechanism) and experimental data (at different given potentials values) in the absence and presence of methanol for the two complexes. For both materials, in the two cases, it is observed a likely direct reaction mechanism (4-electron pathway to water formation). This means that probably there is not a significant production of H2O2, which is undesirable in PEMFC as it degrades the polymer membrane.40 Hence, the catalytic effective area is estimated with eqn (2) considering n = 4, the values are reported in Table 1. All the current data reported (CV, LSV and Tafel plots) were normalized to this effective area.
 | ||
| Fig. 6 Koutecky–Levich plots of the macrocyclic complexes for oxygen reduction in the absence and presence of 2.0 mol L−1 methanol. Electrolyte: 0.5 mol L−1H2SO4. | ||
The kinetic parameters, such as Tafel slope (b), exchange current density (j0) and charge transfer coefficient (α) were obtained from the mass transport corrected Tafel plots (logik vs. E) (Fig. 7) and the Tafel equation (eqn (3)), where η is the overpotential, i is the current and a and b are defined as follows: b = 2.3RT/αF and  .
.
|
η = a + blog[i]
| (3) |
 | ||
| Fig. 7 Tafel plots of the macrocyclic complexes for oxygen reduction in the absence and presence of 2.0 mol L−1 methanol. Electrolyte: 0.5 mol L−1 H2SO4. | ||
The Tafel slope is related to the reaction mechanism and could be estimated directly from the high overpotential region in the Tafel plots. Therefore, the value of α can also be easily calculated from the Tafel slope; this parameter is related to the symmetry of the energy barrier and is a ratio between the effect of potential on the electrochemical free energy of activation and its effect on the electrochemical free energy of the reaction.41,42 Both parameters are shown in Table 1. As can be observed, the Rh complex exhibits similar Tafel slope values to platinum, while the Ru compound shows much higher values, probably indicating similar reaction pathways in the first case. This is supported by the α values, which are also alike for the rhodium compound and platinum, suggesting similar decrements of reaction free energy.41 The two parameters, b and α, are not significantly modified by the presence of methanol in a relatively high concentration, 2 mol L−1.
Finally, one of the most important kinetic parameters is the exchange current density, j0, since it is proportional to the rate constant. The Rh complex shows an exchange current density value close to that of platinum in the absence of methanol, but one nearly an order of magnitude smaller when the alcohol is present. In the case of the Ru complex, j0 is one order of magnitude greater than that of Pt and the presence of methanol does not significantly affect this value, supporting the selective character of this catalyst toward the oxygen reduction. As for other macrocyclic coordination compounds evaluated as catalysts for the ORR, the Fe-phthalocyanine complex reported by Baranton et al. is not stable in an acid medium, the authors explaining this behavior as a demetallation of the macrocycle accompanied by its protonation in the presence of oxygen.16 In contrast, our two materials were completely stable during the electrochemical measurements, an essential condition for their use as ORR electrocatalysts. On the other hand, the Co-cyclam complex informed by Claude et al.32 is inactive toward oxygen unless heat treatments are applied, like most macrocyclic complexes reported to date. In the case of other macrocyclic coordination compounds or nitrogen–carbon composites evaluated as catalysts for the ORR, most reports are in alkaline media,11 probably due to the stability decrease these materials exhibit in acidic electrolytes when they are not previously subjected to heat treatments.14,43,44
Table 1 also collects the open circuit potential values of the novel materials. They all are practically ≥0.8 V/NHE, lower than those of Pt nanoparticles (as usually observed for alternative catalysts), but above most of the macrocyclic complexes reported in acid media and similar to the latest reported values for M–N–C materials11,45
4. Conclusions
The present work reports two transition metal macrocyclic complexes with an important electrocatalytic activity for the oxygen reduction reaction in an acid medium. The novelty and interest of this report lie on several features of the catalytic materials: (1) although two noble elements are being used, Rh and Ru, they are not present as pure metallic particles, like most catalysts of this kind (including Pt), but they are formally oxidized, as Rh(III) and Ru(III), respectively; therefore, common salts are used as metal sources for the syntheses of the complexes, with a consequent cost decrease. (2) The macrocyclic ligand selected, cyclam, has been very well studied and is structurally simple, in contrast with the large and more complex porphyrin and phthalocyanine derivatives reported in the literature. (3) The compounds of this work are synthesized by simple methods in solution, at room temperature, and they do not need to be subjected to high temperatures (800–1000 °C) in order to make them active for the ORR, with the ligand decomposition as a price to pay. Moreover, the simplicity of the synthetic procedure employed should be reflected in an additional economic saving (see point (1)). (4) It is also demonstrated that large conjugations of π (pi) electrons, as occurs in the classical large rings mentioned in point (2), are not essential for macrocyclic metal complexes to perform as effective oxygen reduction electrocatalysts. This finding opens the possibility of numerous nitrogen and perhaps other donor atoms- ligands to be explored in combination with suitable metals as oxygen reduction catalysts. Both complexes reported are likely to follow a direct 4-electron reaction pathway to the formation of water as ORR catalysts. Their open circuit potential values as well as their kinetic parameters are comparable (and in some cases superior) to those of other alternative Pt-free catalysts and Pt itself. In addition, they are resistant to relatively high concentrations of methanol, 2.0 mol L−1. All these properties render the present Rh and Ru materials interesting candidates to be evaluated as cathodes in polymer electrolyte membrane and direct methanol fuel cells.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding from the National Council for Science and Technology (CONACYT, Mexico) through grant SEP-CONACYT A1-S-28734 is gratefully acknowledged. The authors thank R. A. Mauricio-Sánchez, J. E. Urbina-Álvarez and E. Gutiérrez-Arias for technical assistance. The authors are also thankful to Dr V. García-Montalvo (IQ-UNAM) for the elemental combustion analyses. I. L. Vera-Estrada also acknowledges CONACYT for a graduate studies grant.References
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2(5), 864–890, DOI:10.1021/cs3000864.
- O. T. Holton and J. W. Stevenson, Platinum Met. Rev., 2013, 57(4), 259–271, DOI:10.1595/147106713x671222.
- Z. Chen, D. Higgins, A. Yu and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192, 10.1039/c0ee00558d.
- B. C. Ong, S. K. Kamarudin and S. Basri, Int. J. Hydrog. Energy, 2017, 42(15), 10142–10157, DOI:10.1016/j.ijhydene.2017.01.117.
- J. Stacy, Y. N. Regmi, B. Leonard and M. Fan, Renewable Sustainable Energy Rev., 2017, 69, 401–414, DOI:10.1016/j.rser.2016.09.135.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44(8), 2168–2201, 10.1039/c4cs00484a.
- M. Bron and C. Roth, in New and future developments in catalysis: batteries, hydrogen storage and fuel cells, ed. Steven L. Suib, Elsevier B. V., Amsterdam, 2013, ch. 10, pp. 271–305 Search PubMed.
- A. Brouzgou, S. Q. Song and P. Tsiakaras, Appl. Catal., B, 2012, 127, 371–388, DOI:10.1016/j.apcatb.2012.08.031.
- J. K. Dombrovskis and A. E. C. Palmqvist, Fuel Cells, 2016, 16(1), 4–22, DOI:10.1002/fuce.201500123.
- R. Jasinski, Nature, 1964, 201(4925), 1212–1213, DOI:10.1038/2011212a0.
- J. H. Zagal and F. Bedioui, Electrochemistry of N4 Macrocyclic Metal Complexes Volume 1: Energy, Springer International Publishing, Switzerland, 2016 Search PubMed.
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254(23–24), 2755–2791, DOI:10.1016/j.ccr.2010.05.001.
- J. Masa, K. Ozoemena, W. Schuhmann and J. H. Zagal, J. Porphyrins Phthalocyanines, 2012, 16(07n08), 761–784, DOI:10.1142/S1088424612300091.
- J. H. Zagal, F. Bedioui and J. P. Dodelet, N4-Macrocyclic metal complexes, Springer-Verlag, New York, 2006 Search PubMed.
- M. Lefèvre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106(34), 8705–8713, DOI:10.1021/jp020267f.
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J. M. Léger, J. Electroanal. Chem., 2005, 577(2), 223–234, DOI:10.1016/j.jelechem.2004.11.034.
- R. Jiang and D. Chu, J. Electrochem. Soc., 2000, 147(12), 4605, DOI:10.1149/1.1394109.
- C. M. Che, S. S. Kwong, C. K. Poon, T. F. Lai and T. C. W. Mak, Inorg. Chem., 1985, 24(9), 1359–1363, DOI:10.1021/ic00203a019.
- E. Tfouni, K. Q. Ferreira, F. G. Doro, R. S. Da Silva and Z. N. Da. Rocha, Coord. Chem. Rev., 2005, 249(3–4), 405–418, DOI:10.1016/j.ccr.2004.09.009.
- F. Gorzoni, K. Queiroz, Z. Novais, G. Finoto, A. Jesus and E. Tfouni, Coord. Chem. Rev., 2016, 306, 652–677, DOI:10.1016/j.ccr.2015.03.021.
- E. J. Bounsall and S. R. Koprich, Can. J. Chem., 1970, 48(10), 1481–1491, DOI:10.1139/v70-243.
- M. J. Rosales, M. E. Sosa and M. L. Tobe, J. Coord. Chem., 1987, 16(1), 59–65, DOI:10.1080/00958978708079806.
- L. C. Vasconcellos, C. P. Oliveira, E. E. Castellano, J. Ellena and Í. S. Moreira, Polyhedron, 2001, 20(6), 493–499, DOI:10.1016/s0277-5387(00)00621-5.
- B. Bosnich, C. K. Pooh and M. L. Tobe, Inorg. Chem., 1965, 4(8), 1102–1108, DOI:10.1021/ic50030a003.
- E. K. Barefield and F. Wagner, Inorg. Chem., 1973, 12(10), 2435–2439, DOI:10.1021/ic50128a042.
- W. Nam, R. Ho and J. S. Valentine, J. Am. Chem. Soc., 1991, 113(18), 7052–7054, DOI:10.1021/ja00018a062.
- C. A. Stewart, D. A. Dickie and R. A. Kemp, Inorg. Chim. Acta, 2012, 392, 268–276, DOI:10.1016/j.ica.2012.03.025.
- T. N. Huan, E. S. Andreiadis, J. Heidkamp, P. Simon, E. Derat, S. Cobo and M. Fontecave, J. Mater. Chem. A, 2015, 3, 3901–3907, 10.1039/C4TA07022D.
- X. Liang and P. J. Sadler, Chem. Soc. Rev., 2004, 33(4), 246–266, 10.1039/b313659k.
- J. D. Silversides, C. C. Allan and S. J. Archibald, Dalton Trans., 2007, 9, 971–978, 10.1039/b615329a.
- B. H. Kalvelage, A. Mecklenburg, U. Kunz and U. Hoffmann, Chem. Eng. Technol., 2000, 23(9), 803–807, DOI:10.1002/1521-4125(200009)23:9<803::AID-CEAT803>3.0.CO;2-T.
- E. Claude, T. Addou, J. M. Latour and P. Aldebert, J. Appl. Electrochem., 1997, 28(1), 57–64, DOI:10.1023/A:1003297718146.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli The handbook of infrared and raman characteristic frequencies of organic molecules, Academic Press, San Diego, 1991 Search PubMed.
- G. Diaz F, R. Clavijo C, M. Campos-Vallette, M. Saavedra S, S. Diez and R. Muñoz, Vib. Spectrosc., 1997, 15(2), 201–209, DOI:10.1016/S0924-2031(97)00032-5.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons Inc., New Jersey, 2008 Search PubMed.
- H. Schulenburg, M. Hilgendorff, I. Dorbandt, J. Radnik, P. Bogdanoff, S. Fiechter, M. Bron and H. Tributsch, J. Power Sources, 2006, 155(1), 47–51, DOI:10.1016/j.jpowsour.2005.03.238.
- R. H. Castellanos, E. Borja-Arco, A. Altamirano-Gutiérrez, R. Ortega-Borges, Y. Meas and O. Jiménez-Sandoval, J. New Mater. Electrochem. Syst., 2005, 8, 69–75 CAS.
- A. Karschin, I. Katsounaros, S. O. Klemm, J. C. Meier and K. J. J. Mayrhofer, Electrochim. Acta, 2012, 70, 355–359, DOI:10.1016/j.electacta.2012.03.079.
- C. Song and J. Zhang, in PEM Fuel Cell Electrocatalysts and Catalyst Layers, ed. J. Zhang, Springer, London, 2008, ch. 2, pp. 89–134 Search PubMed.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukunda, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. Kimijima and N. Iwashita, Chem. Rev., 2007, 107(10), 3904–3951, DOI:10.1021/cr050182l.
- A. J. Bard and L. Faulkner, Electrochemical methods: Fundamentals and applications, Wiley & Sons, New York, 2001 Search PubMed.
- E. Gileadi, Electrode Kinetics for Chemists, Chemical Engineers and Materials Scientists, Wiley-VCH, New York, 1993 Search PubMed.
- S. Lj. Gojkovic, S. Gupta and R. F. Savinell, J. Electroanal. Chem., 1999, 462(1), 63–72 CrossRef CAS.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Electrochim. Acta, 2010, 55, 2853–2858 CrossRef CAS.
- V. C. Lo, D. Sebastián, M. J. Lázaro, A. S. Aricò and V. Baglio, Catalysts, 2018, 8(12), 650 CrossRef.
- M. L. Barrios-Reyna, J. Uribe-Godínez, J. M. Olivares-Ramírez and O. Jiménez-Sandoval, J. New Mater. Electrochem. Syst., 2016, 19(4), 185–192, DOI:10.14447/jnmes.v19i4.284.
| This journal is © The Royal Society of Chemistry 2020 |