DOI:
10.1039/D0RA02906H
(Paper)
RSC Adv., 2020,
10, 15947-15954
Photo-responsive azobenzene interactions promote hierarchical self-assembly of collagen triple-helical peptides to various higher-order structures†
Received
29th January 2020
, Accepted 2nd April 2020
First published on 21st April 2020
Abstract
Collagen is an essential structural protein in animal tissues and plays key roles in cellular modulation. We investigated methods to discover collagen model peptides (CMPs) that would self-assemble into triple helices and then grow into supramolecular organizations with diverse morphological features, which would be valuable as biomaterials. This challenging undertaking was achieved by placing azobenzene groups on the ends of the CMPs, (GPO)n (n = 3–10), Azo-(GPO)n. In a dilute aqueous solution (80 μM), CD spectra indicated that the Azo-(GPO)n (n > 4) formed triple helices due to strong hydrophobic azobenzene interactions, and that helix stability was increased with the peptide segment length. The resulting triple helices induced a specific azobenzene orientation through turned and twisted configurations as shown by CD spectra. TEM observations for the same solutions disclosed the morphologies for the Azo-CMPs. Azo-(GPO)3, having the shortest peptide segment, showed no nanostructure, both Azo-(GPO)4 and Azo-(GPO)5 provided consistent well-developed nanofiber structures resembling the natural collagen fibers, and Azo-(GPO)ns (n = 6–10) grew into flexible rod-like micelle fibers. In addition, alkyl chain-attached CmAzo-(GPO)5 displayed a toroidal morphology, and Azp-deg-(GPO)5 having a hydrophilic spacer assembled into a bilayer vesicle structure. These diverse morphological features are considered to be due to the characteristics of the pre-organized triple helix units. Photo-isomerization of the azobenzene moiety brought about the disappearance of such characteristic nano-architectures. When the solution concentration was increased up to 1 wt%, only Azo-(GPO)4 and Azo-(GPO)5 spontaneously formed hydrogels exhibiting a satisfactory gel-to-sol transition upon UV irradiation.
Introduction
Collagen, which is known to be the most abundant protein in mammals, is composed of three parallel helices and exists in the extracellular matrix (ECM) as interwoven fiber networks that serve as a scaffold for tissue growth and stability.1–3 Its high natural abundance and good biocompatibility have brought about the development of collagen as a potential biomaterial.4 Although natural collagen has been valued as a useful biomaterial and is conductive to various applications, challenges which have been identified in its reproduction include difficulty in the precise control of scaffold morphology and limited ability to make site-specific chemical modifications to the collagen structure. To overcome these difficulties, numerous strategies have been developed to assemble synthetic collagen model peptides (CMPs) into a higher-order structure.5–10 CMPs with repeating units of Pro-Hyp-Gly (GPO) have been shown to adopt a collagen triple helix structure.11 This core triple helical unit has been used in the formation of CMP fibers via the use of electrostatic interactions,12–14 native chemical ligation,15 cysteine knots,16–18 π–π stacking,19,20 aromatic–proline interaction,21 metal–ligand coordinations,22–24 and cation–π interactions,25 all of which favorably assist the self-assembly of CMPs into micrometer-scale collagen fibers. In the present study, we sought to discover CMPs that would form triple helices and self-assemble into supramolecular structures with diverse morphological features without reorganizing the peptide chains by covalent linkages. This challenging objective was accomplished by attaching an azobenzene group and its derivatives to the N-terminal regions of peptides, (GPO)n (n = 3–10). Azobenzene is known to not only shown hydrophobicity based on its strong stacking capability but also behave as a spectroscopic probe elucidating its interaction mode. Thus the azobenzene moiety would become a useful template to assemble the CMP molecules and to promote them to a higher-order structure. In addition to these characters, azobenzene shows photo-isomerization (photo-switching) accompanying a slight polarity change. Such photo-switching of the azobenzene groups introduced into CMP molecules has been able to control the stability and the structure of collagen triple helices.26–28
One of the key factors in the morphological diversity accomplished through the self-assembly of peptides is now known to be hydrophilic/hydrophobic balance (amphiphilicity), which is based on the peptide sequence. For instance, the peptides composed of Ala and Lys (A3K, A6K and A9K) have provided different morphological assemblies due to differences in amphiphilic balance.29 We have previously described that the triblock and diblock peptides composed of Leu and Lys show β-sheet-based nanofiber, α-helix-based nanoplate and micelle structures which are dependent on the amphiphilicity and block length of the peptides.30 In the case of CMPs, however, since the stability of the collagen triple helix structure is dependent on the peptide length (n), it does not contribute to the hierarchical variation in morphology. In fact, a short CMP of n = 5 (15mer) is unable to form triple helices.31 Substantial stabilization of the triple helical structures can be achieved with the introduction of covalent links between the C-terminal regions of three peptide chains32,33 or by use of a Kemp triacid (KTA) and a TREN (tri(2-aminoethyl) amine) template linked to the N-terminus of three peptide chains.34–37 Triple helical stabilization is considered to be due to the tight alignment of the three termini achieved by tethering the peptide to the self-assembled aggregates. Ideally, a system by which synthetic linear peptide chains self-assemble into desirable higher-order structures is beneficial because some of the covalently linked triple helical peptide compounds makes them difficult to prepare and/or purify owing to their large molecular size. Fields' and Tirrell's groups have developed a noncovalent, self-assembly approach to construct a collagen-like structural motif, in which two long alkyl chains are attached to the N-terminus of a peptide chain mimicking a lipid structure.38–42 Their amphiphilic peptide–lipid has provided a stable triple helical structure even though its peptide length is quite short (12-mer (n = 4)),39 and has formed only a spherical micelle structure.40,41 Stupp's and Hartgerink's groups have also applied the self-assembly strategy of single-alkyl chain peptide amphiphiles to form collagen-like fibers.43,44 By using this strategy, Tong and coworker have shown that an epitope-containing collagen-mimetic amphiphile holds a potential for tissue regeneration applications.45
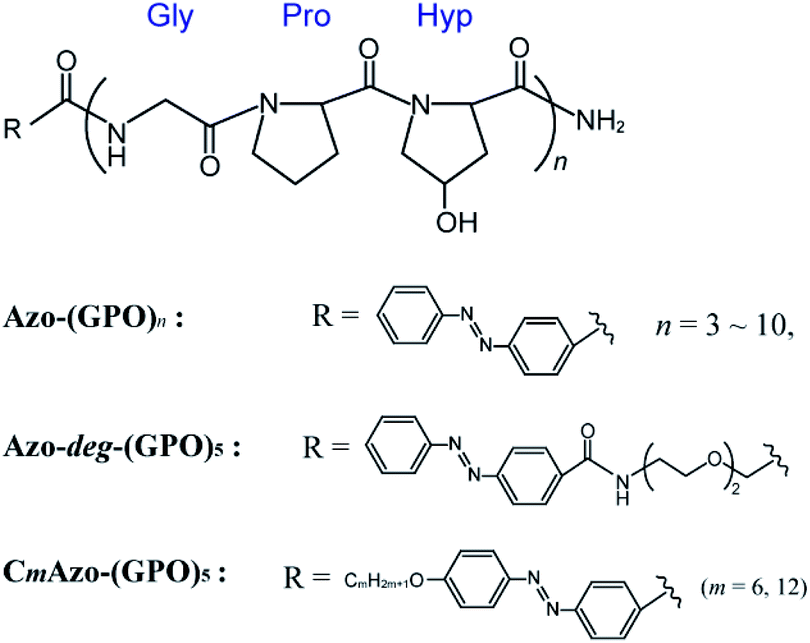
In an effort to strengthen potential hydrophobic interactions between the termini of the triple helices, we designed peptides that contained repeating units of GPO directly terminating with an azobenzene group (Azo-(GPO)n, n = 3–10) or through the use of flexible diethylene glycol (deg) as a spacer (Azo-deg-(GPO)5) (Fig. 1). Peptides terminating in a more hydrophobic azobenzene having a long alkyl chain (CmAzo-(GPO)5, m = 6 and 12) were also designed for comparison. Herein we disclose the hierarchical assembly of these triple helices into various architectures, depending on the peptide chain length (n), the polarity balance (hydrophobic/hydrophilic balance), and photo-isomerization of the azobenzene moiety.
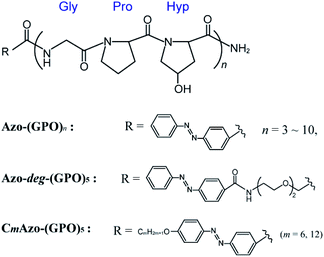 |
| | Fig. 1 Chemical structures of the azobenzene-terminated collagen-mimetic oligopeptides: Azo-(GPO)n (n = 3–10), Azo-deg-(GPO)5, and CmAzo-(GPO)5 (m = 6 and 12). | |
Results and discussion
Design and syntheses of azobenzene-terminated collagen-model peptides (Azo-CMPs)
The hydrophilic/hydrophobic balance of peptides is one of the critical factors to control self-assembling behaviors.29,30 Thus we designed a new CMP whose N-terminus was modified with an azobenzene group. Azobenzene is known to be more hydrophobic than the peptide moieties and shows photo-isomerization, accompanied with a slight change in polarity. A triad repeating sequence of Gly-Pro-Hyp (GPO) was employed as the peptide segment forming the basis of the triple helix as it has been reported to be most stable triplet for collagen triple helices.11 The C-termini are amidic so as to prevent ionization, which can cause phenomena that complicate self-assembly. Fig. 1 displays the basic molecular structures of azobenzene-terminated peptides (Azo-(GPO)n) having various numbers of repeats (n = 3–10), and its derivatives which contain a flexible, hydrophilic diethylene glycol (deg) spacer connecting the (GPO)5 segment with the azobenzene group (Azo-deg-(GPO)5) and a long alkyl chain (CmAzo-(GPO)5, m = 6 and 12). The azobenzene moiety at the N-terminus is expected to show an effective hydrophobic interaction due to its stacking ability in water and to behave as a template that stabilizes collagen triple helical conformation. The additional introduction of the spacer (deg) and the alkyl chains into Azo-(GPO)5 would clarify the role of the azobenzene moiety in self-assembly. Furthermore, the resulting azobenzene aggregate in the N-terminal region of the collagen triple helices must be a driving force for developing into higher-order structures. All of these peptides were prepared by solid phase synthesis using Fmoc chemistry. Characterizations were carried out by using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy (MS) and proton nuclear magnetic resonance (1H NMR) spectroscopy (see ESI†).
Conformational properties of Azo-(GPO)n in water
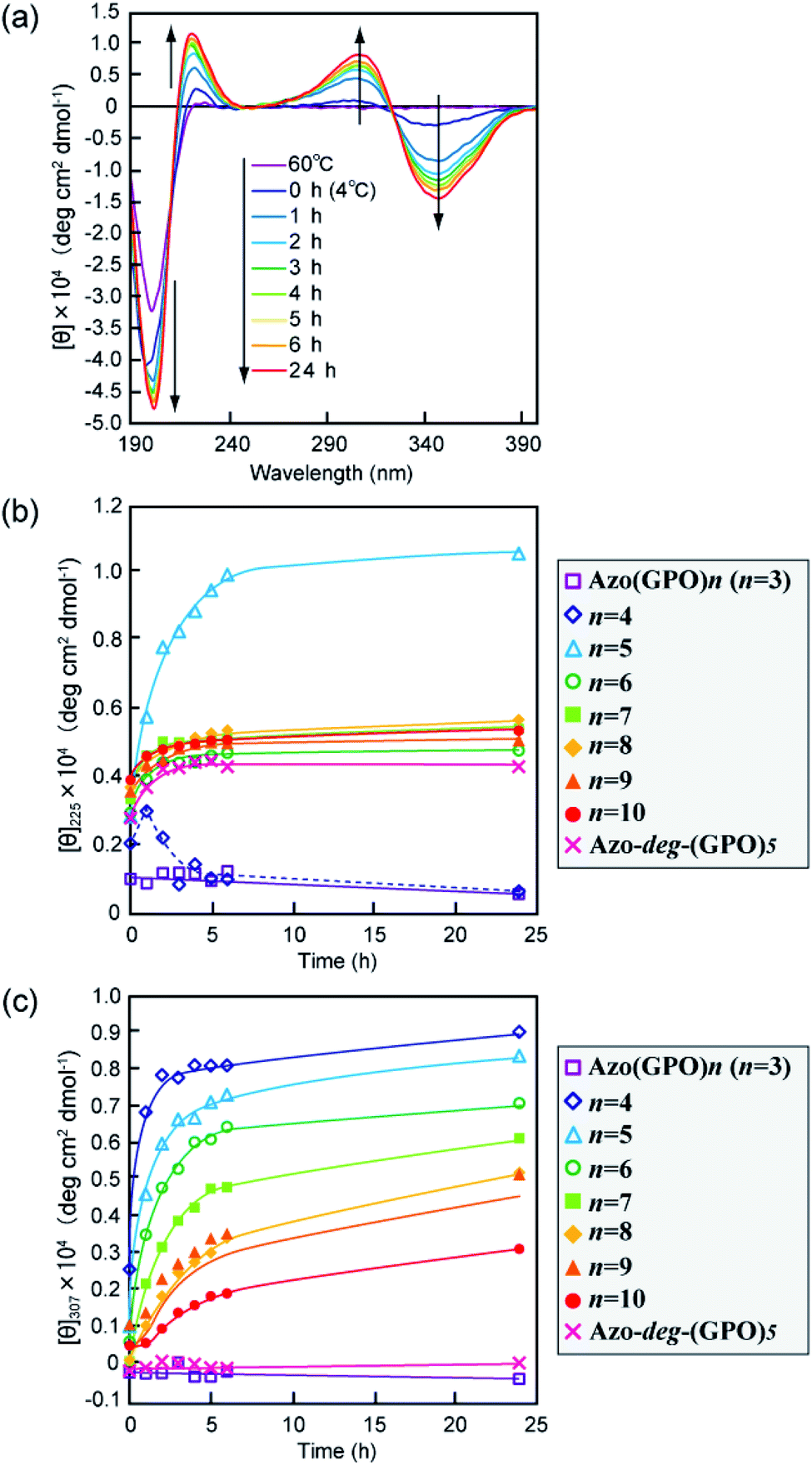
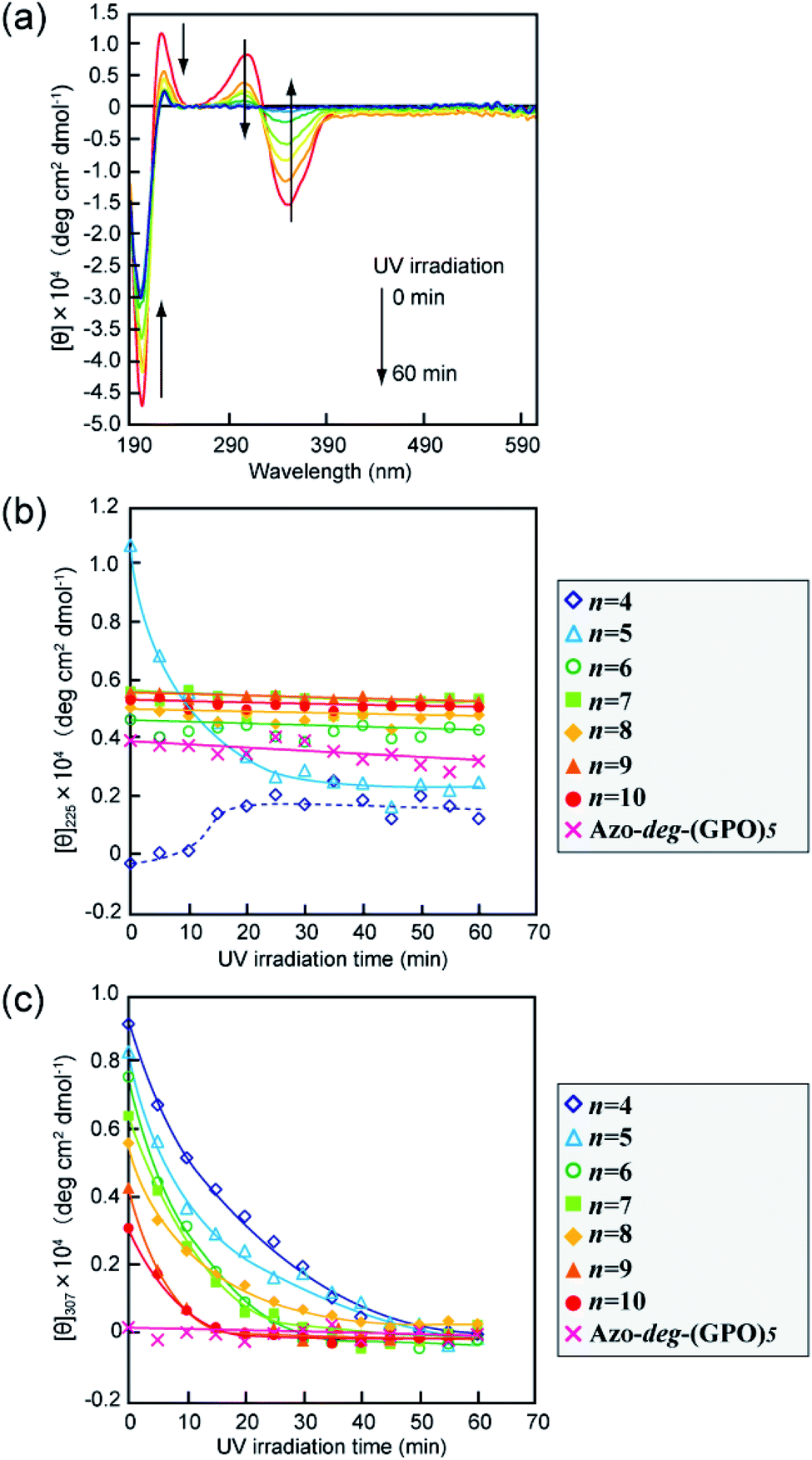
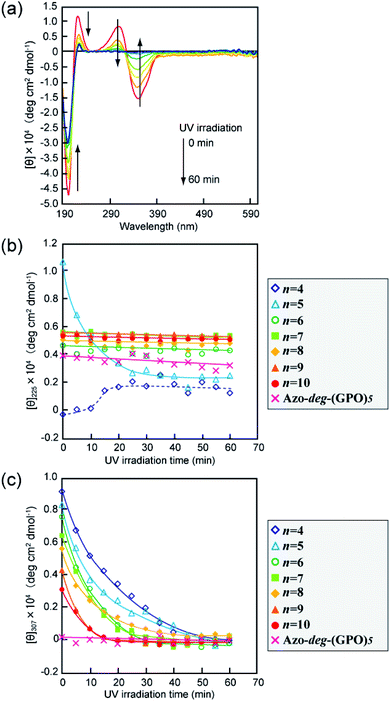
The secondary structures of the Azo-CMPs obtained were first evaluated by measuring the circular dichroism (CD) spectra for their aqueous solutions (containing 5% trifluoroethanol (TFE); [peptide] = 80 μM). Natural collagen has a unique CD spectrum in which a small positive peak appears at around 220 nm and a large trough at approximately 197 nm.46–48 These spectral features have been used as a reference to determine the presence of collagen-like triple helices in water. Fig. 2a shows time dependence of CD spectra measured for Azo-(GPO)5, a typical CMP (and others are shown in Fig. S1, ESI†), in water at 4 °C. The measurement was started when the sample solution was cooled to 4 °C from 60–90 °C, which are the points above which thermal denaturation of the peptides occur. The spectra are indicative of triple helical conformations, where the positive peak is at 225 nm and the trough around 200 nm. These absorbances and the Rpn value (ratio of the ellipticities for positive and negative peaks; the triple helical conformation shows a value from 0.1–0.2) proposed by Goodman et al.34,49 are comparable to known collagen triple helical conformations since the Rpn value was evaluated to be 0.2. In a longer wavelength region around 320 nm, which is a specific absorption band derived from the azobenzene moiety, an induced CD (ICD) effect is clearly observed at 348 nm (negative first Cotton effect) and at 307 nm (positive second Cotton effect), likely due to a specific interaction of azobenzene moieties. Such an observed Cotton effect strongly suggests that an exciton coupling between azobenzene chromophores occurs in a twisted configuration.50 That is to say, the formation of a triple helical structure would induce such a specific orientation of the N-terminal azobenzene groups. In fact, Azo-(GPO)3, with the shortest peptide segment, does not assemble into a triple helical structure and presented no ICD phenomenon (Fig. 2b, c and S1, ESI†). In spite of the formation of a stable triple helical structure, the spacer-containing Azo-deg-(GPO)5 also showed no ICD signal (Fig. 2b, c and S1, ESI†), likely due to the presence of the flexible spacer (deg) which blocked the peptide segments ((GPO)5) from assembling into triple helical structures. These results support the hypothesis that the ICD effect derives from the triple helical structure composed of the peptide segment directly connected with the azobenzene group at the N-terminus. Fig. 2b and c display the time dependency of molar ellipticities at 225 nm due to the triple helical conformation ([θ]225) and at 307 nm due to the ICD of the azobenzene moiety ([θ]307) on the basis of CD spectra for Azo-(GPO)n (n = 3–10) and Azo-deg-(GPO)5. As mentioned above, the [θ]225 and [θ]307 values for Azo-(GPO)3, having the shortest peptide length (n = 3), are very small and almost time independent, meaning that it does not take a triple helical conformation. The time dependency of [θ]225 values for Azo-(GPO)n (n = 6–10) provide very similar profiles; that is, quick increases in [θ]225 values are observed at the beginning, and after incubation for a few hours the values level off between 0.4 and 0.5. The Rpn values were evaluated to be 0.10–0.12 from CD spectra, which are very consistent with those for the collagen triple helix. From the time dependency of [θ]307 values (Fig. 2c), we found that the ICD of the azobenzene moiety increases with incubation time and in particular, its enhancement in the early stage depends strongly on the peptide length (n). Interestingly, the enhancement of ICD becomes much more remarkable for Azo-(GPO)n with shorter peptide segments (n). Such further incremental changes in ICD [θ]307 values after even a few hours of incubation suggest that the triple helical assembly behaves as a precursor to higher-order nano-architectures triggered by specific interactions of N-terminal azobenzene aggregates and/or C-terminal amide groups, as the [θ]225 values due to triple helix formation level off. Furthermore, Azo-(GPO)n with shorter peptide segments could more favorably assemble into a higher-order structure due to easier inter-terminal interactions. In Fig. 2b, we found that the time dependency of [θ]225 values for Azo-(GPO)4 and Azo-(GPO)5 are somewhat unique, compared to the others (Azo-(GPO)6–10). In particular, Azo-(GPO)5 shows an extremely rapid increase in [θ]225 reaching much larger values, and the [θ]225 value of Azo-(GPO)4 increases similar to the others for 1 h and then decreases gradually to that of Azo-(GPO)3, which does not assume the triple helix conformation. On the other hand, since the ICD signals of [θ]307 for both Azo-(GPO)5 and Azo-(GPO)4 are the most markedly enhanced, they can be assumed to assume the triple helical conformation. However, precipitation occurred in the solution of Azo-(GPO)4 after 24 h of incubation, likely due to its growth to a huge aggregate resulting in the unique time dependency of the [θ]225 profile. For alkyl chain-attached CmAzo-(GPO)5 (m = 6 and 12), the same CD experiments were performed, and as a result, CmAzo-(GPO)5 was found to assume a triple helical structure (Rpn > 0.10) and shows the ICD of the specific azobenzene interactions, which were observed for Azo-(GPO)n (n = 5–10). However, the CD spectra showed no time dependency within 24 h, suggesting that rapid self-assembling of CmAzo-(GPO)5 into triple helices and other organizations proceeded because of strong hydrophobic interactions of the alkyl chain-attached azobenzene moiety.
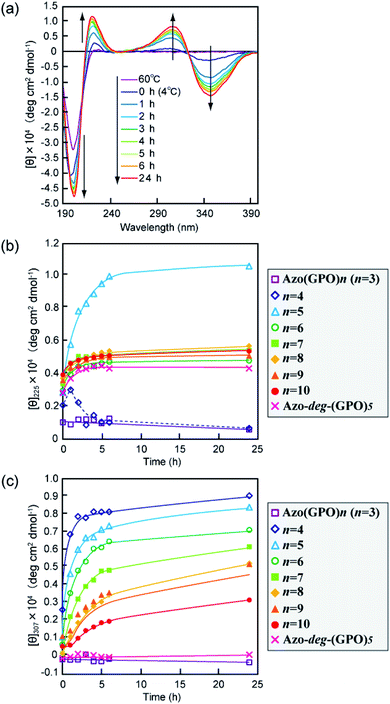 |
| | Fig. 2 (a) Changes in the CD spectra of Azo-(GPO)5 (trans-form) in water upon incubation at 4 °C after the thermal treatment at 60–90 °C, at which the peptides denature. (b and c) Denote time courses of the [θ]225 and [θ]307 values, respectively, for aqueous Azo-(GPO)n (trans-form, n = 3–10) and Azo-deg-(GPO)5 (trans-form) at 4 °C. [peptide] = 80 μM. | |
Morphological observation for Azo-CMPs
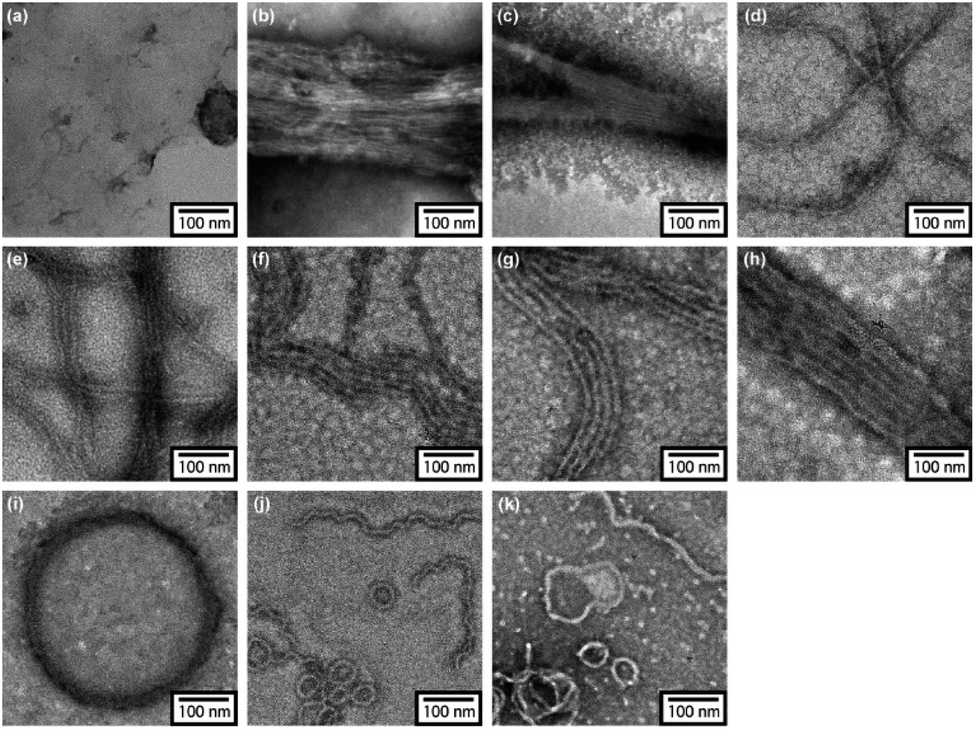
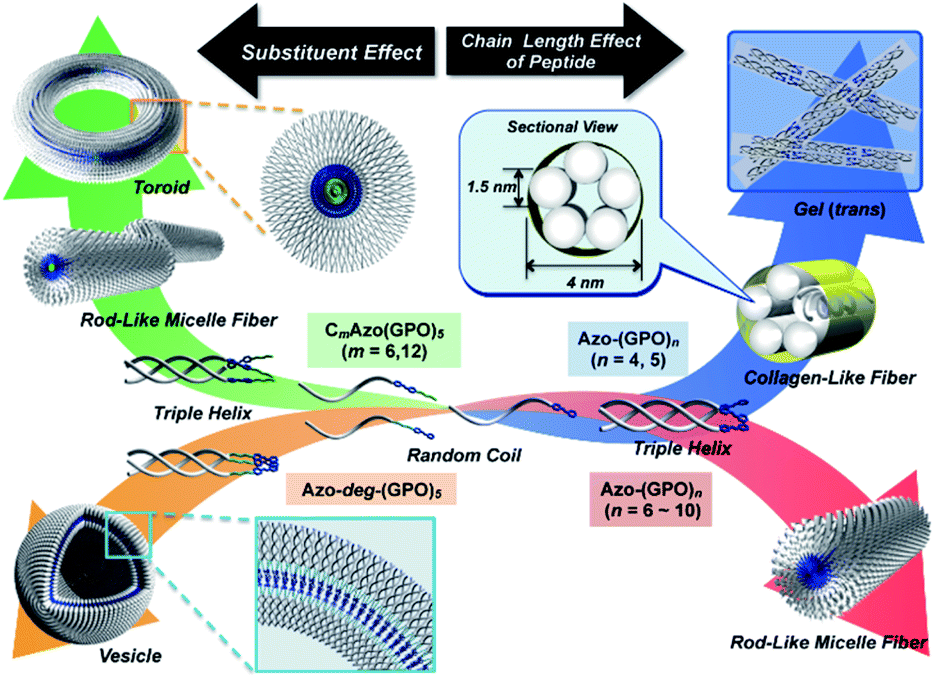
In order to reveal morphological features for these Azo-CMPs, transmission electron microscopy (TEM) observations of the samples after 24 h of incubation were made by negative staining with phosphotungstic acid (Fig. 3). Azo-(GPO)3, with the shortest peptide segment, which did not form a triple helical conformation and was not observed to have any specific nano-structures in the TEM picture (Fig. 3a). Both Azo-(GPO)4 and Azo-(GPO)5, showing peculiar profiles in the time dependency of the [θ]225 value, provided consistent fiber structures with lengths on the order of micro-meters. Closer inspection appeared to show bundles of thinner fibers of approximately 4 nm in width (Fig. 3b and c). This value is similar to the width of the fiber structure of natural collagen51,52 as shown schematically in Fig. 4. The fiber structure is an assembly composed of five triple helix precursors which are elongated one-dimensionally due to interactions between the N-terminal azobenzene aggregates and C-terminal amide groups through hydrogen bonding, and aligned in the same direction. With a further increase in the number of peptide repeats (n = 6–10), nanofibers with homogeneous diameters appear in TEM images. The diameters are observed to increase linearly with lengthening of the peptide segment; that is, in the order of 12 nm (n = 6), 14 nm (n = 7), 16 nm (n = 8), 18 nm (n = 9), and 20 nm (n = 10). Interestingly, these values of diameters are approximately twice as long as those of the theoretical molecular length evaluated by assuming complete triple helical conformation. Thus, we suppose that Azo-(GPO)n (n = 6–10) first formed a triple helical conformation and then grew to rod-like micelle fibers induced by the formation of an azobenzene core through hydrophobic interactions (Fig. 4). Azo-deg-(GPO)5 containing hydrophilic and flexible diethylene glycol (deg) as the spacer formed a single-walled vesicle structure (Fig. 3i), whose thickness is approximately 14 nm. This value corresponds well to twice that of the molecular length of triple helical Azo-deg-(GPO)5. We surmise that the assemblies must be bilayer vesicle structures in which there are hydrophilic triple helices and spacers placed at the periphery of the assembly with azobenzene moiety aggregates at the inner core, as shown in Fig. 4. Alkyl chain-attached CmAzo-(GPO)5 (Fig. 3j and k) shows a characteristic toroidal morphology as well as a turned and twisted fiber structure. By considering the observed width of toroidal rings and fibers (13–14 nm) and the theoretical molecular length of CmAzo-(GPO)5 in triple helical form (6.2 nm and 7.1 nm for m = 6 and m = 12, respectively), CmAzo-(GPO)5 likely forms a rod-like micelle fiber. Such a rod-like micelle fiber can spontaneously transform to a toroidal structure by ring closure. As was mentioned above, Azo-(GPO)n (n = 6–10) also assembled into rod-like micelle fiber structures, but did not assume toroidal ring morphologies. It can be seen from the TEM images that there are considerable differences in flexibility of nanofiber structures between those of Azo-(GPO)n (n = 6–10) and CmAzo-(GPO)5 (m = 6 and 12). Apparently, the nanofibers for the latter seem to be more flexible, and turn and twist more than those of the former. Therefore, such pliability of a nanofiber might be an important factor for assembly into toroidal rings. The attachment of the alkyl chain to the rigid azobenzene moiety likely induces relatively loose assemblies of CmAzo-(GPO)5. The atomic force microscopy (AFM) observations were also performed for the same samples (Fig. S2, ESI†), and the observed morphologies were basically consistent with those of the TEM observations. From these morphological results, we can conclude that the morphological diversity of higher-order assemblies of Azo-CMPs is derived from the characteristics of the pre-organized triple helix unit; that is, through the variation in hydrophilic/hydrophobic balance achieved by changing the peptide length and introducing the soft substituent groups to the rigid azobenzene moiety.
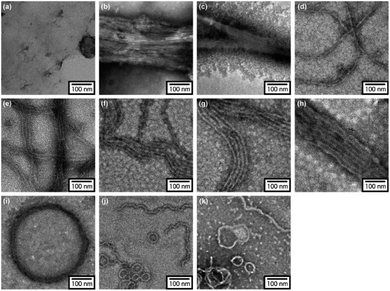 |
| | Fig. 3 TEM images stained by phosphotungstic acid for Azo-(GPO)n (trans-form) (n = 3 (a), 4 (b), 5 (c), 6 (d), 7 (e), 8 (f), 9 (g), and 10 (h)), Azo-deg-(GPO)5 (trans-form) (i), and CmAzo-(GPO)5 (trans-form) (m = 6 (j) and 12 (k)) incubated for 24 h at 4 °C. | |
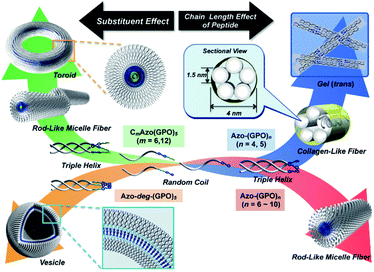 |
| | Fig. 4 Proposed model for a hierarchical self-assembly system from azobenzene-terminated collagen-mimetic oligopeptides. | |
Effects of photo-isomerization of azobenzene on self-assembly
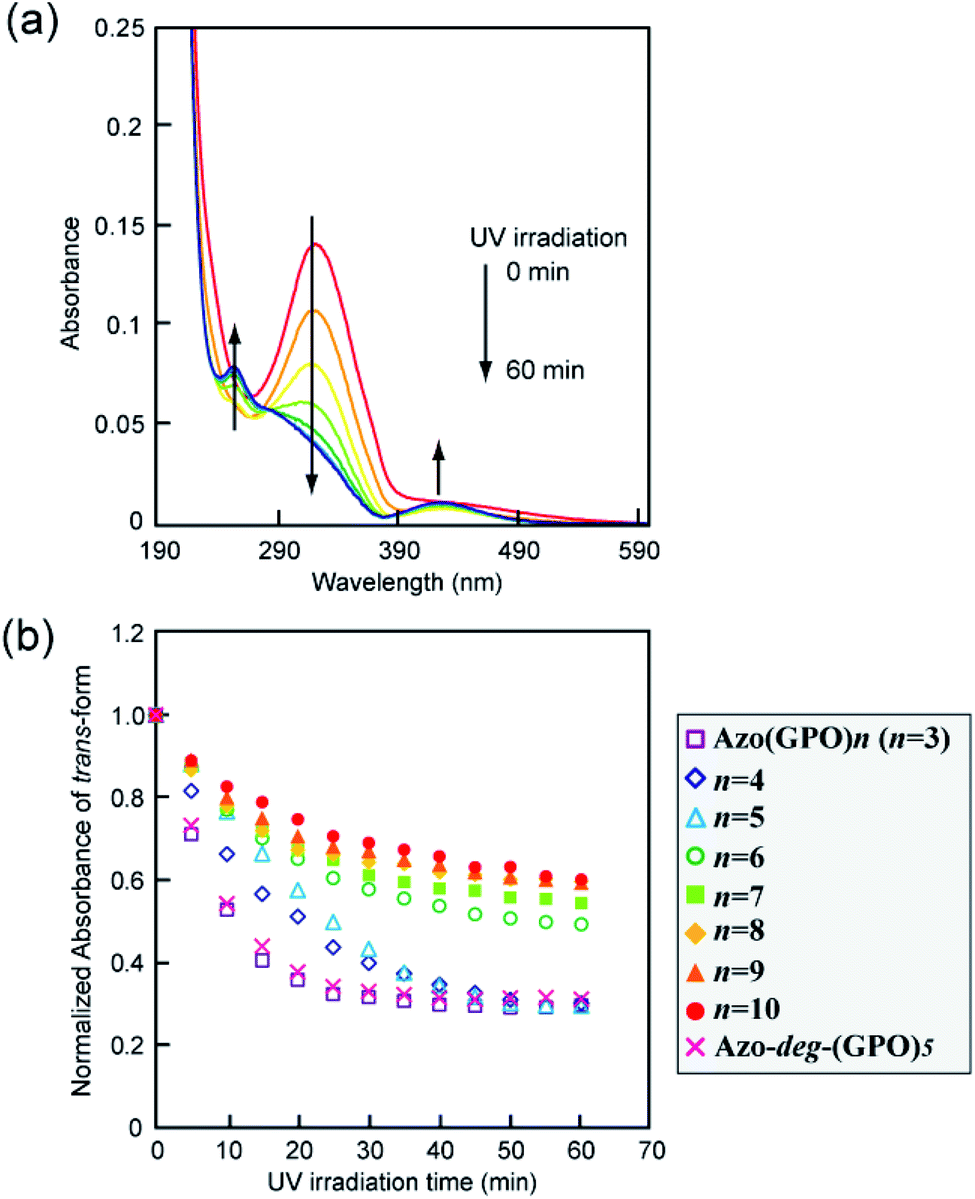
The effects of trans–cis isomerization of the azobenzene moiety on collagen triple helical structures and their self-assemblies were examined spectroscopically and morphologically. Fig. 5a shows representative changes in the absorption spectra of the Azo-(GPO)5 aqueous solution upon UV irradiation at 365 nm and 4 °C. The absorption peak at 320 nm due to the trans-form of the azobenzene was found to decrease with UV irradiation, and at the same time the peak of the cis-isomer at 450 nm increased. After 60 minutes of irradiation, the isomerization reaches a photo-stationary state. Fig. 5b is a plot of the absorbance at 320 nm (trans-form) and the UV irradiation time measured for Azo-(GPO)n (n = 3–10) and Azo-deg-(GPO)5 under the same conditions. The isomerization behavior can be roughly grouped into three types: (I) Azo-(GPO)n (n = 6–10), (II) Azo-(GPO)n (n = 4 and 5), and (III) Azo-(GPO)3 and Azo-deg-(GPO)5. Azo-CMPs belonging to type (I) exhibit the slowest and lowest degree of isomerization, compared with types (II) and (III). Such trends become more remarkable with increasing peptide length (n). Since the peptide length contributes strongly to the stability of triple helical conformation, the stability may lead to a tight stacking of the azobenzene moiety that presumably inhibits the isomerization. In fact, Azo-deg-(GPO)5, having a flexible spacer, shows a relatively rapid decrease in the trans-form with UV irradiation, and only 30 minutes of irradiation is sufficient to reach the photo-stationary state. Similar effective isomerization for Azo-(GPO)3, which does not take a triple helical conformation at all (type (III)), was observed. Therefore, from the isomerization behavior for Azo-(GPO)4 and Azo-(GPO)5 (type (II)), we suggest that the isomerization induces the conformational transition from a loose triple helical structure to a random coil structure. The measurements of the CD spectra also support this conformational transition. Fig. 6a displays changes in the CD spectra with UV irradiation for the Azo-(GPO)5 aqueous solution. The UV irradiation was performed for the sample incubated for 24 h at 4 °C. Both [θ] values of the triple helix conformation at 225 nm and the ICD value of the azobenzene group at 307 nm decrease by UV irradiation, meaning that the photo-isomerization of the azobenzene moiety leads to the decay of the triple helix structure and simultaneously stacking of the twisted azobenzenes. On the other hand, the [θ] values at 225 nm for Azo-(GPO)n (n = 6–10) showed no irradiation time dependency although the [θ] values at 307 nm decrease similarly to those of Azo-(GPO)5 due to the disappearance of the trans-form by isomerization (Fig. 6b and c). This result again suggests that peptide length (n) determines the stability of the triple helix conformation. The triple helix conformation of Azo-deg-(GPO)5, which has a spacer, retains its stability regardless of the isomerization as was expected (Fig. 6b). In the case of alkyl chain-attached CmAzo-(GPO)5 (m = 6 and 12), the triple helices did not decay in spite of the progress of azobenzene photo-isomerization because the hydrophobic interaction of the attached alkyl chain could compensate for the steric and slight polar changes of the azobenzene groups resulting from their isomerization from the trans- to cis-forms. Subsequently, the effect of UV irradiation on the aggregation morphology for all Azo-CMPs was examined by TEM observation (Fig. S3, ESI†). For all of the Azo-CMPs, the cis-form of the azobenzene causes the disappearance of characteristic nano-architectures as observed in the trans-form, although only C12Azo-(GPO)5 retains a partly fragmented flexible fiber structure. Therefore, we conclude that the trans-form is essential to the formation of the characteristic nano-architecture dependent on molecular structures such as the peptide segment length, and the presence of a flexible spacer and alkyl chains.
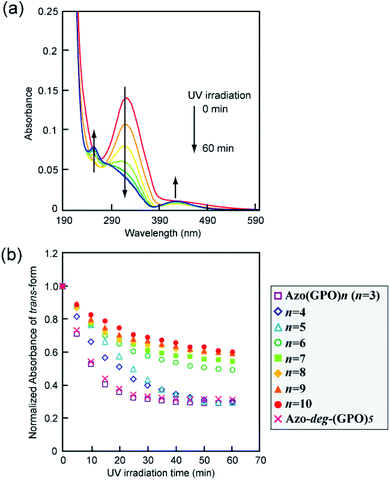 |
| | Fig. 5 (a) Changes in the absorption spectra of Azo-(GPO)5 upon UV irradiation (365 nm) in water at 4 °C. [peptide] = 80 μM. (b) Photo-isomerization processes for Azo-(GPO)n (n = 3–10) and Azo-deg-(GPO)5 in water at 4 °C. | |
 |
| | Fig. 6 (a) Changes in the CD spectra of Azo-(GPO)5 upon UV irradiation (365 nm) in water at 4 °C. [peptide] = 80 μM. (b and c) Denote time courses of the [θ]225 and [θ]307 values, respectively, for Azo-(GPO)n (n = 4–10) and Azo-deg-(GPO)5 in water at 4 °C during UV irradiation. [peptide] = 80 μM. | |
Self-assembly of Azo-CMPs under concentrated conditions
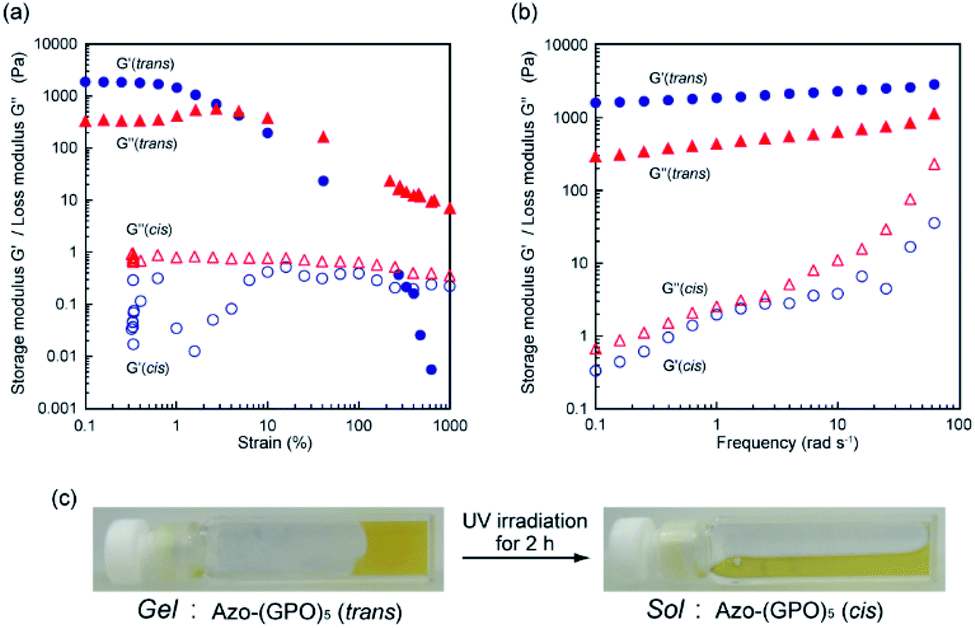
The self-assembly of these Azo-CMPs was further investigated in a concentrated aqueous solution (6 wt%; [peptide] = 8–10 mM). Only Azo-(GPO)4 and Azo-(GPO)5, which self-organize in a dilute aqueous solution similar to natural collagen-like, well-developed fiber structures, formed hydrogels at 6 wt% concentration. C6Azo-(GPO)5 and C12Azo-(GPO)5 were insoluble in water at this concentration due to their higher hydrophobicity. Others such as Azo-(GPO)n (n = 6–10) and Azo-deg-(GPO)5 provided merely fluid, viscous solutions, except for Azo-(GPO)3 whose aqueous solution caused partial precipitation (Fig. S4, ESI†). These results suggest that gelation of these types of CMPs needs a tightly bundled structure of nanofibers as well as a suitable hydrophobic/hydrophilic balance of CMP molecules to generate cross-linking points for network formation. The prepared hydrogel of Azo-(GPO)5 (azobenzene moiety: trans-form) was further characterized by dynamic rheology measurements. A strain sweep experiment was first carried out by measuring the evolution of the moduli while increasing the deformation of the gel. The hydrogel showed a mechanical-responsive gel-to-sol transition when the shear strain amplitude was increased (Fig. 7a). During measurement, the storage (G′) and the loss (G′′) moduli showed a plateau up to a deformation of approximately 1%. Although the amplitude of the strain was increased, both moduli remained almost constant. At a higher strain above 1%, the hydrogel showed a gel-to-sol transition. The dynamic property of the hydrogel was then examined in the linear viscoelasticity by frequency sweep measurements. In the present frequency range, the elastic moduli are constant and the storage moduli are about one order of magnitude higher than the loss moduli (Fig. 7b). Subsequently, the effect of photo-isomerization on the gelation property was examined. Fig. 7c displays photographs of the Azo-(GPO)5 hydrogel before and after UV irradiation for 2 h at 4 °C. The gel-to-sol transition proceeded smoothly. The results of rheological measurements for the sample after 2 h of irradiation also support the hypothesis that the collapse of the gel state arises from the trans-to-cis isomerization of the azobenzene moiety that triggered the disintegration of 3D-network structures (Fig. 7a and b).
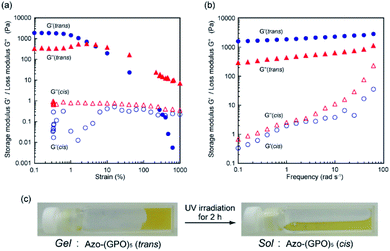 |
| | Fig. 7 (a) Dynamic storage (G′) and loss (G′′) moduli for Azo-(GPO)5-hydrogel (6 wt%) as a function of the strain (frequency: 6 rad s−1) at 4 °C before (●, ▲) and after (○, △) UV irradiation. (b) G′ and G′′ moduli for Azo-(GPO)5-hydrogel (6 wt%) as a function of the frequency (strain: 0.1%) at 4 °C. (c) Photographs of Azo-(GPO)5-hydrogel before and after UV irradiation. | |
Experimental
Materials
N,N-Dimethylformamide (DMF), piperidine, methanol, ethanol, phenol, ethylacetate, acetone, diethyl ether, tetrahydrofuran (THF) hexane, MgSO4 anhydrous, dimethylsulfoxide (DMSO)-d6, and 2,2,2-trifluoroethanol (TFE) were purchased from Nacalai Tesque. Chloroform-d, 4-(phenyazo)benzoic acid and 2,5-dihydroxybenzoic acid (DHBA) were purchased from Sigma Aldrich. N,N′-Diisopropylcarbodiimide (DIPC), dichloromethane (DCM), 4-aminobenzoic acid, 1-bromohexane, 1-bromodecane and 18-crown-6-ether were purchased from Wako Pure Chemical. N-(9-Fluorenylmethoxycarbonyl) (Fmoc)-NH-SAL-MBHA Resin, Fmoc-Hyp(But)-OH, Fmoc-Gly-OH, trifluoroacetic acid (TFA) and 1-hydroxy-7-aza-benzotriazol (HOAt) were purchased from Watanbe Chemical Industries. Fmoc-Pro-OH and Fmoc-8-amino-3,6-dioxaoctanoic acid (Fmoc-deg-OH) were purchased from Peptide Institute. All reagents were used as received.
Measurements
1H NMR spectra were recorded using a JEOL JNM-AL-400 (JEOL Resonance) spectrometer (400 MHz). MALDI-TOF MS analyses were performed on an Autoflex speed (Bluker Daltonics) using DHBA as a matrix. CD spectra were recorded on a J-820 spectropolarimeter (JASCO Ltd.) under N2 atmosphere equipped with a Peltier type thermostatic cell holder coupled with a controller PTC-432L (JASCO Ltd.). Samples were prepared in water to be [peptide] = 80 μM, and incubated at 4 °C after thermo-treatment at 60–90 °C. Experiments were performed in a quartz cell with a 1 mm path length over the range of 190–600 nm at 4 °C. UV-vis spectra were acquired using a V-650 spectrophotometer (JASCO Ltd.) equipped with a Peltier type thermostatic cell holder ETSC-761 (JASCO Ltd.). The AFM images were collected on a SPM-9700 (Shimadzu Co.) operated by tapping using a silicone tip (MPP-1110, tip radius < 1.2 nm, Bruker Nano surfaces). A 10 mL of the aqueous solution of the peptide (80 μM), which was obtained after incubation for 24 h at 4 °C, was placed on freshly cleaved mica. After 10 s, the excess solution was removed by absorption onto filter paper, and the sample was dried at 4 °C. The scanning speed was at a line frequency of 1 Hz, and the original images were sampled at a resolution of 1024 × 1024 points. The TEM images were collected on a JEM 2100F (JEOL Resonance) at 200 kV accelerating voltage. After a small volume of the aqueous sample solution (80 μM, treated for 24 h at 4 °C) was applied to a TEM grid for 20 min, the excess solution was blotted with filter paper and stained with phosphotungstic acid aqueous solution (1 wt%), and the sample was dried in a covered container at room temperature overnight. Photo-isomerization was performed with a Handy UV Lamp SLUV-4 at 365 nm, 4 W (AS ONE Co.). Rheological measurements were performed on a Discover HR-1 TA Instruments equipped with a Peltier device for temperature control. During all rheological measurements, a solvent trap was used to minimize the evaporation. The measurement was carried out using a parallel plate (diameter: 40 mm). The gap was adjusted to be 300 μm to ensure that the geometry is completely filled. The gel samples were loaded and measured after incubation for 24 h at 4 °C.
Syntheses of Azo-CMPs
All Azo-CMPs employed in this study were prepared by SPPS using an Fmoc chemistry. These peptides were synthesized on an Fmoc-NH-SAL-MBHA resin using Fmoc-L-amino acid derivatives [Fmoc-Hyp(But)-OH, Fmoc-Pro-OH, Fmoc-Gly-OH], DIPC, and HOAt in DMF for coupling and piperidine (25%)/DMF for Fmoc removal. Finally, azobenzene derivatives were attached to the N-terminus of the corresponding peptide as follows: 4-(phenylazo)benzoic acid (Azo-(GPO)n), Fmoc-deg-OH and 4-(phenylazo) benzoic acid (Azo-deg-(GPO)5), 4-(4-hexyloxyphenylazo)benzoic acid (see the ESI†) (C6Azo-(GPO)5) or 4-(4-dodecyloxyphenylazo)benzoic acid (see the ESI†) (C12Azo-(GPO)5) were coupled with the corresponding (GPO)n-resins in DMF, including DIPC and HOAt. Treatment with TFA/DCM (v/v 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) cleaved the Azo-CMPs from the resins. After purification, the obtained Azo-CMPs were identified by MALDI-TOF MS and 1H NMR spectroscopies as described in the ESI.†
:1) cleaved the Azo-CMPs from the resins. After purification, the obtained Azo-CMPs were identified by MALDI-TOF MS and 1H NMR spectroscopies as described in the ESI.†
Conclusions
We have introduced the concept of collagen-based supramolecular assemblies having widespread morphological features through the attachment of azobenzene to the short collagen mimetic peptides, Azo-CMPs, via an azobenzene stacking assisted self-assembling trigger. In a dilute solution, we found that such morphological shapes derived from Azo-CMPs such as well-developed nanofibers, rod-like micelle fibers, bilayer vesicles, and toroidal rings. These structures can be individually created by varying the peptide length of triple helices and by incorporating substituents such as an alkyl chain and a flexible spacer, resulting in an appropriate tune of hydrophobic/hydrophilic balance of the Azo-CMP molecules themselves. In a concentrated solution, specific Azo-CMPs (Azo-(GPO)4 and Azo-(GPO)5, which gave well-developed collagen-like fiber structure in a dilute solution) spontaneously formed hydrogels that demonstrated a satisfactory gel-to-sol transition triggered by the trans-to-cis photo-isomerization of azobenzene. The ability to include nanometer to micrometer scale features and versatile morphology variation into designed biomaterial scaffolds may have significant potential for future biomedical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by Grants-in-Aid for Scientific Research (KAKENHI) (26390022, No. 16K05800 and No. 17K04994) from the Japan Society for the Promotion of Science (JSPS), and a MEXT-Supported Program for the Strategic Research Foundation at Private University.
References
- K. Beck and B. Brodsky, J. Struct. Biol., 1998, 122, 17–29 CrossRef CAS PubMed.
- B. Brodsky and A. V. Persikov, Adv. Protein Chem., 2005, 70, 301–339 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- C. H. Lee, A. Singla and Y. Lee, Int. J. Pharm., 2001, 221, 1–22 CrossRef CAS PubMed.
- R. S. Erdmann and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 13957–13959 CrossRef CAS PubMed.
- N. Dai and A. Etzkorn, J. Am. Chem. Soc., 2009, 131, 13728–13732 CrossRef CAS PubMed.
- M. D. Shoulders, J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2006, 128, 8112–8113 CrossRef CAS PubMed.
- A. V. Persikov, J. A. M. Ramshaw, A. Kirkpatrick and B. Brodsky, J. Am. Chem. Soc., 2003, 1252, 11500–11501 CrossRef PubMed.
- D. E. Przybyla and J. Chmielewski, Biochemistry, 2010, 49, 4411–4419 CrossRef CAS PubMed.
- R. Strawn, F. Chen, P. J. Haven, S. Wong, A. Park-Arias, M. D. Leeuw and Y. Xu, Biopolymers, 2018, 109, 23226–23235 CrossRef PubMed.
- A. V. Persikov, J. A. Ramshaw, A. Kirkpatrick and B. Brodsky, Biochemistry, 2000, 39, 14960–14967 CrossRef CAS PubMed.
- S. Rele, Y. Song, R. P. Apkarian, Z. Qu, V. P. Conticello and E. L. Chaikof, J. Am. Chem. Soc., 2007, 129, 14780–14787 CrossRef CAS PubMed.
- L. E. O'Leart, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- I. C. Tanrikulu, A. Forticaux, S. Jin and R. T. Raine, Nat. Chem., 2016, 8, 1008–1014 CrossRef CAS PubMed.
- S. E. Paramonov, V. Gauba and J. D. Hartgerik, Macromolecules, 2005, 38, 7555–7561 CrossRef CAS.
- F. W. Kotch and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3028–3033 CrossRef CAS PubMed.
- C. M. Yamazaki, S. Asada, K. Kitagawa and T. Koide, Biopolymers, 2008, 90, 816–823 CrossRef CAS PubMed.
- O. D. Krishna and K. L. Kiick, Biomacromolecules, 2009, 10, 2626–2631 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E. Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202–2203 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, H. R. Almond Jr, C. A. Maryanoff, K. Balss, M. Breslav, E. Lacy and B. E. Maryanoff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8518–8525 CrossRef PubMed.
- K. Kar, S. Ibrar, V. Nanda, S. P. Kunapuli and B. Brodsky, Biochemistry, 2009, 48, 7959–7968 CrossRef CAS PubMed.
- D. E. Przybyla and J. Chmielewski, J. Am. Chem. Soc., 2008, 130, 12610–12611 CrossRef CAS PubMed.
- M. M. Pires and J. Chmielewski, J. Am. Chem. Soc., 2009, 131, 2706–2712 CrossRef CAS PubMed.
- D. E. Przybyla and J. Chmielewski, J. Am. Chem. Soc., 2010, 132, 7866–7867 CrossRef CAS PubMed.
- C.-C. Chen, W. Hsu, T.-C. Kao and J.-C. Horng, Biochemistry, 2011, 50, 2381–2383 CrossRef CAS PubMed.
- U. Kusebauch, S. A. Cadamuro, H.-J. Musiol, L. Moroder and C. Renner, Chem.–Eur. J., 2007, 13, 2966–2973 CrossRef CAS PubMed.
- S. Samanta and A. Woolley, ChemBioChem, 2011, 12, 1712–1723 CrossRef CAS PubMed.
- V. Kubyshkin, Org. Biomol. Chem., 2019, 17, 8031–8047 RSC.
- H. Xu, J. Wang, S. Han, J. Wang, D. Yu, H. Zhang, D. Xia, X. Zhao, T. A. Waigh and J. R. Lu, Langmuir, 2009, 25, 4115–4123 CrossRef CAS PubMed.
- T. Koga, T. Higuchi, T. Kinoshita and N. Higashi, Chem.–Eur. J., 2006, 12, 1360–1367 CrossRef CAS PubMed.
- S. Sakakibara, K. Inoue, K. Shudo, Y. Kishida, Y. Kobayashi and D. J. Prockop, Biochem. Biophys. Acta, 1973, 303, 198–202 CrossRef CAS.
- H.-P. Germann and E. Heidemann, Biopolymers, 1988, 27, 157–163 CrossRef CAS PubMed.
- C. G. Fields, D. J. Mickelson, S. L. Drake, J. B. McCarthy and G. B. Fields, J. Biol. Chem., 1993, 268, 14153–14160 CAS.
- Y. Feng, G. Melacini, J. P. Taulane and M. Goodman, J. Am. Chem. Soc., 1996, 118, 10351–10358 CrossRef CAS.
- M. Goodman, Y. Feng, G. Melacini and J. P. Taulane, J. Am. Chem. Soc., 1996, 118, 5156–5157 CrossRef CAS.
- G. Melacini, Y. Feng and M. Goodman, J. Am. Chem. Soc., 1996, 118, 10359–10364 CrossRef CAS.
- J. Kwak, A. D. Capua, E. Locardi and M. Goodman, J. Am. Chem. Soc., 2002, 124, 14085–14091 CrossRef CAS PubMed.
- P. Berndt, G. B. Fields and M. Tirrell, J. Am. Chem. Soc., 1995, 117, 9515–9522 CrossRef CAS.
- Y.-C. Yu, P. Berndt, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1996, 118, 12515–12520 CrossRef CAS.
- Y.-C. Yu, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1998, 120, 9979–9987 CrossRef CAS.
- Y.-C. Yu, V. Roontga, V. A. Daragan, K. H. Mayo, M. Tirrell and G. B. Fields, Biochemistry, 1999, 38, 1659–1668 CrossRef CAS PubMed.
- T. Gore, Y. Dori, Y. Talmon, M. Tirrell and H. Bianco-Peled, Langmuir, 2001, 17, 5352–5360 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- H. W. Jun, S. E. Paramonov and J. D. Hartgerink, Soft Matter, 2006, 2, 177–181 RSC.
- J. Luo and Y. W. Tong, ACS Nano, 2011, 5, 7739–7747 CrossRef CAS PubMed.
- S. Sakakibara, Y. Kishida, K. Okuyama, N. Tanaka, T. Ashida and M. Kakudo, J. Mol. Biol., 1972, 65, 371–372 CrossRef CAS PubMed.
- F. R. Brown III, J. P. Carver and E. R. Blout, J. Mol. Biol., 1969, 39, 307–313 CrossRef.
- F. R. Brown III, A. di Corato, G. P. Lorenzi and E. R. Blout, J. Mol. Biol., 1972, 63, 85–99 CrossRef.
- Y. Feng, G. Melacini and M. Goodman, Biochemistry, 1997, 36, 8716–8724 CrossRef CAS PubMed.
- S. Allenmark, Chirality, 2003, 15, 409–422 CrossRef CAS PubMed.
- I. T. Weber, R. W. Harrison and R. V. Iozzo, J. Biol. Chem., 1996, 271, 31767–31770 CrossRef CAS PubMed.
- S. M. Sweeney, J. P. Orgel, A. Fertala, J. D. McAuliffe, K. R. Turner, G. A. Di Lullo, S. Chen, O. Antipova, S. Permul, L. Ala-Kokko, A. Forlino, W. A. Cabral, A. M. Barnes, J. C. Marini and D. S. Antonio, J. Biol. Chem., 2008, 283, 21187–21197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analyses for preparation of Azo-CMPs, Fig. S1–S6: MALDI-TOF MS and 1H NMR spectra, AFM images for Azo-CMPs (trans-form), TEM images for Azo-CMPs after UV irradiation, and photographs of Azo-CMPs (trans-form) in the concentrated situation. See DOI: 10.1039/d0ra02906h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Ryo Yoshikawa and
Tomoyuki Koga
*,
Ryo Yoshikawa and
Tomoyuki Koga