DOI:
10.1039/D0RA03038D
(Paper)
RSC Adv., 2020,
10, 18953-18958
Studies on asymmetric total synthesis of (−)-β-hydrastine via a chiral epoxide ring-opening cascade cyclization strategy†
Received
4th April 2020
, Accepted 11th May 2020
First published on 19th May 2020
Abstract
Herein, facile and enantioselective approaches to synthesize the core phthalide tetrahydroisoquinoline scaffold of (−)-β-hydrastine via both a CF3COOH-catalyzed (86% ee) and KHMDS-catalyzed (78% ee) epoxide ring-opening/transesterification cascade cyclization from chiral epoxide under very mild conditions are described. The key elements include a highly enantioselective epoxidation using the Shi ketone catalyst and an intramolecular CF3COOH-catalyzed cascade cyclization in one pot, and a late-stage C-3′ epimerization under MeOK/MeOH conditions as the key steps to achieve the first total synthesis of (−)-β-hydrastine (up to 81% ee).
Introduction
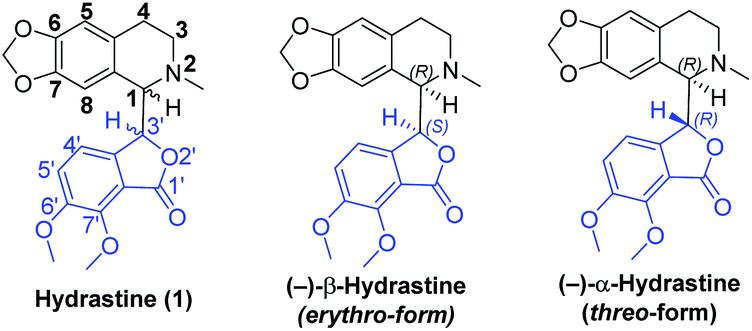
Natural products that feature multiple stereocenters and complex molecular topologies hold a special fascination to chemists. Phthalide tetrahydroisoquinoline alkaloids, such as hydrastine1 (1, Fig. 1), are known to possess interesting and diverse biological properties.2 In light of their fascinating architectural attributes and unique structures, as well as promising biological activities, phthalide tetrahydroisoquinoline alkaloids have been attractive and challenging targets of synthetic and medicinal chemists for decades. However, due to the small quantities of the natural products including hydrastine (1), a comprehensive biological evaluation of this class of alkaloids could not be accomplished. To accelerate further investigation of pharmacological activities and SAR studies, we embarked on the asymmetric total syntheses of this class of alkaloids.
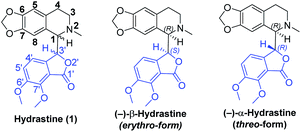 |
| | Fig. 1 The erythro- and threo-form of hydrastine (1). | |
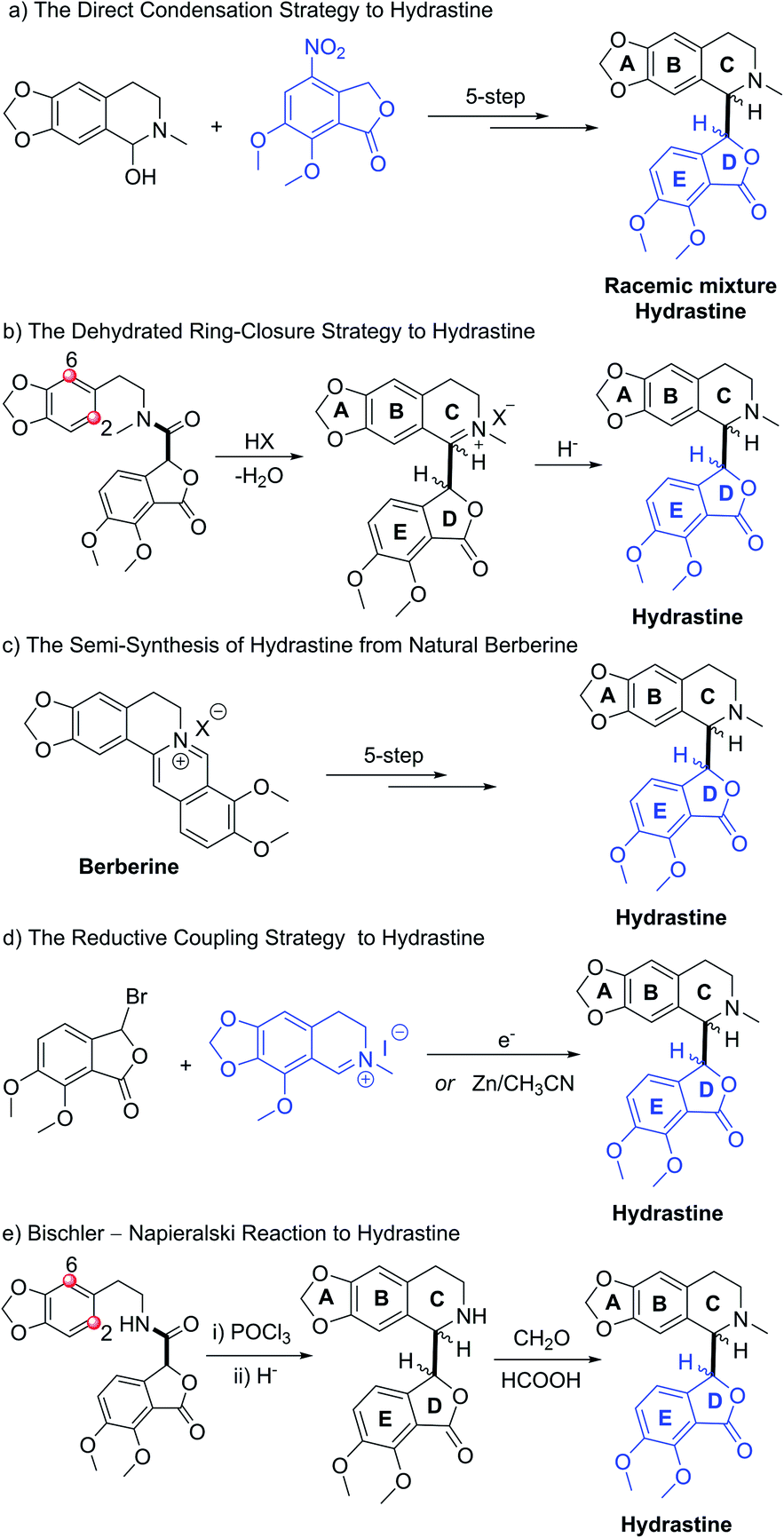
Hydrastine (1), originally isolated from Corydalis longipes3 has been used as an antiseptic,4 as a uterine hemostatic4 and as a potent competitive antagonist at mammalian GABAA receptors (CD50 0.16 mg kg−1, i.v.).5 In 2016, (−)-β-hydrastine [(−)-β-1] was found to exhibit potent antitumor activity against human lung adenocarcinoma cells.6 Structurally, hydrastine (1) comprises a 1,2,3,4-tetrahydroisoquinoline and a phthalide ring and these two units are linked together by two chiral centers at their C-1 and C-3′ carbons to form either erythro- or threo-isomer (Fig. 1).7 Because of unique structural features and interesting biological properties of hydrastine, the design of new protocols for the construction of this privileged scaffold has attracted considerable interest. Since the work for the synthesis of hydrastine (1) by Robinson and co-workers using the direct condensation strategy in 1931 (Scheme 1a),8 several synthetic studies9,10 had been reported for the synthesis of hydrastine (1). Among them, (±)-β-hydrastine and (±)-α-hydrastine were obtained from the substituted β-phenylethylmethylamide of a substituted phthalidecarboxylic acid by dehydrated ring-closure and following reduction in 1950 (Scheme 1b),9 and the semi-synthesis of (±)-β-hydrastine and (±)-α-hydrastine from natural berberine was reported (Scheme 1c).10 In addition, several other approaches11,12 to (±)-β-hydrastine and (±)-α-hydrastine were reported, such as the electrochemical11a and zinc-promoted reductive coupling11b (Scheme 1d). Alternatively, the phthalide tetrahydroisoquinoline skeleton of hydrastine (1) could also be synthesized through the Bischler–Napieralski reaction12 (Scheme 1e) in the similar fashion of the synthesis of noscapine.13 Although these methods successfully achieved the synthesis of hydrastine (1), an inherent drawback associated with all of the above-mentioned strategies was the lack of enantioselectivity. Thus, the efficient, practical and highly enantioselective construction of phthalide tetrahydroisoquinoline structures still constitutes a synthetic challenge. To our best knowledge, no attempt to synthesize (−)-β-hydrastine [(−)-β-1] with high enantioselectivity has been reported.
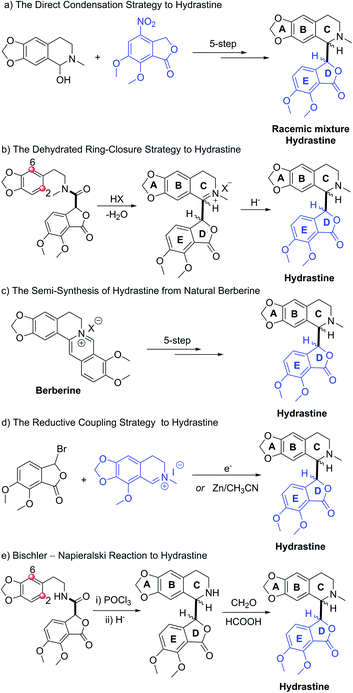 |
| | Scheme 1 The known strategic approaches to hydrastine. (a) The direct condensation strategy. (b) The dehydrated ring-closure strategy. (c) The semi-synthesis strategy. (d) The reductive coupling strategy. (e) The Bischler–Napieralski cyclization strategy. | |
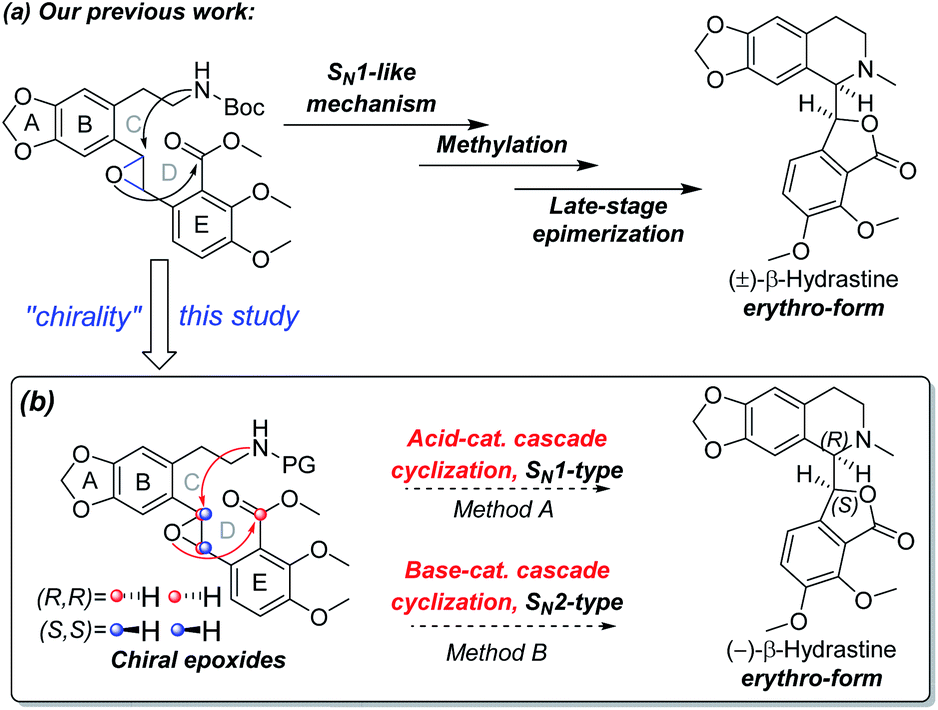
Our research group has been exploring a unified acid-catalyzed epoxide ring-opening/intramolecular transesterification cascade cyclization strategy for synthesis of phthalide tetrahydroisoquinoline alkaloids, in which the C and D rings are constructed simultaneously in one step.14 The method was successfully applied to the diastereoselective total synthesis of (±)-β-hydrastine via an SN1-like mechanism (Scheme 2a).14 In order to investigate further applications of this cascade cyclization strategy for enantioselective total syntheses of phthalide tetrahydroisoquinoline alkaloids, we postulated that the chiral epoxide was utilized as a key stereocontrol element in addition to a reactivity control element to enantioselectively construct the phthalide tetrahydroisoquinoline scaffold based on an SN1-type acid-catalyzed or an SN2-type base-catalyzed cyclization strategy (Scheme 2b). As the Shi ketone catalyst and other variants are synthetically accessible,15,16 we have the ability to generate epoxide substrate bearing the necessary stereochemistry based on the chosen cascade direction. In this paper, we document the success of the cascade cyclization strategy in the form of the first enantioselective total synthesis of (−)-β-hydrastine.
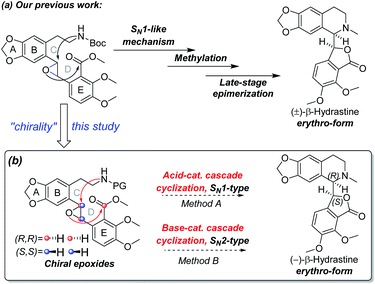 |
| | Scheme 2 (a) Our previous work to the total synthesis of (±)-β-hydrastine. (b) Our proposed synthetic strategy to the total synthesis of (−)-β-hydrastine in this study. | |
Results and discussion
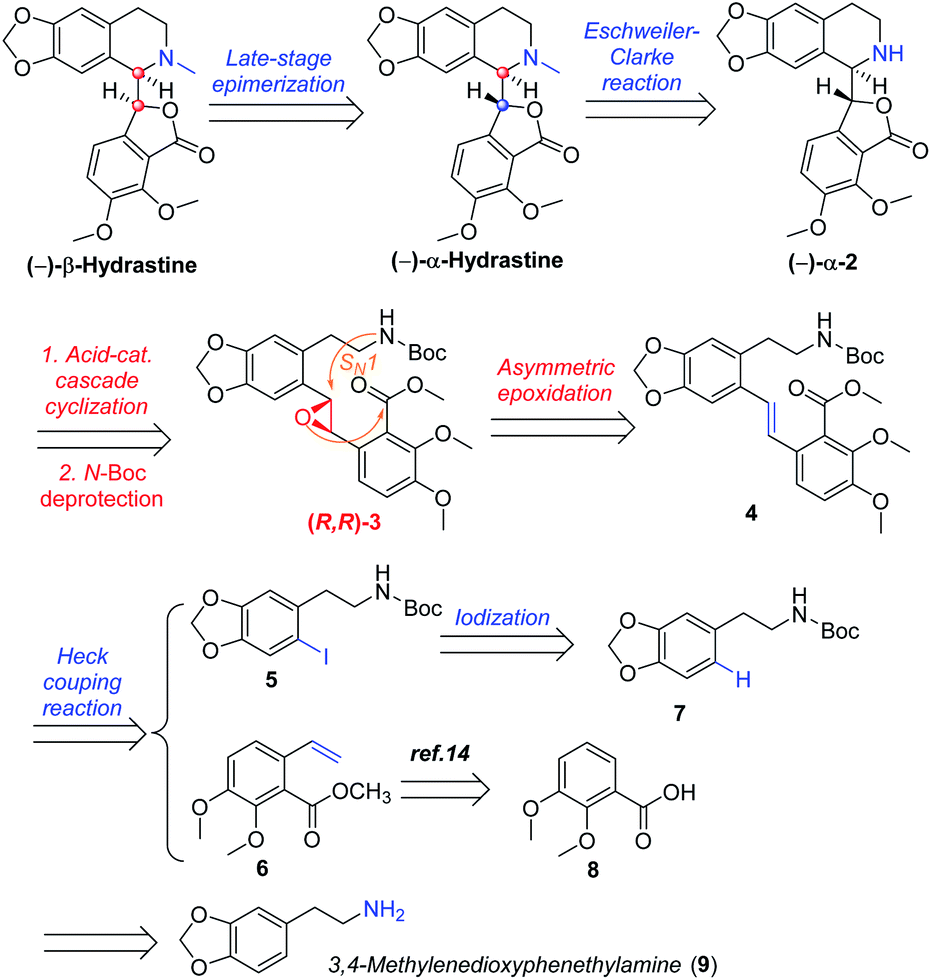
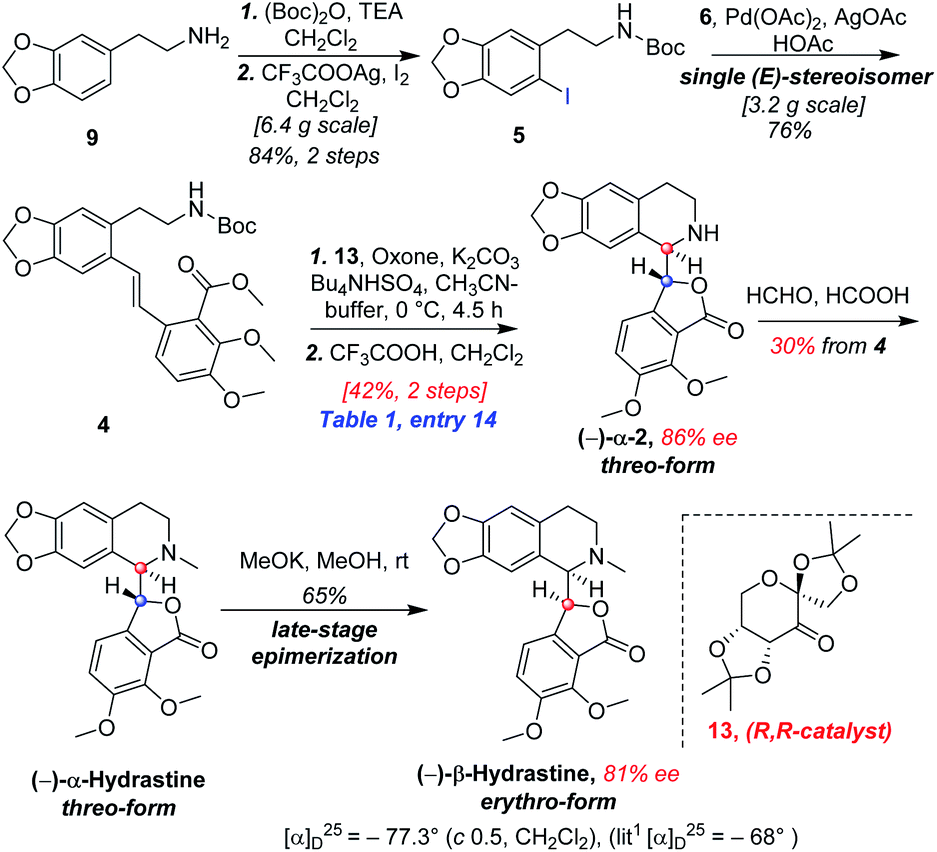
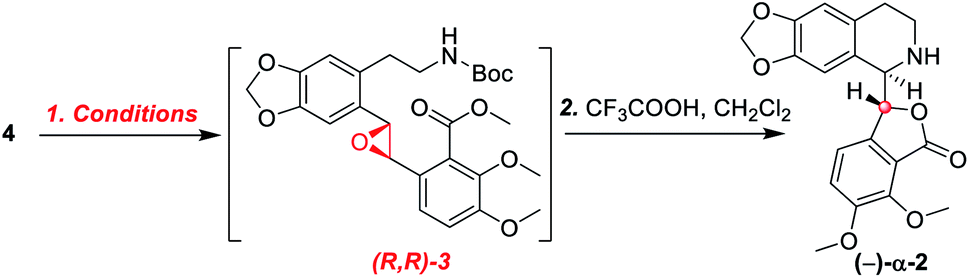
In designing a synthetic route toward (−)-β-hydrastine, we employed the chiral epoxide in order to control both the reactivity and stereoselectivity and envisioned a crucial cascade cyclization transformation, namely, an acid-catalyzed cascade cyclization. Our retrosynthetic analysis of (−)-β-hydrastine was outlined in Scheme 3. The target natural compound (−)-β-hydrastine could be accessed from (−)-α-hydrastine via epimerization at C-3′, and (−)-α-hydrastine could be obtained from (−)-α-2 after methylation. Then, we speculated that the phthalide tetrahydroisoquinoline core of (−)-α-2 could be constructed by an acid-catalyzed epoxide ring-opening/transesterification cascade cyclization of the (R,R)-3, which could be prepared from (E)-stilbene 4 by asymmetric epoxidation with Shi ketone catalyst.15 Moreover, the stilbene 4 might be constructed from iodide 5 and substituted styrene 6 by a Pd-catalyzed Heck coupling reaction.14,17 The iodide 5 could be synthesized by iodization of N-protected phenethylamine 7,14 which could be obtained from commercially available vanillin (9). In addition, the styrene 6 could be derived from commercially available 2,3-dimethoxybenzoic acid (8).
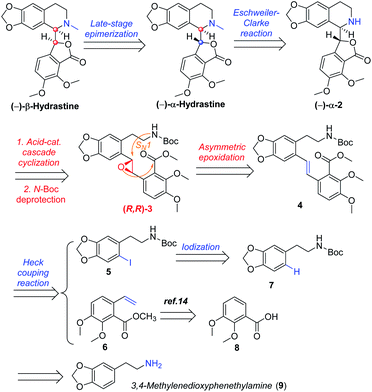 |
| | Scheme 3 Retrosynthetic analysis of (−)-β-hydrastine. | |
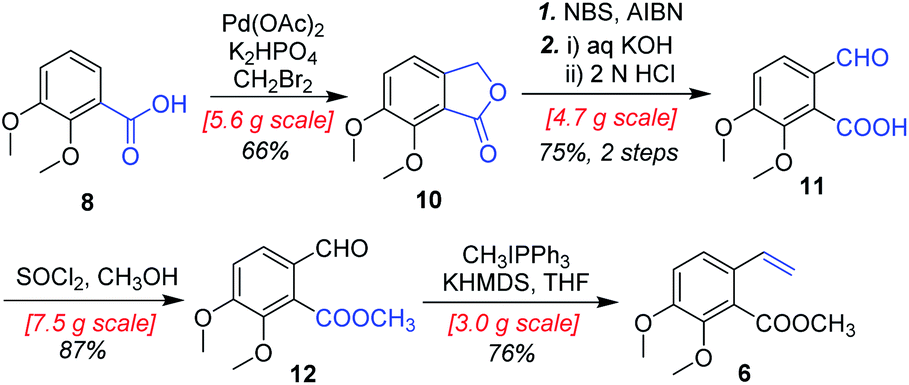
As shown in Scheme 4, our synthesis commenced with the preparation of known (E)-stilbene 4, which was readily prepared from 2,3-dimethoxybenzoic acid (8) in gram scale through a five-step sequence in our previous work14 (Scheme 4).
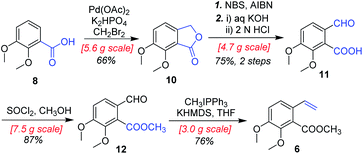 |
| | Scheme 4 Synthesis of substituted styrene 6. | |
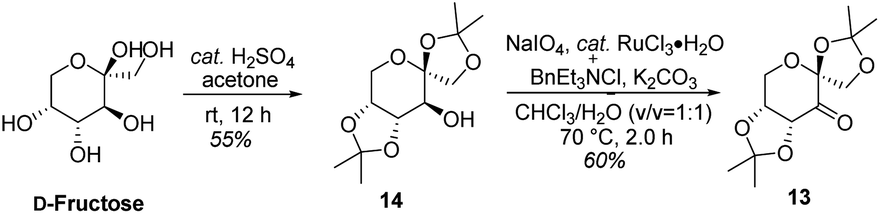
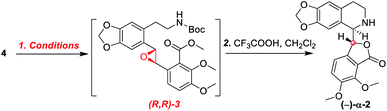
As shown in Scheme 5, we then turned our attention to synthesize the (E)-stilbene 4, the precursor for asymmetric epoxidation, which could be prepared in gram scale from commercially available 9 in three steps in our previously reported manner (Scheme 5).14 With substrate 4 in hand, our next goal was the efficient asymmetric epoxidation with Shi catalyst 13 (Scheme 6)15,16 to obtain (R,R)-3 as the key precursor for the proposed acid-catalyzed cascade cyclization.14 Various conditions were screened with (E)-stilbene 4 (Table 1). Initially, treating (E)-stilbene 4 with ketone 13 in the conditions of our previously reported achiral epoxidation (Table 1, entry 1),14 as well as further changing solvent system17 and increasing the equiv. of ketone 13 (Table 1, entries 2 and 3), failed to give (R,R)-3 (Table 1, entries 1–3). Unfortunately, more extensive investigation of the asymmetric epoxidation reaction using various equiv. of ketone 13, Oxone and K2CO3 in a three-solvent system15,18 revealed that no trace of desired epoxidation product (R,R)-3 was detected by NMR or LC-MS (Table 1, entries 4–8). In addition, although a trace of desired epoxidation product (R,R)-3 was detected when the reactions were conducted at −10 °C for 2 h, the outcome was not substantially improved (Table 1, entries 9–11). The all above-mentioned unsuccessful asymmetric epoxidation with Shi catalyst 13 might be attributed to the heterogeneous reactions. Considering the importance of the reaction solvents, a two-solvent system was researched according to the reported method (Table 1, entry 12),15 resulting in no improvement. However, using the increasing equiv. of ketone 13 (3.0 equiv.) led to the almost complete consuming of (E)-stilbene 4, which still produced a trace of desired (R,R)-3 for the reason that the epoxide (R,R)-3 was postulated to strongly favor the decomposition in the alkaline reaction environment at room temperature for 24 h (Table 1, entry 13). Accordingly, subsequent screening studies revealed that the yield of epoxide (R,R)-3 could be drastically improved by proper selection of equiv. of ketone 13, reaction temperatures and reaction times (Table 1, entries 14 and 15). Consequently, optimal yield (42% for two steps) and enantiomeric excess (ee, 86%) of (−)-α-2 were obtained via epoxide (R,R)-3 which was produced from (E)-stilbene 4 by an epoxidation reaction with 3.0 equiv. of ketone 13 at 0 °C for 4.5 h (Table 1, entry 14), and following CF3COOH-catalyzed cascade cyclization and N-Boc deprotection in one pot.14 Additionally, other methods of asymmetric epoxidation, using 30% H2O2 as oxidant19 (Table 1, entries 16 and 17), did not improve yield and enantiomeric excess. With highly enantioselective (−)-α-2 in hand, we were pleased to synthesize (−)-β-hydrastine [(−)-β-1] using already established conditions for two transformations.14 The N-methylation of (−)-α-2 occurred under classic Eschweiler–Clarke reaction conditions,14 thus yielding (−)-α-hydrastine [(−)-α-1]. Remarkably, without purification of (−)-α-2, (−)-α-hydrastine was prepared from 4 in the three-step yield of 30% (see Scheme 5). Finally, a late-stage epimerization of (−)-α-1 at C-3′ under MeOK/MeOH conditions furnished (−)-β-hydrastine [(−)-β-1] with 81% ee, which was in agreement with authentic reference standards for this natural product1 (see the ESI†).
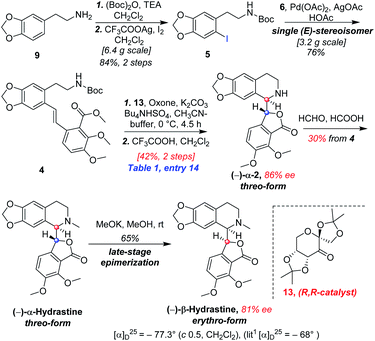 |
| | Scheme 5 Total synthesis of (−)-β-hydrastine in method A. | |
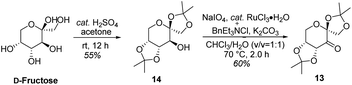 |
| | Scheme 6 Synthesis of (R,R)-catalyst 13. | |
Table 1 Optimization of asymmetric epoxidation of (E)-stilbene 4 for the acid-catalyzed cascade cyclization
|
| Entrya |
Ketone 13 (equiv.) |
Conditionsb |
Yieldc (%) |
eed (%) |
Substrate 4 (0.1 mmol). Condition A: Oxone (5.6 equiv.), NaHCO3 (18.9 equiv.), CH3CN–CH2Cl2–H2O (v/v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:5), 0 °C, 48 h. Condition B: Oxone (5.0 equiv.), NaHCO3 (15.5 equiv.), Bu4NHSO4 (5 mol%), CH3CN–aq. Na2EDTA (4 × 10−4 M) (v/v = 1.5:1), 0 °C, 2.0 h. Condition C: Oxone (2.02 equiv.), K2CO3 (4.04 equiv.), Bu4NHSO4 (5 mol%), CH3CN–DMM–0.05 M aq. Na2HPO4–0.05 M aq. KH2PO4 (pH = 7.0) (v/v/v = 1:2:1), 0 °C, 24 h. Condition D: Oxone (1.38 equiv.), K2CO3 (5.8 equiv.), Bu4NHSO4 (5 mol%), CH3CN–DMM–0.05 M Na2B4O7·10H2O of aq. Na2EDTA (4 × 10−4 M) solution (v/v/v = 1:2:2), 0 °C, 1.5 h. Condition E: Oxone (1.38 equiv.), K2CO3 (5.8 equiv.), Bu4NHSO4 (5 mol%), CH3CN–0.05 M Na2B4O7·10H2O of aq. Na2EDTA (4 × 10−4 M) solution (v/v = 3:2), 0 °C to rt, 24 h. Condition F: Oxone (4.6 equiv.), K2CO3 (18.6 equiv.), Bu4NHSO4 (5 mol%), CH3CN–0.05 M Na2B4O7·10H2O of aq. Na2EDTA (4 × 10−4 M) solution (v/v = 3:2, 25 mL), 0 °C, 4.5 h. Condition G: 30% H2O2 (30 equiv.), CH3CN–1.0 M K2CO3 in 4 × 10−4 M of EDTA (v/v = 2:1), 0 °C, 36 h. Condition H: 30% H2O2 (30 equiv.), CH3CN–EtOH–CH2Cl2 (v/v/v = 1:1:2), 2.0 M K2CO3 in 4 × 10−4 M of EDTA, 0 °C, 36 h. Isolated yield of (−)-α-2. The enantiomeric excess was determined by chiral HPLC (Chiralpak IH). The epoxide (R,R)-3 was not detected by TLC and the starting material 4 was recovered. Oxone (1.38 equiv.) and K2CO3 (5.8 equiv.) were used. The reactions were stopped after 2 h for −10 °C. Most of the starting material 4 was recovered. Substrate 4 (1.0 mmol). Most of the epoxide (R,R)-3 was decomposed. Substrate 4 (0.3 mmol). The reactions were stopped after 8 h for 0 °C. Substrate 4 (0.4 mmol). DMM = dimethoxymethane. :4:5), 0 °C, 48 h. Condition B: Oxone (5.0 equiv.), NaHCO3 (15.5 equiv.), Bu4NHSO4 (5 mol%), CH3CN–aq. Na2EDTA (4 × 10−4 M) (v/v = 1.5:1), 0 °C, 2.0 h. Condition C: Oxone (2.02 equiv.), K2CO3 (4.04 equiv.), Bu4NHSO4 (5 mol%), CH3CN–DMM–0.05 M aq. Na2HPO4–0.05 M aq. KH2PO4 (pH = 7.0) (v/v/v = 1:2:1), 0 °C, 24 h. Condition D: Oxone (1.38 equiv.), K2CO3 (5.8 equiv.), Bu4NHSO4 (5 mol%), CH3CN–DMM–0.05 M Na2B4O7·10H2O of aq. Na2EDTA (4 × 10−4 M) solution (v/v/v = 1:2:2), 0 °C, 1.5 h. Condition E: Oxone (1.38 equiv.), K2CO3 (5.8 equiv.), Bu4NHSO4 (5 mol%), CH3CN–0.05 M Na2B4O7·10H2O of aq. Na2EDTA (4 × 10−4 M) solution (v/v = 3:2), 0 °C to rt, 24 h. Condition F: Oxone (4.6 equiv.), K2CO3 (18.6 equiv.), Bu4NHSO4 (5 mol%), CH3CN–0.05 M Na2B4O7·10H2O of aq. Na2EDTA (4 × 10−4 M) solution (v/v = 3:2, 25 mL), 0 °C, 4.5 h. Condition G: 30% H2O2 (30 equiv.), CH3CN–1.0 M K2CO3 in 4 × 10−4 M of EDTA (v/v = 2:1), 0 °C, 36 h. Condition H: 30% H2O2 (30 equiv.), CH3CN–EtOH–CH2Cl2 (v/v/v = 1:1:2), 2.0 M K2CO3 in 4 × 10−4 M of EDTA, 0 °C, 36 h. Isolated yield of (−)-α-2. The enantiomeric excess was determined by chiral HPLC (Chiralpak IH). The epoxide (R,R)-3 was not detected by TLC and the starting material 4 was recovered. Oxone (1.38 equiv.) and K2CO3 (5.8 equiv.) were used. The reactions were stopped after 2 h for −10 °C. Most of the starting material 4 was recovered. Substrate 4 (1.0 mmol). Most of the epoxide (R,R)-3 was decomposed. Substrate 4 (0.3 mmol). The reactions were stopped after 8 h for 0 °C. Substrate 4 (0.4 mmol). DMM = dimethoxymethane. |
| 1 |
3.0 |
A |
NDe |
— |
| 2 |
3.0 |
B |
NDe |
— |
| 3 |
5.0 |
B |
NDe |
— |
| 4 |
0.3 |
C |
NDe |
— |
| 5 |
0.3 |
D |
NDe |
— |
| 6 |
1.6 |
Df |
NDe |
— |
| 7 |
2.0 |
Df |
NDe |
— |
| 8 |
3.0 |
Df |
NDe |
— |
| 9 |
3.0 |
Df,g |
Traceh |
— |
| 10 |
1.6 |
Df,g |
Traceh |
— |
| 11 |
1.6 |
Dg |
Traceh |
— |
| 12i |
0.3 |
E |
NDe |
— |
| 13 |
3.0 |
E |
Tracej |
— |
| 14k |
3.0 |
F |
42 |
86 |
| 15k |
1.0 |
Fl |
24 |
79 |
| 16m |
3.0 |
G |
25 |
58 |
| 17m |
3.0 |
H |
40 |
71 |
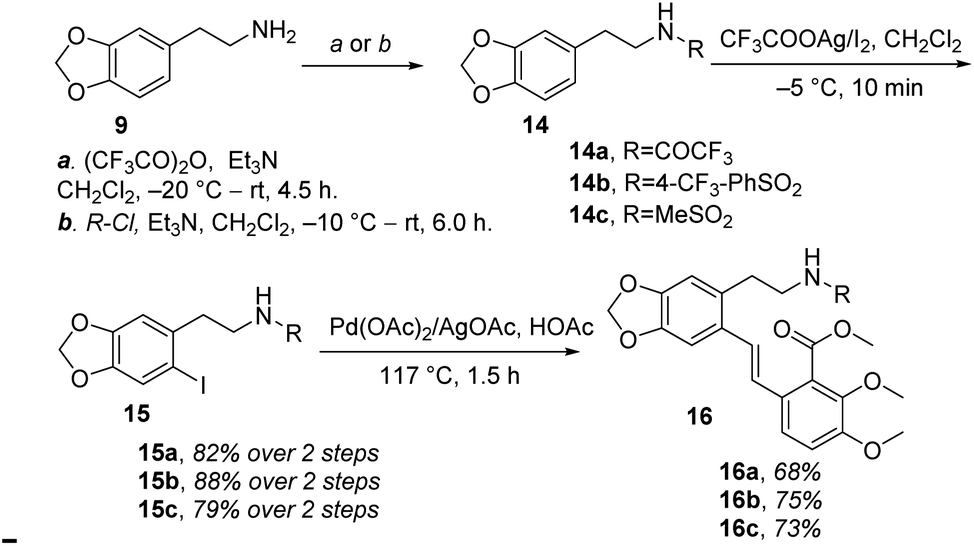
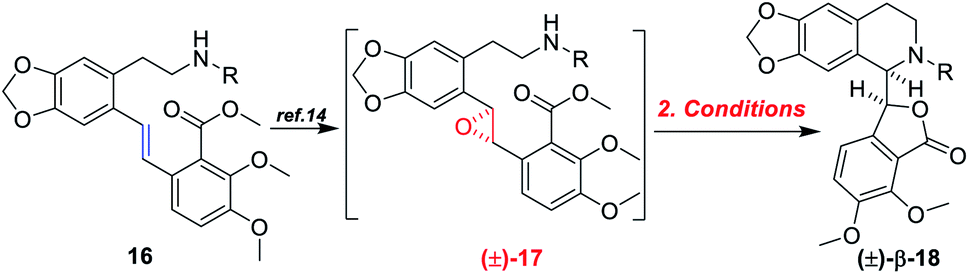
Therefore, (−)-β-hydrastine had been synthesized with a high enantiomeric excess (81% ee) by using CF3COOH-catalyzed cascade cyclization reaction and C-3′ epimerization under MeOK/MeOH conditions as the key steps (Scheme 5). Inspired by the ring-opening reaction of epoxides in basic conditions via an SN2-type mechanism,20 we also assumed that the core phthalide tetrahydroisoquinoline scaffold with the R and S configurations at C-1 and C-3′ could be enantioselectively structured based on a base-catalyzed epoxide ring-opening/transesterification cascade cyclization strategy in order to avoid the late-stage epimerization reaction at C-3′. Correspondingly, the chiral epoxide with (S,S) configurations was chosen as the substrate to direct the cascade reaction under basic conditions. To test the working hypothesis, initially, we prepared a series of racemic model substrates [(±)-17] through DMDO-mediated epoxidation14 from the corresponding (E)-stilbenes (16), which were subjected to base-catalyzed cascade cyclization model reactions. Similarly, the precursors 16 for epoxidation were prepared in three steps, namely, N-protection, I2/CF3COOAg-mediated iodination and following Pd-catalyzed Heck coupling reaction (Scheme 7).
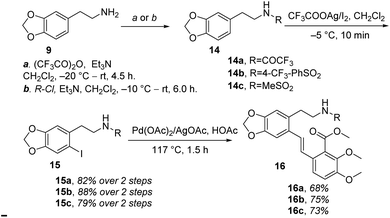 |
| | Scheme 7 Syntheses of (E)-stilbenes 16a–c. | |
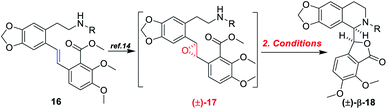
With the (E)-stilbenes 16a–c in hand, we focused our attention on the base-catalyzed cascade cyclization model reactions. Initially, we chosen trifluoroacetyl21 as the N-protection group to examine the reactivity in the presence 60% NaH21 in 1,4-dioxane, unfortunately, leading to no desired cyclization product (±)-β-18a (Table 2, entry 1). After that, we further optimized the reaction conditions of the model reaction, and the results were summarized in Table 2. Using (±)-17a as the model substrate, firstly, extensive screening of different bases (KHMDS, LDA, and n-BuLi), solvents (THF and 1,4-dioxane), and reaction temperatures resulted in no desired (±)-β-18a or the decomposition of (±)-17a (Table 2, entries 2–7). In our opinion, it was probably due to that the electron-withdrawing ability of trifluoroacetyl group was not strong enough to fulfill the deprotonation process of amino in the presence of strong bases, and then failing to facilitate desired cascade cyclization reaction. In light of the above reason, the 4-(trifluoromethyl)benzenesulfonyl, a much stronger electron-withdrawing group, was used and the corresponding (±)-17b was subjected to the model reactions (Table 2, entries 8–10). To our delight, the desired cyclization product (±)-β-18b was generated in 11% yield when the reaction was carried out by action of 1.0 M KHDMS in THF at 0–65 °C for 12 h (Table 2, entry 8). Encouraged by this result, we further optimized the reaction conditions by changing reaction solvent and temperature. As displayed in Table 2, the reaction solvent had little effect on the yield of (±)-β-18b (Table 2, entry 9). A decrease in the reaction time from 12 h to 5 h resulted in a three-time increase in yield for the same reaction solvent and temperature (Table 2, entry 10). In addition, considering that the steric hindrance of 4-(trifluoromethyl)benzenesulfonyl might lead to lower yields, a mesyl-protected model substrate (±)-17c was prepared for further investigation of model reactions (Table 2, entries 11–17). The results revealed that no (±)-β-18c was delivered from (±)-17c in both KHMDS and NaHMDS conditions (Table 2, entries 11–13). However, the model substrate (±)-17c was conducted to 60% NaH in 1,4-dioxane, resulting in the desired (±)-β-18c in 15% yield (Table 2, entry 14). Moreover, attempts were made to improve the yields by extending the reaction time, lowering the reaction temperature and decreasing the equiv. of NaH, but the improvement was not significant (Table 2, entries 16–17). Finally, the optimal conditions were determined to be the model substrate (±)-17b in THF at 0–65 °C for 5 h in the presence of KHMDS (1.5 equiv.) with stirring (Table 2, entry 10).
Table 2 Optimization of racemic model substrates [(±)-17] for the base-catalyzed cascade cyclization
|
| Entrya |
17 |
Base (equiv.) |
Solvent |
Temp. (°C) |
T (h) |
Yieldb (%) |
| The substrate 16 (0.1 mmol) was dissolved in solvent and then the base was added in dropwise. Isolated yield of (±)-β-18 in two steps. Most of the epoxide (±)-17 was decomposed. The (±)-β-18 was not detected by TLC and the epoxide (±)-17 was recovered. NaHMDS = sodium bis(trimethylsilyl)amide. KHMDS = potassium bis(trimethylsilyl)amide. LDA = lithium diisopropylamide. |
| 1 |
17a |
60% NaH (2.5) |
1,4-Dioxane |
0–65 |
12 |
—c |
| 2 |
17a |
KHMDS (1.2) |
1,4-Dioxane |
0–65 |
12 |
NDd |
| 3 |
17a |
KHMDS (1.2) |
1,4-Dioxane |
0–84 |
12 |
—c |
| 4 |
17a |
KHMDS (1.2) |
Dry THF |
0–65 |
12 |
NDd |
| 5 |
17a |
KHMDS (1.2) |
Dry THF |
0 to rt |
12 |
NDd |
| 6 |
17a |
LDA (1.2) |
Dry THF |
0–65 |
12 |
—c |
| 7 |
17a |
n-BuLi (1.2) |
Dry THF |
−78 to rt |
12 |
—c |
| 8 |
17b |
KHMDS (1.2) |
Dry THF |
0–65 |
12 |
11 |
| 9 |
17b |
KHMDS (1.5) |
1,4-Dioxane |
0–65 |
12 |
10 |
| 10 |
17b |
KHMDS (1.5) |
Dry THF |
0–65 |
5.0 |
30 |
| 11 |
17c |
KHMDS (1.5) |
Dry THF |
0–65 |
12 |
NDd |
| 12 |
17c |
NaHMDS (1.5) |
Dry THF |
0–65 |
12 |
NDd |
| 13 |
17c |
KHMDS (1.5) |
1,4-Dioxane |
0–84 |
12 |
—c |
| 14 |
17c |
60% NaH (2.5) |
1,4-Dioxane |
0–65 |
12 |
15 |
| 15 |
17c |
60% NaH (2.5) |
1,4-Dioxane |
0–65 |
24 |
10 |
| 16 |
17c |
60% NaH (2.5) |
1,4-Dioxane |
0–60 |
12 |
16 |
| 17 |
17c |
60% NaH (1.5) |
1,4-Dioxane |
0–60 |
5.0 |
19 |
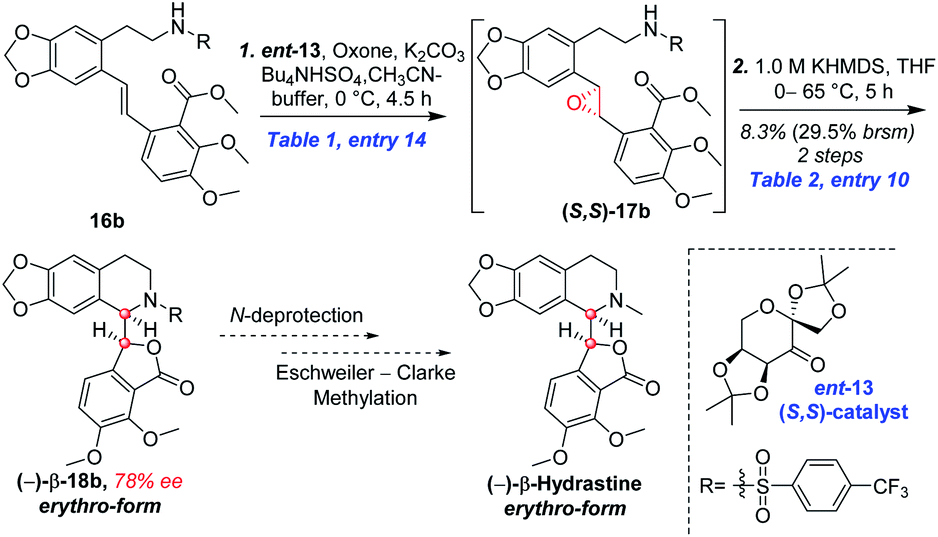
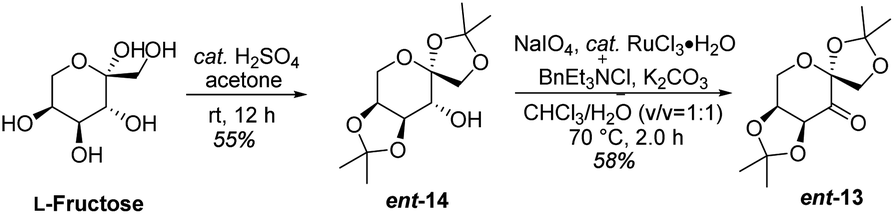
Based on the abovementioned results, the (E)-stilbene 16b was smoothly transformed to phthalide-tetrahydroisoquinoline-containing scaffold (−)-β-18b in 8% (30% brsm) yield over two steps with the enantiomeric excess (ee) of 78% (Scheme 8), where asymmetric epoxidation of 16b with Shi catalyst ent-13 (ref. 15 and 16) (Scheme 9) delivered (S,S)-17b, and the resulting epoxide underwent KHMDS-catalyzed cascade cyclization to give corresponding (−)-β-18b. After that, the total synthesis of (−)-β-hydrastine would be completed through the N-deprotection and the known methylation14 reaction. Regarding to the late-stage N-deprotection of (−)-β-18b, several conditions, such as Mg/MeOH,22 SmI2/THF,23 Na-naphthalide/THF,24 had been screened, but failing to afford (−)-β-2 (see the ESI†) and further studies on the late-stage N-deprotection are still in progress.
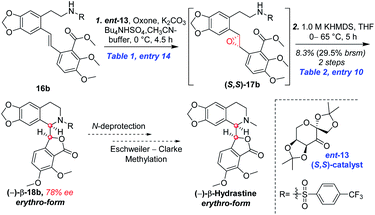 |
| | Scheme 8 Synthesis of the phthalide tetrahydroisoquinoline core of (−)-β-hydrastine in method B. | |
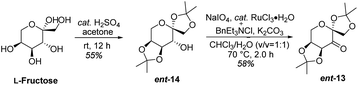 |
| | Scheme 9 Synthesis of (S,S)-catalyst ent-13. | |
Conclusions
In conclusion, we have completed an efficient and stereocontrolled approach to total synthesis of the phthalide tetrahydroisoquinoline alkaloid (−)-β-hydrastine [(−)-β-1]. The key elements include a high enantioselective epoxidation using the Shi ketone catalyst and an intramolecular acid-catalyzed cascade cyclization in one pot, and a late-stage epimerization. Furthermore, it should be noted that the facile and enantioselective construction of the core phthalide tetrahydroisoquinoline scaffold was made possible by the CF3COOH-catalyzed or KHMDS-catalyzed epoxide ring-opening cascade cyclization from chiral epoxide under very mild conditions. In this reaction, the phthalide tetrahydroisoquinoline scaffold was enantioselectively constructed with one C–N bond and one C–C bond formed simultaneously. Efforts toward the asymmetric total syntheses of other phthalide tetrahydroisoquinoline alkaloids, such as (−)-α-noscapine, and the structure-related alkaloids, are currently ongoing in our laboratory. To conclude, the chemistry described herein would serve as an efficient alternative strategy to synthesize biologically active phthalide tetrahydroisoquinoline alkaloids and their derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the Program for Innovative Research Team of the Ministry of Education and the Program for Liaoning Innovative Research Team in University.
References
- G. Blasko, D. J. Gula and M. Shamma, J. Nat. Prod., 1982, 45, 105–122 CrossRef CAS.
-
(a) S. Y. Yin, Y. M. Kim, J. J. Lee, C. M. Jin, Y. J. Yang, S.-K. Park, S. K. Yoo and M. K. Lee, Biol. Pharm. Bull., 2007, 30, 1547–1550 CrossRef CAS PubMed;
(b) S. Y. Yin, Y. M. Kim, J. J. Lee, C. M. Jin, Y. J. Yang, J. J. Ma, M. H. Kang, M. Kai and M. K. Lee, Neuropharmacology, 2004, 47, 1045–1052 CrossRef CAS PubMed;
(c) M. K. Lee, Y. H. Zhang, J. S. Shin and S. S. Lee, Med. Sci. Res., 1997, 25, 619–620 CAS.
-
(a) R. N. Jha, M. B. Pandey, A. K. Singh, S. Singh and V. P. Singh, Nat. Prod. Res., 2009, 23, 250–255 CrossRef CAS PubMed;
(b) M. Goel, U. P. Singh, R. N. Jha, V. B. Pandey and M. B. Pandey, Folia Microbiol., 2003, 48, 363–368 CrossRef CAS PubMed.
- K. Goswami, V. Yadava, V. Padmanabhan and S. Banerjee, Acta Crystallogr., 1993, 49, 504–506 CrossRef PubMed.
- J. Huang and G. A. R. Johnston, Br. J. Pharmacol., 1990, 99, 727–730 CrossRef CAS PubMed.
- B. Guo, X. Li, S. Song, M. Chen, M. Cheng, D. Zhao and F. Li, Oncol. Rep., 2016, 35, 2246–2256 CrossRef CAS PubMed.
-
(a) M. Shamma and V. S. Georgiev, Tetrahedron Lett., 1974, 15, 2339–2342 CrossRef;
(b) K. Iwasa, M. Kamigauchi, M. Sugiura and N. Takao, J. Nat. Prod., 1987, 50, 1083–1088 CrossRef CAS.
- E. Hope, F. L. Pyman, F. G. P. Remfry and R. Robinson, J. Chem. Soc., 1931, 236–247 RSC.
- R. D. Haworth, A. R. Pinder and R. Robinson, Nature, 1950, 165, 529 CrossRef CAS.
-
(a) J. L. Moniot and M. Shamma, J. Org. Chem., 1979, 44, 4337–4342 CrossRef CAS;
(b) J. L. Moniot and M. Shamma, J. Am. Chem. Soc., 1976, 98, 6714–6715 CrossRef CAS PubMed.
-
(a) T. Shono, Y. Usui and H. Hamaguchi, Tetrahedron Lett., 1980, 21, 1351–1354 CrossRef CAS;
(b) T. Shono, H. Hamaguchi, M. Sasaki, S. Fujita and K. Nagami, J. Org. Chem., 1983, 48, 1621–1628 CrossRef CAS;
(c) K. Orito, M. Miyazawa, R. Kanbayashi, M. Tokuda and H. Suginome, J. Org. Chem., 1999, 64, 6583–6596 CrossRef CAS PubMed.
- L. Na, D. Zhao, S. Song, L. Sun and M. Cheng, Acta Chim. Sin., 2011, 69, 356–361 CAS.
- Z. Varga, G. Blasko, G. Dornyei and C. Szantay, Acta Chim. Hung., 1991, 128, 831–837 CAS.
- J. Li, Y. Liu, X. Song, T. Wu, J. Meng, Y. Zheng, Q. Qin, D. Zhao and M. Cheng, Org. Lett., 2019, 21, 7149–7153 CrossRef CAS PubMed.
-
(a) Z.-X. Wang, Y. Tu, M. Frohn, J.-R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS;
(b) Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806–9807 CrossRef CAS.
-
(a) J. Kang, G. J. Lim, S. K. Yoon and M. Y. Kim, J. Org. Chem., 1995, 60, 564–577 CrossRef CAS;
(b) S. Mio, Y. Kumagawa and S. Sugai, Tetrahedron, 1991, 47, 2133–2144 CrossRef CAS.
- D. Shabashov and O. Daugulis, Org. Lett., 2006, 8, 4947–4949 CrossRef CAS PubMed.
- B. Wang, X.-Y. Wu, O. A. Wong, B. Nettles, M.-X. Zhao, D. Chen and Y. Shi, J. Org. Chem., 2009, 74, 3986–3989 CrossRef CAS PubMed.
-
(a) L. Shu and Y. Shi, Tetrahedron, 2001, 57, 5213–5218 CrossRef CAS;
(b) L. Shu and Y. Shi, Tetrahedron Lett., 1999, 40, 8721–8724 CrossRef CAS.
-
(a) T. Angelini, F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Tetrahedron Lett., 2010, 51, 1566–1569 CrossRef CAS;
(b) N. Oguni, Y. Miyagi and K. Itoh, Tetrahedron Lett., 1998, 39, 9023–9026 CrossRef CAS;
(c) D. L. Fox, A. A. Robinson, J. B. Frank and R. N. Salvatore, Tetrahedron Lett., 2003, 44, 7579–7582 CrossRef CAS.
- D. Knight, S. L. E. Overman and G. Pairaudeau, J. Am. Chem. Soc., 1995, 117, 5776–5788 CrossRef.
-
(a) C. Qi, F. Hasenmaile, V. Gandon and D. Lebœuf, ACS Catal., 2018, 8, 1734–1739 CrossRef CAS;
(b) M. Nagamoto, T. Yanagi, T. Nishimura and H. Yorimitsu, Org. Lett., 2016, 18, 4474–4477 CrossRef CAS PubMed.
- V. Pace, J. P. Rae and D. J. Procter, Org. Lett., 2014, 16, 476–479 CrossRef CAS PubMed.
- N. Chakor, S. Dallavalle, L. Scaglioni and L. Merlini, Eur. J. Org. Chem., 2010, 6217–6223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, NMR spectra and HPLC chromatograms for products. See DOI: 10.1039/d0ra03038d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Dongmei Zhao
abc,
Dongmei Zhao