
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel 1′-homo-N-2′-deoxy-α-nucleosides: synthesis, characterization and biological activity†
Alejandro Carneroa,
Virginia Martín-Nievesa,
Yogesh S. Sanghvib,
Olivia O. Russelc,
Leda Bassitc,
Raymond F. Schinazic,
Susana Fernández *a and
Miguel Ferrero*a
*a and
Miguel Ferrero*a
aDepartamento de Química Orgánica e Inorgánica, Universidad de Oviedo, 33006-Oviedo, Asturias, Spain. E-mail: mferrero@uniovi.es; fernandezgsusana@uniovi.es
bRasayan Inc., 2802 Crystal Ridge Road, Encinitas, CA 92024-6615, USA
cCenter for AIDS Research, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30322, USA
First published on 21st April 2020
Abstract
For the first time, a series of novel 1′-homo-N-2′-deoxy-α-nucleosides containing natural nucleobases as well as 5-fluoro and 5-iodopyrimidine analogs have been synthesized in an efficient manner. Additionally, a high yield protocol for the assembly of a dimeric scaffold containing two sugar moieties linked to the N-1 and N-3 positions of a single pyrimidine base has been accomplished. The structures of the novel homonucleosides were established by a single crystal X-ray structure of 1′-homo-N-2′-deoxy-α-adenosine and NMR studies. The biological activity of these 1′-homo-N-2′-deoxy-α-nucleosides as antiviral (HIV-1 and HBV) and cytotoxic studies was measured in multiple cell systems. The unique structure and easy accessibility of these compounds may allow their use in the design of new nucleoside analogs with potential biological activity and as a scaffold for combinatorial chemistry.
1. Introduction
Nucleosides are building-blocks of nucleic acids, where these molecules play a key role in the genetic process of all living organisms. Natural nucleosides offer a unique framework for the design of novel therapeutic drugs due to their central function in biological processes. As a result, over thirty chemically modified nucleosides and their analogs are on the market for the treatment of cancer and viral infections.1 Undoubtedly nucleoside scaffolds have been an excellent starting point for introducing chemical modifications leading to the discovery of biologically functional molecules. In nature, nucleosides appear mainly as β-anomers and rarely as the α-anomers. More recently the syntheses of α-anomeric nucleosides have attracted wide attention due to their unique properties and potential applications.2 Additionally, the position of the glycosidic bond could be altered by inserting another carbon atom to create 1′-homo-N-nucleoside structures. This structural motif imparts several unique features such as the absence of an anomeric effect, enzymatic and chemical stability of the glycosidic bond, conformational flexibility, extra lipophilicity and increased distance between the 5′-hydroxyl group and the base.3 In our on-going quest for the design and synthesis of novel nucleoside analogs, we embarked on the synthesis of less common modification 1′-homo-N-2′-deoxy-α-nucleosides where two features are incorporated in a single molecule. To the best of our knowledge, this is the first report on the synthesis of 1′-homo-N-2′-deoxy-α-nucleosides containing all four natural bases in an efficient manner. Additionally, herein we describe the synthesis of a bifunctional scaffold where two sugar units were linked to a single pyrimidine base.Considering the importance of nucleoside analogs as bioactive motif, we reasoned that new methods for the synthesis of 1′-homo-N-2′-deoxy-α-nucleosides are warranted. Hitherto, two major methods were used to prepare 1′-homo-N-nucleosides: (i) introduction of the nucleobase by nucleophilic displacement of a leaving group at C-1′ exocyclic methyl group of the pentofuranose ring; and (ii) de novo construction of the nucleobase by condensation reactions from methylamino derivatives at C-1′.4
Following these routes, 1′-homo-N-bicyclic carbonucleosides 1 (Fig. 1),5 1′-homo-N-bicyclico-[2.2.1]heptane nucleosides 2,6 emissive 1′-homo-N-nucleosides 3,7 and 1′-homo-N-locked nucleoside 48 were synthesized. Also, α-nucleosides analogs such as arabinonucleosides 5 or 2′-deoxy-α-nucleosides 6 are easily formed due to reduced steric hindrance compared with β-ribonucleosides.2,9 Additionally, α-nucleosides are used to prepare α-oligonucleotides which leads to more stable duplexes.10
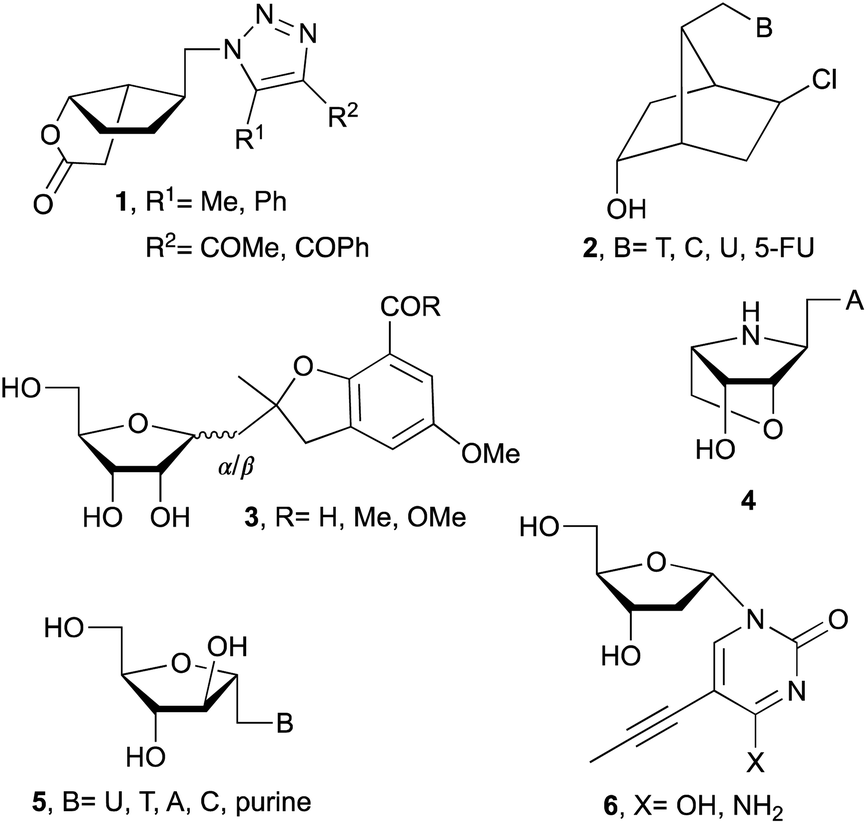 | ||
| Fig. 1 Structures of 1′-homo-N-nucleosides and/or α-nucleosides. | ||
Related with the research described in this manuscript, β-ribo11 and α-ribo12 1′-homo-N-nucleosides (7 and 8, respectively) have been prepared with variable success due to the use of multistep process and undesired side reactions. Whereas, fewer examples are available related to the synthesis of 1′-homo-N-2′-deoxynucleosides. For example, β-deoxy derivatives (9) were described by Saladino and Crucianelli,13a Doboszewski,13b,c and Herdewijn.13d In relation to the closest work described here, that is, the synthesis of α-deoxy 1′-homo-N-2′-nucleosides only Beaucage and co-workers14 have reported the synthesis with one nucleobase, thymine (10c).
Our pursuit for the design of novel nucleosides and the development of their synthesis protocols,8,15 here we wish to report the synthesis of 1′-homo-N-2′-deoxy-α-nucleosides 10 with both purine and pyrimidine nucleobases. Installation of bioactive 5-fluoro and 5-iodo uracil derivatives is also demonstrated to access new structures and test their biological activities. Eight modified nucleosides reported herein were tested for their biological activity as antiviral against HIV-1 and HBV. Their cytotoxicity assay is also included in this study.
2. Results and discussion
Synthesis of 1′-homo-N-2′-deoxy-α-nucleosides
Beaucage and coworkers reported on the synthesis of 1′-homo-α-thymidine where pyrimidine ring was constructed via an acyclic analog.14 This approach is not practical for the synthesis of multiple analogs containing both natural and unnatural nucleobases. Therefore, we wish to develop a new synthetic route which permits the synthesis of any 1′-homo-α-nucleoside analog from a single precursor such as sugar derivative 11. To prepare the 1′-homo-N-2′-deoxy-α-nucleoside derivatives 10, we envisioned a synthetic approach wherein the nucleobase was introduced by nucleophilic displacement of a leaving group in sugar precursor 11, readily obtained from the α-cyano sugar derivative 12 (Fig. 2 and 3).16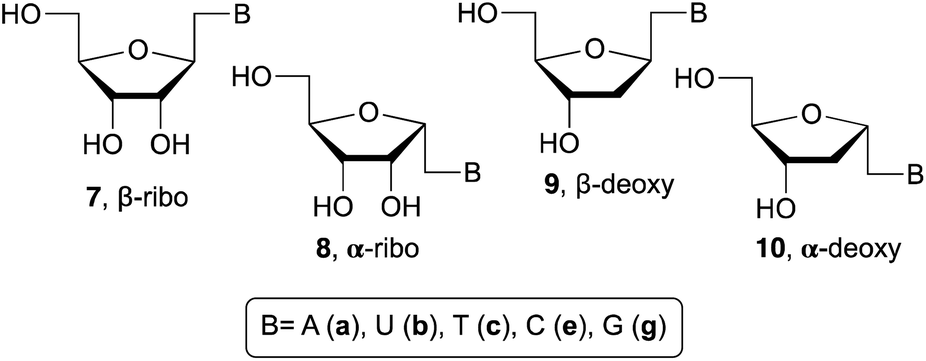 | ||
| Fig. 2 Structures of α/β-ribo and α/β-2′-deoxy 1′-homo-N-nucleosides. | ||
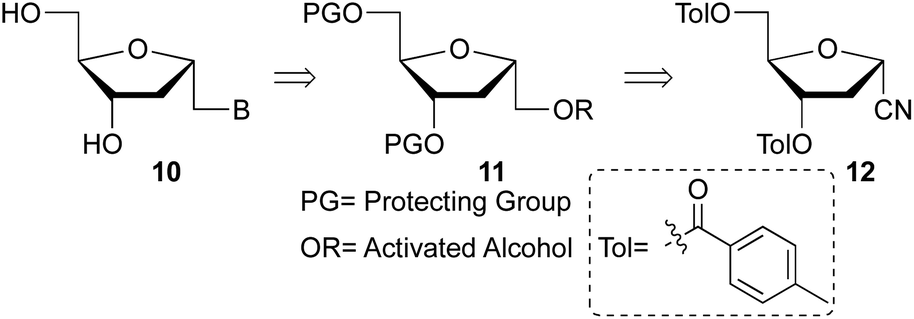 | ||
| Fig. 3 Retrosynthetic analysis to 1′-homo-N-2′-deoxy-α-nucleosides. | ||
Therefore, nitrile 12 was converted into the corresponding methyl ester by treatment with potassium hydroxide in MeOH/H2O. Under these conditions, removal of the toluoyl protecting groups is also observed, affording compound 13 in 75% isolated yield after chromatography (Scheme 1). One-pot deprotection of toluoyl groups, hydrolysis of nitrile and in situ esterification demonstrates a highly efficient protocol compared to the literature report for the corresponding β-anomer.16b
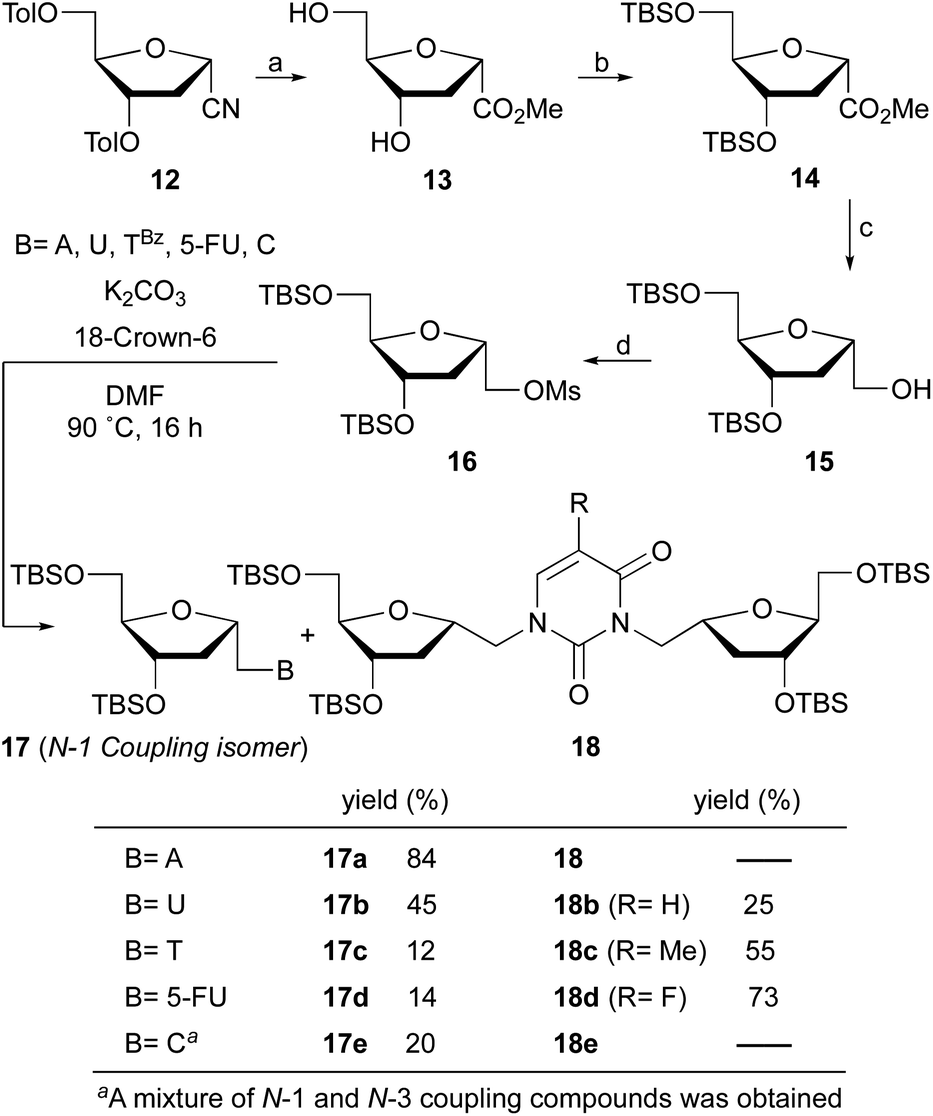 | ||
| Scheme 1 Reaction conditions: (a) KOH, MeOH/H2O, rt, 3 h (75%); (b) TBSCl, imidazole, CH2Cl2, 50 °C, 5 h (85%); (c) LiAlH4, THF, −45 °C, 30 min (90%); (d) MsCl, DMAP, Py, 0 °C to rt, 24 h (95%). | ||
Protection of the hydroxyl groups with tert-butyldimethylsilyl chloride (TBSCl) in the presence of imidazole gave silyl ether derivative 14. The reduction of ester 14 with lithium aluminum hydride in THF at −45 °C afforded after 30 min alcohol 15 in 90% yield. It is noteworthy that higher reaction temperatures during reduction leads to deprotection of the silyl groups and low temperature is critical to the successful outcome. Transformation of the alcohol into the mesylate 16 was achieved in excellent yield with methanesulfonyl chloride and catalytic DMAP in pyridine.
With mesylate 16 in hand, the stage was set for rapid synthesis of α-homonucleosides 10. Next, we opted for the base mediated displacement reaction using K2CO3 and 18-crown-6 in DMF. To our delight, the coupling of unprotected adenine base with mesylate 16 under these conditions at 90 °C furnished 1′-homo-N-2′-deoxy-α-adenosine (17a) in 84% yield after column chromatography purification. With the guanine base, the reaction of mesylate 16 under similar conditions furnished a complex mixture of products.
Under identical reaction conditions, uracil, a pyrimidine base when coupled with mesylate 16 furnished a mixture of two products. The expected 1′-homo-N-2′-deoxy-α-uridine (17b) was isolated in 45% yield. The second product was characterized as a dimer 18b, in which the uracil is linked with two deoxyribose moieties at the N-1 and N-3 positions of the base. The structure of dimer 18b was supported by spectral data.
Interestingly, the coupling of thymine base with mesylate 16 furnished two products where desired homonucleoside 17c was isolated in 12% yield and dimer 18c in 55% yield (Scheme 1). Similarly, the coupling of 5-fluorouracil base with mesylate 16 resulted in the formation of dimer 18d in 73% yield as a major product along with 17d in 14% yield. This observation is unprecedented where a dimer is obtained as a major product and offers a new synthetic pathway to the assembly of unique nucleoside scaffolds.
The synthesis of 1′-homo-N-2′-deoxy-α-cytidine was also complicated due to the formation of two products. Reaction of 16 with cytosine gave a mixture of N-1 (compound 17e) and N-3 (see Fig. 4b) coupled nucleosides. The structure of these products was established by homonuclear (COSY) and heteronuclear (HSQC, HMBC) correlations. In the 13C–1H HMBC experiment for 17e, a correlation cross-peak was observed between the H6 proton of the base and the methylene carbon at 6′-position. However, in the N-3 derivative this cross-peak does not exist.
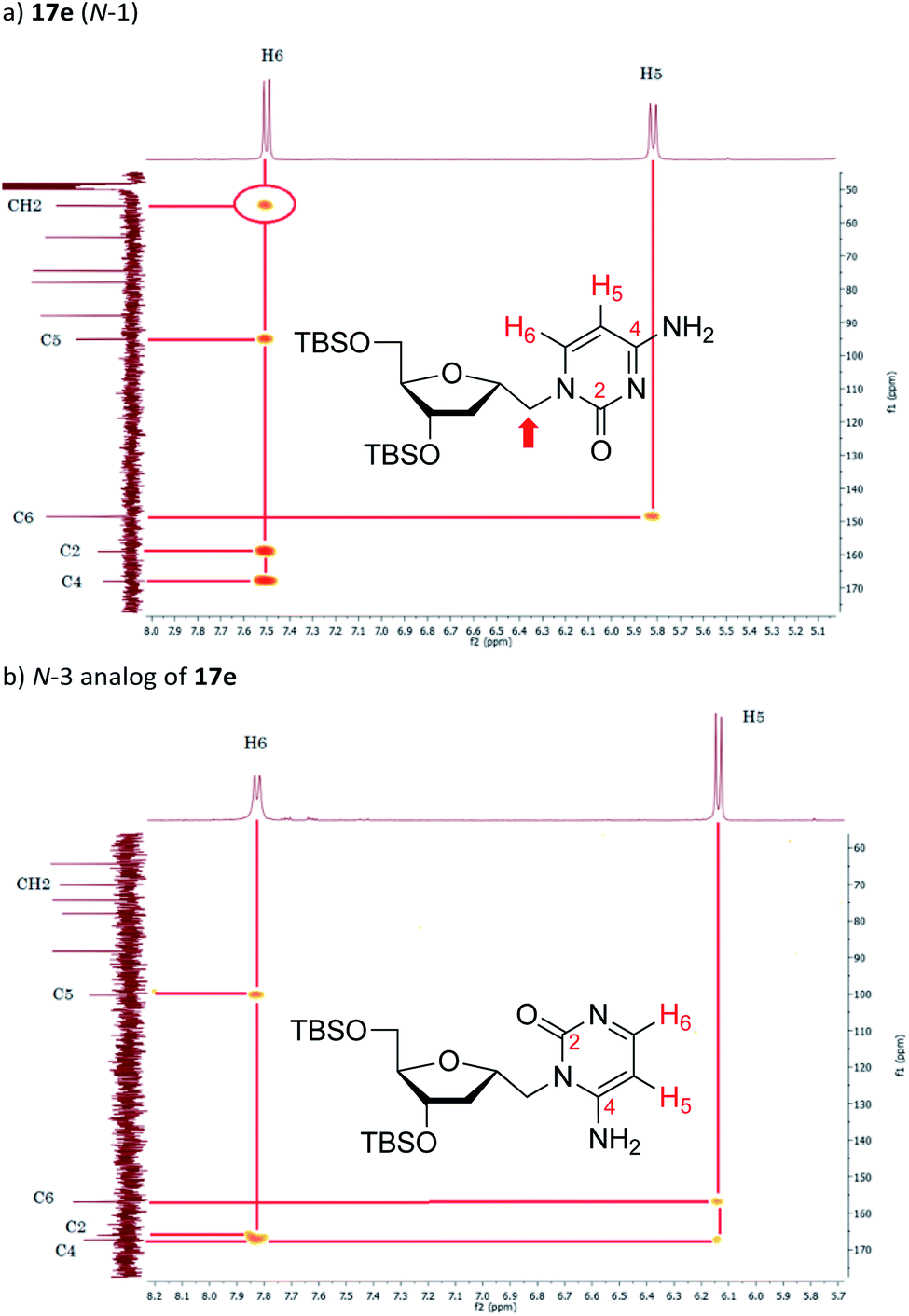 | ||
| Fig. 4 HMBC spectra enlargements of: (a) 17e and (b) its N-3 analog. | ||
In an attempt to improve the yield of the desired pyrimidine coupled products, other strategies were tested. Glycosylation under Vorbrüggen's conditions17 required acetate 19 (Scheme 2), which was easily prepared from 15 by treatment with acetic anhydride, Et3N, and DMAP in CH2Cl2. Reaction of 19 with silylated bases such as uracil or 5-fluorouracil or 5-iodouracil in the presence of trimethylsilyl triflate failed to provide the coupled product, while deprotection of the TBS groups was observed.
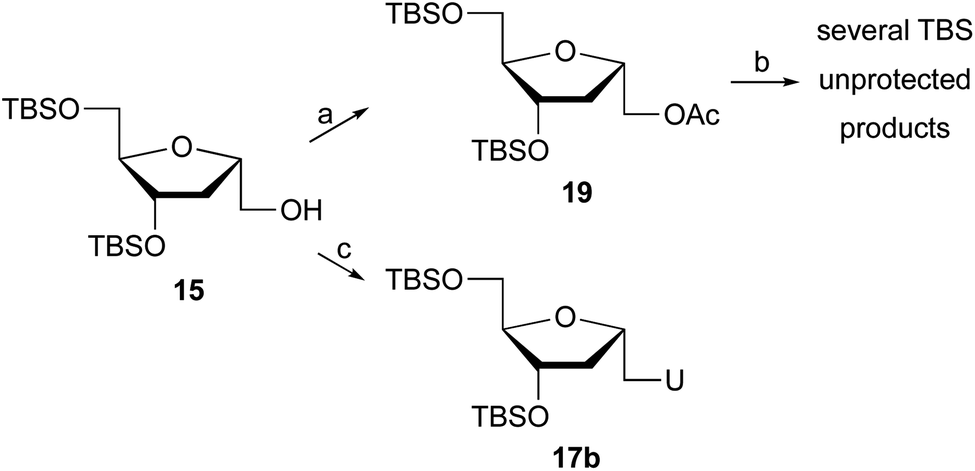 | ||
| Scheme 2 Reaction conditions: (a) Ac2O, Et3N, DMAP, CH2Cl2, rt, 2 h (90%); (b) (i) Base (U, 5-FU, 5-IU), BSA, MeCN, 80 °C, 1 h, (ii) TMSOTf, 0 °C to 80 °C; (c) Ph3P, DIAD, uracil, THF, −10 °C to 25 °C, 24 h (15%). | ||
The Mitsunobu reaction is an important technique in nucleoside synthesis for the direct coupling of alcohols with bases under mild conditions. Thus, treatment of 15 with uracil, Ph3P and DIAD generated 1′-homo-N-2′-deoxy-α-uridine (17b), but with only 15% yield. The low yield is due, in part, to the difficulty of isolating the product during chromatography, since several byproducts are obtained post Mitsunobu reaction.
Since alternative coupling protocol failed to improve the outcome of the reaction and the formation of desired product, we revisited the first protocol offering a viable option. This time, we replaced the mesylate with tosylate hoping that we might observe better coupling efficiency and overall yield.
The synthesis of tosylate 20 (Scheme 3) was straightforward by the reaction of alcohol 15 with p-toluensulfonyl chloride (TsCl) and catalytic DMAP in pyridine. Next, we performed the coupling reaction of 20 with the corresponding nucleobase in the presence of K2CO3 and 18-crown-6. The reaction of 20 with adenine proceeds with excellent yield, giving rise to α-homonucleoside 17a with a 91% isolated yield, slightly higher than when the reaction was carried out with mesylated 16. Significant improvement was observed when 20 reacted with uracil or cytosine, isolating 1′-homo-N-2′-deoxy-α-uridine (17b) and 1′-homo-N-2′-deoxy-α-cytidine (17e) with 65% and 69% yield, respectively, versus 45% and 20% yield obtained from the mesylate derivative. However, the coupling of the tosylated derivative 20 with thymine led exclusively to the dimer 18c. To the best of our knowledge, this is first report of dimer 18c synthesis in high yield. Next, in order to control the regioselectivity of the coupling with thymine, the N-3-position of the base was protected. The reaction was carried out according to the procedure described by Ludek and Meier.18 For that, thymine was allowed to react with benzoyl chloride in acetonitrile-pyridine to give N3-benzoylthymine. Reaction of 20 and N3-benzoylthymine under the reaction conditions indicated above afforded 1′-homo-N-2′-deoxy-α-thymidine (17c) in 30% yield, although the major product of the reaction was still the dimer 18c, probably due to the in situ deprotection of the base during the coupling. Nevertheless, the formation of the desired product 17c has been considerably improved when N3 protected thymine was utilized.
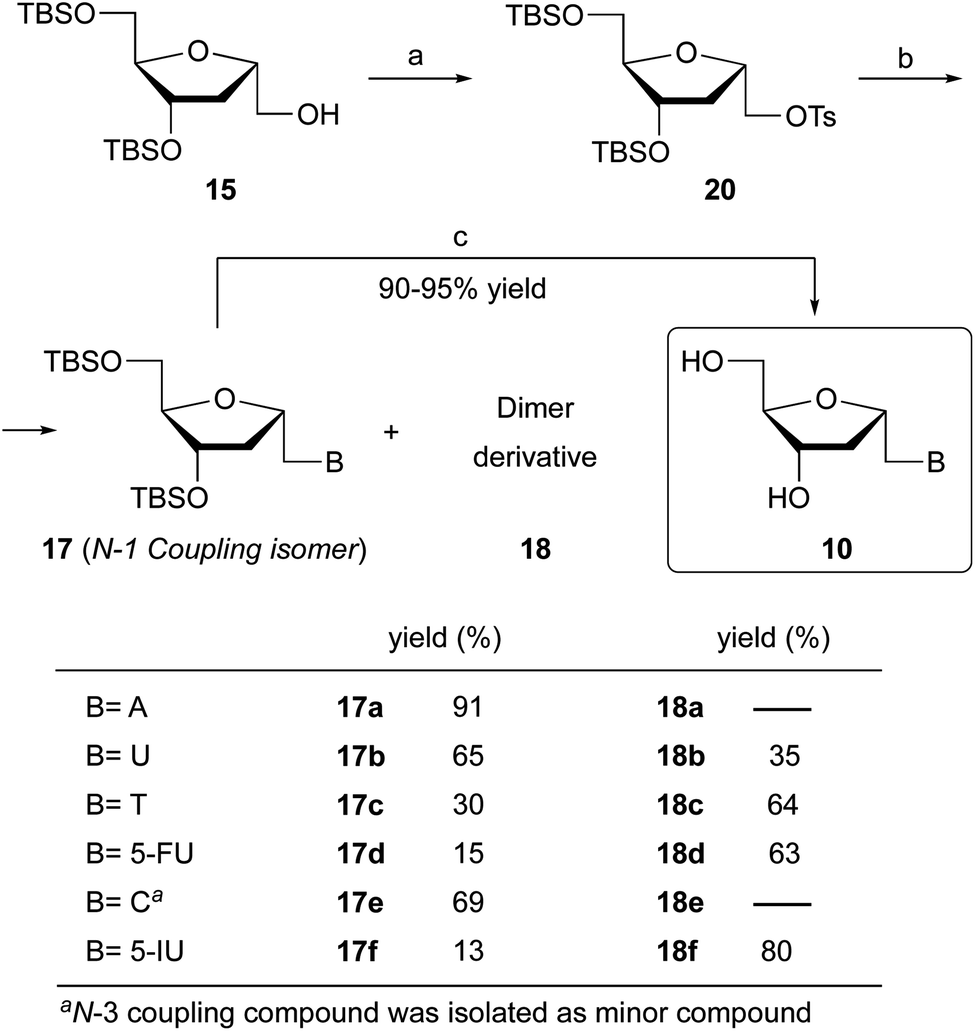 | ||
| Scheme 3 Reaction conditions: (a) TsCl, DMAP, Py, 0 °C to rt, 9 h (90%); (b) B = A, U, TBz, 5-FU, C, 5-IU, K2CO3, 18-crown-6, DMF, 90 °C, 7–8 h; (c) TBAF, THF, 25 °C, 2 h (90–95%). | ||
The coupling of 20 and 5-fluorouracil or 5-iodouracil was performed next. The major product was the dimer derivative but the N-1 coupled analog was also formed in 15% and 13% yield, respectively. In both cases, the N-3 protection of the base did not improve the yield of the N-1 substituted product. Multiple products were observed when 20 was coupled with the guanine base.
Finally, the treatment of compounds 17a–f with tetrabutylammonium fluoride (TBAF) in THF afforded the 1′-homo-N-2′-deoxy-α-nucleosides 10a–f in 90–95% yield. The structure of all derivatives was established by NMR homonuclear (COSY) and heteronuclear (HSQC, HMBC) correlations. Unambiguous confirmation of the structure of the synthesized compounds was obtained by a single crystal X-ray analysis of 10a (Fig. 5).
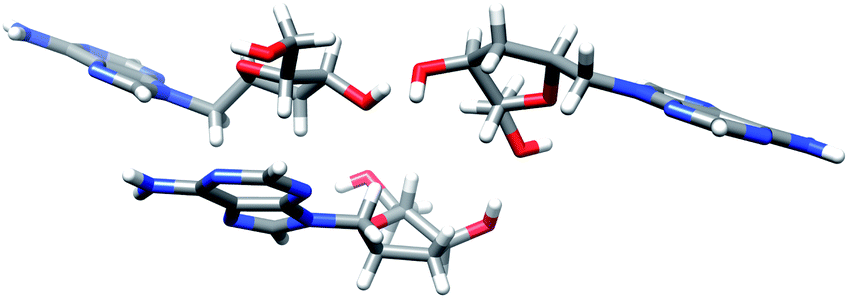 | ||
| Fig. 5 X-ray structure of 1′-homo-N-2′-deoxy-α-adenosine (10a). The structure shows three molecules per asymmetric unit. | ||
Single crystal X-ray data for compound 10a is given in Table 1. Homo-N-2′-deoxynucleoside analog 10a crystallized as colourless small crystals. Fig. 5 is a Chimera drawing,19 which corresponds to asymmetric unit X-ray diffraction studies of compound 10a.20 Note that the sugar conformation is North type with the 2′-exo and 3′-endo configurations. The structure shows three molecules per asymmetric unit linked by H-bonds, that is, six molecules per crystal.
| Formula | C11H15N5O3 |
| Molecular weight | 265.28 |
| Crystal system | Monoclinic |
| Space group | P21 |
| a, Å | 15.8711 (6) |
| b, Å | 8.04111 (18) |
| c, Å | 16.4342 (5) |
| Z | 6 |
| Final R | 0.035 |
| Reflections | 4865 |
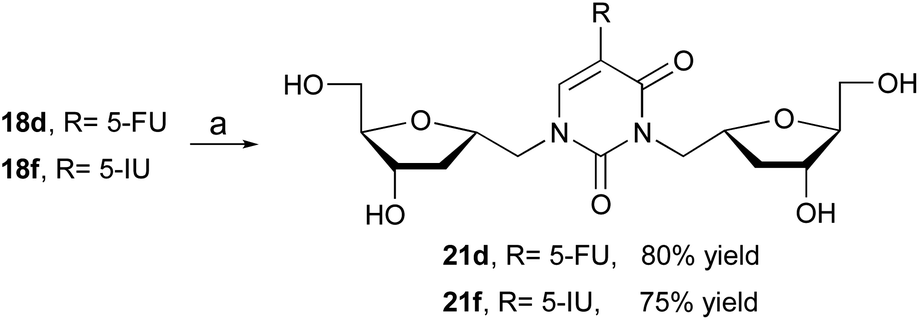
The formation of various dimers 18 is synthetically important because it offers an access to the assembly of bifunctional 2′-deoxynucleoside analogs. Literature search for reports on similar structures has been limited to two examples. First report by Prystas and Sorm21 describing the formation of N1,N3-diribofuranosyl derivative as a byproduct during ribosylation of 6-methyl-4-methoxy-2(1H)-pyrimidinone with a ribofuranosyl chloride derivative. In the second example, Zhang and co-workers22 reported the isolation of a similar derivative in the synthesis of isonucleosides. In order to further investigate the importance of this bifunctional nucleoside, we have evaluated the biological activity of dimeric compounds 21 (Scheme 4). For that, desilylation of 18d and 18f with TBAF in THF furnished the desired derivatives 21d and 21f in 80% and 75% yield, respectively.
 | ||
| Scheme 4 Reaction conditions: (a) TBAF, THF, 25 °C, 2 h. | ||
Biological evaluation
3. Experimental
General
TLC plates were visualized with 2.5% p-anisaldehyde, 3.5% sulfuric acid, and 1% acetic acid in ethanol. Pure α-cyano-sugar 12 is commercially available16a and TBz was prepared as previously reported.18 Unless otherwise specified column chromatography was performed over silica 60 Å (230–400 mesh).General procedure for desilylation of 17. Synthesis of 10
TBAF (6 eq., 1.0 M in THF) was added dropwise to a stirred solution of 17 (1 eq.) in anhydrous THF (0.1 M) at 0 °C. After 5 min, the ice bath was removed, and the reaction mixture was stirred at room temperature for 2 h. Next, solvents were evaporated, and the residue was purified by column chromatography (gradient eluent 15–20% MeOH/CH2Cl2 for 10a–c, 10e; gradient eluent 5–20% MeOH/CH2Cl2 for 10d and 10f) to afford 10a–f in 90–95% yield.1,2-Dideoxy-1α-(methoxycarbonyl)-D-ribofuranose (13)
To a solution of 12 (500 mg, 1.3 mmol) in MeOH (15 mL) at room temperature were added 3.1 mL of a 2.5 M KOH solution (25% MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O). The progress of the reaction was monitored by TLC (10% MeOH/CH2Cl2) until no further reaction was apparent (3 h). Next, the mixture was neutralized with DOWEX 50WX8 hydrogen form (50–100 mesh). After removal of the resin by filtration, the solution was evaporated, and the residue was purified by column chromatography (5% MeOH/CH2Cl2) to afford 13 as a yellowish oil in 75% yield. Rf: 0.22 (5% MeOH/CH2Cl2); 1H NMR (300.13 MHz, MeOH-d4): δ 2.11 (dt, 1H, H2, J = 13.2, 3.9 Hz), 2.47 (ddd, 1H, H2, J = 13.2, 8.8, 6.2 Hz), 3.59 (m, 2H, H5), 3.73 (s, 3H, Me), 3.99 (q, 1H, H4, J = 4.2 Hz), 4.23 (dt, 1H, H3, J = 6.3, 3.6 Hz), 4.60 (dd, 1H, H1, J = 8.8, 4.2 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ 39.7 (C2), 52.5 (Me), 63.1 (C5), 72.7 (C3), 77.3 (C1), 88.6 (C4), 175.6 (C
:H2O). The progress of the reaction was monitored by TLC (10% MeOH/CH2Cl2) until no further reaction was apparent (3 h). Next, the mixture was neutralized with DOWEX 50WX8 hydrogen form (50–100 mesh). After removal of the resin by filtration, the solution was evaporated, and the residue was purified by column chromatography (5% MeOH/CH2Cl2) to afford 13 as a yellowish oil in 75% yield. Rf: 0.22 (5% MeOH/CH2Cl2); 1H NMR (300.13 MHz, MeOH-d4): δ 2.11 (dt, 1H, H2, J = 13.2, 3.9 Hz), 2.47 (ddd, 1H, H2, J = 13.2, 8.8, 6.2 Hz), 3.59 (m, 2H, H5), 3.73 (s, 3H, Me), 3.99 (q, 1H, H4, J = 4.2 Hz), 4.23 (dt, 1H, H3, J = 6.3, 3.6 Hz), 4.60 (dd, 1H, H1, J = 8.8, 4.2 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ 39.7 (C2), 52.5 (Me), 63.1 (C5), 72.7 (C3), 77.3 (C1), 88.6 (C4), 175.6 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) ppm; HRMS (ESI+, m/z): calcd for C7H12NaO5 [M + Na]+: 199.0577, found: 199.0578.
O) ppm; HRMS (ESI+, m/z): calcd for C7H12NaO5 [M + Na]+: 199.0577, found: 199.0578.
3,5-Bis-O-(tert-butyldimethylsilyl)-1,2-dideoxy-1α-(methoxycarbonyl)-D-ribofuranose (14)
TBSCl (904.3 mg, 6 mmol) was added to a solution of 13 (210 mg, 1.2 mmol) and imidazole (408.5 mg, 6 mmol) in anhydrous CH2Cl2 (4 mL) at room temperature. The reaction mixture was stirred at 50 °C for 5 h, then diluted with water/ice, and extracted with CH2Cl2. The organic layers were dried (Na2SO4) and evaporated in a vacuum. The residue was purified by column chromatography (10% EtOAc/hexane) to yield 14 as a viscous liquid in 85% yield. Rf: 0.65 (20% EtOAc/hexane); 1H NMR (300.13 MHz, MeOH-d4): δ 0.088 (s, 3H, Si–Me), 0.095 (s, 3H, Si–Me), 0.098 (s, 3H, Si–Me), 0.102 (s, 3H, Si–Me), 0.87 (s, 9H, Si–tBu), 0.92 (s, 9H, Si–tBu), 2.16 (dt, 1H, H2, J = 13.0, 2.4 Hz), 2.39 (ddd, 1H, H2, J = 13.0, 8.9, 5.2 Hz), 3.53 (dd, 1H, H5, J = 11.0, 5.8 Hz), 3.67 (dd, 1H, H5, J = 11.0, 3.8 Hz), 3.71 (s, 3H, O–Me), 4.01 (ddd, 1H, H4, J = 5.7, 3.7, 2.0 Hz), 4.36 (dt, 1H, H3, J = 5.2, 2.1 Hz), 4.59 (dd, 1H, H1, J = 8.9, 2.7 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ −5.3 (Si–CH3), −5.2 (Si–CH3), −4.7 (Si–CH3), −4.6 (Si–CH3), 18.7 (SiCMe3), 19.2 (SiCMe3), 26.2 (3CH3–tBu), 26.4 (3CH3–tBu), 39.9 (C2), 52.5 (Me), 64.3 (C5), 74.2 (C3), 77.7 (C1), 89.5 (C4), 175.4 (CO) ppm; HRMS (ESI+, m/z): calcd for C19H40NaO5Si2 [M + Na]+: 427.2306, found: 427.2302.
3,5-Bis-O-(tert-butyldimethylsilyl)-1,2-dideoxy-1α-(hydroxymethyl)-D-ribofuranose (15)
LiAlH4 (44 mg, 1.16 mmol) was added to a solution of 14 (360 mg, 0.89 mmol) in anhydrous THF (6 mL) at −45 °C, and the reaction was stirred for 30 min at the same temperature. Next, the mixture was diluted with THF and MeOH, the residue was filtered on Celite and concentrated to afford 15 as a viscous liquid (90% yield), which was sufficiently pure for the next step. Rf: 0.34 (20% EtOAc/hexane); 1H NMR (300.13 MHz, MeOH-d4): δ 0.082 (s, 3H, Si–Me), 0.084 (s, 3H, Si–Me), 0.10 (s, 6H, Si–Me), 0.91 (s, 9H, Si–tBu), 0.92 (s, 9H, Si–tBu), 1.73 (ddd, 1H, H2, J = 12.8, 5.9, 4.8 Hz), 2.24 (ddd, 1H, H2, J = 12.9, 7.5, 6.5 Hz), 3.54 (dd, 1H, H6, J = 11.4, 4.6 Hz), 3.62 (m, 3H, 2H5 + H6), 3.81 (q, 1H, H4, J = 4.1 Hz), 4.13 (m, 1H, H1), 4.40 (ddd, 1H, H3, J = 6.4, 4.7, 3.9 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ −5.3 (Si–CH3), −5.2 (Si–CH3), −4.6 (Si–CH3), −4.5 (Si–CH3), 18.8 (SiCMe3), 19.2 (SiCMe3), 26.3 (3CH3–tBu), 26.4 (3CH3–tBu), 37.9 (C2), 64.4 (C5), 66.0 (C6), 74.4 (C3), 80.9 (C1), 87.9 (C4) ppm; HRMS (ESI+, m/z): calcd for C18H40NaO4Si2 [M + Na]+: 399.2357, found: 399.2358.3,5-Bis-O-(tert-butyldimethylsilyl)-1,2-dideoxy-1α-(((methylsulfonyl)oxy)methyl)-D-ribofuranose (16)
To a solution of 15 (135 mg, 0.36 mmol) in anhydrous pyridine (0.65 mL) at 0 °C were added MsCl (62 μL, 0.8 mmol) and catalytic DMAP (5 mg). The solution was stirred at room temperature for 24 h. Then, the solvent was removed in a vacuum, and the residue was dissolved in water and extracted with Et2O. The organic layers were dried (Na2SO4) and evaporated in a vacuum. The residue was purified by column chromatography (20% EtOAc/hexane) to yield 16 as a viscous liquid in 95% yield. Rf: 0.42 (20% EtOAc/hexane); 1H NMR (300.13 MHz, MeOH-d4): δ 0.08 (s, 6H, Si–Me), 0.11 (s, 6H, Si–Me), 0.92 (s, 18H, Si–tBu), 1.75 (dt, 1H, H2, J = 13.0, 3.9 Hz), 2.32 (m, 1H, H2), 3.09 (s, 3H, Ms–Me), 3.61 (dq, 2H, H5, J = 11.0, 4.6 Hz), 3.89 (q, 1H, H4, J = 4.3 Hz), 4.20 (m, 1H, H6), 4.39 (m, 3H, H1 + H3 + H6) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ −5.3 (Si–CH3), −5.2 (Si–CH3), −4.62 (Si–CH3), −4.57 (Si–CH3), 18.8 (SiCMe3), 19.2 (SiCMe3), 26.3 (3CH3–tBu), 26.4 (3CH3–tBu), 37.5 (Ms–Me), 37.6 (C2), 64.4 (C5), 73.4 (C6), 74.4 (C3), 78.0 (C1), 88.6 (C4) ppm; HRMS (ESI+, m/z): calcd for C19H42NaO6SSi2 [M + Na]+: 477.2133, found: 477.2134.General procedure for coupling of 20 with nucleobases. Synthesis of 17 and 18
To a solution of 20 in anhydrous DMF (0.1 M) was added K2CO3 (1.5 eq. for A, C, 5-FU, and 5-IU; 1.2 eq. for U; 2.1 eq. for TBz), 18-crown-6 (1.5 eq.), and the nucleobase (1.2 eq. for A, U, C, and TBz; 1.5 eq. for 5-FU and 5-IU). The mixture was stirred at 90 °C and the reaction progress followed by TLC (5% MeOH/CH2Cl2) until no further reaction was apparent (7–8 h). Solvents were evaporated and the residue was purified by column chromatography (5% MeOH/CH2Cl2) to yield 17 (or 18). Yields are indicated in Scheme 3.3,5-Bis-O-(tert-butyldimethylsilyl)-1,2-dideoxy-1α-((acetoxy)methyl)-D-ribofuranose (19)
To a solution of 15 (44 mg, 0.12 mmol) in anhydrous CH2Cl2 (1.2 mL) at room temperature was added Et3N (50 μL, 0.36 mmol), Ac2O (170 μL, 1.8 mmol) and catalytic DMAP (2 mg). The solution was stirred for 2 h. Solvents were evaporated, and the residue purified by column chromatography (10% EtOAc/hexane) to afford 19 in 90% yield as a viscous liquid. Rf: 0.65 (20% EtOAc/hexane); 1H NMR (300.13 MHz, MeOH-d4): δ 0.081 (s, 3H, Si–Me), 0.085 (s, 3H, Si–Me), 0.10 (s, 6H, Si–Me), 0.91 (s, 9H, Si–tBu), 0.92 (s, 9H, Si–tBu), 1.71 (dt, 1H, H2, J = 13.1, 4.5 Hz), 2.05 (s, 3H, Ac–Me), 2.28 (dt, 1H, H2, J = 13.6, 6.9 Hz), 3.63 (qd, 2H, H5, J = 11.0, 4.5 Hz), 3.85 (q, 1H, H4, J = 3.9 Hz), 4.05 (dd, 1H, H6, J = 10.9, 3.7 Hz), 4.24 (m, 1H, H6), 4.27 (m, 1H, H1), 4.40 (dt, 1H, H3, J = 6.2, 3.9 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ −5.3 (Si–CH3), −5.2 (Si–CH3), −4.6 (Si–CH3), −4.5 (Si–CH3), 18.8 (SiCMe3), 19.2 (SiCMe3), 20.8 (Ac–CH3), 26.3 (3CH3–tBu), 26.4 (3CH3–tBu), 38.1 (C2), 64.4 (C5), 68.1 (C6), 74.4 (C3), 78.1 (C1), 88.4 (C4), 172.6 (CO) ppm.
3,5-Bis-O-(tert-butyldimethylsilyl)-1,2-dideoxy-1α-((tosyloxy)methyl)-D-ribofuranose (20)
To a solution of 15 (100 mg, 0.26 mmol) in anhydrous pyridine (0.5 mL) at 0 °C were added TsCl (111.3 mg, 0.58 mmol) and catalytic DMAP (5 mg). The solution was stirred at room temperature for 9 h. Solvents were evaporated, and the residue was purified by column chromatography (15% EtOAc/hexane) to yield 20 as a colourless viscous liquid in 90% yield. Rf: 0.60 (20% EtOAc/hexane); 1H NMR (300.13 MHz, MeOH-d4): δ 0.040 (s, 3H, Si–Me), 0.042 (s, 3H, Si–Me), 0.05 (s, 3H, Si–Me), 0.06 (s, 3H, Si–Me), 0.84 (s, 9H, Si–tBu), 0.90 (s, 9H, Si–tBu), 1.64 (dt, 1H, H2, J = 13.3, 3.4 Hz), 2.21 (ddd, 1H, H2, J = 13.7, 8.1, 5.8 Hz), 2.45 (s, 3H, Ts–Me), 3.46 (dd, 1H, H5, J = 10.9, 5.6 Hz), 3.57 (dd, 1H, H5, J = 10.9, 3.8 Hz), 3.69 (m, 1H, H4), 3.98 (dd, 1H, H6, J = 10.0, 4.0 Hz), 4.12 (dd, 1H, H6, J = 10.0, 7.6 Hz), 4.27 (m, 1H, H1), 4.33 (m, 1H, H3), 7.43 (d, 2H, Harom, J = 8.5 Hz), 7.79 (d, 2H, Harom, J = 8.4 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ −5.3 (Si–CH3), −5.2 (Si–CH3), −4.6 (2Si–CH3), 18.7 (SiCMe3), 19.2 (SiCMe3), 21.6 (CH3–Ts), 26.3 (3CH3–tBu), 26.4 (3tBu–CH3), 37.6 (C2), 64.4 (C5), 73.6 (C6), 74.5 (C3), 77.9 (C1), 88.7 (C4), 129.1 (2Carom), 131.0 (2Carom), 134.4 (Cipso), 146.3 (Cipso) ppm; HRMS (ESI+, m/z): calcd for C25H47O6SSi2 [M + H]+: 531.2626, found: 531.2632.General procedure for desilylation of 18d and 18f. Synthesis of 21d and 21f
A procedure analogous to that described for the synthesis of 10 was used.Antiviral assays
000 cells per well in collagen-coated 96-well plates. Test compounds or 3TC (control) were added to HepAD38 cells to a final concentration of 10 μM. Real-time PCR for HBV DNA lasted 7 days. On day 7, total DNA was purified from supernatant using commercially available kit (DNeasy 96 Blood & Tissue kit, Qiagen). The HBV DNA was amplified in a real-time PCR assay using LightCycler 480 (Roche) as described elsewhere.23c All samples were tested in triplicate. Analysis: the concentration of compound that inhibited HBV DNA replication by 50% (EC50) was determined by linear regression.4. Conclusions
We have synthesized a series of novel 1′-homo-N-2′-deoxy-α-nucleoside analogs of A, U, T, 5-FU, C, and 5-IU via coupling of a nucleobase and a tosylated intermediate sugar precursor readily obtained from a cyano derivative, which is easily accessible on large-scale. Displacement reactions took place in the presence of K2CO3 and 18-crown-6 in DMF at elevated temperature. The structure of all derivatives was established by NMR spectroscopy and confirmation was obtained by a single crystal X-ray analysis of 1′-homo-N-2′-deoxy-α-adenosine. It is worthy of mention that only the thymine derivative has been previously described synthesized via a more complex route. In addition to the desired compounds we have obtained the corresponding dimer derivatives resulting from the coupling of the sugar moiety at the N-1 and N-3 positions of the base. The occurrence of these dimers is very interesting since few derivatives of this type have been reported in the literature, primarily as byproducts. Eight homonucleosides, including dimers, were tested as antiviral agents. These compounds did not exhibit selective antiviral activity against HIV-1 and HBV. Also, none of them exhibited apparent cytotoxicity in all four cells types tested. Although the results obtained from the preliminary biological screening led to no hit, we believe these novel α-nucleosides may serve as important building-blocks for therapeutic oligonucleotides.10 This study established a facile path for the synthesis of multifunctional nucleoside analogs that may elicit biological activity and make an attractive scaffold for drug discovery using the combinatorial approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Spanish Ministerio de Ciencia e Innovación (MICINN, Project CTQ2014-55015-P) and Principado de Asturias (Project IDI/2018/000181) are gratefully acknowledged. This work was also supported in part by NIH grants 1-R01-AI-132833 (to R. F. S.) and 5P30-AI-50409 (CFAR) (to R. F. S.).References
- (a) E. De Clercq, Curr. Opin. Microbiol., 2005, 8, 552–560 CrossRef CAS PubMed; (b) S. Rachakonda and L. Cartee, Curr. Med. Chem., 2004, 11, 775–793 CrossRef CAS PubMed; (c) Recent Advances in Nucleosides: Chemistry and Chemotherapy, ed. C. K. Chu, Elsevier Science, New York, 2002 Search PubMed; (d) Nucleoside Analogs in Cancer Therapy, ed. B. D. Chenson, M. J. Keating and W. Plunkett, Marcel Dekker, New York, 1997 Search PubMed; (e) Nucleosides and Nucleotides as Antitumor and Antiviral Agents, ed. C. K. Chu and D. C. Baker, Plenum Press, New York, 1993 Search PubMed; (f) Chemistry of Nucleosides and Nucleotides, ed. L. B. Townsend, Plenum Press, New York, 1988 Search PubMed.
- See a recent review on α-nucleosides: G. Ni, Y. Du, F. Tang, J. Liu, H. Zhao and Q. Chen, RSC Adv., 2019, 9, 14302–14320 RSC.
- A. E. Wróblewski, I. E. Glowacka and D. G. Piotrowska, Eur. J. Med. Chem., 2016, 118, 121–142 CrossRef PubMed.
- C. Lamberth, Org. Prep. Proced. Int., 2002, 34, 149–167 CrossRef CAS.
- D. González-Calderón, M. G. Mejía-Dionicio, M. A. Morales-Reza, J. G. Aguirre-de Paz, A. Ramírez-Villalva, M. Morales-Rodríguez, A. Fuentes-Benítes and C. González-Romero, Bioorg. Chem., 2016, 69, 1–6 CrossRef PubMed.
- C. I. Tanase, C. Draghici, A. Hanganu, L. Pintilie, M. Maganu, A. Volobueva, E. Sinegubova, V. V. Zarubaev, J. Neyts, D. Jochmans and A. V. Slita, Molecules, 2019, 24, 2446 CrossRef CAS PubMed.
- Y. Okada, K. Shimada, Y. Kitano and K. Chiba, Eur. J. Org. Chem., 2014, 1371–1375 CrossRef CAS.
- S. Martínez-Montero, S. Fernańdez, Y. S. Sanghvi, J. Chattopadhyaya, M. Ganesan, N. G. Ramesh, V. Gotor and M. Ferrero, J. Org. Chem., 2012, 77, 4671–4678 CrossRef PubMed.
- (a) P. Busca, I. McCort, T. Prangé and Y. Le Merrer, Eur. J. Org. Chem., 2006, 2403–2409 CrossRef CAS; (b) P. Angibeaud, J. Defaye and H. Franconie, Carbohydr. Res., 1980, 78, 195–204 CrossRef CAS.
- F. Morvan, J. Zeidler and B. Rayner, Tetrahedron, 1998, 54, 71–82 CrossRef CAS.
- (a) D. C. Pryde, D. S. Middleton, P. T. Stephenson, P. Wainwright, A. Maddaford, X. Zhang, D. Leese, R. Glen, J. Hart, N. Forrest and T. Guyot, Tetrahedron Lett., 2011, 52, 6415–6419 CrossRef CAS; (b) J. Farkas, Collect. Czech. Chem. Commun., 1971, 36, 3043–3046 CrossRef CAS; (c) J. A. Montgomery and K. Hewson, J. Heterocycl. Chem., 1970, 7, 443–445 CrossRef CAS; (d) M. Bobek and J. Farkas, Collect. Czech. Chem. Commun., 1969, 34, 1684–1689 CrossRef CAS.
- M. W. Winkley, Carbohydr. Res., 1973, 31, 245–254 CrossRef CAS.
- (a) R. Saladino, V. Neri, P. Checconi, I. Celestino, L. Nencioni, A. T. Palamara and M. Crucianelli, Chem.–Eur. J., 2013, 19, 2392–2404 CrossRef CAS PubMed; (b) B. Doboszewski, Nucleos Nucleot. Nucleic Acids, 2009, 28, 875–901 CrossRef CAS PubMed; (c) B. Doboszewski, Nucleosides Nucleotides, 1997, 16, 1049–1052 CrossRef CAS; (d) N. Hossain, N. Blaton, O. Peeters, J. Rozenski and P. Herdewijn, Tetrahedron, 1996, 52, 5563–5578 CrossRef CAS.
- (a) C. L. Scremin, J. H. Boal, A. Wilk, L. R. Philips and S. L. Beaucage, Bioorg. Med. Chem. Lett., 1996, 6, 207–212 CrossRef CAS; (b) J. H. Boal, A. Wilk, C. L. Scremin, G. N. Gray, L. R. Philips and S. L. Beaucage, J. Org. Chem., 1996, 61, 8617–8626 CrossRef CAS.
- (a) A. Carnero, S. Pérez-Rentero, A. Alagia, A. Aviñó, Y. S. Sanghvi, S. Fernández, M. Ferrero and R. Eritja, RSC Adv., 2017, 7, 9579–9586 RSC; (b) A. Carnero, Y. S. Sanghvi, V. Gotor, S. Fernández and M. Ferrero, Org. Process Res. Dev., 2015, 19, 701–709 CrossRef CAS; (c) S. Martínez-Montero, S. Fernández, Y. S. Sanghvi, E. A. Theodorakis, M. A. Detorio, T. R. Mcbrayer, T. Whitaker, R. F. Schinazi, V. Gotor and M. Ferrero, Bioorg. Med. Chem., 2012, 20, 6885–6893 CrossRef PubMed; (d) S. Martínez-Montero, S. Fernández, Y. S. Sanghvi, V. Gotor and M. Ferrero, Eur. J. Org. Chem., 2012, 5483–5490 CrossRef; (e) S. Martínez-Montero, S. Fernández, Y. S. Sanghvi, V. Gotor and M. Ferrero, Org. Biomol. Chem., 2011, 9, 5960–5966 RSC; (f) T. Rodríguez-Pérez, S. Fernández, Y. S. Sanghvi, M. Detorio, R. F. Schinazi, V. Gotor and M. Ferrero, Bioconjugate Chem., 2010, 21, 2239–2249 CrossRef PubMed; (g) S. Martínez-Montero, S. Fernández, Y. S. Sanghvi, V. Gotor and M. Ferrero, J. Org. Chem., 2010, 75, 6605–6613 CrossRef PubMed; (h) T. Rodríguez-Pérez, S. Fernández, S. Martínez-Montero, T. González-García, Y. S. Sanghvi, V. Gotor and M. Ferrero, Eur. J. Org. Chem., 2010, 1736–1744 CrossRef.
- (a) Pure α-cyano-sugar 12 is available from Sapala Organics Pvt. Ltd, India, http://www.sapalaorganics.com; (b) T. H. Smith, M. A. Kent, S. Muthini, S. J. Boone and P. S. Nelson, Nucleosides Nucleotides, 1996, 15, 1581–1594 CrossRef CAS.
- H. Vorbrüggen and C. Ruh-Pohlenz, Handbook of Nucleoside Synthesis, Wiley Interscience, 2001 Search PubMed.
- O. R. Ludek and C. Meier, Eur. J. Org. Chem., 2006, 941–946 CrossRef CAS.
- Molecular graphics and analyses were performed with the UCSF Chimera package (http://www.rbvi.ucsf.edu/chimera). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
- CCDC 1980305 contains the supplementary crystallographic data for compound 10a.
- M. Prystas and F. Sorm, Collect. Czech. Chem. Commun., 1969, 34, 2316–2347 CrossRef CAS.
- Z. J. Yang, H. W. Yu, J. M. Min, L. T. Ma and L. H. Zhang, Tetrahedron Asymmetry, 1997, 8, 2739–2747 CrossRef CAS.
- (a) R. F. Schinazi, J.-P. Sommadossi, V. Saalmann, D. L. Cannon, M.-W. Xie, G. C. Hart, G. A. Smith and E. F. Hahn, Antimicrob. Agents Chemother., 1990, 34, 1061–1067 CrossRef CAS PubMed; (b) R. F. Schinazi, A. McMillan, D. Cannon, R. M. Mathis, R. M. Lloyd, A. Peck, J.-P. Sommadossi, M. St. Clair, J. Wilson, P. A. Furman, G. Painter, W.-B. Choi and D. C. Liotta, Antimicrob. Agents Chemother., 1992, 36, 2423–2431 CrossRef CAS PubMed; (c) L. J. Stuyver, S. Lostia, M. Adams, J. S. Mathew, B. S. Pai, J. Grier, P. M. Tharnish, Y. Choi, Y. Chong, H. Choo, C. K. Chu, M. J. Otto and R. F. Schinazi, Antimicrob. Agents Chemother., 2002, 46, 3854–3860 CrossRef CAS PubMed; (d) M. S. Belen'kii and R. F. Schinazi, Antiviral Res., 1994, 25, 1–11 CrossRef.
- S. K. Ladner, M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo, C. Seeger and R. W. King, Antimicrob. Agents Chemother., 1997, 41, 1715–1720 CrossRef CAS PubMed.
- T.-C. Chou and P. Talalay, Adv. Enzym. Regul., 1984, 22, 27–55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1D and 2D spectra. CCDC 1980305. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03254a |
| This journal is © The Royal Society of Chemistry 2020 |