
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsosorbide bis(methyl carbonate) synthesis from isosorbide and dimethyl carbonate: the key role of dual basic–nucleophilic catalysts†‡
José R. Ochoa-Gómez *,
Leire Lorenzo-Ibarreta,
Cristina Diñeiro-García and
Olga Gómez-Jiménez-Aberasturi
*,
Leire Lorenzo-Ibarreta,
Cristina Diñeiro-García and
Olga Gómez-Jiménez-Aberasturi
TECNALIA, Basque Research and Technology Alliance (BRTA), Parque Tecnológico de Alava, Leonardo Da Vinci 11, 01510 Miñano (Alava), Spain. E-mail: jrochoag@telefonica.net
First published on 18th May 2020
Abstract
Isosorbide bis(methyl carbonate) (IBMC) is a scarcely studied green chemical with potential applications in the manufacturing of non-isocyanate polyurethanes and bisphenol A-free polycarbonates. Its synthesis by transesterification of isosorbide with dimethyl carbonate (DMC) is very negatively influenced by the presence of small amounts of acidic impurities in isosorbide when heterogeneous inorganic carbonates such as potassium and cesium carbonates are used as catalysts. In this paper it is shown that the problem can be solved by using homogeneous catalysts consisting of nitrogenated bases and superbases having a suitable dual nucleophilic–basic character and able to form a highly reactive acyl intermediate with the electrophilic reactant DMC. Cycloaliphatic secondary and tertiary amines, guanidines and amidines covering a nucleophilicity parameter (N) range between 13.58 and 20.58 in either acetonitrile or dichloromethane, and a pKa range in acetonitrile between 15.68 and 26.02 have been tested in batchwise mode. Highly active catalysts leading to hydroxyl conversions of 84–93% require a minimum N of 16 and a pKa ranging from 18.0 to 26.0. Within this pKa range, N must increase by about 0.5–0.6 units per each unit the pKa falls to keep the catalytic activity, indicating that nucleophilicity has approximately twice as much influence as basicity on the catalytic activity. One guanidine (TBD), one amidine (DBN) and three cycloaliphatic secondary amines (N-methylpyrrolidine, quinuclidine and DABCO) have been found to be excellent catalysts at 5 mol% vs. ISO. The side reaction leading to oligomer formation is not avoided, with oligomers, mainly the dimer, affording 6 wt% of the crude product independently of hydroxyl-conversion and catalyst type.
Introduction
The shift from a non-sustainable petroeconomy to a sustainable bioeconomy is a current unstoppable worldwide megatrend leading within the Chemical Industry to multiple actions to find new safe and sustainable processes to manufacture molecules and materials already well stablished in the worldwide market. One of such actions is the search for synthetic routes to manufacture polyurethanes without using isocyanates, i.e. the so-called NIPUs, such as that involving the reaction between dicarbonates and diamines.In this context, molecules such as IBMC can play an important role in the next future. Its two linear carbonate end groups make IBMC a potentially useful monomer for the synthesis of sustainable biobased polymers by polycondensation, such as the above mentioned NIPUs and also polycarbonates by reacting with diols (Scheme 1). IBMC can be obtained from ISO which is a non-toxic biomass-derived chemical currently obtained by a double dehydration of sorbitol,1 which in turn is industrially synthesized by hydrogenation of glucose resulting from polysaccharides such as starch and cellulose.2 It is believed that its rigid bicyclic aliphatic structure could allow ISO and molecules derived thereof, such as IBMC, to be an alternative to the toxic MDI and bisphenol A in the manufacturing of polyurethanes and polycarbonates, respectively.3
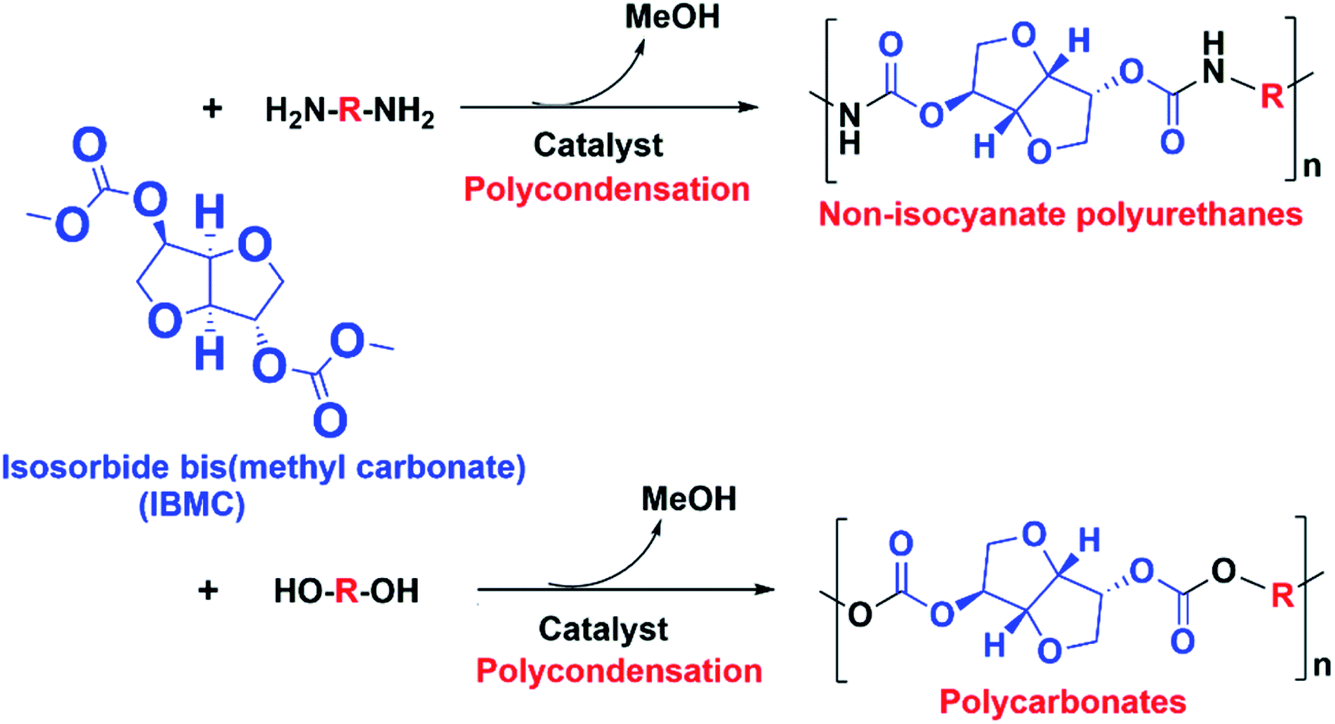 | ||
| Scheme 1 Possible IBMC-derived NIPUs and polycarbonates. | ||
However, IBMC is a scarcely explored molecule so that before its industrial introduction both a validation of its manufacturing process at pilot scale in an industrially relevant environment and a demonstration of the usefulness of IBMC-derived polymers in some market sectors are required. Both issues are the main objectives of the VIPRISCAR project, funded by the BBI-JU under the umbrella of the H2020 EU framework program for research and innovation.4
As far as we know there are two IBMC manufacturing processes reported. The first one is by reacting ISO with a highly toxic chloroformate ester.5 Therefore, in the VIPRISCAR project the second method, transesterification of ISO with DMC, has been chosen because it is a safe and environmental compatible process avoiding toxic reactants, in close alignment with the green chemistry rules. The eco-friendly nature of ISO has been stated before, while DMC is also an eco-friendly chemical which is increasingly gaining acceptance due to its versatility as both solvent and reactant.6 A general overview of the desired reaction and side-reactions is depicted in Scheme 2.
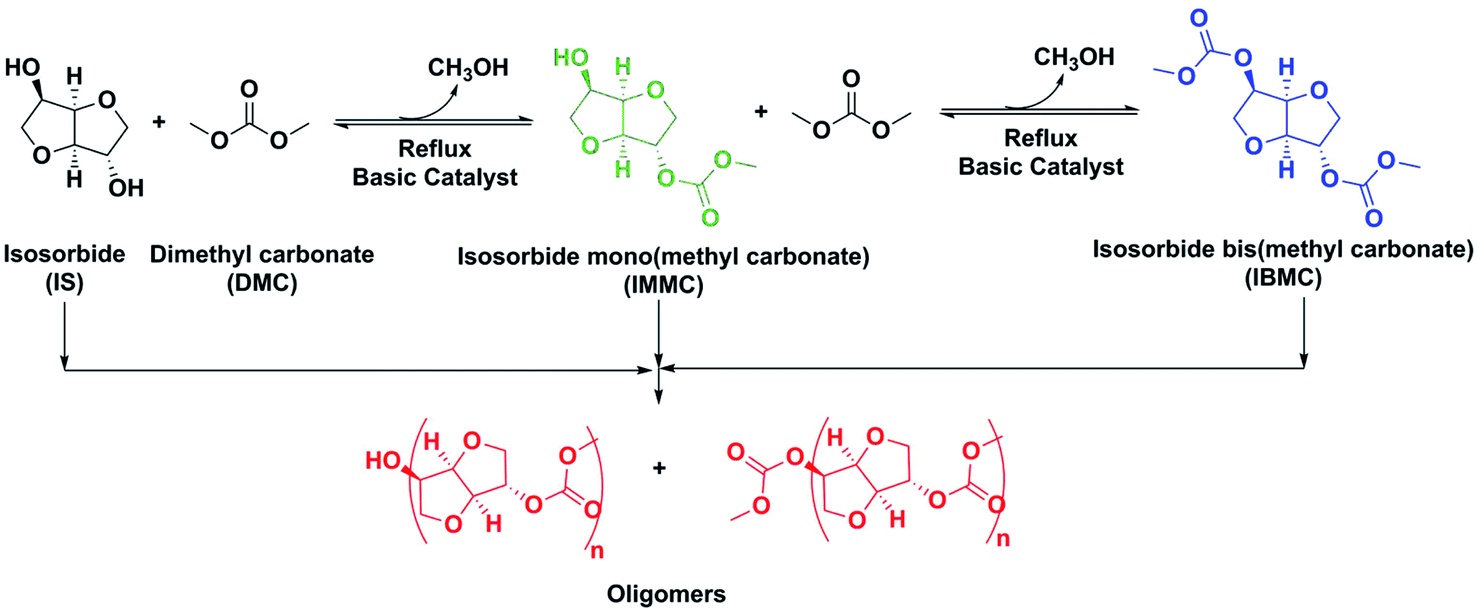 | ||
| Scheme 2 Synthesis of IBMC and oligomers thereof. Only the 2-exo isomer of IMMC is shown. | ||
This synthesis using basic heterogeneous catalysts, preferably K2CO3, LiOH and KOH, was first reported by Fuertes et al.7 with some additional few data provided later by Aricò et al.8 The synthesis is carried out at reflux in a large excess of DMC. Full ISO and IMMC intermediate conversions are achieved by shifting the equilibrium to the right by continuously distilling a mixture of methanol/DMC. Unfortunately, ISO has two hydroxyl non-equivalent moieties: OH-endo and OH-exo. OH-endo in a tetrahydrofuran cycle is linked by a hydrogen bridge to the oxygen atom in the other tetrahydrofuran cycle, while OH-exo is not, which results in different reactivities.9 This means that the synthesis of IBMC by transesterification with DMC is a two-step 1-pot reaction. Although ISO is quickly converted into IMMC in the first step, the different OH groups reactivities make the life of the IMMC molecules in the reaction medium relatively long thereby increasing the probability of reaction with both IBMC and another IMMC molecules resulting in oligomers formation. Although these oligomers are not an obstacle to obtain polycarbonates and NIPUs because, provided that they are methoxycarbonyl-ended, they can also polymerize like IBMC, their presence in high amounts is not desired because they do not allow a fine tuning of the target polymer structure. Oligomers formation was high in the above-mentioned process developed by Fuertes et al., requiring a costly purification procedure.
An interesting approach to IBMC from ISO and DMC was recently reported by Qian et al. using a series of 4-substituted phenolate ILs as transesterification catalysts.10 Very interestingly, they suggest a reaction mechanism in which the dual basic-nucleophilic behavior of the catalysts enhances their activities. They report a 99% selectivity to IBMC but their results are based on GC-MS measurements which do not allow neither the quantification of oligomers nor its detection provided they are obtained in an amount below 10 wt% relative to the total amount of crude isolated product (see ESI†).
The amount of oligomers was drastically reduced by means of an improved transesterification procedure developed by our group involving the use of potassium or cesium carbonate as a catalyst in amounts ≤ 2 mol% vs. ISO, with the latter being the preferred catalyst.11,12 Therefore, this was the process selected to be scaled-up in the VIPRISCAR project. However, during experimentation an unexpected result was observed: ISO reactivity was dependent of supplier and batch number. When found above a specific but low amount, acidic impurities contained in ISO, probably resulting from an inefficient removal of the acidic catalyst (typically sulfuric acid) used in the dehydration of sorbitol, influence strongly and negatively the catalyst activity. This paper describes the search for catalysts able to deal with acidic commercial ISO and show how some cyclic tertiary amines, amidines and guanidines show a high catalytic activity to convert acidic-ISO into IBMC when used in catalytic amounts, provided that they have a dual basic–nucleophilic character properly balanced.
Experimental
Materials
ISO was from four suppliers: Alfa-Aesar, Acros, Carbosynth, and Roquette 99% pure. All other reagents and catalysts were supplied by Sigma-Aldrich and were at least 98% pure. When using cesium or potassium carbonates as catalysts it is very important that both catalysts and ISO are water-free to avoid catalyst agglomeration during reaction leading to a catastrophic lost of their activities. Inorganic carbonates can be dried at 105 °C while ISO at 65 °C overnight, if needed.Synthetic procedure and experimental set up
Catalyst screening was carried out batchwise by reacting ISO (2 g) and DMC under reflux (90 °C at the beginning of the reaction and decreasing as reaction proceeds to reach values of about 84–88 °C under the reaction procedure herein used and depending on the hydroxyl conversion obtained) at a DMC/ISO MR of 30 in the presence of the desired amount of catalyst in an Eyela multireactor (Picture P1 in ESI†) consisting of 5 tube-shaped reactors, each one able to work with up to 60 mL of reaction mixture at different temperatures under the same magnetic stirring. It is important to highlight that the synthesis of IBMC herein reported is a transesterification, so that it is ruled by equilibrium. That means that a 100% hydroxyl moieties conversion can be only achieved if the coproduct MeOH is continuously removed from the reaction medium by distillation to shift reaction equilibrium to right. And this is not possible in the reactions herein reported because they were designed in batchwise mode and aimed at looking for catalysts able to keep their activity in the presence of acidic impurities in ISO. That is, this paper is not related to process optimization but to catalyst activity.Reactions were monitored by following the disappearance of the OH-band of ISO and IMMC at 3360–3394 cm−1 (it is continuously shifted from 3360 to 3394 cm−1 as reaction proceeds) over time by ATR-FTIR using a Bruker ALPHA Platinum-ATR-FTIR Spectrometer, till its intensity was constant over time. The disappearance of the OH-band is accompanied with the continuous increase in intensity of a band at 1750–1755 cm−1, typical of carbonyl groups in linear organic carbonates and corresponding to in IMMC + IBMC. Spectra at the beginning (corresponding to pure ISO) and after completion of a reaction are shown in Fig. S1 (see ESI†). All spectra were recorded at 65 °C after very quick evaporation of the solvent at the same temperature following the deposition of 1–2 drops of reaction medium on the sample holder.
Analytical procedures
GPC measurements were carried out using a Polymer Laboratories equipment model PL-GPC-50 Plus fitted with a refractive index detector. Eluent was tetrahydrofuran and polystyrene standards from Easy Cal PS-2 were used to obtain a molar mass versus elution time calibration line with a 300 × 7.5 mm Resipore column (particle size 3 μm, Mw range: 200–400000 g mol−1) at 40 °C. Injection volume was 20 μL, flow rate 1 mL min−1 and the concentration of samples was 2000–3000 ppm in THF for detecting unambiguously all oligomer species. A typical GPC chromatogram showing the main species involved in the reaction is depicted in Fig. S2 (ESI†), wherein it is shown that IBMC, IMMC and ISO eluate together.GC-MS measurements were carried out using a Gas Chromatograph Agilent GC 7890B, a MS detector 5977A and a HP5-ms column of 30 m, 0.25 mm and 0.25 μm. Analytical conditions: samples dissolved in DCM (1350 ppm) after DMC evaporation; injector temperature, 300 °C; detector temperature, 250 °C; split, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20; injection volume, 1 μL; oven temperature was started at 40 °C (5 min) and it was increased at 5 °C min−1 up to 300 °C and kept at this temperature for 5 min; injection mode: scan; carrier gas: helium at 1 mL min−1; mass range: 33.0–550.0; running time, 36 min. A typical GC chromatogram of an incomplete reaction is depicted in Fig. S3 (ESI†), wherein it can be seen how oligomers are also detected by GC-MS, mainly if its content is higher than 10 wt%. However, they cannot be quantified by this technique because only a small fraction is evaporated in the GC system due to their high boiling points. This can be seen undoubtedly in Fig. S4 (ESI†).
:20; injection volume, 1 μL; oven temperature was started at 40 °C (5 min) and it was increased at 5 °C min−1 up to 300 °C and kept at this temperature for 5 min; injection mode: scan; carrier gas: helium at 1 mL min−1; mass range: 33.0–550.0; running time, 36 min. A typical GC chromatogram of an incomplete reaction is depicted in Fig. S3 (ESI†), wherein it can be seen how oligomers are also detected by GC-MS, mainly if its content is higher than 10 wt%. However, they cannot be quantified by this technique because only a small fraction is evaporated in the GC system due to their high boiling points. This can be seen undoubtedly in Fig. S4 (ESI†).
COH in % were determined by ATR-FTIR using eqn (1):
| COH = (1 − (At-(3360–3394)/A0)) × 100 | (1) |
In a batch reaction, the crude product after reaction completion consists of a mixture of IMMC, IBMC and oligomers (predominantly dimer plus some trimer) and, only at low conversions, of unreacted ISO. Its composition can be determined by combining GPC results with those from GC-MS. From GPC the areas (%) corresponding to the sum of ISO, IMMC and IBMC (A1) and oligomers (A2 = sum of the areas of all peaks corresponding to oligomers, usually dimer + trimer) are obtained. From GC-MS, the areas (%) of ISO (A3), IMMC (A4) and IBMC (A5) relative to the total area of these species are also obtained. Then, the composition (wt%) of isolated crude product is calculated as follows:
- ISO = A1 × A3/100
- IMMC = A1 × A4/100
- IBMC = A1 × A5/100
- Oligomers = A2.
The target product, IBMC, was also characterized by NMR. Data of its 1H-NMR and 13C-NMR spectra were provided elsewhere.12
Results and discussion
Catalyst deactivation by acidic impurities in ISO
The strong negative influence of acidic impurities is evidenced for the heterogeneous K2CO3 catalyst in Fig. 1, where conversions versus time for several loads using ISO-2.81 are compared with those of ISO-3.42 at 2 mol% K2CO3. A 70% OH-conversion is obtained in 120 min with both ISO-7.60 and ISO-3.42 at catalysts load of 1.5 and 2 mol%, respectively, while 0% conversion is obtained with isosorbide-2.81 up to 8 mol% catalyst, a 100% decrease. Catalyst activity is restored by increasing its load but to match the reactivity of ISO-3.42 an amount 12.5-fold higher is needed!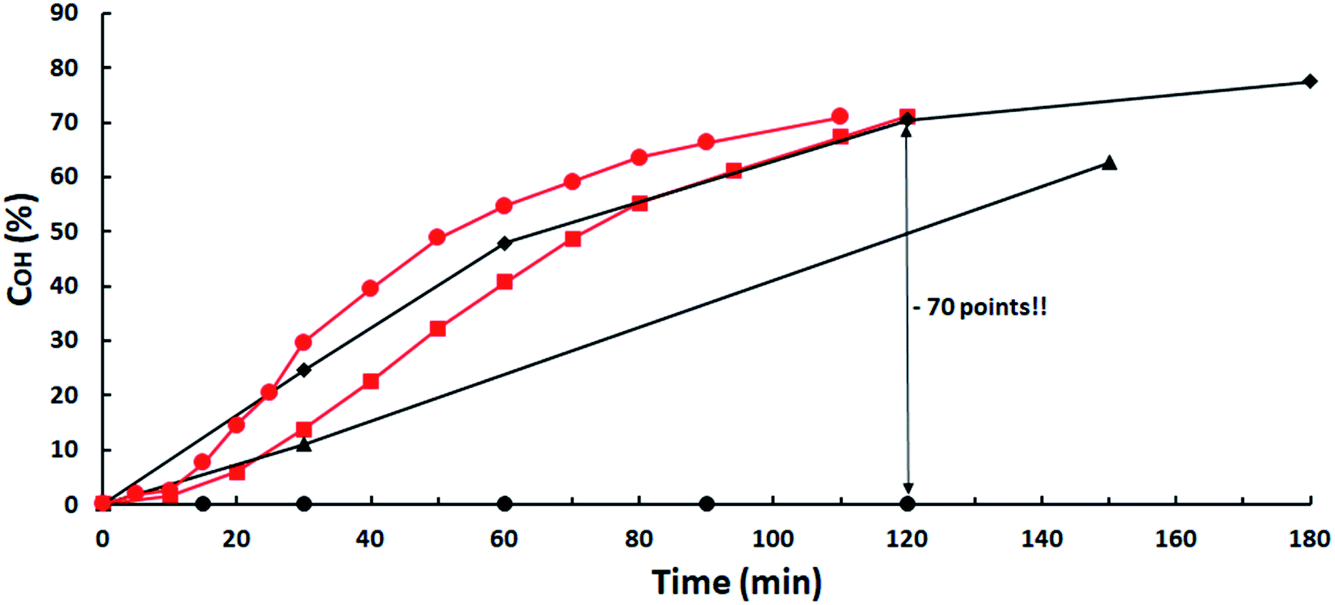 | ||
Fig. 1 Influence of ISO acidity on K2CO3 catalytic activity. ISO-7.60: ( ) 1.5 mol% K2CO3; ISO-3.42: ( ) 1.5 mol% K2CO3; ISO-3.42: ( ) 2 mol%; ISO-2.81: ( ) 2 mol%; ISO-2.81: ( ) 8 mol%; ( ) 8 mol%; ( ) 16 mol%; ( ) 16 mol%; ( ) 25 mol%. ) 25 mol%. | ||
Situation is even worst when Cs2CO3, a catalyst acting partially as a homogeneous one,12 is used as shown in Fig. 2. COH decreases from 90% with ISO-7.60/1.5 mol% Cs2CO3 vs. ISO and ISO-3.42/2 mol% Cs2CO3 to almost 0% with ISO-2.81 and 7.5 mol% Cs2CO3. Catalyst is quickly deactivated below an IISO acidity of 3.42. Activity is partially restored by increasing catalyst load but, even at 15 mol%, a 55.5% decrease (49 points) in OH-conversion is produced relative to that with ISO-2.81.
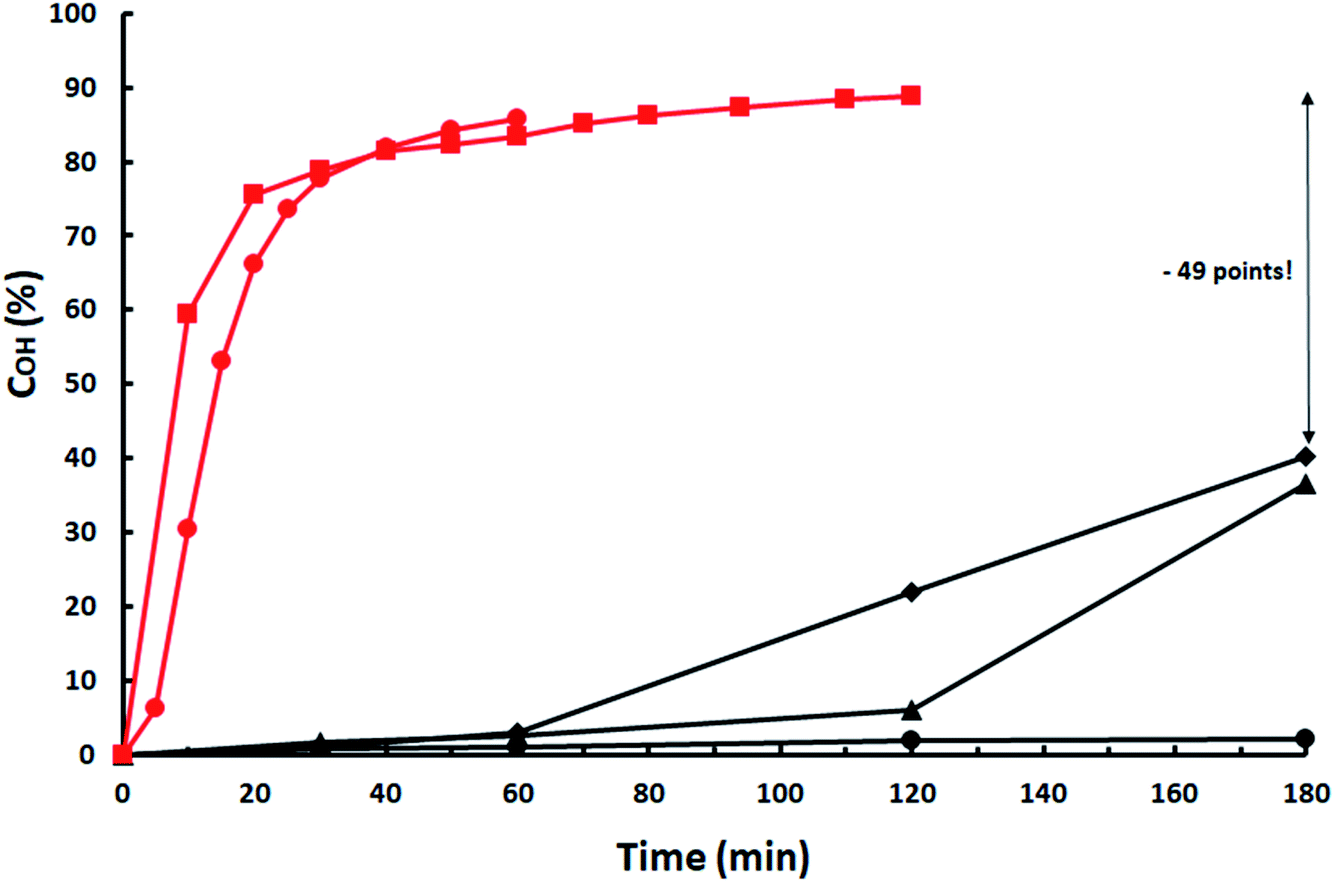 | ||
Fig. 2 Influence of ISO acidity on Cs2CO3 catalytic activity. ISO-7.6: ( ) 1.5 mol% Cs2CO3; ISO-3.42: ( ) 1.5 mol% Cs2CO3; ISO-3.42: ( ) 2 mol% Cs2CO3; ISO-2.81: ( ) 2 mol% Cs2CO3; ISO-2.81: ( ) 7.5 mol%; ( ) 7.5 mol%; ( ) 10 mol%; ( ) 10 mol%; ( ) 15 mol%. ) 15 mol%. | ||
A pH of 2.81 at 1 M ISO concentration means that the proton concentration is 1.549 × 10−3 M and the proton moles per gram of ISO is 1.06 × 10−5. This amount can be neutralized with 0.53 × 10−5 mol carbonate/g-ISO, i.e., 3.9% of the total amount of catalyst used (2 mol% catalyst vs. ISO = 1.37 × 10−4 mol-catalyst/g-ISO). Therefore, the obvious conclusion would be that acidic impurities in ISO are not the cause, or the unique cause, of catalyst deactivation. We have not found an explanation for this fact, but the fact is that there is a strong relationship between the ISO acidity and the catalyst activity which increases as the ISO acidity decreases. Therefore, the measurement of the pH of a 1 M aqueous solution of an ISO material is a good quality control to determine its suitability for the synthesis of IBMC.
A first approach to restore catalyst activity was ISO purification by crystallization. It restored catalyst activity in a large extent, but it was discarded because a low 45% crystallization yield was obtained in both acetone and methyl ethyl ketone leading to much higher manufacturing costs. A better 83% crystallization yield was obtained from DMC, but ISO acidic impurities removal was insufficient.
Catalysts suitable to deal with acidic ISO
At first, an obvious choice to find catalysts able to keep their activities working in real catalytic amounts would be to look for within superbases soluble in the reaction medium. It could be expected that the high basicity of superbases,13 together with the easy availability of the catalytic sites of the homogeneous catalysts could effectively neutralize the small amount of acidic impurities in ISO sacrificing only an economically acceptable amount of catalyst. And this works when, for instance, a well known and very effective superbase in a lot of different types of reactions, such as TBD,14 a guanidine, is used as shown by results depicted in Fig. 3. OH-conversion is 5.5% when ISO-2.81 and 2 mol% TBD vs. ISO is used, 83.5 points lower than that with ISO-3.42 at the same catalyst concentration. However, COH increases swiftly with TBD concentration matching that of ISO-3.42 at a 4 mol% TBD concentration and surpassing it at a 5 mol% TBD concentration, i.e., with a 2.5-fold increase in catalyst concentration. So, it could be concluded that basicity is the key parameter ruling the catalyst activity. The higher the basicity the higher the activity by means of hydroxyl moiety activation by hydrogen abstraction from the OH moieties in ISO leading to the highly nucleophilic isosorboxide anions which subsequently attack the electrophilic carbon of the carbonyl group in DMC causing methanol removal and resulting first on IMMC formation and subsequently on IBMC synthesis.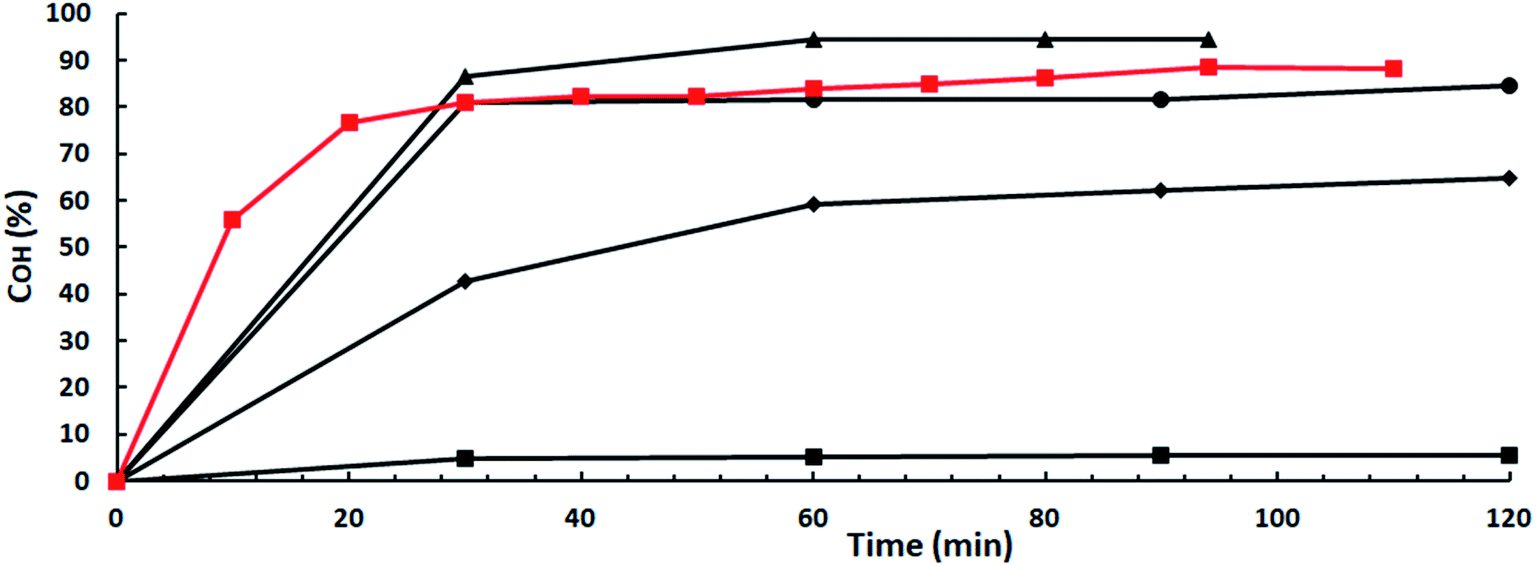 | ||
Fig. 3 OH-conversions vs. time for TBD as a catalyst: ISO-3.42: ( ) 2 mol% TBD vs. ISO; ISO-2.81: ( ) 2 mol% TBD vs. ISO; ISO-2.81: ( ) 2 mol%; ( ) 2 mol%; ( ) 3 mol%; ( ) 3 mol%; ( ) 4 mol% ( ) 4 mol% ( ) 5 mol%. ) 5 mol%. | ||
To check this hypothesis several reactions were carried with ISO-2.81 and different nitrogenated bases and superbases at 5 mol% vs. ISO. Maximum OH-conversions and the reaction time at which they were obtained are given in Table 1, together with catalyst basicities in ACN as given by the pKa of their conjugated acids.
| Catalyst | pKab (ACN) | trc (min) | COH-max (%) |
|---|---|---|---|
| a Reaction conditions: 2 g ISO-2.81; DMC/ISO MR: 30; reflux; batchwise; catalyst concentration: 5 mol% vs. ISO-2.81.b Taken from ref. 15.c Reaction time needed to achieve de maximum OH-conversion. | |||
| TBD | 26.02 | 50 | 93.0 |
| DBU | 24.31 | 180 | 29.8 |
| MTBD | 25.47 | 240 | 35.2 |
| DABCO | 18.29 | 60 | 84.4 |
| TEA | 18.83 | 120 | 0.1 |
OH-Conversion decreases dramatically from 93% in 50 min to 29.8% in 180 min when TBD is substituted by the superbase DBU, an amidine. The decrease in conversion is even very significant when MTBD, a guanidine-type superbase with a basicity very similar to that of TBD, is used. However, DABCO, a bicyclic tertiary diamine, having a basicity 106.02 lower than that of DBU, 107.73 lower than that of TBD and 107.18 lower than that of MTBD, leads to a high 84.4% OH-conversion in 60 min. Then, it could be expected that TEA, an aliphatic amine with a basicity slightly higher than DABCO, would lead to a similar conversion than DABCO. However, after 120 min of reaction OH-conversion was practically zero. So, catalyst activity in this reaction is not ruled by basicity, at least exclusively. There must be another very influential parameter.
A common feature of the above highly active catalysts, TBD and DABCO, is that they have a high nucleophilic character as measured by the nucleophilicity parameter N,16 whose values for a lot of organic chemicals can be found in the Prof. H. Mayr's Database of Reactivity Parameters.17 The higher N (is value is logarithmic) the higher the nucleophilicity. Relationship between N values for bases given in Table 1 and maximum OH-conversions are given in Table 2. According to N values, the highly active TBD and DABCO catalysts are stronger nucleophiles than catalysts with a moderate catalytic activity, such as the superbases MTBD and DBU. However, TEA, with no catalytic activity, has a nucleophilic parameter lower than that of DABCO but about one order of magnitude higher than that of the superbase TBD. Therefore, its lack of catalytic activity must be ascribed to its low basicity, 107-fold lower than that of TBD. Consequently, it must be concluded that both catalyst basicity and catalyst nucleophilicity play a role in the reaction herein reported and that a suitable combination of nucleophilicity and basicity is required to become a good catalyst.
To try to confirm this hypothesis we analyzed the catalytic activities of another 6 nitrogenated bases using ISO-2.81. The results together with those for the previous nitrogenated bases are given in Table 3, while the variations of COH with time for each reaction are depicted in Fig. S5 (ESI†). All N values are given in ACN, except for those not available in such solvent: MTBD and TBD (in DCM). Despite nucleophilicity is solvent dependent, the values given in Tables 2 and 3 can be considered as comparable for the purposes of this discussion as shown by the available values for several chemicals. For instance, N values in ACN and DCM, respectively, are 17.10 and 17.30 for TEA, 16.80 and 16.50 for N-methylmorpholine, 18.72 and 18.90 for N-methylpiperidine, and 20.59 and 20.60 for N-methylpyrrolidine.
| Entry | Catalyst | Structure | N1 | pKa | tr (min) | WGF3 | COH (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: ISO-2.81: 2 g; batch (30 mL); reflux; DMC/ISO-2.81 molar ratio: 30. Catalyst concentration: 5 mol% vs. ISO-2.81. Reaction time (tr) till constant OH-conversion was achieved.b pKa values in ACN; 1N values in ACN except for 2 in DCM; 3Within Geometric Form in Fig. 5.c Ref. 18.d Ref. 15.e Ref. 13. | |||||||
| 1 | N-Methylmorpholine | 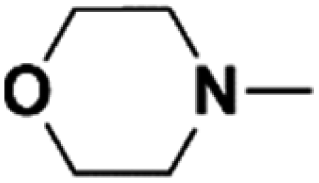 |
16.80 | 15.68c | 120 | C | 0 |
| 2 | TEA | 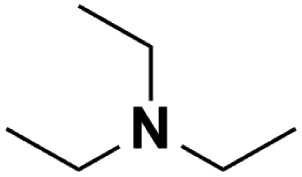 |
17.10 | 18.83d | 120 | C | 0.1 |
| 3 | N-Methylpiperidine | 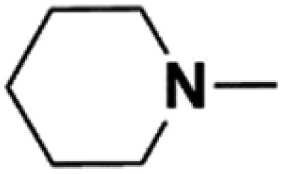 |
18.72 | 18.24d | 120 | C | 0.5 |
| 4 | DMAP | 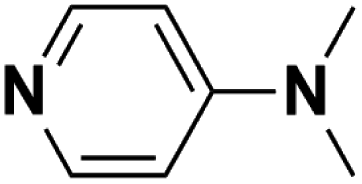 |
15.51 | 17.96d | 60 | C | 9.6 |
| 5 | DBU | 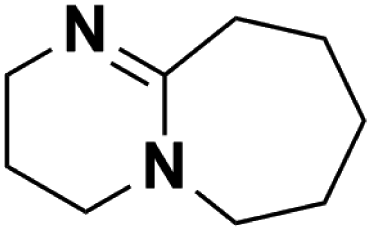 |
15.29 | 24.31d | 180 | B | 29.8 |
| 6 | MTBD | 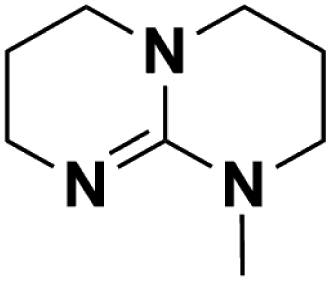 |
14.432 | 25.47d | 240 | B | 35.2 |
| 7 | DBN | 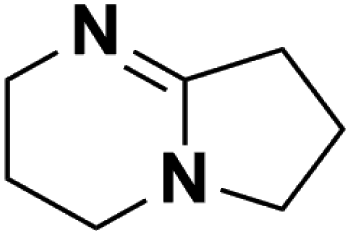 |
16.28 | 23.79e | 240 | A | 76.0 |
| 8 | DABCO | 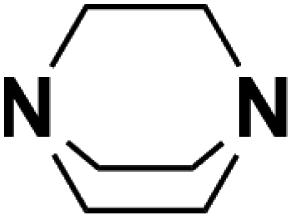 |
18.80 | 18.29d | 60 | A | 84.4 |
| 9 | Quinuclidine | 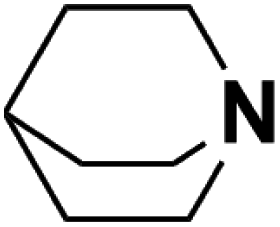 |
20.54 | 19.70d | 60 | A | 85.4 |
| 10 | N-Methylpyrrolidine | 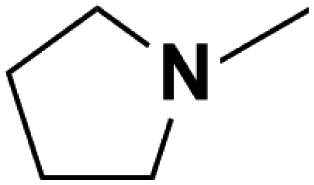 |
20.59 | 18.42c | 240 | A | 89.2 |
| 11 | TBD | 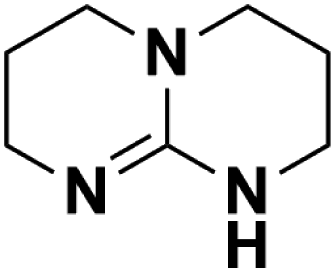 |
16.162 | 26.02d | 50 | A | 93.2 |
The key role of catalyst basicity can be clearly seen from results depicted in Fig. 4, in which OH-conversion versus pKa has been plotted for two catalyst groups consisting of catalysts with similar nucleophilicity. The first one consists of three catalysts (DAMP, DBN and TBD) having an average N of 15.98 with a low RSD of 2.59%. As it can be seen, conversion increases swiftly and linearly with basicity, from 9.6% for pKa 17.96 (DMAP) to 93.2% for pKa 26.02 (TBD). Importantly, the correlation is very good as shown by the determination coefficient R2 = 0.9944. The second one is composed by six catalysts (the previous three plus N-methylmorpholine, triethylamine and DBU) having an average N of 16.19 with a something higher but still low RSD of 4.35%. OH-conversion also increases very quickly with pKa. The curve has been fitted to a second-degree polynomial equation and, although the determination coefficient (R2 = 0.8083) is not so good than before, it is high enough (correlation coefficient r = 0.8990) to conclude that a direct correlation exists. The poorer correlation relative to the previously one with three catalysts is due to the higher RSD for nucleophilicity parameter considering its higher influence on catalytic activity than pKa as it will set forth herein later.
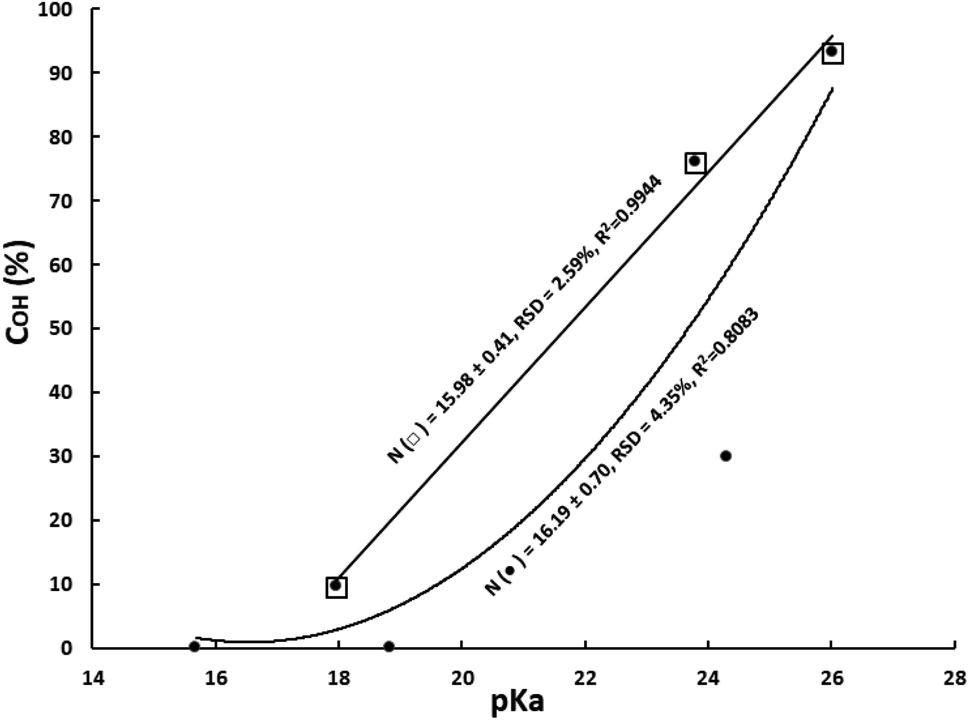 | ||
| Fig. 4 Variation of OH-conversion with pKa. (□): entries 4, 7 and 11 in Table 3; (●) entries 1, 2, 4, 5, 7 and 11. | ||
For a clearer analysis of the pKa–N interdependent influence on the catalytic activities of these catalysts, both values for each catalyst versus the OH-conversion achieved with them have been plotted in Fig. 5.
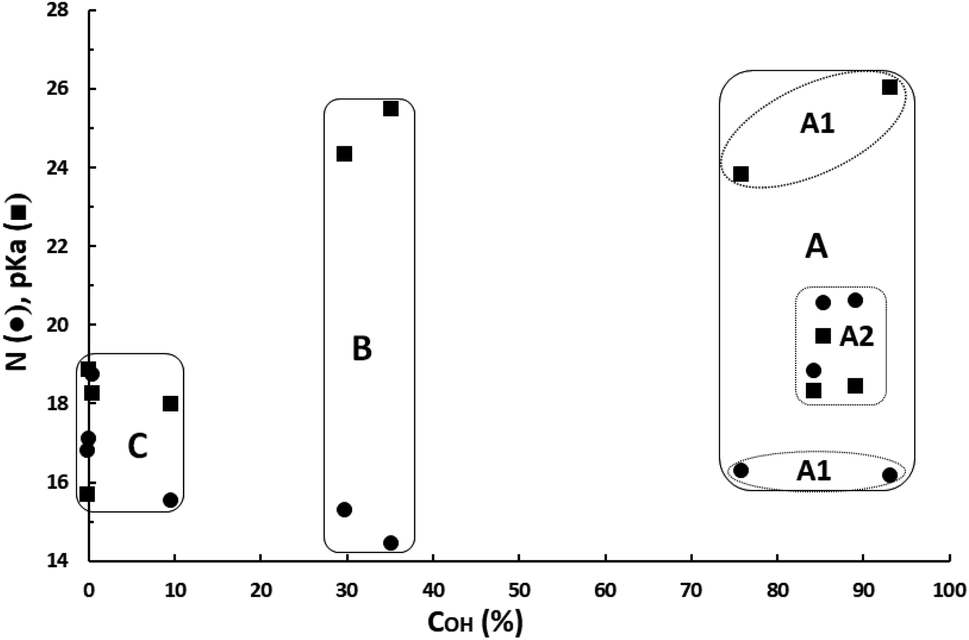 | ||
| Fig. 5 Correlations between nucleophilicity (N), basicity (pKa) and OH-conversion (COH). | ||
Catalysts can be classified in three clusters:
- Cluster A: very active superbases catalysts having a pKa between 18.29 and 26.02 (∼18.0–26.0), a N value between 16.16 and 20.59 (∼16.0–20.6) and leading to COH between 76% and 93%. This cluster consists of two subgroups: A1 consisting of two superbases (TBD and DBN) having a pKa between 23.79 and 26.02 and a N value slightly higher than 16; and A2 consisting of three basic catalysts (N-methylpyrrolidine, DABCO and quinuclidine) having a pKa between 18.29 and 19.70, and a N value between 18.80 and 20.59. According to the classical definition of superbase (pKa ≥ pKa of proton sponge = 18.62 in ACN) the basicities of this last subgroup are just in the border of superbases.
- Cluster B: moderately active superbases catalysts having a pKa between 24.31 and 25.47 (∼24.0–25.5), a N value between 14.43 and 15.29 (∼14.0–15.5) and leading to COH between 30% and 35%.
- Cluster C: catalysts with none or very low catalytic activity having a pKa between 15.68 and 18.83 (∼15.5–19.0), a N value between 15.51 and 18.72 (∼15.5–19.0) and leading to COH below 10%.
Despite basicities of the two superbases belonging to cluster B are like those of superbases of subgroup A1 (TBD and DBN) in cluster A, they lead on average to a COH 55 points lower than that for these catalysts. This can be only explained because they have a nucleophilic power (average N of 14.86) ∼23-fold lower than that for TBD and DBN (average N of 16.22). Catalysts of subgroup A2 (N-methylpyrrolidine, DABCO and quinuclidine) in cluster A have on average a basicity of about 105.5-fold lower and a nucleophilicity of about 103.7-fold higher than the first subgroup in the same cluster, indicating that from the catalytic activity standpoint the negative effect of the decrease in basicity is compensated by the positive effect of the increase in nucleophilicity and suggesting that nucleophilicity has a higher weight on catalytic activity than basicity. It can be concluded, on one hand, that bases and superbases with a pKa in ACN between about 18.0–26.0 must have a minimum N of about 16 to be good catalysts for the reaction herein studied. And, on the other hand, that the catalytic activity can be kept high when pKa decreases if N increases. Comparing average pKa–N data for subgroups A1 and A2, it can be deduced that N must increase by ∼0.60 units per each unit the pKa falls, in order to keep the catalyst activity constant.
Excluding N-methylpiperidine, results for catalysts in cluster C show that basic nitrogenated chemicals having both pKa and N values below 17–18 will not have any catalytic activity in the reaction herein studied. N-Methylpiperidine shows an anomalous catalytic behavior because its pKa–N values are practically equal to those for DABCO (cluster A, subgroup A2), a very good catalyst. A plausible explanation is that the three alkyl groups bonded to each nitrogen in DABCO are “tied-back” in a bicyclic structure leaving more loose the unpaired electrons of the nucleophilic nitrogen atoms to attack the electrophilic carbon in DMC, while the unpaired electrons of the nitrogen in N-methylpiperidine are more constrained, more sterically hindered, due to the methyl group. In fact, as stated by Mayr's group, the equation used for developing the reactivity parameters, with N being one of them, scale “does not include steric effects and, therefore, can only be used for semiquantitative predictions of rate constants”.19 The increased nucleophilicity of N-methylpyrrolidine, similar in structure to N-methylpiperidine but with a high catalytic activity in the synthesis of IBMC, can be explained as due to its smaller cycle size that overcomes steric hindrance in this reaction.
From the above discussion is deduced that in this reaction nucleophilicity has a higher weight than basicity on the catalytic activity of the nitrogenated bases studied. A further support for this conclusion can be obtained by fitting the OH-conversions data in Table 1, excluding the “anomalous” N-methylpiperidine base, to a linear equation of two independent variables, N and pKa. The resulting eqn (2) is as follows:
| COH = −430.76 + 17.52N + 8.66pKa | (2) |
Having a determination coefficient R2 = 0.7827 and a multiple correlation coefficient r of 0.8847, enough high for concluding that nucleophilicity has approximately twice as much influence as basicity on the catalytic activity. The equation predicts that a strong superbase such as 1,1,3,3-tetramethylguanidine (pKa = 23.35 in ACN, N = 13.58 in DMC) with pKa–N values out of the clusters plotted in Fig. 5 is not a suitable catalyst for this reaction (experimental COH = 2.3% in 120 min, predicted one = 9.4%).
Proposed reaction mechanism
According the above discussion, if properly balanced the dual basic-nucleophilic character of the catalyst drives the reaction according to the simplified mechanism proposed in Scheme 3: The catalyst basicity, if suitable, increases the nucleophilic character of hydroxyl groups in ISO by proton abstraction, while the catalyst nucleophilicity, if suitable, allows its reaction with DMC leading to a highly reactive acyl-base intermediate 1 in which the carbon in the carbonyl moiety becomes strongly electrophilic. Consequently, the activated hydroxyl groups in isosorbide react easily and quickly with the acyl-base intermediate 1 by attacking its highly electrophilic carbon atom regenerating the catalyst and introducing methoxycarbonyl moieties into isosorbide resulting in IBMC and methanol with catalyst regeneration. An alternative mechanism could be one in which the abstraction of the protons from the hydroxyl groups in ISO is not done by the nitrogenated catalysts but by the strongly basic methoxide ion being the anion in the acyl-base intermediate 1. However, this approach would mean that the catalytic activity of the nitrogenated base lies exclusively on its nucleophilicity with its basicity playing no role, which is not true as it has been demonstrated herein above (see Fig. 4).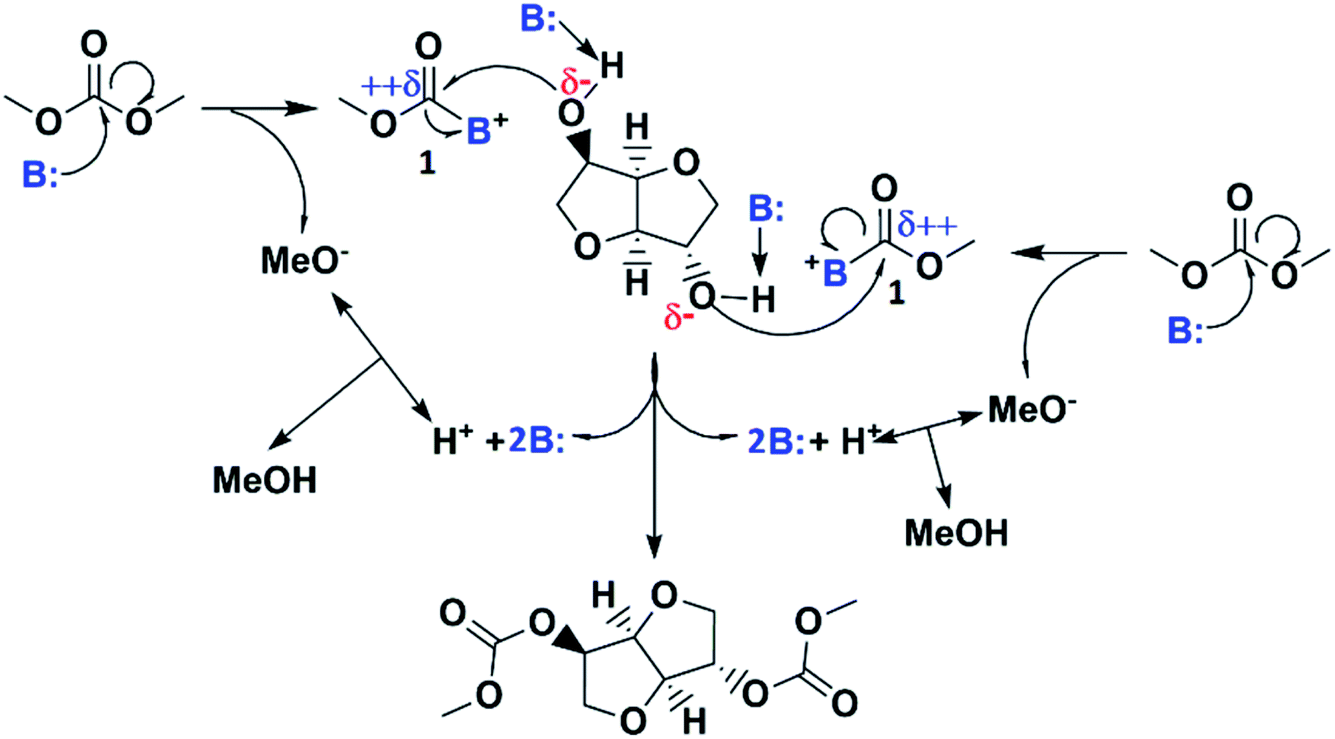 | ||
| Scheme 3 Simplified proposed reaction mechanism. | ||
This proposed reaction mechanism caused by the high activity of catalysts with a dual and suitable basic–nucleophilic character is supported by the above mentioned proposed mechanism for IBMC synthesis from ISO and DMC using 4-substituted phenolate ILs as catalysts,10 and also by other mechanisms reported in literature for other reactions. Thus, evidence of the acyl transfer activity of TBD has been reported, e.g., in the transesterification of vinyl acetate with benzyl alcohol in which N-acetyl-TBD was formed, together with acetaldehyde, as a stable and neutral compound by reaction of vinyl acetate and TBD in a 1:1 MR, compound which was in turn converted into benzyl acetate with regeneration of TBD after addition of benzyl alcohol to the reaction medium.20 Other example is the aminolysis of esters with primary amines as described by Kiesewetter et al.,14 in which TBD reacts reversibly with esters to yield an acyl-TBD intermediate that acylates amines to generate the amides with TBD regeneration. These authors report also that the analogous bicyclic guanidine 1,4,6-triazabicyclo[3.3.0]oct-4-ene (TBO, N = 14.44 in CH2Cl2,16 52.5-fold lower than that of TBD) is a much less active acylation catalyst than TBD concluding that the higher reactivity of TBD is due to both its higher basicity and nucleophilicity than TBO as well as the high reactivity of the acyl-TBD intermediate. Likewise, Sabot et al., reported the high efficiency of TBD in the aminolysis of esters with primary and secondary amines unlike MTBD despite both have similar basicities.21 The efficiency of TBD was ascribed to its activity as a bifunctional nucleophilic organocatalyst, with the nucleophilicity of TBD being the key difference between both superbases because it is 53.7-fold higher than that of MTBD, as reported in Table 1.
Likewise, evidence of the acyl transfer activity has been reported for DBU for the synthesis of N-heteroaryl unsymmetrical ureas,22 and for the N-carbonylation of N-heteroaromatics,23 and also for DABCO in other reactions in which DMC is involved and it was attributed to the nucleophilic character of this bicyclic tertiary diamine. Thus, Shieh et al. proposed the formation of an acyl intermediate such as 1 in Scheme 3, as a key intermediate in the N-methylation of indoles catalyzed by DABCO using DMC as a methylating agent.24 The clear interaction between DABCO and DMC has been shown by Munshi et al. who reported the synthesis of a DABCO-DMC IL by refluxing DABCO and DMC in a DMC/DABCO MR of 6 for 5 h.25 Finally, further reactions involving an interaction between DMC and the nitrogenated organocatalysts DBU, TBD and DABCO as an intermediate step to the final product can be found in a recent review by Tundo et al.26
An important issue derived from the proposed reaction mechanism and the above discussion is that having a suitable N–pKa values is not enough for a catalyst to have a high catalytic activity in the synthesis of IBMC. In addition, it must be able to form a highly reactive intermediate, such as 1 in Scheme 3, with the reactant having the electrophilic center, i.e., an unstable intermediate which can easily evolve to the target chemical. This explains why some nitrogenated bases, such as the cycloaliphatic secondary amines pyrrolidine, perhydroazepine, piperidine and piperazine, having N–pKa values predicting moderate to good catalytic activities according to eqn (2) have null or near to null catalytic activities, as shown by results given in Table 4.
| Base | Structure | N1 | pKa | COH-p (%) | COH-exp (%) |
|---|---|---|---|---|---|
| a Reaction conditions: ISO-2.81: 2 g; batch (30 mL); reflux; DMC/ISO-2.81 molar ratio: 30. Catalyst concentration: 5 mol% vs. ISO-2.81. Reaction time (tr) till constant OH-conversion was achieved = 120 min.b pKa values in ACN; 1N values in ACN except for 2in DCM; 1Within that geometric form in Fig. 5.c Ref. 15.d Calculated as given in ESI. | |||||
| Pyrrolidine | 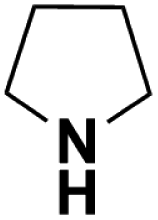 |
18.52 | 19.62c | 63.7 | 1.0 |
| Perhydroazepine | 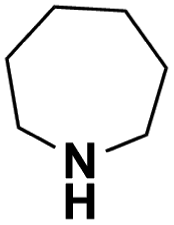 |
18.292 | 19.31d | 57.0 | 1.0 |
| Piperidine | 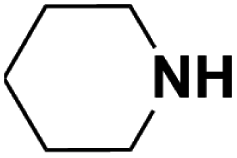 |
17.35 | 19.35c | 40.8 | 4.1 |
| Piperazine | 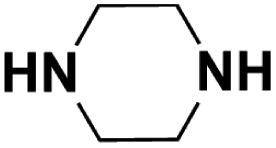 |
17.222 | 18.69c | 32.8 | 6.1 |
Indeed, it is known that secondary amines react with DMC resulting in stable urethanes,26 which in our opinion is the cause of their either null or very poor catalytic performance in the conversion of ISO into IBMC. However, they act as catalysts in other reactions in which form unstable and highly reactive intermediates driving the reactions to completion. For instance, piperidine catalyzes the Knoevenagel condensation reaction of acetylacetone with benzaldehyde. The latter reacts with piperidine yielding a carbinolamine which decomposes via hydroxide ion elimination to yield an iminium cation. The hydroxide ion deprotonates the central methylene group in acetylacetone, forming an enolate that attacks the iminium cation and leads to an unstable addition intermediate which finally evolves to the target product with regeneration of piperidine catalyst.27 A similar mechanism involving the reaction between a cycloaliphatic secondary amine catalyst and an aldehyde to yield a carbinolamine intermediate which decomposes to an iminium cation, which in turn reacts with the enolate of the aldehyde to yield an unstable intermediate, which finally evolves to the target chemical with regeneration of the catalyst, is proposed by Ishikawa et al. for the pyrrolidine-catalyzed homo-aldol condensation reactions of aldehydes.28 Finally, the piperidine- and pyrrolidine-catalyzed cycloaddition of 1-isopropyl-2-phenylaziridine with phenyl isothiocyanate to yield the desired thiazolidin-2-ylidene, in which a highly reactive urea-type intermediate is formed by reaction of the catalyst and the isothiocyanate in a first step, which in a second step reacts with the azidirine to yield the desired product with catalyst regeneration constitutes another example of a highly reactive reactant-catalyst intermediate needed to complete the desired reaction.29
Oligomer formation
Compositions of isolated crude products after solvent evaporation for reactions carried out with the very active catalysts belonging to cluster A in Fig. 5 are given in Table 5. Despite it is generally known that DMC leads to methoxycarbonylations at 90 °C whereas at temperatures > 120–150 °C methylations occur,6a the quantitative O-methylation of isosorbide with DMC at 90 °C, the temperature of IBMC synthesis, using sodium methoxide in stoichiometric excess as a base has been reported.30 In the present study, a complete analysis of the product composition was only carried for reactions carried out with the highly active catalysts reported in Table 5 and no methylation product was detected by GC-MS as well as for reactions catalyzed by potassium and cesium carbonate.12 For the other catalysts with low to moderate yields (entries 4, 5 and 6 in Table 3) the formation of methoxycarbonyls chemicals was obvious as show by the continuous growth of the carbonyl band at 1750–1755 cm−1 in the ATR-FTIR spectra.For reactions reported in Table 5 oligomers consisted exclusively of dimer and trimer, with the dimer affording for at least 95% of total oligomers. Therefore, data for dimer contents are reported as the sum of dimer plus trimer. The variation of IBMC and IMMC contents with OH-conversions follow a logic pattern according to the reaction nature as depicted in Scheme 2: IMMC content decreases at the same rate than that of IBMC increases when OH-conversion increases. In fact, the slopes and determination coefficients of the linear fittings for IBMC vs. COH and IMMC vs. COH are 2,17 and 0.9914, and 2,11 and 0.9904, respectively. IMMC is largely being converted into IBMC as OH-conversion increases independently of the catalyst type. Seemingly, within the OH-conversion range and with the type of catalysts analyzed the dimer content is constant as shown by the RSD (19.3%) of its average value (6.0 wt%) which is acceptable considering that the analytical error always increases when the analyte content is low.
Therefore, none of the catalysts studied (1 guanidine, 1 amidine and three cycloaliphatic tertiary amines) avoid the formation of oligomers. This issue, out of the scope of this paper, must be treated under a kinetic perspective looking for conditions able to minimize the reaction rate of the oligomerization reactions relative to that of IBMC formation, such as the diminution in catalyst load11,12 and/or a careful control of the other reaction parameters.
Conclusions
The parameters influencing the catalytic activity of catalysts suitable for the synthesis of IBMC by transesterification with DMC in batchwise mode at reflux temperature has been studied. The catalytic performance is strongly and negatively affected by the presence of acidic impurities in ISO. The typical heterogeneous potassium and cesium carbonate catalysts are deactivated when ISO acidity is 2.81 (pH of a 1 M aqueous solution of ISO) such that amounts much higher than the catalytic ones are needed to restore their activity. The problem can be overcome by using homogeneous catalysts consisting of nitrogenated bases and superbases if they have a suitable dual nucleophilic–basic character and are able to form a highly reactive acyl intermediate with the electrophilic reactant DMC. Cycloaliphatic secondary and tertiary amines, guanidines an amidines covering a nucleophilicity parameter (N) range between 13.58 and 20.58 in either ACN or DMC, and a pKa range in ACN between 15.68 and 26.02 have been tested. Highly active catalysts leading to hydroxyl conversions of 84–93% require a minimum N of 16 and a pKa ranging from 18.0 to 26.0. A catalyst having a N of 16 requires a pKa between 23.5 and 26.0 to show high catalytic activity. Within this pKa range, N must increase by about 0.5–0.6 units per each unit the pKa falls to keep the catalytic activity, indicating that nucleophilicity has approximately twice as much influence as basicity on the catalytic activity.One guanidine (TBD), one amidine (DBN) and three cycloaliphatic secondary amines (N-methylpyrrolidine, quinuclidine and DABCO) have been found to be excellent catalysts at 5 mol% vs. ISO. Cycloaliphatic secondary amines, such as, e.g., perhydroazepine and pyrrolidine, despite having the suitable nucleophilicity–basicity combination, show none or very poor catalytic activity due to the formation of stable urethanes by reaction with DMC.
The side reaction leading to oligomer formation is not avoided, with oligomers, mainly the dimer, affording for 6 wt% of the crude product independently of hydroxyl-conversions and catalyst-type.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| ACN | Acetonitrile |
| COH (%) | Conversion(s) of total hydroxyl groups contained by ISO and IMMC, as obtained by ATR-FTIR |
| IBMC | Isosorbide bis(methyl carbonate) (l,4:3,6-dianhydro-2,5-bis-O-(methoxy-carbonyl)-D-glucitol) |
| IL(s) | Ionic liquid(s) |
| IMMC | Isosorbide mono(methyl carbonate) |
| ISO | Isosorbide (1,4:3,6-dianhydro-D-glucitol), ISO-X (X = 2.81, 3.42, 7.60). ISO providing a pH of X in 1 M aqueous solution |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DBN | 1,5-Diazabicyclo[4.3.0]non-5-ene |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DMAP | 4-(Dimethylamino)pyridine |
| DMC | Dimethyl carbonate |
| MDI | Methylene diphenyl diisocyanate |
| MR | Molar ratio |
| MTBD | 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene |
| NIPUs | Non-isocyanate polyurethanes |
| RSD | Relative standard deviation (%) |
| TBD | Triazabicyclodecene |
| TEA | Triethylamine |
Acknowledgements
This paper is a part of the research carried out within the VIPRISCAR project which has received funding from the Bio Based Industries Joint Undertaking (JU) under the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No. 790440. The JU receives support from the European Union's Horizon 2020 research and innovation programme and the Bio Based Industries Consortium. Thanks are due to research teams involved in VIPRISCAR project for valuable discussion during project meetings: Dr Stefaan De Wildeman et al. (B4Plastics, Belgium), Dr Yaying Chen et al. (Exergy, UK), Dr Daniel Klein et al. (Jowat SE, Germany), Mrs Elena Benedetti et al. (AEP Polymers, Italy), Dr Erasmo Cadena et al. (Vertech Group, France), Dr José-María Cuevas (Gaiker IK4, Spain) and Dr Sandra Medel (Leitat, Spain). Thanks are also due to Mr Robert Kirschbaum (SakuragiConsult) and Mr Alan Patrick Giles (Rawlings Giles), members of the VIPRISCAR's Advisory Board for valuable advices about market issues. Special acknowledgment is due to Dr Soraya Prieto (Tecnalia) for smooth project management.References
- (a) M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed; (b) C. Dussenne, T. Delaunay, V. Wiatz, H. Wyart, I. Suisse and M. Sauthier, Green Chem., 2017, 19, 5332–5344 RSC.
- J. R. Ochoa-Gómez and T. Roncal, in Production of Platform Chemicals from Renewable Resources, ed. Z. Fang, R. L. Smith Jr and X. Qi, Springer Book Series - Biofuels and Biorefineries, Singapore, 2017, ch. 9, vol. 7, pp. 265–310 Search PubMed.
- A. M. Nelson and T. E. Long, Polym. Int., 2012, 61, 1485–1491 CrossRef CAS.
- VIPRISCAR: Validation of an industrial process to manufacture isosorbide bis(methyl carbonate) at pilot level. https://vipriscar.eu/ Search PubMed.
- T. Abe, S. Saibara, H. Katayama, S. Aoyama, K. Aoi, M. Yokoe, and M. Okada, US Pat. 7455935B2, 2008.
- (a) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed; (b) P. Tundo, Pure Appl. Chem., 2001, 73, 1117–1124 CAS; (c) G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC; (d) M. Selva, A. Perosa, D. Rodríguez-Padrón and R. Luque, ACS Sustainable Chem. Eng., 2019, 7, 6471–6479 CrossRef CAS.
- P. Fuertes, M. Ibert, E. Josien, P. Tundo and F. Aricò, US Pat. 8339601B2, 2013.
- F. Aricò, S. Evaristo and P. Tundo, ScienceOpenResearch, 2014 DOI:10.14293/S2199-1006.1.sorchem.ab3r7e.v2.
- (a) J. S. Brimacombe, A. B. Foster, M. Stacey and D. H. Whiffen, Tetrahedron, 1958, 4, 351–360 CrossRef CAS; (b) R. U. Lemieux and A. G. McInnes, Can. J. Chem., 1960, 88, 136–140 CrossRef; (c) J. C. Goodwin, J. E. Hodge and R. Weisleder, Carbohydr. Res., 1980, 79, 133–141 CrossRef CAS; (d) K. W. Buck, J. M. Duxbury, A. B. Foster, A. R. Perry and J. M. Webber, Carbohydr. Res., 1966, 2, 122–131 CrossRef CAS; (e) P. Che, F. Lu, X. Nie, Y. Huang, Y. Yang, F. Wang and J. Xu, Chem. Commun., 2015, 51, 1077–1080 RSC.
- W. Qian, X. Tan, Q. Su, W. Cheng, F. Xu, L. Dong and S. Zhang, ChemSusChem, 2019, 12, 1169–1178 CrossRef CAS PubMed.
- J. R. Ochoa-Gómez, S. Gil-Río, B. Maestro-Madurga, L. Lorenzo-Ibarreta, and O. Gómez-Jiménez-Aberasturi, US Pat. 9540390 B2, 2017; J. R. Ochoa-Gómez, S. Gil-Río, B. Maestro-Madurga, L. Lorenzo-Ibarreta, and O. Gómez-Jiménez-Aberasturi, Jpn. Pat. 6130516B2, 2017; J. R. Ochoa-Gómez, S. Gil-Río, B. Maestro-Madurga and L. Lorenzo-Ibarreta, and O. Gómez-Jiménez-Aberasturi, Eur. Pat. 2949654B1, 2017.
- J. R. Ochoa-Gómez, S. Gil-Río, B. Maestro-Madurga, O. Gómez-Jiménez-Aberasturi and F. Río-Pérez, Arabian J. Chem., 2019, 12, 4764–4774 CrossRef.
- T. Ishikawa, Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, John Wiley & Sons, Wiltshire, 2009 Search PubMed.
- M. K. Kiesewetter, M. D. Scholten, N. Kirn, R. L. Weber, J. L. Hedrick and R. M. Waymouth, J. Org. Chem., 2009, 74, 9490–9496 CrossRef CAS PubMed.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 2019, 6735–6748 CrossRef CAS.
- H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938–957 CrossRef.
- https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/, last access 2020/03/11.
- K. T. Leffek, P. Pruszynski and k. Thanapaalasingham, Can. J. Chem., 1989, 67, 590–595 CrossRef CAS.
- https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank2/pages/show_page/7, last access 2020/03/28.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- C. Sabot, K. A. Kmar, S. Meunier and Ch. Mioskowski, Tetrahedron Lett., 2007, 48, 3863–3866 CrossRef CAS.
- M. Carafa, V. Mele and E. Quaranta, Green Chem., 2012, 14, 217–225 RSC.
- M. Carafa, F. Iannone, V. Mele and E. Quaranta, Green Chem., 2012, 14, 3377–3385 RSC.
- W.-C. Shieh, S. Dell, A. Bach, O. Repič and T. J. Blacklock, J. Org. Chem., 2003, 68, 1954–1957 CrossRef CAS PubMed.
- M. K. Munshi, S. M. Gade, V. H. Rane and A. A. Kelkar, RSC Adv., 2014, 4, 32127–32133 RSC.
- P. Tundo, M. Musolino and F. Aricò, Green Chem., 2018, 20, 28–85 RSC.
- E. V. Dalessandro, H. P. Collin, L. Gustavo, L. Guimarães, M. S. Valle and J. R. Pliego Jr, J. Phys. Chem. B, 2017, 121, 5300–5307 CrossRef CAS PubMed.
- T. Ishikawa, E. Uedo, S. Okada and S. Saito, Synlett, 1999, 4, 450–452 CrossRef.
- M. Sengoden, M. Vijay, E. Balakumar and T. Punniyamurthy, RSC Adv., 2014, 4, 54149–54157 RSC.
- P. Tundo, F. Aricò, G. Gauthier, L. Rossi, A. E. Rosamilia, H. A. Bevinakatti, R. L. Sievert and C. P. Newman, ChemSusChem, 2010, 3, 566–570 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03552a |
| ‡ Part of this article was presented by José R. Ochoa-Gómez as a Keynote speaker in the Global Chemical Engineering and Chemistry Conference Expo, Valencia (Spain), March 25th–26th, 2019. |
| This journal is © The Royal Society of Chemistry 2020 |