
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetite ultrafine particles/porous reduced graphene oxide in situ grown onto Ni foam as a binder-free electrode for supercapacitors
Yingling Mao *ab,
Benhu Zhoua and
Sen Penga
*ab,
Benhu Zhoua and
Sen Penga
aCollege of Science, ShaoYang University, ShaoYang, 422000, China. E-mail: yinglingmao@126.com
bHunan Provincial Key Laboratory of Grids Operation and Control on Multi-Power Sources Area, ShaoYang, 422000, China
First published on 1st June 2020
Abstract
Here, we report a simple and green electrochemical route to fabricate a porous network of a Fe3O4 nanoparticle-porous reduced graphene oxide (p-rGO) nanocomposite supported on a nickel-foam substrate, which is directly used as a binder-free charge storage electrode. Through this method, pristine Fe3O4 NPs/Ni, p-rGO/Ni and Fe3O4 NPs@p-rGO/Ni electrodes are fabricated and compared. In the fabricated Fe3O4 NPs@p-rGO/Ni electrode, the porous rGO sheets served as a conductive network to facilitate the collection and transportation of electrons during the charge/discharge cycles, improving the conductivity of magnetite NPs and providing a larger specific surface area. As a result, the Fe3O4 NPs@p-rGO/Ni exhibited a specific capacitance of 1323 F g−1 at 0.5 A g−1 and 79% capacitance retention when the current density is increased 20 times, where the Fe3O4 NPs/Ni electrode showed low specific capacitance of 357 F g−1 and 43% capacity retention. Furthermore, the composite electrode kept 95.1% and 86.7% of its initial capacitances at the current densities of 1 and 4 A g−1, respectively, which were higher than those of a Fe3O4/NF electrode at similar loads (i.e. 80.4% and 65.9% capacitance retentions at 1 and 4 A g−1, respectively). These beneficial effects proved the synergistic contribution between p-rGO and Fe3O4. Hence, such ultrafine magnetite particles grown onto a porous reduced GO network directly imprinted onto a Ni substrate could be a promising candidate for high performance energy storage aims.
1. Introduction
Electrochemical capacitors, also known as supercapacitors, have the potential to emerge as promising energy storage devices due to positive characteristics such as short charging time, long cycle life, and high power and energy density.1 Among the different components of an electrochemical capacitor, the electrode material is of great importance. However, between various electrode materials utilized for this purpose, transition metal oxides have been proved to be favourable electrode materials for supercapacitors.2 For example, hydrous ruthenium dioxide (RuO2·nH2O) has been found to have great potential for manufacturing advanced electrode materials because of its outstanding properties, which include high theoretical specific capacitance (720 F g−1) as well as high electrical conductivity.3 However, large-scale applications of this material for supercapacitors are definitely hindered by its poisonous effect, scarcity, and high cost. Hence, design of a desirable low-cost electrode material with a high cycling stability and high energy density is crucial for electrochemical energy storage. Therefore, researchers are searching for cheaper and more environmentally-friendly metal oxides with improved characteristics such as nickel oxide, cobalt oxide, manganese oxide, vanadium oxide and iron oxides.4–6 Among these compounds, iron-based materials have been receiving considerable attention because of their low cost, earth abundance, high theoretical capacitance and eco-friendly nature.7 It is worth mentioning that iron oxides have been extensively considered as the negative electrode materials for supercapacitor.8 However, iron oxide electrodes still suffer from relatively low capacitance (<300 F g−1) and long-cycle life in aqueous electrolyte,8 originating from their low diffusion coefficient for electron transfer as well as low electrical conductivity and also structural deterioration during consecutive charge/discharge cycles. As known, the incorporation iron oxide nanoparticles into carbonaceous materials (such as nanotubes, graphene and graphene oxide) can significantly enhance the electrochemical performance of composite material in comparison to single iron oxide component.9Graphene is an intriguing two-dimensional carbon nanomaterial that is widely utilized to design fascinating electrode materials for supercapacitors because of its ultra-thin nanostructure, excellent electrical conductivity, and mechanical stability along with very high theoretical specific surface area (2700 m2 g−1).10,11 Recently, researchers have reported extended works on the application of graphene-based/iron oxide composites as electrochemical materials.12–14 In the graphene-based/iron oxide hybrids, electrochemical performances are improved due to the synergistic effects between graphene and iron oxide-based structures. In these systems, graphene has offered not only a conductive backbone to facilitate electron transport, but also guarantees mechanical stability that maintains the integrity of the electrode during the electrochemical processes.15 In addition, graphene with metal oxide can be an economic and promising route to attain high supercapacitive performance. In this regard, magnetite/reduced graphene oxide (Fe3O4/rGO) has received increasing research interest due to the structural stability, large surface area, excellent electrical conductivity, strong magnetic properties as well as wide electrochemical potential window.16,17 However, there are some works reporting the preparation of Fe3O4/rGO applied in supercapacitor and lithium-ion batteries.18–23 For example, Devi et al.19 synthesized Fe3O4/rGO by using a one-step chemical reduction method, and a maximum specific capacitance was achieved at 416 F g−1 with capacitance retention of 88.57% for Fe3O4/rGO nanocomposite. Beyazay et al.20 reported free-standing N-doped reduced GO papers decorated with magnetite nanoparticles prepared by chemical reduction followed by thermal annealing, and stable capacitive behavior was observed for the fabricated iron oxide/N-doped rGO electrode with showing 203 F g−1 at 0.5 mA cm−2. Madhuvilakku et al.21 reported Fe3O4 and Fe3O4/rGO nanoflowers synthesized by facile biogenic reductant route and mentioned that the presence of synergic between components in Fe3O4/rGO shorten ion diffusion path length and promotes capacitances delivered by Fe3O4/rGO/Ni foam, where this capability was correlated to the increased electrode surface area, facilitated electron transfer kinetics and highly stability Fe3O4/rGO matrix. Three-dimensional (3D) reduced graphene oxide nanosheets (rGO NSs) containing iron oxide nanoparticles (Fe3O4 NPs) have been also fabricated by one-pot microwave approach,22 and the prepared 3D hybrid electrode has shown good cycling stability with a retention ratio of 91.4% after starting up to 9600 cycles. Zhao et al.23 prepared Fe3O4/reduced graphene oxide (RGO/Fe3O4) nanocomposite through in situ hydrothermal grown on Fe foil, and observed an excellent electrochemical performance with a high capability of 337.5 mF cm−2 at 20 mA cm−2 and a superior cyclability with only 2.3% capacity loss from the 600th to the 2000th cycle.
Reviewing the literature revealed that the Fe3O4/rGO hybrids have been prepared through solvothermal, sol–gel, hydrothermal and hydrolysis methods. These synthesis methods are rather complex and difficult to control, and usually need to toxic reductive agents for chemical reduction of graphene oxide (i.e. hydrazine). So, it is necessary to explore a simpler and more adopted synthesis method of iron oxide-based composites with enhanced electrochemical performance. Here, we introduce a facile electrochemical/electrophoretic platform for direct grown of iron oxide onto porous reduced graphene oxide, which co-deposited on the Ni foam (i.e. Fe3O4@rGO/Ni foam) as binder-free high-performance electrode for energy storage applications i.e. supercapacitor usage. The developed electrochemical-based strategy for the preparation of Fe3O4/rGO composites is carried out without using any reductive agent and subsequent heating process. Notably, electrochemical deposition i.e. cathodic electrodeposition has been reported that is an efficient, simple and inexpensive route for synthesis of nanostructured metal hydroxides and oxides,24,25 but this deposition strategy has not been applied for preparation of Fe3O4–porous rGO composites. The developed procedure is based on the simultaneously electrophoretic deposition of graphene oxide sheets followed Ni foam by in situ cathodic formation of Fe3O4 nanoparticles onto the rGO sheets. Using this method, two types of binder-free electrodes i.e. Fe3O4@Ni foam and Fe3O4@rGO/Ni foam electrodes were respectively prepared in electrolyte containing iron salts and iron salts/graphene oxide suspension, and the prepared electrodes were characterized using XRD, IR, BET, Raman, DTA-TG, TEM and FE-SEM techniques. The charge storage capabilities of the fabricated electrodes were evaluated through electrochemical tests of cyclic voltammetry (CV), EIS and galvanostatic charge–discharge (GCD), and the results were compared to evaluate the effects of porous reduced graphene oxide on the supercapacitive ability of Fe3O4 electrode material.
2. Experimental procedure
2.1. Materials
All materials were of analytical purity and utilized as received without any purification. All solutions prepared in DI water (18 MΩ). H2SO4 (98%, Merck), KMnO4 (99.8%, Sigma-Aldrich), H2O2 (30%, Sigma-Aldrich), NaNO3 (99.9%, Merck) and Na2SO4 (99.9% Merck) were used as the reactant materials.2.2. Preparation of porous graphene oxide (p-GO)
Graphite oxide was synthesized from natural graphite by a modified Hummers method.26,27 Briefly, 3 g of graphite powder was added to 69 mL of concentrated H2SO4 with stirring in an ice bath, followed by addition of 1.5 g of NaNO3 into the mixture. While maintaining continuous stirring, 9 g of KMnO4 was added gently to the suspension. The rate of addition was controlled carefully to prevent the raising of the temperature to more than 20 °C. Then, the ice-bath was removed and the temperature of the suspension was brought to 35 °C, where it was maintained at this temperature for 30 min. Subsequently, 137 mL of deionized water was slowly added into the mixture, causing an increase in temperature to 98 °C, and the mixture was maintained at this temperature for 15 min. An additional 140 mL of H2O was added to dilute the suspension, and 10 mL of 30% H2O2 was injected into the solution to completely react with the excess KMnO4. Then, the resulting mixture was filtered and washed with 10% HCl solution, deionized water and ethanol in successive to remove other ions trapped within the graphene oxide nanosheets. The collected material was dried in vacuum at 60 °C overnight to obtain brown GO powder.2.3. Preparation of pristine Fe3O4 and Fe3O4@p-rGO composite
The schematic view of the preparation procedure is shown in Fig. 1. A typical two-electrode containing electrochemical cell was used in the deposition experiments. In fact, the electrosynthesis set-up was composed of a Ni foam/Ni foil (surface area = 2 cm2) centered between two parallel graphite anodes. The electrodeposition runs were carried out by an electrochemical workstation system (Potentiostat/Galvanostat, Model: GW-2201) in galvanostatic mode with applying a current density of 10 mA cm−2. The deposition time and bath temperature were 30 min and 40 °C, respectively. In the electrodeposition of pristine Fe3O4 sample, an additive-free solution of 400 mg Fe(III) nitrate and 200 mg Fe(II) chloride in 200 mL of deionized water was used. For synthesis of Fe3O4/rGO composite, the same electrochemical set up was used, and only the composition of electrolyte was changed. The electrodeposition bath was prepared as follow: first, 50 mg p-GO was dispersed in 200 mL deionized water without addition using sonication for 1 h. Then, 400 mg of Fe(III) nitrate and 200 mg of Fe(II) chloride were added into p-GO dispersed solution and stirred for 4 h. The resulted suspension was used as the electrodeposition bath in the electrodeposition of Fe3O4/p-rGO composite. For deposition of the Fe3O4/p-rGO composite onto Ni foam support, current density of 10 mA cm−2 was applied into the above mentioned two-electrode system for 30 min (Fig. 1). After deposition, the nickel foam support was washed several times with the deionized water and dried at 80 °C for 4 h. Both fabricated electrodes (i.e. pristine Fe3O4@Ni foam and Fe3O4@p-rGO/Ni foam) were directly used as working electrodes in the electrochemical tests. The powder forms of both samples were also synthesized through the same deposition route with only substitution of nickel foam by nickel foil. The obtained pristine oxide and composite powders were characterized by different analyses.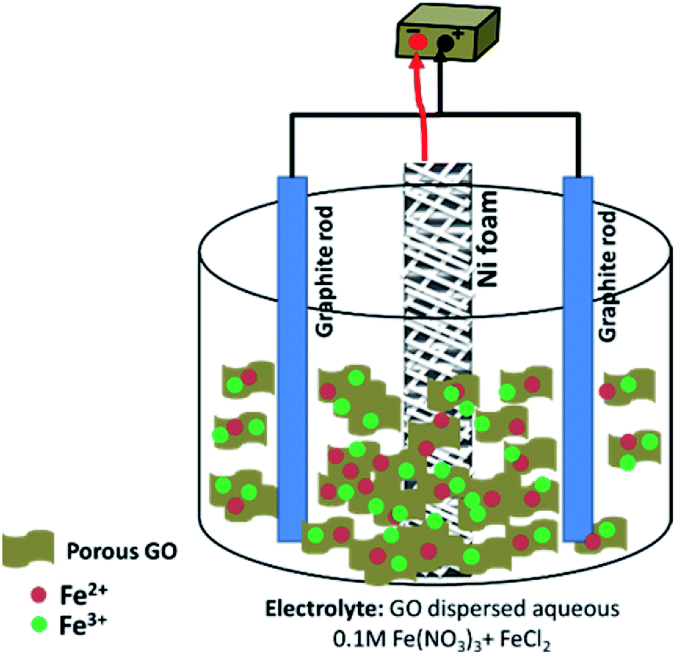 | ||
| Fig. 1 Schematic view of the electrochemical set-up. | ||
2.4. Characterization analyses
The FE-SEM images of the prepared powders were provided through field-emission scanning electron microscopy (FE-SEM, Mira 3-XMU with accelerating voltage of 30 kV). The crystal structures of the prepared powders were determined by X-ray diffraction (XRD, Phillips PW-1800) using a Cu Kα radiation. Transmission electron microscopy (TEM, model Zeiss EM900) with an accelerating voltage of 80 kV was also used to better observation of the sample morphologies. TGA/DSC analyses were performed via STA-1500 analyzer and studied in N2 atmosphere within the temperature range of 25–600 °C by applying a heating rate of 5 °C min−1. Raman data was provided by Takram instrument model: P50C0R10 with Nd:YAG laser (532 nm). The measurement of specific surface area for the prepared samples was obtained through measuring N2 adsorption–desorption isotherms at 77 K with a Quanta-chrome NOVA-2200e system. FTIR spectra were recorded using a Bruker Vector 22 FT-IR spectrometer within the wave numbers of 400–4000 cm−1.2.5. Electrochemical tests
Cyclic voltammetry (CV), galvanostatic charging/discharging (GCD) as well as electrochemical impedance spectroscopy (EIS) were used for estimation of electrochemical performances of the prepared electrodes. These tests were done using an electrochemical station i.e. AUTOLAB®, Eco Chemie, PGSTAT 30. A three-electrode system was assembled for the CV, EIS and GCD tests, which includes the working electrode (i.e. the deposited active material onto nickel foam), reference electrode (Ag/AgCl, saturated KCl) and the counter electrode (platinum wire). All the electrochemical tests were carried out in Na2SO3 (2 M) aqueous solution. The mass loadings of pristine Fe3O4 and Fe3O4/rGO onto the Ni foam were about 6.5 mg and 5.2 mg, respectively. The CVs of the fabricated working electrodes were recorded in the potential range of −1.0 to +0.1 V vs. Ag/AgCl at the potential sweeps of 2, 5, 10, 25, 50 and 100 mV s−1. The GCD curves were recorded at the different current loads of 0.5, 1, 2, 4, 6, 8 and 10 A g−1 within a potential range of −1.0 to 0 V vs. Ag/AgCl. The EIS was conducted in the frequency range between 100 kHz and 0.01 Hz with applying perturbation of 5 mV at open-circuit potential. The specific capacitances of the fabricated electrodes were calculated from two methods: (i) eqn (1) (CV curves) and (ii) eqn (2) (galvanostatic charge/discharge curves):
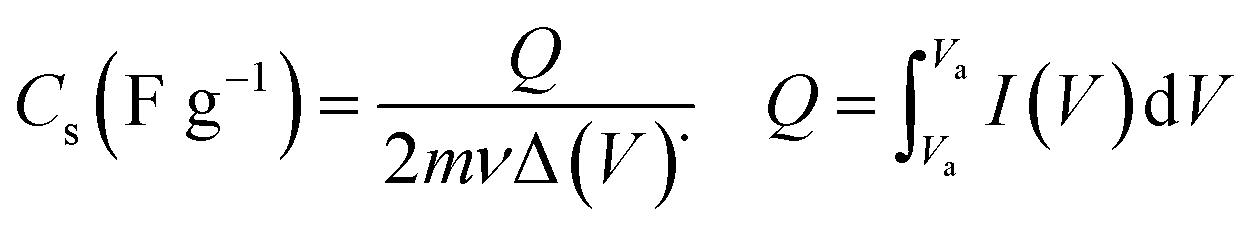 | (1) |
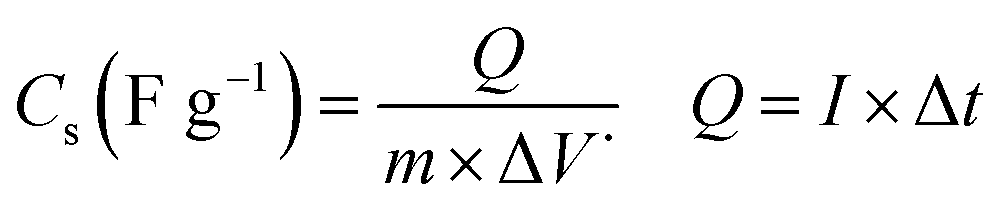 | (2) |
In these formula, Cs is the specific capacitance in F g−1 unit, m is the weight of the deposited material on the electrode in g unit, ν is the scan rate of CV curves in V s−1, and ΔV (Va − Vc) represents the potential window (V). Q in CV curves stands for the total charge stored within the enclosed CV profile and the denominator 2 is used to deplete the effect of charge half-cycle, I is the current in A unit and Δt is the discharge time in s unit at the determined current density.
3. Results and discussion
3.1. Structural and morphological characterizations
Fig. 2 presents the X-ray diffraction patterns of both electro-synthesized iron oxide and its composite along with GO in powder state. Fig. 2a shows the XRD pattern of graphene oxide powder. In the XRD pattern of graphene oxide acquired by the improved Hummers' method, the (001) crystal plane of GO was obvious, which is a characteristic peak of GO demonstrating the successful exfoliation of graphite sheets by using oxidizing agents used in Hummers' method.27 In both XRD patterns of Fe3O4@rGO/NF and Fe3O4/NF, the two sharp diffraction peaks at about 44.5° and 51.8° are ascribed to the nickel foam support.28 The presence of more four diffraction peaks for both electrodes rather than peaks related to nickel foam substrate, are corresponded to the (220), (311), (400) and (511) planes of cubic Fe3O4 (JCPDS no. 65-3107). Hence, both electro-synthesized samples have magnetite crystal structure and results revealed that presence of porous graphene does not change the crystal structure of the deposited magnetite material.29,30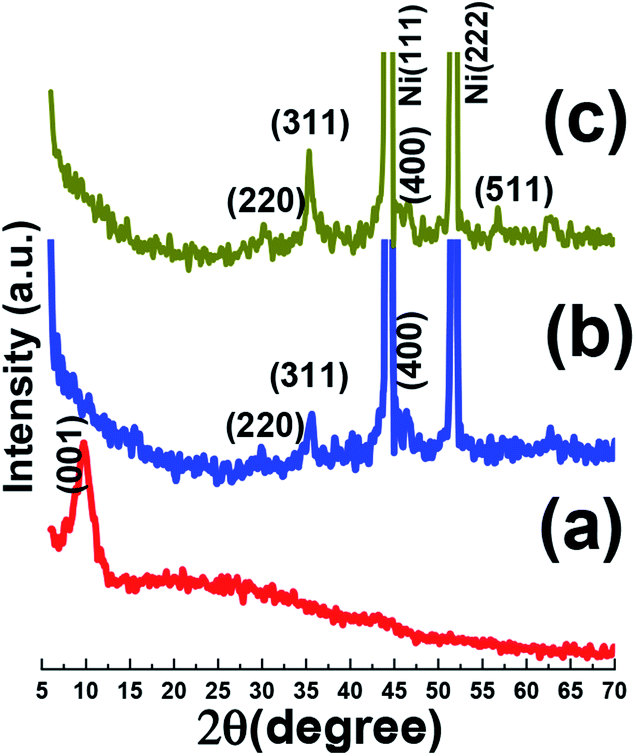 | ||
| Fig. 2 XRD patterns data of (a) the synthesized GO powder, and (b) the Fe3O4/Ni foam and(c) Fe3O4@ reduced graphene oxide composite/Ni foam. | ||
The evolution of the chemical composition of the prepared samples was done by FT-IR, as shown in Fig. 3. For comparison, IR spectrum of pristine graphene oxide (GO) was also provided. IR spectrum of the Fe3O4 sample (Fig. 3a), showed the broad IR peak from 550 to 650 cm−1 relating to Fe–O bonds.31 The broad absorption peak centered at about 3429 cm−1 was assigned the O–H stretching vibration. Furthermore, the IR band at about 3447 cm−1 is related to the deformation vibrations hydroxyl groups connected to the surface of Fe3O4 sample.31,32 Fig. 3b presents the IR bands of pristine graphene oxide. Graphene oxide contains a broad range of oxygen functional groups. The characteristic peaks related to the vibration modes of functional groups of GO are observed in Fig. 3b, i.e. O–H bonding at 3405 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding at 1723 cm−1, C–O carboxy bonding at 1242 cm−1, C–O epoxy bonding at 1175 cm−1 and C–O alkoxy bonding at 1061 cm−1.33 Also, strong CC stretching deformation of a honeycomb carbon network is seen at 1628 cm−1. However, the Fe3O4@p-rGO IR data proved that most oxygen-containing functionalities in the GO were absent even still there are some residual oxygen functional groups on the rGO surface with weaker sharpness after the electrochemical reduction through applying constant cathodic current. The peak at about 1630 cm−1 in rGO shows strong band, this suggests the recovery of sp2 lattice.33,34
O bonding at 1723 cm−1, C–O carboxy bonding at 1242 cm−1, C–O epoxy bonding at 1175 cm−1 and C–O alkoxy bonding at 1061 cm−1.33 Also, strong CC stretching deformation of a honeycomb carbon network is seen at 1628 cm−1. However, the Fe3O4@p-rGO IR data proved that most oxygen-containing functionalities in the GO were absent even still there are some residual oxygen functional groups on the rGO surface with weaker sharpness after the electrochemical reduction through applying constant cathodic current. The peak at about 1630 cm−1 in rGO shows strong band, this suggests the recovery of sp2 lattice.33,34
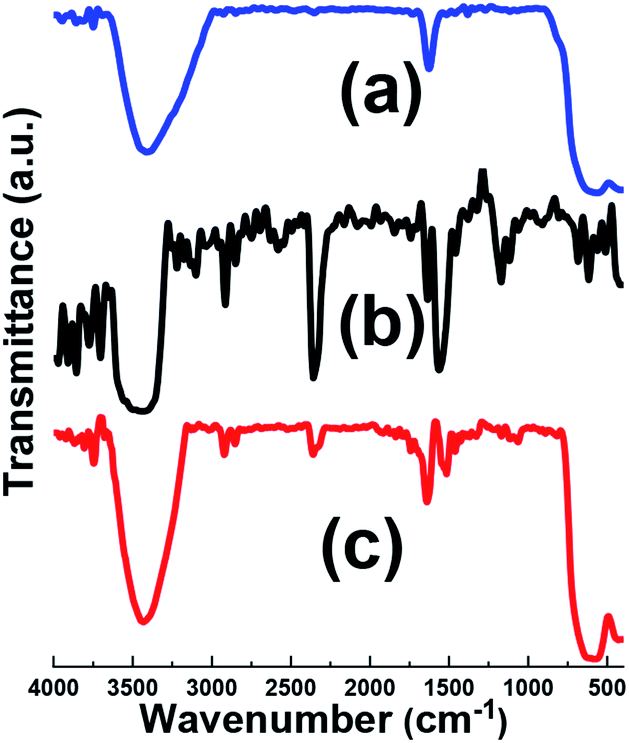 | ||
| Fig. 3 IR spectra of (a) pristine Fe3O4, (b) GO and (c) Fe3O4@p-rGO composite. | ||
Raman spectroscopy is a powerful instrument to determine the structure and electronic properties of carbon materials. Raman spectra of graphene oxide sample has been shown in Fig. 4a. Th graphene oxide showed three bands at 1350.7, 1613, and 2783 cm−1 well known as D, G and 2D bands, respectively. The G band is corresponded to the vibration of graphitic sp2 carbon domains, while the D band relates to the sp3-hybridized carbon and utilized to show defects correlated with vacancies and grain boundaries within carbonaceous materials. The peak positioned at about 2800 cm−1 is ascribed to the two phonon inter-valley double resonance mechanism while another bands at 2970 and 3189 cm−1 are ascribed to the D + D′ and 2D′ peaks and these peaks related to the intra-valley scattering due to lattice defects and overtone of the phonon modes that give rise to D′ bands, respectively.35–37 It is known that ID/IG ratio gives the degree of defects present in GO sample which was as high as 0.9.35 Fig. 4b, shows Raman spectra of pristine Fe3O4 sample. This spectrum showed five active modes of (Ag + Eg + 3T2g) and all these peaks were observed for Fe3O4 at wave numbers under 1000 cm−1. The band at about 1400 cm−1 is related to the 2nd order of scattering.38–40 Comparing with pristine GO sample (Fig. 4c), the D/G ratio of composite sample becomes higher and the D band becomes broader, suggesting a higher level of disorder of the graphene layers by the introduction of Fe3O4 in composite sample.35,36 It is also worthnoting that the movement of G band to lower energy region endorse that GO sheets are converted to graphitic structures.37 On the other hand, it was observed that the intensity of D band also increased which is desirable for improvement of ionic/electronic transfer that could leading improvement in electrochemical performance of composite sample.41 Thus, the Raman data, in good agreement with the XRD and FTIR achievements, further proved the fabrication of the Fe3O4@p-rGO composite.
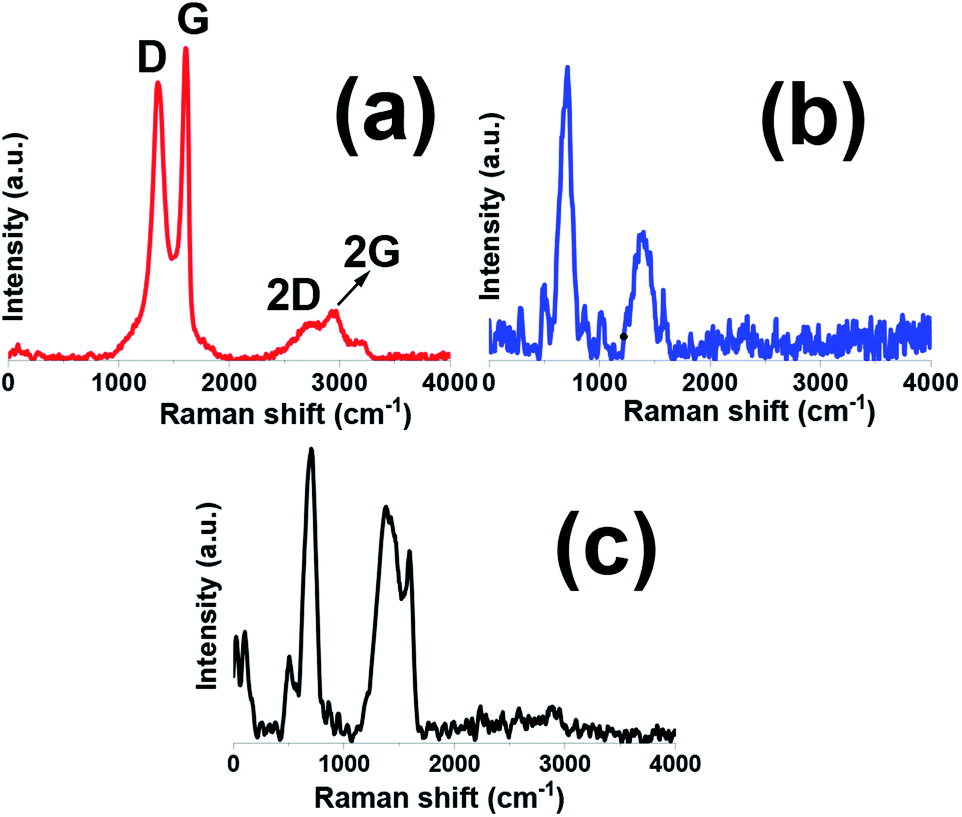 | ||
| Fig. 4 Raman spectra of (a) GO, (b) pristine Fe3O4 and (b) Fe3O4@p-rGO composite. | ||
Fig. 5a shows the morphology of prepared GO material by modified Hummer's method. It is clearly seen that graphene oxide has thin sheet-like texture, which randomly aggregated. More detailed observation demonstrating the GO sample has sheet-like structure with aggregated morphology and wrinkles are obvious from higher magnified FE-SEM image presented in Fig. 5b. The aggregated morphology was related to the strong π–π interaction between two adjacent graphene oxide nanosheets. EDS elemental analysis implicating the presence of carbon and oxygen elements in the GO substance revealing the purity of the synthesized graphene oxide (as shown in inset of Fig. 5b). The high oxygen content in the EDS results means that the oxidation of graphite has been occurred to high level (inset of Fig. 5b). TEM was taken from GO to specify the detailed morphology of this materials and its corresponding images are displayed in Fig. 5c and d. TEM images manifested the GO sheets with wrinkles on its surface which could be advantageous for better interaction with various ions and materials. Moreover, TEM observation clearly shows that GO is composed of single to few layers of nanosheets.
 | ||
| Fig. 5 (a and b) FE-SEM images, EDS data (inset in Fig. 4b), and (c and d) TEM images of the prepared graphene oxide. | ||
Further characterization of samples was done through FE-SEM observations. Nickel foam substrate widely utilized as electrode support for various applications owing to its open structure as well as its three-dimensional structure with a pore size of 200–300 μm as revealed by FE-SEM observations in Fig. 6a and b. Such a unique structure could provide high surface for better precipitation of various materials in its surface. Also, the EDS elemental analysis of Ni foam showed the presence of only nickel element since its surface treated chemically to remove possible created nickel oxide prior to using as substrate for deposition of electroactive materials. The morphology of the as-prepared magnetite and its related EDS elemental analysis are shown in Fig. 6c–e. Low magnified image implicated the nearly homogenous deposition of iron oxide on the surface of foam support (Fig. 6c).
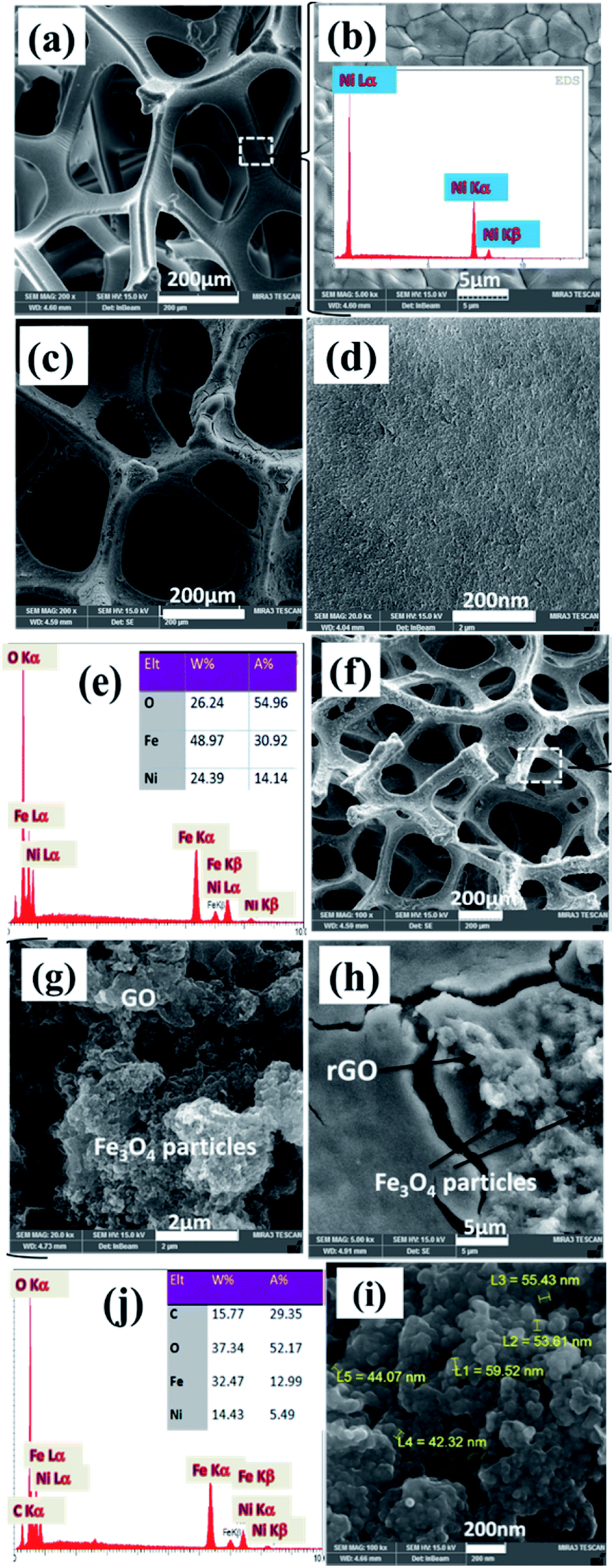 | ||
| Fig. 6 FE-SEM images and EDS data of (a and b) blank Ni foam, and the fabricated (c–e) pristine Fe3O4@Ni foam and (f–j) Fe3O4/p-rGO@Ni foam. | ||
However, higher magnified FE-SEM image showed that the as-prepared iron oxide has nanoparticle morphology which created a nearly densely pack film on the surface of nickel foam support (Fig. 6d). The EDS spectrum showing the atomic and weight percentage of iron oxide electrochemically deposited on the nickel foam are shown in Fig. 6e. The presence of nickel element related to the nickel foam support. Also, the iron and oxygen atomic percentage at the investigated area of EDS were 31% and 55%, respectively. The morphology of the fabricated composite and also its EDS spectra are seen in Fig. 6f–j. Low magnified FE-SEM image showed the completely homogenous film on the surface of nickel foam (Fig. 6f). However, at lower magnified, FE-SEM images for this sample, two types of morphologies (i.e. particle and sheet) are detectable in Fig. 6g and h. And FE-SEM images of composite are totally different from FE-SEM images of pristine Fe3O4/NF (Fig. 6c and d), which are corresponding to the Fe3O4 and rGO, respectively. In fact, complete uniform texture (i.e. particles grown on sheets) is seen for the fabricated composite (Fig. 6h). This observation implicated the stable and regular conditions for electrophoretic deposition of GO sheets on the nickel foam cathode and also electrochemical formation of iron oxide NPs on the rGO sheets (Fig. 6h and i). However, Fe3O4 nanoparticles have diameters within the range of 50 nm (Fig. 6i). The energy-dispersive X-ray (EDS) results of the composite sample (Fig. 6j) showed the existence of Fe, O, Ni and C elements. In fact, the composite is composed of 32.5 wt% Fe, 15.5 wt% C and 37.5 wt% O element along with 14.5 wt% of Ni originating from foam support. With comparison of EDS results, it was revealed that the presence of C element and high percentage of O element in the composite powder confirming the formation of Fe3O4@p-rGO deposit on the surface of nickel foam.
Fig. 7 shows TEM images of Fe3O4 nanoparticles and also Fe3O4@rGO in powder forms. Transmission image of Fe3O4 showed particle morphology of this material (Fig. 7a), which is consistent with its FE-SEM images (Fig. 6c and d). The observed aggregation of nanoparticles is owing to their magnetic properties which could attract each other that concluded nanoparticles with aggregated morphology (Fig. 7a). However, TEM image of composite material showed the presence of both graphene nanosheets with some pores on its surface along with particles which nearly homogenously distributed on the surface of the electrochemically reduces graphene oxide (Fig. 7b). The more precise view through TEM observations revealed that the Fe3O4 nanoparticles have diameters within several nanometers (Fig. 7c) rather than tens of nanometers detected by FE-SEM image (Fig. 6i).
 | ||
| Fig. 7 TEM images of the synthesized (a) pristine Fe3O4 and (b and c) Fe3O4/p-rGO. | ||
TGA/DSC analyses of the prepared samples were also conducted within the room temperature to 700 °C under nitrogen atmosphere (Fig. 8). The highest weight loss for one sample is related to the graphene oxide sample one which showed nearly 5% weight loss during the TG analysis. However, all samples showed two exothermic DSC peaks at about 100–200 °C and 200–300 °C which correlated with weight losses in TG curves (Fig. 8b), implicating the removal of physically and crystallized water molecules within the samples. Furthermore, another sharp exothermic peak in DSC curves of graphene oxide and composite sample (Fig. 8a) is ascribed to the removal of labile oxygen containing functionalities within these two samples.42–45 As a whole, due to thermal stability of the iron oxide within the temperature range, the TG and DSC profiles of two GO and Fe3O4@rGO samples are similar.
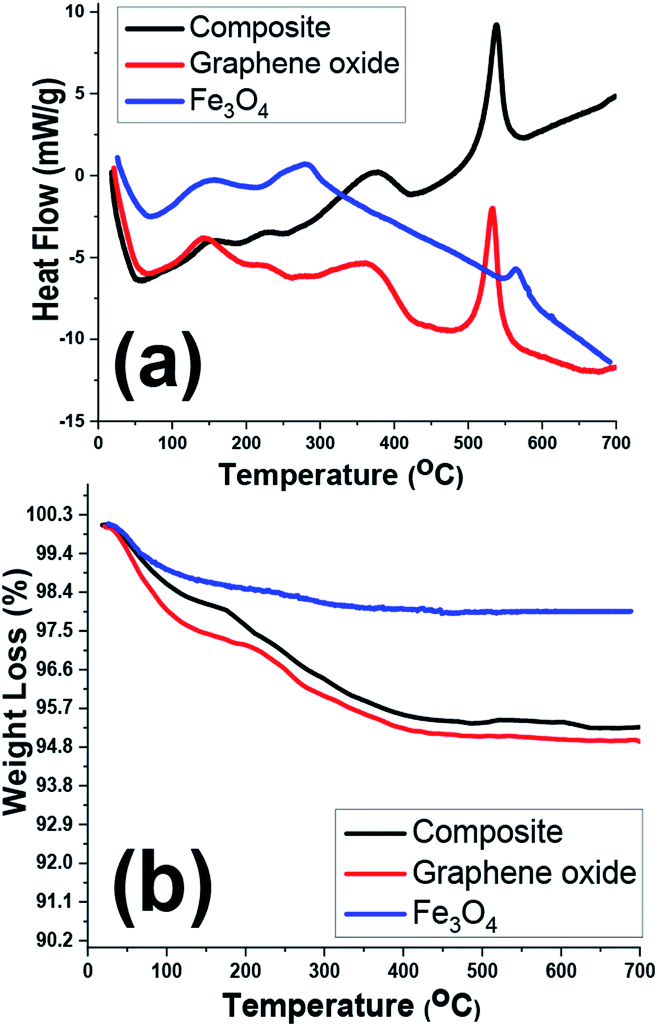 | ||
| Fig. 8 (a) DSC and (b) TG curves of GO, pristine Fe3O4 and Fe3O4@p-rGO composite. | ||
BET–BJH analyses of the scrubbed Fe3O4 and Fe3O4@rGO powders were taken to estimate their efficient active surface area and mean pore diameters as well as total pore volume. Fig. 9 displays N2 adsorption–desorption isotherm plots of samples along with their corresponding pore size and total pore volume. The pure Fe3O4 sample shows Brunauer–Emmett–Teller (BET) surface area of around 39 m2 g−1, which declares its proper surface area (Fig. 9a). Furthermore, the pore diameter and also total pore volume of the samples were determined from the Barrett–Joyner–Halenda (BJH) distribution curves. However, the Fe3O4 sample portrays total pore volume and pore size of 0.038 cm3 g−1 and 2.45 nm, respectively (Fig. 9b and c). Meanwhile, the high levels of the pores fall into the size ∼2.5 nm, revealing the presence of mesoporous benign of Fe3O4. The introducing of rGO into the Fe3O4 nanoparticles matrix leads in significant increment of specific surface area to 329 m2 g−1 and hysteresis loop (Fig. 9d), which is higher than that of pure Fe3O4 sample (Fig. 9a). In the case of hybrid material, it shows mean pore size of 2.86 nm as well as total pore volume of 0.462 cm3 g−1 (Fig. 9e and f) which are higher than those of Fe3O4 sample. The higher specific surface area as well as enhanced pore size and pore volume of hybrid powder rather than the presence of reduced graphene oxide could be attributed to the interaction between iron salts and graphene oxide nanosheets in primarily suspension which results in precipitation of more porous film on the nickel foam support as revealed by TEM image (Fig. 7c). Moreover, the BJH analysis showed the presence of mesoporous structure for both samples including Fe3O4 and Fe3O4@rGO the hybrid material which could be helpful for better mass transfer between electrode surfaces and electrolyte solution.
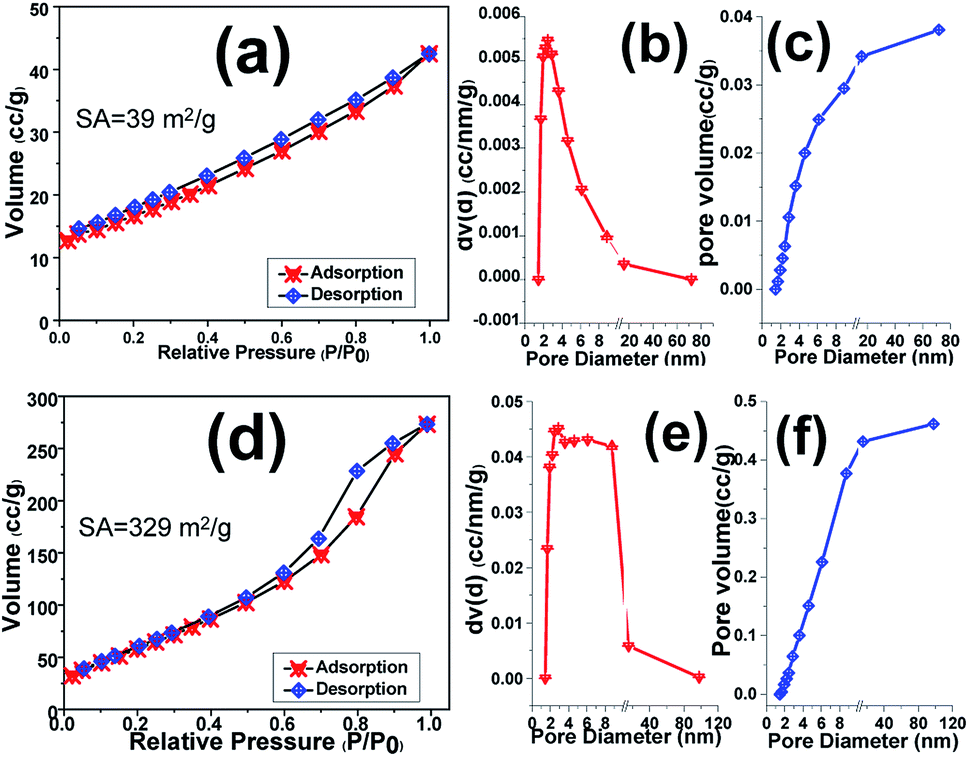 | ||
| Fig. 9 BET and BJH pore size distribution curves of (a–c) pristine Fe3O4 NPs and (d–f) Fe3O4@p-rGO composite. | ||
3.2. Electrochemical characterizations
The electrochemical performances of the fabricated electrodes were attained in 2 M Na2SO3 solution, which utilized as the electrolyte. Fig. 10a showed the CV curves of the pristine Fe3O4/NF, rGO/NF and Fe3O4@rGO/NF electrodes at 5 mV s−1 within the potential range of −1.0 to +0.1 V (vs. Ag/AgCl). In the case of rGO/NF, it was observed that CV curve containing a redox peak which is attributed to oxygen containing functionalities faradaic reaction and is in good compromise with previous sections demonstrating the partial reduction of oxygen containing groups by electrochemical process. However, both other electrodes consisting of iron oxide nanoparticles show pseudocapacitance shape. A clear distortion from EDLC behavior was obvious in the CV curves (Fig. 10b and c) which originating from the pseudocapacitive of the redox reaction among the active material and electrolyte ions. The capacitance mechanism is because of the reduction/oxidation of the SO32− anions adsorbed onto the Fe3O4 along with the oxidation/reduction reaction between Fe2+ and Fe3+ through possible intercalations along with the electrolyte:46–49| 2SO32− + 3H2O + 4e− ↔ S2O32− + 6OH− | (3) |
| S2O32− + 3H2O + 8e− ↔ 2S− + 6OH− | (4) |
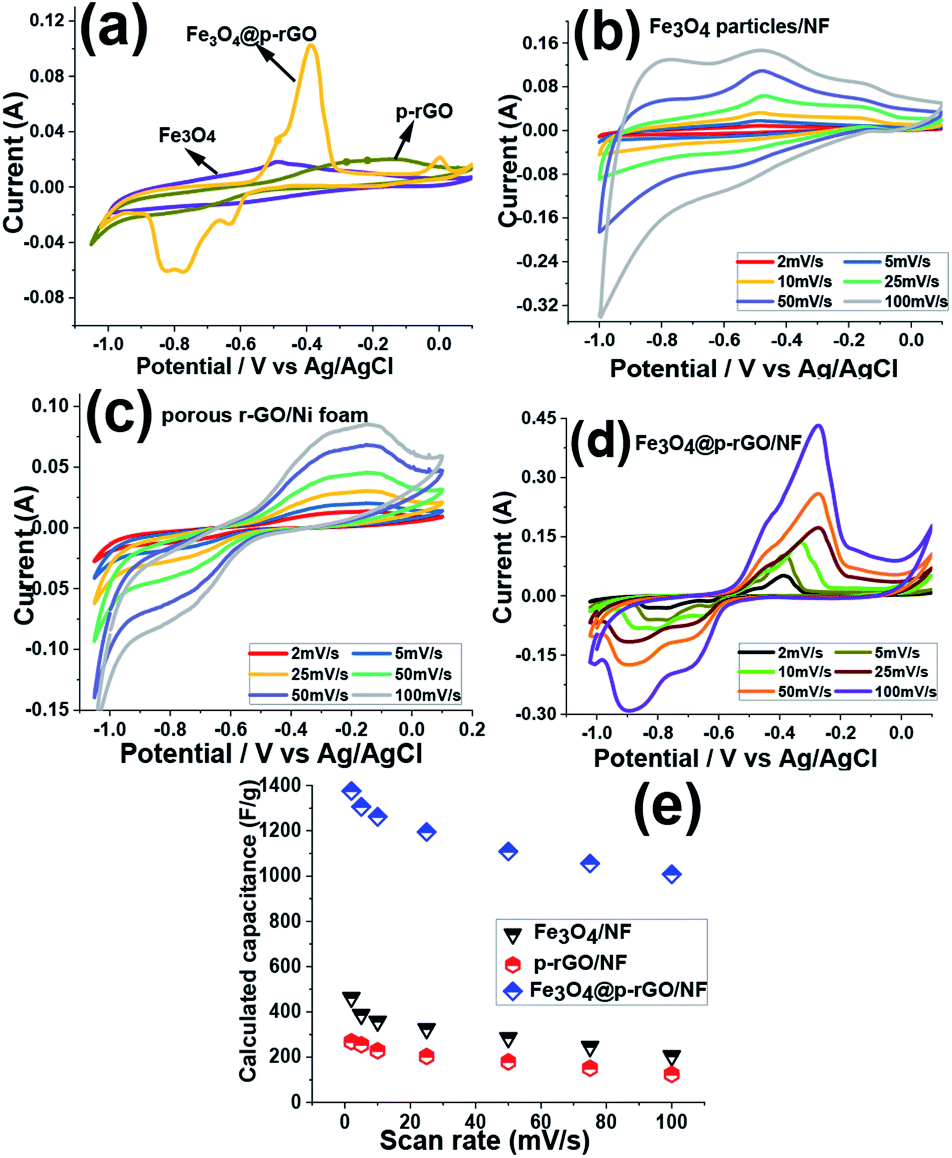 | ||
| Fig. 10 (a) Comparison of the capacitive behavior of the fabricated NF electrodes at the scan rate of 5 mV s−1, (b–d) CVs of Fe3O4/NF, rGO/NF and Fe3O4@p-rGO/NF at the scan rates of 2–100 mV s−1, (e) the calculated capacitances values vs. scan rate for the fabricated electrodes. | ||
Moreover, it is seen that the integrated area of CV curve for Fe3O4@rGO/NF electrode is higher than those of the pure Fe3O4/NF and rGO/NF electrodes, which manifests the higher electrochemical reaction activity of the composite electrode in comparison to two other electrodes. This enhancement indicated simultaneous contribution of charge storage between rGO and Fe3O4 components within the composite material resulted in the both faradic and electric double layer capacitor processes in the composite material.
Such a synergy could be helpful for increasing the electrochemical performance of composite material in comparison to single component electrodes. Hence, the rGO/Fe3O4 electrode provides higher capacitance. The CVs of Fe3O4/NF, rGO/NF and Fe3O4@p-rGO/NF electrodes were recorded at various scan rates of 2–100 mV s−1 and the results are presented in Fig. 10b–d. All electrodes clearly showed pseudocapacitive behavior rather than EDLC behavior. The pure iron oxide deposited onto nickel foam support portrayed a highest specific capacitance value of 463 F g−1 at the sweep rate of 2 mV s−1 whereas rGO/NF and composite electrodes showed specific capacitances of 268 F g−1 and 1376 F g−1 at the same scan rate. The higher specific capacitance of composite material is due to the presence of rGO part of the composite electrode, where rGO rather than providing the conductive substrate for Fe3O4 particles, it could increase the specific surface are. Hence, the fabricated composite electrode could provide higher specific capacitance when compared to other pristine electrodes. The specific capacitances of each electrode were calculated through eqn (1). The pristine Fe3O4 electrode could bring specific capacitance values of 463, 397, 348, 315, 285, 246 and 205 F g−1 at the scan rates of 2, 5, 10, 25, 50, 75 and 100 mV s−1, respectively (Fig. 10e). The pristine rGO/NF electrode displayed capacitance values of 268, 255, 228, 196, 179, 151 and 124 F g−1 at the scan rates of 2, 5, 10, 25, 50, 75 and 100 mV s−1, respectively. Also, it was obtained that the composite material including Fe3O4 and rGO components was able to deliver the high specific capacitances of 1376, 1306, 1262, 1195, 1109, 1056 and 1008 F g−1 at the scan rates of 2, 5, 10, 20, 50, 75 and 100 mV s−1, respectively (Fig. 10e). These values indicated that the Fe3O4@rGO/NF electrode shows excellent electrochemical performance in comparison to the other electrodes prepared in this article. Such an observation is relating to this fact that the capacitive characteristics of Fe3O4 nanoparticles is improved in the presence of rGO, which is ascribed to the good physical contact between the Fe3O4 NPs onto electrochemically reduced graphene oxide sheets and also the high interfacial area amongst the two components and the high surface area along with mesoporous texture of the composite as indicated by BET results (Fig. 9c and d).
Fig. 11a and b show the charge–discharge plots (−1 to 0 V vs. Ag/AgCl) of Fe3O4/NF, rGO/NF and Fe3O4@rGO/NF electrodes recorded at two current densities of 0.5 and 6 A g−1. The GCD curves for all electrodes showed the battery-type electrochemical behavior, as witnessed by the plateaus at the charge/discharge curves, which are in good arrangement with CV profiles of these electrodes (Fig. 10b–d). Furthermore, GCD curves demonstrated the Fe3O4@rGO/NF electrode has higher charge–discharge capability at both current densities as confirmed by its longer charge and discharge times, in comparison to the pristine Fe3O4/NF and rGO/NF electrodes, which proved better performance of composite electrode. Fig. 11c–e show the GCD curves of prepared electrodes at various current densities ranging from 0.5 to 10 A g−1. The GCD profiles of the composite electrode are shown in Fig. 11e, where the presence of plateaus at both charge/discharge branches completely confirmed its pseudocapacitor behavior. The SC values achieved for electrodes were calculated through eqn (2), and the obtained SC values were plotted in Fig. 11f. In the case of pristine iron oxide, the SC values were found to be 357 F g−1, 326 F g−1, 192 F g−1, 253 F g−1, 214 F g−1, 177 F g−1 and 156 F g−1 at the respective discharging loads of 0.5, 1, 2, 4, 6, 8 and 10 A g−1, respectively (Fig. 11f). In the case of rGO/NF electrode, the SC values of 211, 204, 196, 184, 175, 162 and 142 F g−1 were obtained at similar current densities to pristine iron oxide/nickel foam support (Fig. 11f). However, the Fe3O4/p-rGO nanocomposite electrode was found to be capable to provide specific capacitance of 1323, 1275, 1215, 1148, 1109, 1087 and 1044 F g−1 at the current densities of 0.5, 1, 2, 3, 5, 7 and 10 A g−1, respectively (Fig. 11f). The values are very close to those values acquired from CVs (Fig. 10e), dedicating the good capacitive behavior of the prepared electrodes. Generally, as the current density increases, the specific capacity decreases owing to this fact that the gradual voltage drop and inadequate active sites within the electrode material. In fact, by increasing the current density some portions of electrode material are not accessible for diffusion of electrolyte ions and hence electrochemical redox reactions which leading to decay in specific capacitances for all electrodes.50,51
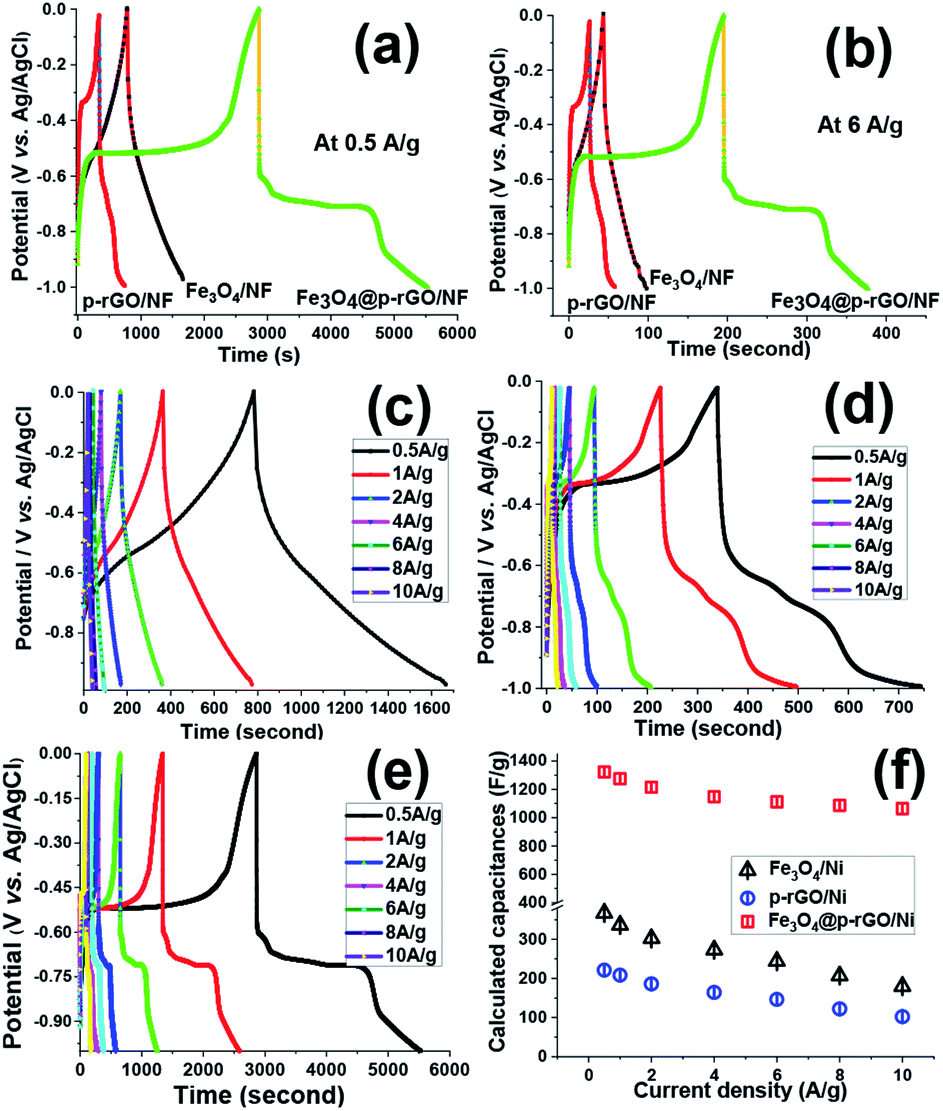 | ||
| Fig. 11 (a and b) GCD profiles for the deposited active materials onto NF current collector at the current loads of 0.5 A g−1 and 6 A g−1 and the GCD profiles of (c) Fe3O4/NF, (d) rGO/NF and (e) Fe3O4@rGO/NF electrodes at various current loads within the range of 0.5–10A g−1 and (f) calculated specific capacitance values against current density for all electrodes. | ||
The Fe3O4 nanoparticles supported rGO nanosheets provided more rapid ion transportation and were advantageous for charge storage of composite electrode. The capacity retentions for the Fe3O4/NF, p-rGO/NF and Fe3O4@p-rGO/NF were estimated to be ≈43%, ≈68% and ≈79% at the discharge current increased as high as 20 times from 0.5 to 10 A g−1. These findings indicated the positive synergy assistances between Fe3O4 and porous rGO, which come from; (1) uniformly anchoring the Fe3O4 particles onto porous rGO sheets (as confirmed by TEM observations in Fig. 7b and c), (2) suppressing the re-stacking of porous rGO by incorporation of Fe3O4 particles into the porous rGO sheets, (3) providing the highly conductive electronic network for the redox process of Fe3O4 particles by porous rGO sheets, and (4) hindering the volume change and agglomeration of iron oxide by porous rGO (as confirmed by BET data in Fig. 9). This cooperation enhances the Fe3O4@rGO interface area, and utilization of active materials, and also provides more accessible ionic electronic transport pathways and improves the poor electrical properties and charge transfer pathways of Fe3O4, which results the good supercapacitive performance for the fabricated Fe3O4@rGO/NF electrode. Table 1 provides the cycling stabilities reported for iron oxide–graphene oxide based composites in literature. From comparing the listed data, it is verified that the charge storage ability of our prepared composite is comparable with the reported Cs data for the Fe3O4–rGO composite electrodes in the literature.
| Fe3O4 structure type | Electrolyte | Current density (A g−1) | Specific capacitance (F g−1) | Capacity retentions (%) | Ref. |
|---|---|---|---|---|---|
| Fe3O4/Fe–CNTs | 2 M Na2SO3 | 1 | 1065 | 82.5% after 1000 | 52 |
| Fe3O4 NPs | 2 M KOH | 2.5 | 1075 | 95.3% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
53 |
| Fe3O4–rGO–MoO3 | 2 M KOH | 2.5 | 1666 | 95.1 after 5000 | 53 |
| Fe3O4 NRs/NH2–rGO | 1 M Na2SO4 | 1 | 972 | 99.7% after 1000 | 54 |
| Fe3O4/RGO | 0.5 M Na2SO4 | 1 | 154 | 97% after 500 cycles | 55 |
| FeOOH nanoparticles | 2 M KOH | 1 | 1066 | 91% after 10000 cycles |
56 |
| K-doped FeOOH/Fe3O4/SS | 2 M KOH | 1 | 1296 | 126% after 8000 cycles | 57 |
| N-doped carbon supporting Fe3O4 | 6 M KOH | 1 | 206 | 95% after 5000 cycles | 58 |
| Fe3O4 NPs | 2 M Na2SO3 | 0.5 | 196 | 94% after 3000 cycles | 59 |
| α-Fe2O3/rGO | 2 M KOH | 1 | 970 | 75% after 2000 cycles | 60 |
| Mn3O4–Fe2O3/Fe3O4@rGO | 1 M KOH | 1 | 590.7 | 64.5% after 1000 cycles | 61 |
| Pure Fe3O4 | 2 M Na2SO3 | 1 | 336 | 80.4% after 1000 cycles | This work |
| Fe3O4/rGO | 2 M Na2SO3 | 1 | 1275 | 95.1% after 4000 cycles | This work |
The cycling stabilities of the fabricated binder-free electrodes were analyzed via continuous 4000 consecutive charge/discharge cycles at the current densities of 1 and 4 A g−1 in a 2 M Na2SO3 as electrolyte media and the results presented in Fig. 12. The calculated data revealed that the pristine iron oxide electrode loosed around 19.6% and 34.1% of its initial capacitances (Fig. 12b and d) after 4000 cycles at the discharge of 1 to 4 A g−1, respectively (Fig. 12a and c). In fact, the capacity stabilities of 80.4% and 65.9% were measured for the pristine Fe3O4/NF electrode at the current load of 1 to 4 A g−1, respectively. While, the composited electrode (i.e. Fe3O4@rGO/NF), retains 95.1% and 86.7% of its primary capacitances (Fig. 12a and c). The specific capacitance values of the deposited iron oxide nanoparticles and Fe3O4@rGO and composite sample onto Ni foam reduced from 269 F g−1 to 179 F g−1, and from 1215 F g−1 to 1006 F g−1 after 4000 charge–discharging at the currents of 1 and 4 A g−1 in 2 M Na2SO3 electrolyte, respectively (Fig. 12b and d). These findings indicated that the Fe3O4/rGO composite could provide excellent cycling abilities (i.e. higher capacitance values and lower capacity decay) within the 4000 continues charge–discharging at both current loads of 1 A g−1 and 4A g−1. The exceptional cycling stability of the composite electrode is attributed to the high structural stability of the composite electrode on the nickel foam support resulted from the direct deposition via a simple and facile methodology along with reduced volume expansion of composite material which normally occurred for iron oxide nanostructures. The unique morphology is also helpful for such a high specific capacitance retention (as seen in Fig. 4). It is also worth mentioning that the presence of rGO with high specific surface area and porous texture improves specific capacitance and also cycle stability of the composite electrode. From the data listed in Table 1, it is also confirmed that our Fe3O4/p-rGO composite system shows comparable performance to those in the literature and even portrays better performance than them further revealing the excellent capacitance behavior of the composite material.
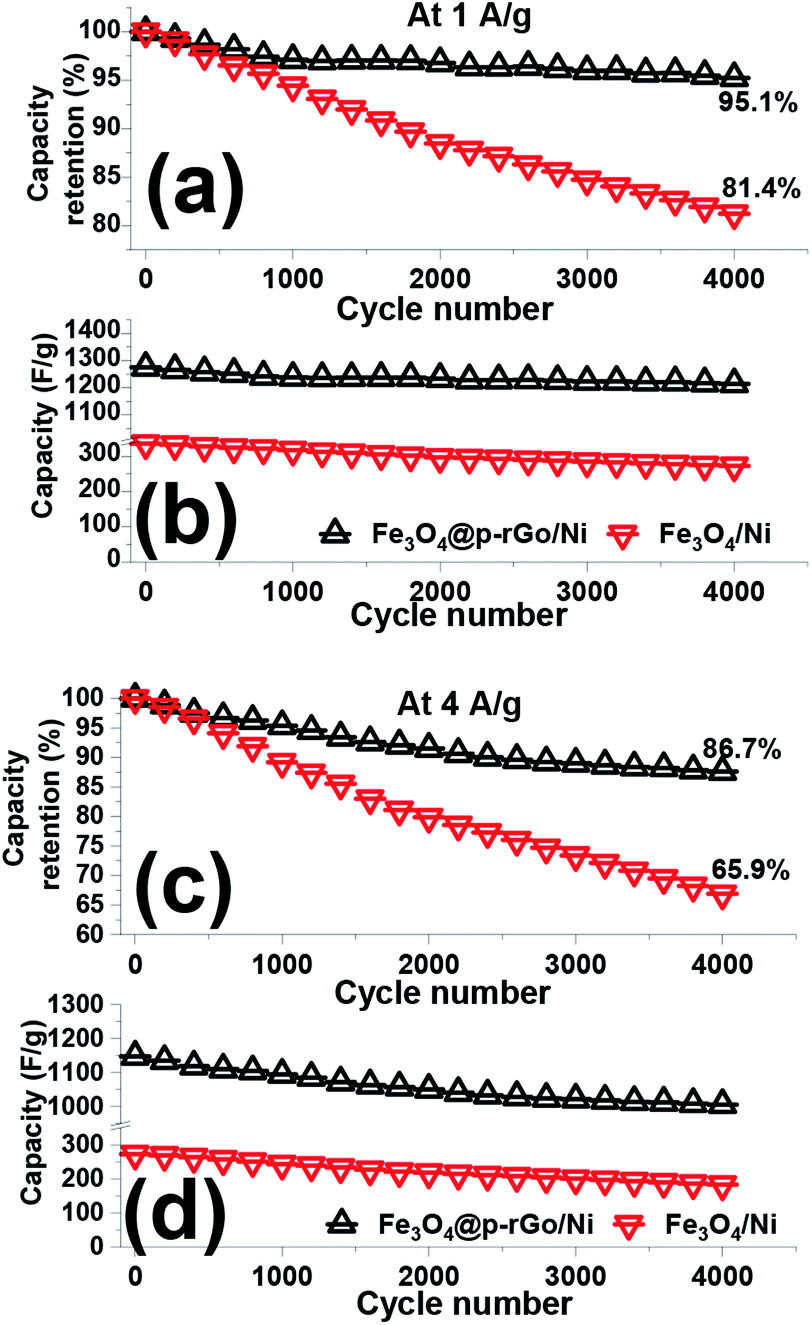 | ||
| Fig. 12 (a and c) Capacity loss and (b and d) capacitance values and for the fabricated Fe3O4/NF and Fe3O4@p-rGO/NF electrodes within 4000 cycling at the discharge loads of 1 A g−1 and 4 A g−1. | ||
As a powerful technique utilized for estimation of electrochemical processes at the electrode/electrolyte interface, EIS tests were done for both fabricated electrodes and their Nyquist plots were recorded. The Nyquist plots of pristine Fe3O4/NF and Fe3O4@rGO/NF in 2 M Na2SO3 are presented in Fig. 13. As it is obvious from Nyquist plots, blending of Fe3O4 and rGO in the Fe3O4/rGO electrode resulted in reducing in both of the real and imaginary section of the impedance. The series resistance (Rs), electron transfer resistance (Rct), double layer capacitance (CPE1) and pseudocapacitance (CPE2) could be utilized as components of impedance data (as indicated in inset of Fig. 13). CPE element was used to show the inhomogeneity of electrode surface in each case instead of double layer capacitance.60,61 The Rs value of the composite electrode is very low demonstrating its fair electrical conductivity. However, composite electrode has lower Rs value of 0.24 Ω as compared with Rs value of Fe3O4/NF electrode.
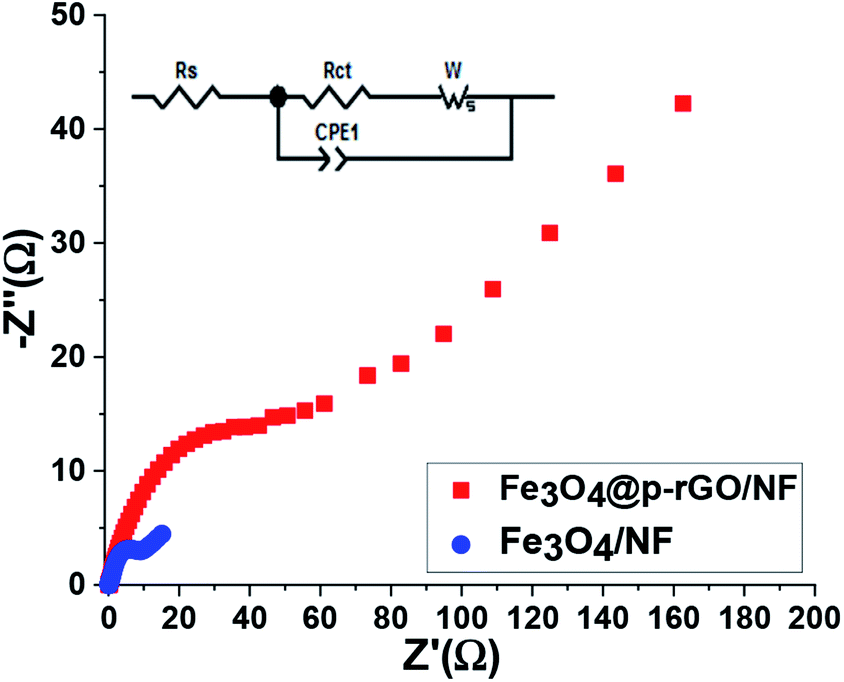 | ||
| Fig. 13 The Nyquist plots for Fe3O4/NF and Fe3O4@p-rGO/NF electrodes. The inset shows the corresponding equivalent circuit pattern. | ||
The low Rs value results in low internal resistance which is beneficial for high rate performance of composite electrode. The significant difference between two electrodes occurred within the high to low frequency region. Moreover, it was also seen that at very low frequency range, the composite electrode showed steeper line with less length because of more facilitated mass transfer through composite electrode in comparison to iron oxide electrode as evidenced by FE-SEM images and electrochemical results (Fig. 6, 10–12). However, at high frequencies, the observed semicircle is ascribed to the charge transfer resistance which was very lower in diameter for composite electrode in comparison to pristine Fe3O4. These findings endorse the effective synergy among Fe3O4 and rGO carbonaceous materials, which causes by the presence of highly electrically conductive rGO porous nanosheets for the redox reaction occurred at the electrode/electrolyte interface for Fe3O4 particles. Furthermore, such a synergy could prevent the volume change and agglomeration of the Fe3O4 particles during charge/discharge cycles due to the presence of rGO.
4. Conclusions
In summary, a novel and facile co-deposition strategy (i.e. EPD/ECD) was introduced for the fabrication of Fe3O4@rGO composite onto Ni foam substrate. The FE-SEM and TEM observations confirmed the simultaneous electrophoretic deposition of rGO sheets on the cathode surface and electrochemical formation of iron oxide particles on the deposited rGO sheets. Based on these observations, a detailed formation mechanism was proposed and explained. The electrochemical evaluations through cyclic voltammetry and galvanostatic charge–discharging proved the high supercapacitive ability of the fabricated Fe3O4@rGO composite. Based on the obtained results, this one-pot EPD/ECD procedure is proposed as facile synthesis method for fabrication of binder-free iron oxide/carbonous hydride electrodes for supercapacitor applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Youth Project of Hunan Provincial Department of Education (No. 18B428) and General Project of Hunan Provincial Department of Education (No. 18C0796).Notes and references
- P. K. Panda, A. Grigoriev, Y. K. Mishra and R. Ahuja, Nanoscale Adv., 2020, 2, 70–108 RSC.
- P. Zhang, F. Wang, S. Yang, G. Wang, M. Yu and X. Feng, Energy Storage Mater., 2020, 28, 160–187 CrossRef.
- L. Y. Chen, Y. Hou, J. L. Kang, A. Hirata, T. Fujita and M. W. Chen, Adv. Funct. Mater., 2013, 3, 851–856 CAS.
- B. Patil, C. Park and H. Ahn, RSC Adv., 2019, 9, 33643–33652 RSC.
- M. A. A. Mohd Abdah, N. H. Nabilah Azman, S. Kulandaivalu and Y. Sulaiman, Mater. Des., 2020, 186, 108199 CrossRef.
- X. Xu, K. S. Hui, K. N. Hui, H. Wang and J. Liu, Mater. Horiz., 2020, 7, 1246–1278 RSC.
- Y. Zeng, M. Yu, Y. Meng, P. Fang, X. Lu and Y. Tong, Adv. Funct. Mater., 2016, 6, 1601053 Search PubMed.
- V. D. Nithya and N. SabariArul, J. Power Sources, 2016, 327, 297–318 CrossRef CAS.
- C. Guan, J. Liu, Y. Wang, L. Mao, Z. Fan, Z. Shen, H. Zhang and J. Wang, ACS Nano, 2015, 9, 5198–5207 CrossRef CAS PubMed.
- E. C. Vermisoglou, P. Jakubec, A. Bakandritsos, M. Pykal, S. Talande, V. Kupka, R. Zbořil and M. Otyepka, Chem. Mater., 2019, 31, 4698–4709 CrossRef CAS PubMed.
- W. C. Ng, C. Y. Tan, B. H. Ong and A. Matsuda, Procedia Eng., 2017, 184, 587–594 CrossRef.
- Z. Wang and C. J. Liu, Nano Energy, 2015, 11, 277–293 CrossRef CAS.
- Y. H. Kim and S. J. Park, Curr. Appl. Phys., 2011, 11, 462–466 CrossRef.
- R. Bhujel, S. Rai, U. Deka and B. P. Swain, J. Alloys Compd., 2019, 792, 250–259 CrossRef CAS.
- X. Zhao, Y. Jia and Z. H. Liu, J. Colloid Interface Sci., 2019, 536, 463–473 CrossRef CAS PubMed.
- V. D. Nithya and N. Sabari Arul, J. Mater. Chem. A, 2016, 4, 10767–10778 RSC.
- H. A. Ghalya, A. G. El-Deen, E. R. Souayc and N. K. Allam, Electrochim. Acta, 2019, 310, 58–69 CrossRef.
- X. Shi, S. Zhang, X. Chen, T. Tang and E. Mijowska, J. Alloys Compd., 2017, 726, 466–473 CrossRef CAS.
- N. A. Devi, S. Nongthombam, S. Sinha, R. Bhujel, S. Rai, W. I. Singh, P. Dasgupta and B. P. Swaina, Diamond Relat. Mater., 2020, 104, 107756 CrossRef CAS.
- F. E. Sarac Oztuna, O. Unal, H. Y. Acar, U. Unal and T. Beyazay, ChemElectroChem, 2019, 6, 3774–3781 CrossRef.
- R. Madhuvilakku, S. Alagar, R. Mariappan and S. Piraman, Sens. Actuators, B, 2017, 253, 879–892 CrossRef CAS.
- R. Kumar, R. K. Singh, A. R. Vaz, R. Savu and S. A. Moshkalev, ACS Appl. Mater. Interfaces, 2017, 9, 8880–8890 CrossRef CAS PubMed.
- C. Zhao, X. Shao, Y. Zhang and X. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 30133–30142 CrossRef CAS PubMed.
- M. Aghazadeh and M. R. Ganjali, J. Mater. Sci., 2018, 53, 295–308 CrossRef CAS.
- M. Aghazadeh and M. R. Ganjali, Ceram. Int., 2018, 44, 520–529 CrossRef CAS.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- F. Tuz, J. Jee-Wook and L. Woo-Gwang Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef.
- S. Shahrokhian, R. Mohammadi and E. Asadian, Int. J. Hydrogen Energy, 2016, 41, 17496–17505 CrossRef CAS.
- Y. L. Ren, H. Y. Wu, M. M. Lu, Y. J. Chen, C. L. Zhu, P. Gao, M. S. Cao, C. Y. Li and Q. Y. Ouyang, ACS Appl. Mater. Interfaces, 2012, 4, 6436–6442 CrossRef CAS PubMed.
- Y. Dong, Z. Yang, Q. Sheng and J. Zheng, Colloids Surf., A, 2018, 538, 371–377 CrossRef CAS.
- S. Mallick, P. P. Jana and C. Retna Raj, ChemElectroChem, 2018, 5, 2348–2356 CrossRef CAS.
- A. Ben-Refael, I. Benisti and Y. Paz, Catal. Today, 2020, 340, 97–105 CrossRef CAS.
- E. Andrijanto, S. Shoelarta, G. Subiyanto and S. Rifki, AIP Conf. Proc., 2016, 1725, 020003 CrossRef.
- D. He, Z. Peng, W. Gong, Y. Luo, P. Zhao and L. Kong, RSC Adv., 2015, 5, 11966–11972 RSC.
- M. Mohandoss, S. S. Gupta, A. Nelleri, T. Pradeep and S. M. Maliyekkal, RSC Adv., 2017, 7, 957–963 RSC.
- Y. Sun, W. Zhang, H. Yu, C. Hou, D. Li, Y. Zhang and Y. Liu, J. Alloys Compd., 2015, 638, 182–187 CrossRef CAS.
- A. Jorio, L. G. Cançado and L. M. Malard, Vibrations in graphene, Cambridge University Press, 2017, pp. 71–89 Search PubMed.
- N. Zhang, C. Chen, X. Yan, Y. Huang, J. Li, J. Ma and D. H. L. Ng, Electrochim. Acta, 2017, 223, 39–46 CrossRef CAS.
- H. Liu, M. Jia, Q. Zhu, B. Cao, R. Chen, Y. Wang, F. Wu and B. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 26878–26885 CrossRef CAS PubMed.
- S. Majumder, M. Sardar, B. Satpati, S. Kumar and S. Banerjee, J. Phys. Chem. C, 2018, 122, 21356–21365 CrossRef CAS.
- T. Qi, J. Jiang, H. Chen, H. Wan, L. Miao and L. Zhan, Electrochim. Acta, 2013, 114, 674–680 CrossRef CAS.
- H. L. Guo, P. Su, X. Kang and S. K. Ning, J. Mater. Chem. A, 2013, 1, 2248–2255 RSC.
- Y. Li, T. Jing, G. Xu, J. Tian, M. Dong, Q. Shao, B. Wang, Z. Wang, Y. Zheng, C. Yang and Z. Guo, Polymer, 2018, 149, 13–22 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, X. Chen, L. Leng, H. Wang, H. Li and G. Zeng, Chem. Eng. J., 2015, 262, 597–606 CrossRef CAS.
- M. A. Majeed Khan, W. Khan, M. Ahamed and A. N. Alhazaa, Mater. Sci. Semicond. Process., 2019, 99, 44–53 CrossRef.
- M. Aghazadeh and M. R. Ganjali, J. Mater. Sci.: Mater. Electron., 2018, 29, 2291–2300 CrossRef CAS.
- M. Aghazadeh, J. Mater. Sci.: Mater. Electron., 2017, 28, 18755–18764 CrossRef CAS.
- X. Yu, M. Wang, A. Gagnoud, Y. Fautrelle, R. Moreau and X. Li, Electrochim. Acta, 2017, 248, 150–159 CrossRef CAS.
- Q. Wu, R. Jiang and H. Liu, Ceram. Int., 2020, 46, 12732–12739 CrossRef CAS.
- K. Jiang, B. Sun, M. Yao, N. Wang, W. Hu and S. Komarneni, Microporous Mesoporous Mater., 2018, 265, 189–194 CrossRef CAS.
- H. Fang, F. Meng, J. Yan, G. Chen, L. Zhang, S. Wu, S. Zhang, L. Wang and Y. Zhang, RSC Adv., 2019, 9, 20107–20112 RSC.
- J. Sun, P. Zan, X. Yang, L. Ye and L. Zhao, Electrochim. Acta, 2016, 215, 483–491 CrossRef CAS.
- A. J. Khan, M. Sufyan Javed, M. Hanif, Y. Abbas, X. Liao, G. Ahmed, M. Saleem, S. Yune and Z. Liu, Ceram. Int., 2020, 46, 3124–3131 CrossRef CAS.
- F. Zhu, Y. Wang, Y. Zhang and W. Wang, Int. J. Electrochem. Sci., 2017, 12, 7197–7204 CrossRef CAS.
- S. Ghasemi and F. Ahmadi, J. Power Sources, 2015, 289, 129–137 CrossRef CAS.
- K. Asare Owusu, L. Qu, J. Li, Z. Wang, K. Zhao, C. Yang, K. M. Hercule, C. Lin, C. Shi, Q. Wei, L. Zhou and L. Mai, Nat. Commun., 2017, 8, 14264 CrossRef PubMed.
- H. Luo, K. Tao and Y. Gon, Dalton Trans., 2019, 48, 2491 RSC.
- X. Zhu, D. Hou, H. Tao and M. Li, J. Alloys Compd., 2020, 821, 153580 CrossRef CAS.
- M. Aghazadeh, I. Karimzadeh and M. R. Ganjali, J. Mater. Sci.: Mater. Electron., 2017, 28, 13532–13539 CrossRef CAS.
- L. Chen, D. Liu and P. Yang, RSC Adv., 2019, 9, 12793–12800 RSC.
- R. Kumar, S. M. Youssry, K. Zay Ya, W. Kian Tan, G. Kawamura and A. Matsuda, Diamond Relat. Mater., 2020, 101, 107622 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |