
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile way to improve zeolite Y-based catalysts' properties and performance in the isobutane–butene alkylation reaction†
Shunli Zhou,
Chengxi Zhang *,
Yongxiang Li*,
Bingzhang Shao,
Yibin Luo and
Xingtian Shu
*,
Yongxiang Li*,
Bingzhang Shao,
Yibin Luo and
Xingtian Shu
State Key Laboratory of Catalytic Material and Reaction Engineering, Research Institute of Petroleum Processing, Sinopec, Beijing 100083, China. E-mail: zhangcx.ripp@sinopec.com; liyx.ripp@sinopec.com
First published on 5th August 2020
Abstract
Different amounts of SiO2 were added to the Al2O3 binders to investigate the binder effect on zeolite Y-based catalysts. The added SiO2 improved the mesopore volume and acidity of the catalysts. Characterization results showed that the catalysts' acid amount increased with increasing the SiO2 amount in the binder, which achieved maximum value when 12% SiO2 was added to the binder. The doped SiO2 in Al2O3 binders improved the Al2O3 phase transformation temperature, which is crucial for Al species to break out of the phase energy and migrate into the zeolite. The lifetime of catalyst Y-Al2O3-12SiO2 is 3.7 times higher than that of Y-Al2O3-0SiO2, and the selectivity of the target products simultaneously improved by 7 percentage point. This work should bring some inspiration to the design and application of zeolite-based catalysts.
Introduction
Zeolite-based catalysts play important roles in producing our daily necessary chemicals such as fuel, medicines, cloth and so on.1–3 The broad applications of zeolites originate from their well-arranged channels and their tailored acidity, which cause them to perform excellently in various kinds of catalytic processes.3 In industrial reactors, zeolites need to be shaped into scale-up catalysts. Zeolites should be dispersed in the binders to improve the mechanical properties, thermal stability and reduce the cost of the catalyst.4,5 Binders bring significant effects to the scale-up catalysts as compared with the pure zeolite.6 However, numerous research efforts have focused on the zeolite performance but little research has been devoted to investigating the effects of binders on the catalysts.7,8Isobutane–butene alkylation is an important reaction in producing gasoline with a high octane number.9 The constraints of aromatics and the sulfur content in gasoline have become increasingly severe to reduce vehicle exhaust pollution.10 Isobutane–butene alkylate is highly desired because of its high octane number and environmental advantages, including low or no contents of sulfur, benzene, aromatics, and alkenes.11 Current industrial alkylation processes use hydrofluoric (HF) or sulfuric acid (H2SO4) as the catalysts.12 This leads to serious safety and environmental risks from the transport and storage of the concentrated liquid acids. The equipment corrosion and the process of dealing with the acid–oil sludges produced as by-products are also big problems. To solve these problems and reduce the environmental impact, zeolite-based catalysts are ideal catalysts to replace these liquid acids. However, it is still difficult to broadly commercialize the process because of the rapid deactivation of zeolite-based catalysts.13 The chemisorption capacity of butene is approximately two orders of magnitude higher than that of isobutane for zeolite-based catalysts.14 This leads to the intensive formation of oligomerization products, which would be transformed into coke covering the acid sites and blocking the zeolite channels.14–16
Many efforts have been made to figure out the isobutane–butene alkylation mechanism of zeolite Y-based catalysts.17 To prolong the catalysts' lifetime and improve the selectivity of the target products, the pore structure and acidity of the zeolite should be carefully modified. A high density and strength of Brønsted acid are crucial for improving the selectivity of the target products,13 while intensified acidity and optimized pore structure are beneficial for the lifetime.14
Modification of the Brønsted acid strength and construction of a hieratical pore structure could be achieved by post-synthesis or the dealumination of zeolite. However, this would damage the zeolite's framework, and thus reduce the acid amount. Recently, it has been found that the acidity of catalysts could be tailored by the interaction between zeolite and binders.6–8,18 Detailed understanding and tuning of the binder effects would be meaningful for the industrial application of the scaled-up catalysts. Many researchers have found that the Al2O3 binder could increase catalysts' acid amounts owing to the migration of Al species from the binder to the zeolite,6,8 and the immigrated Al species could enhance the Brønsted acid strength.2 It would be important to increase the amount of immigrated Al species for further improvement of the catalysts' acidity.
Here, we synthesized an aqueous colloidal SiO2 and doped it into the Al2O3 binder to formulate the zeolite into scaled up catalysts. The interactions between the binder and zeolite Y are discussed. The structure and acidity of the catalysts were analyzed by NH3-Temperature Program Desorption (NH3-TPD), X-ray diffraction (XRD), magic angle spinning nuclear magnetic resonance (MAS NMR) and infrared spectroscopy (IR spectroscopy), and the relationship between the binder and catalysts' properties are discussed. The isobutane–butene alkylation reaction was chosen as a probe reaction, and the effects of the catalysts' properties on the alkylation activity and stability were investigated.
Experimental
Materials
Ethyl orthosilicate (AR) was purchased from Innochem Chemical Works. HNO3 (66 wt%) was purchased from Sinopharm Chemical Reagent Co. Ltd. Al2O3 (boehmite) and zeolite Y, were purchased from Sinopec Catalyst. Co. LTD. Deionized water was produced in-house.Preparation of the catalysts
Ethyl orthosilicate was added to water and was hydrolysed into aqueous colloidal SiO2 at room temperature. The colloidal SiO2 was added to boehmite with a specific ratio (0%, 4%, 8%, 12% and 16%, dry mass ratio, based on the weight loss at 600 °C) as the binder of the catalyst. Zeolite and binder were added in a ratio of 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 (mass ratio). HNO3 was added to water and then added to the powder to form a paste, then the paste was added to the extruder. The extrudate was dried in an oven at 120 °C for 12 h and calcined at 600 °C for 4 h. The catalysts were named Y-Al2O3-0SiO2, Y-Al2O3-4SiO2, Y-Al2O3-8SiO2, Y-Al2O3-12SiO2 and Y-Al2O3-16SiO2, respectively.
:15 (mass ratio). HNO3 was added to water and then added to the powder to form a paste, then the paste was added to the extruder. The extrudate was dried in an oven at 120 °C for 12 h and calcined at 600 °C for 4 h. The catalysts were named Y-Al2O3-0SiO2, Y-Al2O3-4SiO2, Y-Al2O3-8SiO2, Y-Al2O3-12SiO2 and Y-Al2O3-16SiO2, respectively.
Physicochemical characterization
To characterize the XRD patterns of the catalysts, an EMPYREAN powder diffractometer (PANalytical Corporation, Netherland) using Cu Kα radiation (λ = 0.154 nm) at a scanning rate of 0.02°·min−1 from 5–35° was employed. The relative crystallinity of the catalysts was calculated from the standard sample in the laboratory. In situ XRD was employed to characterize the Al2O3-SiO2 samples, and the method is as follows: the XRD patterns were recorded every 20 °C from 310 °C to 430 °C with a heating rate of 5 °C min−1. The scanning area was focused at 47.5–53° at a scanning rate of 0.02°·min−1. The relative crystallinity of γ-Al2O3 was calculated from the relative peak area at 49.5°.An X-ray Fluorescence (XRF) spectrometer (ZSX Primus II, Rigaku) was employed to investigate the chemical compositions of the samples.
Coke formed in catalysts were evaluated by thermal gravimetric analysis (TGA, Pyris 1, PerkinElmer, US). The samples were held at 50 °C for 10 min, then heated from 50 °C to 1000 °C at 10 °C min−1 under an air atmosphere to remove the coke.
Nitrogen adsorption–desorption isotherms were carried out on a Micromeritics ASAP2420 instrument. Fresh samples were outgassed under vacuum for 9 h at 330 °C to desorb the impurities prior to the adsorption measurements. The Brunauer–Emmett–Teller (BET) equation was employed to calculate the surface area. The total pore volume was evaluated from the volume adsorbed at p/p0 = 0.99, whereas the micropore surface area and the micropore volume were determined by the t-plot method.
29Si MAS NMR experiments were performed on a Bruker AVANCE III 500 WB spectrometer at a resonance frequency of 99.3 MHz using a 7 mm double-resonance MAS probe. The magic-angle spinning speed was 5 kHz in all experiments, and a typical π/6 pulse length of 1.8 μs was adopted for 29Si resonance. The chemical shift of 29Si was referenced to TMS.
27Al MAS NMR experiments were performed on a Bruker AVANCE III 600 WB spectrometer at a resonance frequency of 156.4 using a 4 mm double-resonance MAS probe at a sample spinning rate of 12 kHz. The chemical shift of 27Al was referenced to 1 M aqueous Al(NO3)3. 27Al MAS NMR spectra were recorded by the small-flip angle technique using a pulse length of 0.4 μs (<π/12) and a recycle delay of 1 s.
The peak area of the 29Si spectrum was integrated and the framework Si/Al ratio was estimated, the formula is as follows:
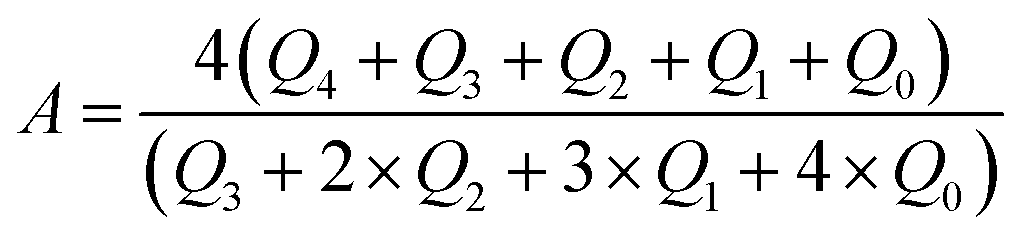 | (1) |
NH3-TPD experiments were carried out on a Chemstar TPx (Quantachrome, America) unit equipped with a thermal conductivity detector. Here, 150 mg catalyst was ground into a 20–40 mesh before use, then degassed in a He stream at 550 °C for 1 h. After cooling to 100 °C, the sample was put in a mixed gas flow of 5% NH3 and 95% He (40 ml min−1) for the sufficient adsorption of NH3. After desorbing the physically absorbed NH3, the TPD profiles were recorded from 150 °C to 250 °C at a constant heating rate of 10 °C min−1 under He flow (40 ml min−1), and then the flow was kept for 30 min at 250 °C. The temperature was increased to 350 °C, 450 °C and 550 °C using the same method. The desorbed ammonia was monitored by a thermal conductivity detector, and the peak area was integrated and calibrated to calculate the desorbed amount of NH3 under different temperatures. The desorbed amount of NH3 was defined as the number of acid sites; the acid sites at 100–250 °C were defined as weak acids, the acid sites at 250–450 °C were defined as medium acid sites, and the 450–550 °C acid sites were defined as strong acid sites, and the sum acidity was the total acidity of the catalysts.
The IR spectra of skeleton hydroxy-groups of the catalysts were recorded as follows: 20 mg catalyst was ground into a powder and pressed into the mold under 20 MPa. The sample was placed into the in situ device and degassed, heated to 450 °C for 120 min, then cooled to room temperature and the spectra recorded. The acidity of the catalysts was determined from the IR spectrum of adsorbed pyridine. The sample was dehydrated at 450 °C for 2 h. The sample was saturated with pyridine at room temperature for 10 min then degassed for 30 min to desorb the physically adsorbed pyridines, and then heated to 200 °C and 350 °C, separately, for 30 min to desorb the chemisorbed pyridine before collecting the spectra. The absorption peak at 1540 cm−1 was assigned to the Brønsted acid (B acid) and the peak at 1450 cm−1 was assigned to the Lewis acid (L acid). The acid sites were estimated by the integration of the peak area and calculated with the following formula:
| CB = 1.88ABR2/W | (2) |
| CL = 1.42ALR2/W | (3) |
Alkylation reaction
Isobutane–butene alkylation was carried out on a fixed-bed reactor under high pressure to evaluate the catalysts' activity. Here, 5 g of catalyst (20–40 mesh) was loaded into the reactor, then heated up and held at 500 °C for 2 h under N2 atmosphere. After cooling to 75 °C, a mixture of isobutane and trans-2-butene (simplified as butene) with a mass ratio of 250:1 was fed, and the WHSV was 100 g h−1. The products were automatically collected through a six-port valve with a sample loop into a gas chromatograph (Agilent, 7890B) with two FID detectors.
Definition and calculation formula of butene conversion are as follows: butene conversion refers to the amount of butene that has been reacted at a certain moment with respect to the total butene entering the reactor at that moment, defined as X. The calculation formula is
| X = (1 − AC4t/AC40) × 100% | (4) |
Selectivity refers to the mass fraction of a component in the alkylate. The selectivity is defined as
| SCn = δCn × 100% | (5) |
The catalysts' lifetime was defined as the time on stream from the beginning to the time that the conversion of butene started to be less than 100%.
The rate of hydrogen transfer reaction was correlated with the n-butane amount generated at the initial stage.21,22 The hydrogen transfer rate was calculated according to the n-butane amount formed in the first 2 h; the formula is as follows:
| Hydrogen transfer rate = WHSV × (A2Cn/A2Ci) × 100% | (6) |
Results and discussion
Catalyst characterization
The texture properties of the catalysts and binders are listed in Table 1. It can be seen from Table 1 that the specific surface areas of the catalysts were similar. Compared with zeolite Y, the specific surface area of the catalysts decreased, which might be attributed to binder dilution and unit cell destruction under calcination. The specific surface area of different catalysts changed slightly. The micropore area of the catalysts was nearly unchanged as SiO2 was added, and the corresponding micropore volume of the catalysts was also kept steady. It showed that the mesopore volume of pure Al2O3 and SiO2 were several times higher than that of zeolite Y. The mesopore volume of the catalysts was much larger than the theoretical volume. The increased mesopore volume may have been formed by the skeleton collapse caused by dealumination during calcination. The presence of SiO2 in the binder greatly increased the mesopore volume of the catalysts. This could be attributed to the changed in Al2O3 stacking due to the addition of SiO2. The colloidal SiO2 doped in Al2O3 underwent condensation during calcination and left a mesoporous network.23 However, it was shown that the mesopore volume of the catalysts did not increase with the increasing SiO2 amount in the binder.| Catalyst | Stotal (m2 g−1) | Smicro (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) practical/theoreticalb |
|---|---|---|---|---|---|
| a Theoretical mesopore volume.b Theoretical results were calculated with an equation: V = 85% × Y + 15% (x wt% × SiO2 + (100 − x) wt% × γ-Al2O3), x was the SiO2 amount in the binder. | |||||
| Y-Al2O3-0SiO2 | 621 | 549 | 0.370 | 0.255 | 0.115/0.071a |
| Y-Al2O3-4SiO2 | 622 | 548 | 0.411 | 0.252 | 0.159/0.074a |
| Y-Al2O3-8SiO2 | 624 | 542 | 0.407 | 0.250 | 0.157/0.078a |
| Y-Al2O3-12SiO2 | 639 | 559 | 0.402 | 0.247 | 0.155/0.081a |
| Y-Al2O3-16SiO2 | 644 | 564 | 0.412 | 0.257 | 0.155/0.085a |
| Y | 710 | 681 | 0.363 | 0.315 | 0.048 |
| γ-Al2O3 | 290 | 211 | 0.349 | 0.149 | 0.200 |
| SiO2 | 545 | — | 0.771 | — | 0.771 |
The physicochemical properties of the catalysts are shown in Table 2. Compared with zeolite Y, the relative crystallinity of the catalysts declined to about 70%, which may have been caused by the binder dilution and unit cell destruction during calcination. The unit cell of the catalysts shrank, which was attributed to dealumination under calcination. However, it could be seen that the unit cell shrinkage was inhibited with the increasing addition of SiO2.
| Catalyst | Cell parameter (nm) | Crystallinity (%) | SiO2 (%) | Al2O3 (%) |
|---|---|---|---|---|
| Y-Al2O3-0SiO2 | 2.447 | 65.9 | 68.1 | 31.1 |
| Y-Al2O3-4SiO2 | 2.450 | 65.7 | 67.1 | 32.0 |
| Y-Al2O3-8SiO2 | 2.451 | 69.4 | 67.0 | 32.2 |
| Y-Al2O3-12SiO2 | 2.452 | 69.0 | 70.3 | 28.9 |
| Y-Al2O3-16SiO2 | 2.452 | 71.9 | 73.0 | 26.2 |
| Y | 2.457 | 92.1 | 80.1 | 19.7 |
| Al2O3 | — | — | — | 99.9 |
| SiO2 | — | — | 99.9 | — |
The NH3-TPD results are shown in Table 3. Compared with Y-Al2O3-0SiO2, the presence of SiO2 in the binder improved the total acid amount of the catalysts. The total acid amount of the catalysts increased with the increasing SiO2 amount, reaching the maximum when 12% SiO2 was added in the binder. The amount of medium acid also increased with the increasing SiO2 content, but the weak and strong acids remained relatively steady. In order to verify whether the increased acid amount was caused by the binder, the acidity of the binders before and after the addition of SiO2 were tested. The results showed that the binder's acid amount increased slightly after the addition of SiO2, which indicated that the increased acid sites of the catalysts were not contributed by the binder, but were caused by the interaction between the zeolite and the binders.
| Catalysts | Acid amount (μmol g−1) | |||
|---|---|---|---|---|
| Weak | Medium | Strong | Total | |
| Y-Al2O3-0SiO2 | 774 | 530 | 86 | 1390 |
| Y-Al2O3-4SiO2 | 943 | 617 | 88 | 1648 |
| Y-Al2O3-8SiO2 | 958 | 664 | 77 | 1699 |
| Y-Al2O3-12SiO2 | 981 | 739 | 73 | 1793 |
| Y-Al2O3-16SiO2 | 957 | 795 | 44 | 1796 |
| Al2O3 | 308 | 206 | 67 | 582 |
| Al2O3-12SiO2 | 315 | 236 | 89 | 640 |
The MAS NMR spectra of the catalysts are shown in Fig. 1. There are mainly five peaks in the 27Al spectra, the peak at 0 ppm was attributed to the peak of the six-coordinate Extra-Framework Alumina (EFAL) formed by the calcination of the catalyst. The displacements at 9 ppm and 68 ppm were attributed to γ-Al2O3 in the binder. The displacement peak at 60 ppm was attributed to the tetra-coordinated framework aluminum in the zeolite.24 The 55 ppm peak was attributed to the twisted four-coordinated Al species. There were no differences in the Al spectra before and after the addition of SiO2. This showed that the catalysts' γ-Al2O3 peak area at 9 ppm decreased with the increasing SiO2 content. There may be two reasons: (i) the Al2O3 amount decreased as the SiO2 content increased; (ii) the presence of SiO2 promoted Al migration into the zeolite.
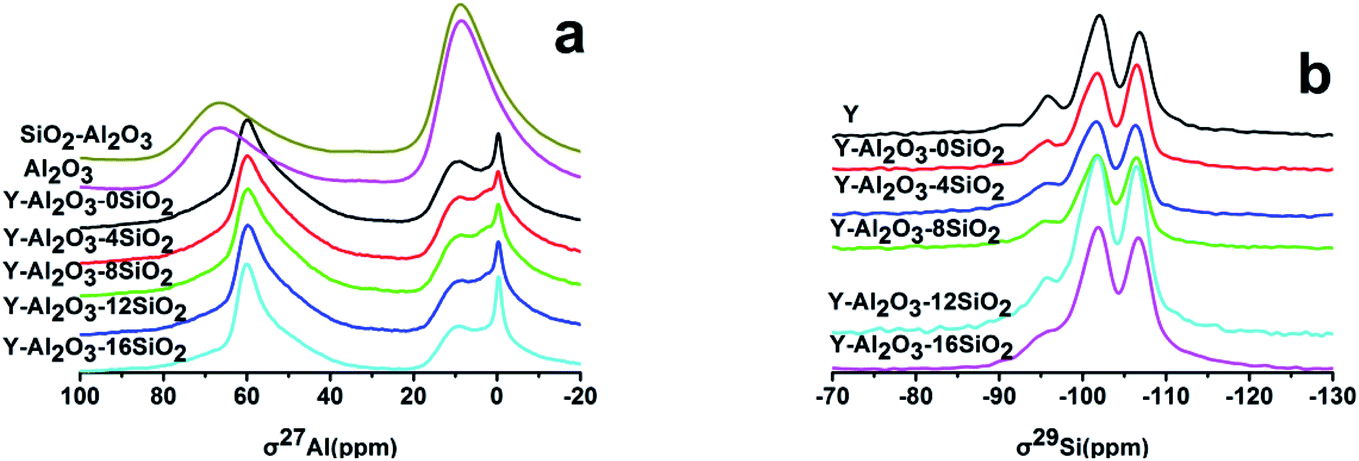 | ||
| Fig. 1 The MAS NMR spectra of the catalysts. (a) 27Al spectra; (b) 29Si spectra. | ||
The MAS NMR 29Si spectrum of each catalyst is shown in Fig. 1b. The displacements at −106 ppm, −102 ppm, −95 ppm and −92.3 ppm were to Si(4Si) (Q4), Si(3Si, 1Al) (Q3), Si(2Si, 2Al) (Q2) and Si(1Si, 3Al) (Q1), respectively.19 The catalysts' peak areas were integrated and the framework Si/Al ratios of the catalysts were estimated and are listed in Table 4. It was shown that the framework Si/Al ratio was 4.48 in zeolite Y, but when scaled up with Al2O3, the framework Si/Al ratio changed to 7.01; this was attributed to the dealumination under high temperature calcination. However, the Si/Al ratio gradually decreased with the increasing SiO2 content, which indicated that the added SiO2 prevented dealumination, which was consistent with the XRD results.
| Catalysts | Q4 | Q3 | Q2 | Q1 | Si/Al |
|---|---|---|---|---|---|
| Y | 10 | 20.4 | 3.01 | 1.63 | 4.48 |
| Y-Al2O3-0SiO2 | 10 | 7.56 | 1.72 | 0 | 7.01 |
| Y-Al2O3-4SiO2 | 10 | 7.03 | 1.94 | 0.21 | 6.65 |
| Y-Al2O3-8SiO2 | 10 | 6.78 | 2.08 | 0.15 | 6.68 |
| Y-Al2O3-12SiO2 | 10 | 10.03 | 2.32 | 0.18 | 6.20 |
| Y-Al2O3-16SiO2 | 10 | 8.3 | 1.88 | 0.23 | 6.65 |
The IR spectra in the OH-stretching region of the catalysts are shown in Fig. 2. The absorption peaks at 3604 cm−1 and 3667 cm−1 were attributed to the absorption peak of non-framework aluminum (Al(OH)n(3−n)+) at the ionic position; the absorption peaks at 3565 cm−1, 3626 cm−1 and 3743 cm−1 were assigned to the skeleton hydroxyl groups in the sodalite cage, supercage, and the surface hydroxyl group or terminal hydroxyl group of SiO2, respectively.24–27 It could be seen that Y-Al2O3-0SiO2 showed no obvious absorption peak at 3743 cm−1, but the catalysts with SiO2 had a strong absorption peak that was attributed to the Si–OH of SiO2. After the addition of SiO2, the hydroxyl peak at 3626 cm−1 gradually increased with the increasing SiO2 content. Compared with Y-Al2O3-0SiO2, the catalysts with SiO2 in the binder also showed absorption enhancement at 3604 cm−1 and 3667 cm−1. The results indicated that the presence of SiO2 helped the Al species to migrate to the ionic positions of zeolite, and the presence of SiO2 improved the hydroxyl group amounts in the supercage. The spectra showed that all the peaks reached the maximum when 12% SiO2 was added to the binder.
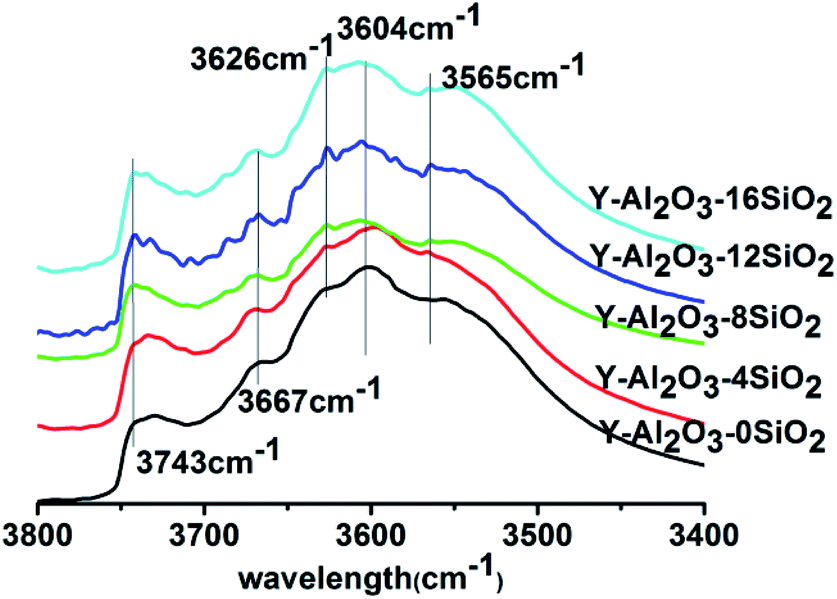 | ||
| Fig. 2 The IR spectra of the catalysts in the OH-stretching region. | ||
Table 5 shows the acidity distribution based on the pyridine adsorption IR (py-IR) of the catalysts. The acid sites below 200 °C and 350 °C were assigned to total acid and strong acid, respectively. It was shown that the acidity of the catalysts varied with the SiO2 amounts. The total and strong acid amounts increased with the increasing SiO2 amount, which achieved the maximum when 12% SiO2 was added in the binders. The B/L value of Y-Al2O3-12SiO2 was twice that of Y-Al2O3-0SiO2 for both the total acid and the strong acid. The total Brønsted acid amount increased with the increasing SiO2 amount, which was attributed to the new acid sites created by the interaction between the binder and the terminal Si–OH. The increase in the total Lewis acid amount was attributed to the increased EFAL at the ionic position. The EFAL at the ionic position would also enhance the strength of the nearby Brønsted acid sites, which would lead to the improved B/L value at 350 °C.
| Catalysts | 200 °C/(μmol g−1) | B/L | 350 °C/(μmol g−1) | B/L | ||||
|---|---|---|---|---|---|---|---|---|
| B acid | L acid | Total acid | B acid | L acid | Strong acid | |||
| Y-Al2O3-0SiO2 | 177 | 303 | 480 | 0.58 | 103 | 125 | 228 | 0.82 |
| Y-Al2O3-4SiO2 | 216 | 308 | 524 | 0.70 | 155 | 153 | 308 | 1.01 |
| Y-Al2O3-8SiO2 | 245 | 297 | 542 | 0.82 | 215 | 154 | 369 | 1.40 |
| Y-Al2O3-12SiO2 | 267 | 286 | 553 | 0.93 | 228 | 145 | 373 | 1.57 |
| Y-Al2O3-16SiO2 | 248 | 301 | 549 | 0.82 | 206 | 157 | 363 | 1.31 |
Alkylation performance
The catalytic performances of the catalysts were evaluated in the isobutane–butene alkylation reaction, and the results are shown in Fig. 3. The results showed that the catalysts' lifetimes increased with the increasing SiO2 amount, which achieved the maximum when 12% SiO2 was added. The lifetime of Y-Al2O3-12SiO2 was 3.7 times higher than that of Y-Al2O3-0SiO2. Interestingly, it was found that the selectivity of the target product C8 shared the same trend as lifetime, and Y-Al2O3-12SiO2 acquired the highest C8 selectivity. This could be due to the catalyst's higher hydride transfer rate. The results are consistent with the pyridine IR results; the Brønsted acid amount shared the same trend as lifetime and C8 selectivity. As competing reactions and products, the trends of the selectivity variation of the side products C5–C7 and C9+ were the opposite of the C8 selectivity. The C5–C7 and C9+ selectivities both had a minimum value when 12% SiO2 was added in the binder.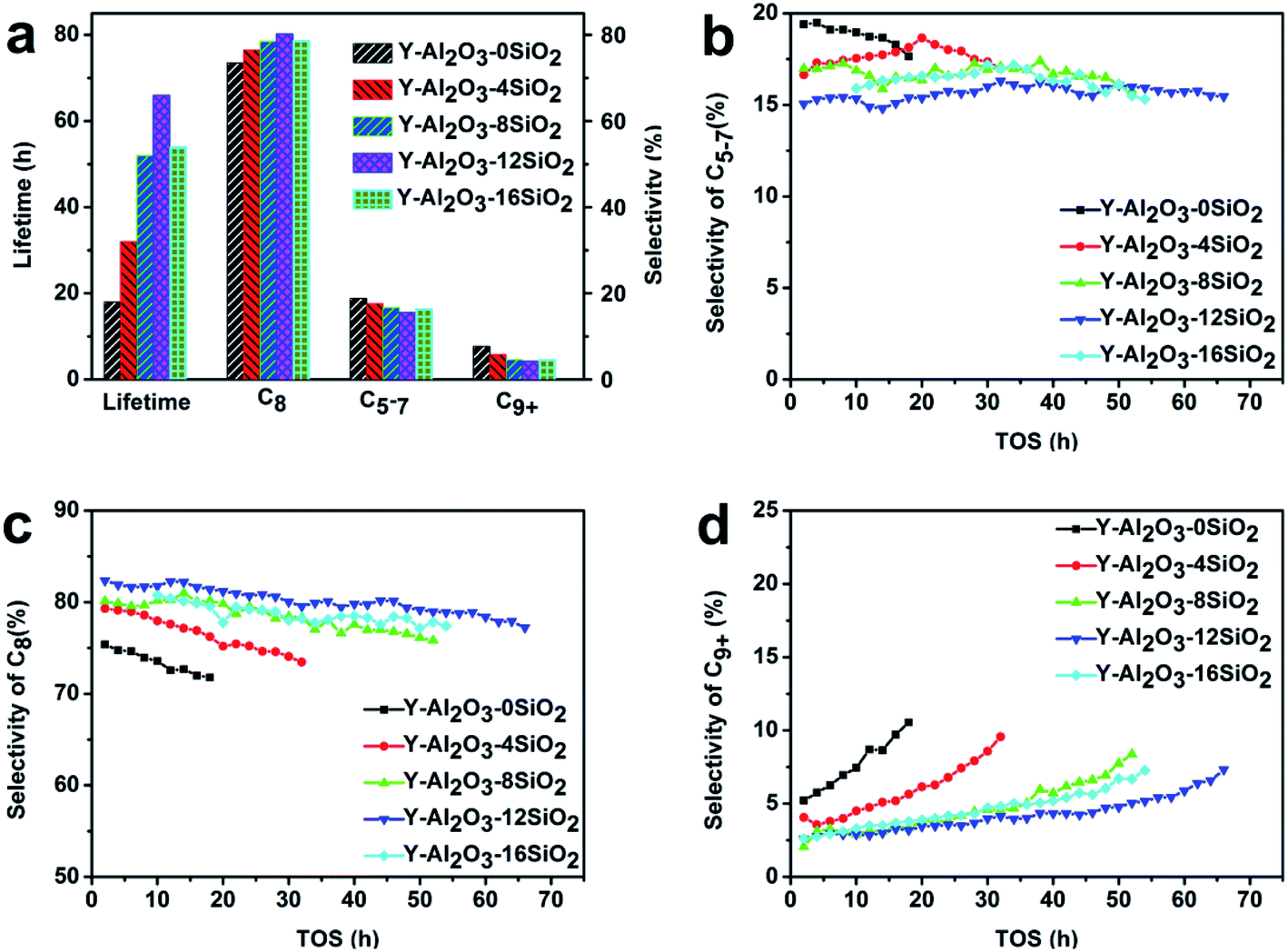 | ||
| Fig. 3 The catalytic performance of the catalysts in alkylation. (a) Lifetime and selectivity distribution. (b) Selectivity of C5–C7. (c) Selectivity of C8. (d) Selectivity of C9+. | ||
In order to verify whether the improved lifetime was caused merely by the binder alone, the binder with 12% SiO2 was tested on alkylation. The results showed that the conversion of the pure binder at 1 h was 70%, and the conversion quickly reduced to zero at 3 h, which indicated that the binder was inactive. NH3-TPD also confirmed that the addition of SiO2 did not bring about a significant increase in the acid amount. It could be concluded that doping SiO2 in Al2O3 binders did not increase the acidity of the binder.
The average yields of alkylates of each catalyst are listed in Table 6. It was shown that the alkylate yield increased with the increasing SiO2 addition, which reached a maximum at 12% SiO2 loading amount. The yield of H2SO4 alkylation was 93.10 and the yield of HF alkylation was 91.50.28 It could be concluded that the alkylate yield of the catalysts with SiO2 added to the binder is comparable to liquid-acid alkylation.
| Catalysts | Yield (%) |
|---|---|
| Y-Al2O3-0SiO2 | 89.5 |
| Y-Al2O3-4SiO2 | 93.5 |
| Y-Al2O3-8SiO2 | 93.6 |
| Y-Al2O3-12SiO2 | 94.5 |
| Y-Al2O3-16SiO2 | 94.0 |
Discussion
The zeolite–binder interaction would affect the catalyst activity through the influence on porosity and acidity.5,7,29 The interaction between the zeolite and different binders would affect the pore structure of the catalysts, which would change the diffusion rate of the reaction and production molecules. The acidity improvement was attributed to the Al migration from the binder to the zeolite through the following ways: (i) Al species reacted with terminal Si-hydroxyl generating new Si–O–Al bond and acid sites.5,30 (ii) Al species immigrated to the ionic position of the zeolite, which could enhance the acid strength, as proposed in many works.2,31 The migrated ionic position of Al(OH)n(3−n)+ would also protect the neighboring ionic bond-linked Al atom from dealumination under high-temperature conditions. Chen et al. found that Al migration occurred during calcination and was accomplished below 450 °C when the Al2O3 phase transformation took place.30 The Al migration process required high temperatures to break out the boehmite phase during the phase transformation process.32 We propose that doping with SiO2 could postpone the Al2O3 phase transformation, which would increase the transformation temperature and increase the amount of Al migration. In situ XRD was employed to characterize the phase transformation process of the samples; the XRD patterns are shown in Fig. 4, and the relative crystallinities of γ-Al2O3 at different temperatures are shown in Table 7. The phase transform temperature of the Al2O3-12SiO2 sample was higher than that of the pure Al2O3 sample. The relative crystallinity of γ-Al2O3 at different temperatures decreased with the increasing SiO2 amounts in Al2O3. The results confirmed that the presence of SiO2 indeed increased the transformation temperature of the binder, which was crucial for Al species migration.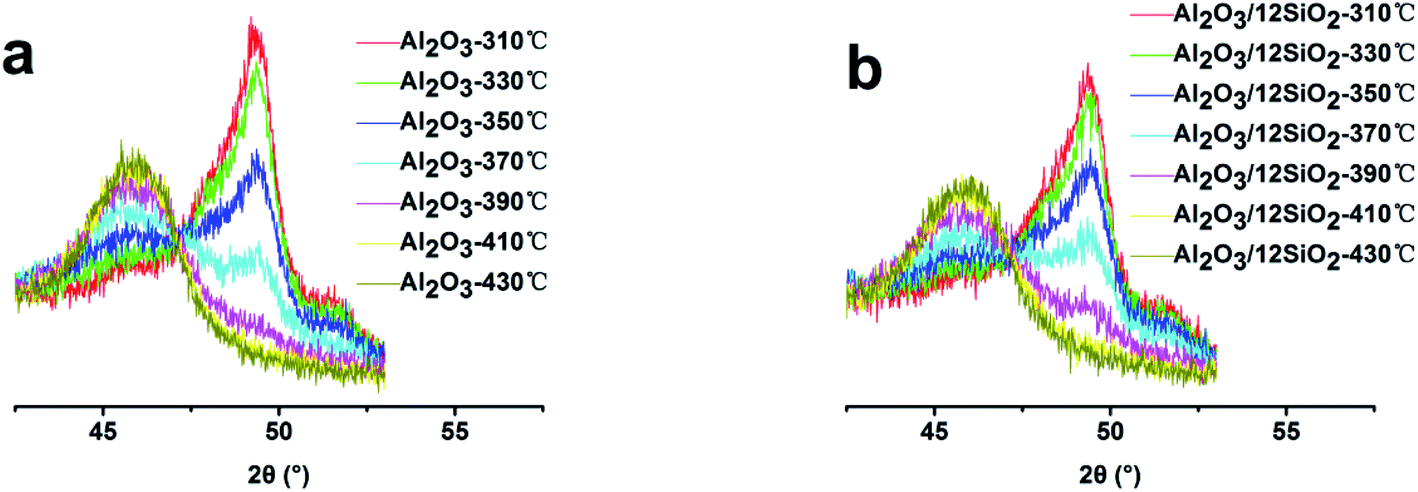 | ||
| Fig. 4 The in situ XRD pattern of the binders. (a) Al2O3; (b) Al2O3/12SiO2. | ||
| Sample | Relative crystallinity (%) | ||||
|---|---|---|---|---|---|
| 330 °C | 350 °C | 370 °C | 390 °C | 410 °C | |
| Al2O3-0SiO2 | 13.0 | 27.4 | 69.8 | 91.8 | 100 |
| Al2O3-4SiO2 | 11.1 | 22.3 | 59.7 | 89.7 | 100 |
| Al2O3-8SiO2 | 9.8 | 25.6 | 57.1 | 89.0 | 100 |
| Al2O3-12SiO2 | 10.1 | 16.7 | 55.3 | 87.4 | 95 |
| Al2O3-16SiO2 | 8.8 | 19.4 | 56.2 | 88.8 | 100 |
The XRD results showed that the unit cell size of the catalysts increased with the increasing SiO2 amount. The increased SiO2 promoted Al migration, which formed the ionic position of Al(OH)n(3−n)+, which could protect the zeolite's framework from dealumination. The MAS NMR Al spectra showed that the γ-Al2O3 area of the catalyst decreased with the addition of SiO2, which verified the increased Al migration with increasing SiO2 amount. The MAS NMR Si spectra confirmed the Al migration and the protection of the zeolite's framework from dealumination, which is consistent with the XRD and NH3-TPD results. The pyridine adsorption IR results showed that the presence of SiO2 improved the amount and strength of the Brønsted acid sites. Interestingly, all of the characterization results showed that it achieved the maximum value when 12% SiO2 was added in the binder. In summary, it was concluded that when 12% SiO2 was added in the binder, the Al species that immigrated into the zeolite reached the maximum, which maximized the preservation of the zeolite's framework. The continued increase of SiO2 would decrease the Al2O3 content, which would in turn reduce the Al amount that could migrate, thus the acidity was reduced when the SiO2 amounts added were higher than 12%.
The alkylation process is shown in Fig. 5.21 The catalytic process started with the formation of the C4 carbenium ion (C4+) via alkene protonation, then the C4+ would react with butene and form the C8 carbenium ion (C8+). The initiation process was then taken over by propagation reaction cycles. The propagation reaction is governed by three steps, which are oligomerization, isomerization of the C8+ and hydride transfer between isobutane and carbenium ion intermediate (C8+). The oligomerization results in macromolecules such as C12, C16 and so on.33 These macromolecules produce coke or are cracked into small molecules like C5–7. The coke blocks the channels or is adsorbed on active sites leading to the fast deactivation of the catalysts.34,35 The hydride transfer and isomerization always happen at the same time, which can produce C8 isomers and new C4+. The key elementary reactions that influence the product distribution and catalyst lifetime are the hydride transfer and oligomerization.17 Hydride transfer reactions should be fast enough to compete with oligomerizations to obtain high target product selectivity and lifetime. Brønsted acid sites are the most valuable for alkylation.17,36 Corma et al. found that C8 selectivity and catalyst lifetime are in proportion to the amount of Brønsted acid sites.13
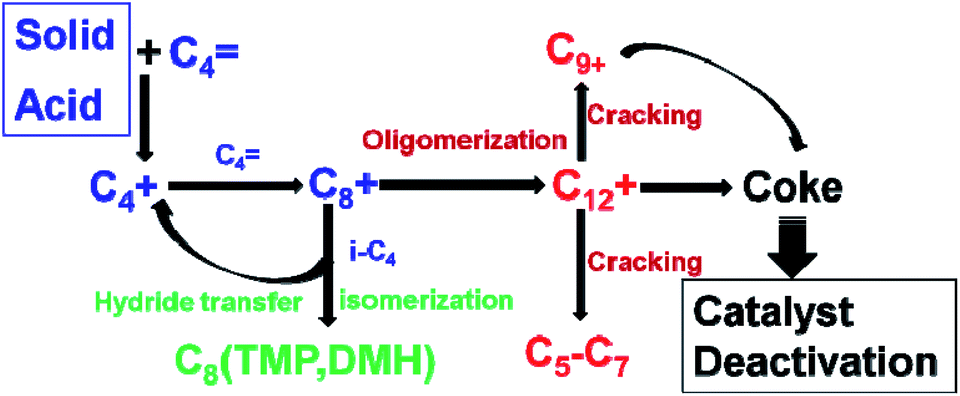 | ||
| Fig. 5 Scheme showing isobutane–butene alkylation. | ||
The catalysts' hydrogen transfer rates are listed in Table 8, which shows that the hydrogen transfer rate is coincident with the catalysts' lifetime and C8 selectivity, and the results are consistent with previous studies.13,33
| Catalysts | Hydrogen transfer rate (g h−1) |
|---|---|
| Y-Al2O3-0SiO2 | 0.0165 |
| Y-Al2O3-4SiO2 | 0.0242 |
| Y-Al2O3-8SiO2 | 0.0269 |
| Y-Al2O3-12SiO2 | 0.0345 |
| Y-Al2O3-16SiO2 | 0.0287 |
The TGA curves of Y-Al2O3-0SiO2 and Y-Al2O3-12SiO2 deactivated catalysts are shown in Fig. 6. The weight loss below 200 °C was caused by dehydration, and the weight loss higher than 200 °C was attributed to the coke in the catalysts.37 The coke content was calculated from TGA curves and are listed in Table 9. The coke amount of Y-Al2O3-12SiO2 was lower than that of Y-Al2O3-0SiO2. The coke deposition in the catalysts was attributed to oligomerization, which is competitive against the hydrogen transfer reaction. The hydrogen transfer rate of Y-Al2O3-12SiO2 is higher than that of Y-Al2O3-0SiO2 and would, therefore, repress oligomerization and decrease the formation of macromolecules that would result in coke. The coke formed would cover the acid sites or block zeolite channels, causing the catalysts' fast deactivation.
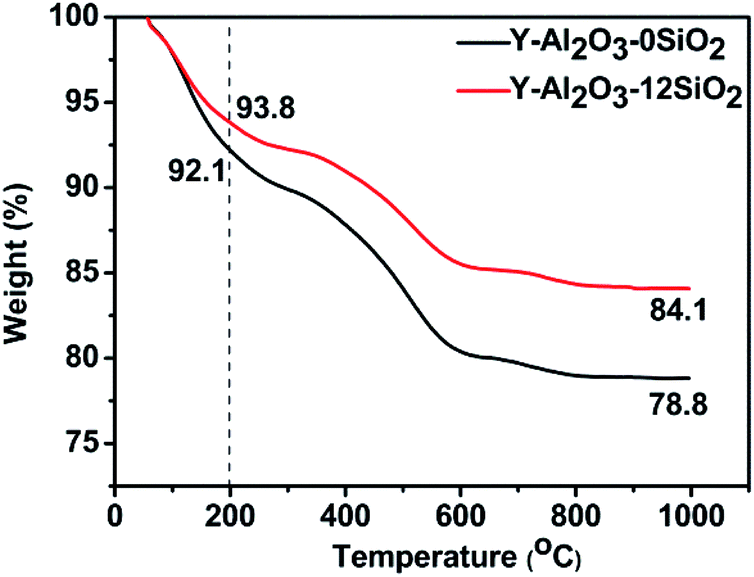 | ||
| Fig. 6 The TGA curves of the catalysts. | ||
| Catalysts | Coke content (%) |
|---|---|
| Y-Al2O3-0SiO2 | 13.3 |
| Y-Al2O3-12SiO2 | 9.7 |
Conclusions
We have shown that SiO2 doped in the Al2O3 binders could improve the zeolite Y-based catalysts' acidity and catalytic properties. The catalysts' acidities were improved with increasing the SiO2 amount, and the best doping amount was 12%. It has been shown that the presence of SiO2 increased the mesopore volume. Isobutane–butene alkylation was carried out to evaluate the catalysts' catalytic activities. The lifetimes and the selectivities of target products increased with the increasing SiO2 amounts in the binder and achieved the maximum when 12% SiO2 was added. Acidity and catalytic property change principles were proposed. It was verified that the added SiO2 increased the phase transformation temperature of Al2O3, which was important for Al species migration from binders to the zeolite. The migrated Al species improved the catalysts' acidities and activities. The catalytic performance was enhanced by simply tuning the composition of the binder, and the results should provide inspiration for the design and applications of zeolite-based catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFB0306502) and Sinopec Research Program (117020-1, 118017-1).Notes and references
- S. Mintova, M. Jaber and V. Valtchev, Chem. Soc. Rev., 2015, 44, 7207–7233 RSC.
- A. Primo and H. Garcia, Chem. Soc. Rev., 2014, 43, 7548–7561 RSC.
- A. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS.
- S. Mitchell, N. L. Michels and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 6094–6112 RSC.
- G. T. Whiting, F. Meirer, M. M. Mertens, A. J. Bons, B. M. Weiss, P. A. Stevens, E. de Smit and B. M. Weckhuysen, ChemCatChem, 2015, 7, 1312–1321 CrossRef CAS PubMed.
- U. J. Etim, P. Bai, Y. Wang, F. Subhan, Y. Liu and Z. Yan, Appl. Catal., A, 2019, 571, 137–149 CrossRef CAS.
- N. L. Michels, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2014, 4, 2409–2417 CrossRef CAS.
- G. T. Whiting, S. H. Chung, D. Stosic, A. D. Chowdhury, L. I. van der Wal, D. Fu, J. Zecevic, A. Travert, K. Houben, M. Baldus and B. M. Weckhuysen, ACS Catal., 2019, 9, 4792–4803 CrossRef CAS.
- C. Sievers, J. S. Liebert, M. M. Stratmann, R. Olindo and J. A. Lercher, Appl. Catal., A, 2008, 336, 89–100 CrossRef CAS.
- W. Shen, Y. Gu, H. Xu, D. Dubé and S. Kaliaguine, Appl. Catal., A, 2010, 377, 1–8 CrossRef CAS.
- W. Zheng, D. Li, W. Sun and L. Zhao, Chem. Eng. Sci., 2018, 186, 209–218 CrossRef CAS.
- Y. Li, R. Liu, G. Zhao, Z. Zhou, J. Zhang, C. Shi, X. Liu, X. Zhang and S. Zhang, Fuel, 2018, 216, 686–696 CrossRef CAS.
- A. Corma, A. Martinez and C. Martinez, J. Catal., 1994, 146, 185–192 CrossRef CAS.
- H. Wang, X. Meng, G. Zhao and S. Zhang, Green Chem., 2017, 19, 1462–1489 RSC.
- Z. Liu, X. Yi, G. Wang, X. Tang, G. Li, L. Huang and A. Zheng, J. Catal., 2019, 369, 335–344 CrossRef CAS.
- T. Hamzehlouyan, M. Kazemeini and F. Khorasheh, Chem. Eng. Sci., 2010, 65, 645–650 CrossRef CAS.
- C. Liu, R. A. van Santen, A. Poursaeidesfahani, T. J. H. Vlugt, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2017, 7, 8613–8627 CrossRef CAS PubMed.
- S. S. David, E. G. Wiliam, C. Pochen, N. M. Joseph, M. L. Rudolph, T. W. C. Cynthia and D. C. Clarence, J. Catal., 1985, 93, 471–474 CrossRef.
- Z. Yan, D. Ma, J. Zhuang, X. Liu, X. Liu, X. Han, X. Bao, F. Chang, L. Xu and Z. Liu, J. Mol. Catal. A: Chem., 2003, 194, 153–167 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- H. Zhang, J. Xu, H. Tang, Z. Yang, R. Liu and S. Zhang, Ind. Eng. Chem. Res., 2019, 58, 9690–9700 CrossRef CAS.
- K. Yoo, E. C. Burckle and P. G. Smirniotis, J. Catal., 2002, 211, 6–18 CrossRef CAS.
- J. Lefevere, L. Protasova, S. Mullens and V. Meynen, Mater. Des., 2017, 134, 331–341 CrossRef CAS.
- L. E. Sandoval-Díaz, J. A. González-Amaya and C. A. Trujillo, Microporous Mesoporous Mater., 2015, 215, 229–243 CrossRef.
- A. Feng, Y. Yu, L. Mi, Y. Cao, Y. Yu and L. Song, Microporous Mesoporous Mater., 2019, 280, 211–218 CrossRef CAS.
- M. Gackowski, K. Tarach, Ł. Kuterasiński, J. Podobiński, B. Sulikowski and J. Datka, Microporous Mesoporous Mater., 2019, 281, 134–141 CrossRef CAS.
- Y. Matsunaga, H. Yamazaki, T. Yokoi, T. Tatsumi and J. N. Kondo, J. Phys. Chem. C, 2013, 117, 14043–14050 CrossRef CAS.
- A. Corma and A. Martinez, Catal. Rev., 1993, 35, 483–570 CrossRef CAS.
- X. Du, X. Kong and L. Chen, Catal. Commun., 2014, 45, 109–113 CrossRef CAS.
- N. Y. Chen, M. C. Liu, S. C. Yang, H. S. Sheu and J. R. Chang, Ind. Eng. Chem. Res., 2015, 54, 8456–8468 CrossRef CAS.
- R. V. Jasra, B. Tyagi, Y. M. Badheka, V. N. Choudary and T. S. G. Bhat, Ind. Eng. Chem. Res., 2003, 42, 3263–3272 CrossRef CAS.
- P. Amol, Z. Ł. Amrute, S. Hannah, W. Claudia and S. Ferdi, Science, 2019, 366, 485–489 CrossRef PubMed.
- A. Feller, J. Catal., 2003, 220, 192–206 CrossRef CAS.
- F. A. Diaz-Mendoza, L. Pernett-Bolano and N. Cardona-MartõÂnez, Thermochim. Acta, 1998, 312, 47–61 CrossRef CAS.
- K. Ren, J. Long, Q. Ren, Y. Li and Z. Dai, Mol. Simulat., 2017, 43, 1348–1355 CrossRef CAS.
- S. Zhou, C. Zhang, Y. Li, Y. Luo and X. Shu, Ind. Eng. Chem. Res., 2020, 59, 5576–5582 CrossRef CAS.
- Y. Zhang, M. Li, E. Xing, Y. Luo and X. Shu, Catal. Commun., 2019, 119, 67–70 CrossRef CAS.
Footnote |
| † This paper is dedicated to Prof. Xingtian Shu on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2020 |