
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe shiny side of copper: bringing copper(I) light-emitting electrochemical cells closer to application†
Sarah Keller a,
Alessandro Prescimonea,
Maria-Grazia La Placab,
José M. Junquera-Hernándezb,
Henk J. Bolinkb,
Edwin C. Constablea,
Michele Sessolo*b,
Enrique Ortí*b and
Catherine E. Housecroft*a
a,
Alessandro Prescimonea,
Maria-Grazia La Placab,
José M. Junquera-Hernándezb,
Henk J. Bolinkb,
Edwin C. Constablea,
Michele Sessolo*b,
Enrique Ortí*b and
Catherine E. Housecroft*a
aDepartment of Chemistry, University of Basel, BPR 1096, Mattenstrasse 24a, CH-4058 Basel, Switzerland. E-mail: catherine.housecroft@unibas.ch
bInstituto de Ciencia Molecular, Universidad de Valencia, 46980 Paterna, Valencia, Spain. E-mail: michele.sessolo@uv.es; enrique.orti@uv.es
First published on 16th June 2020
Abstract
Heteroleptic [Cu(P^P)(N^N)][PF6] complexes, where N^N is 5,5′-dimethyl-2,2′-bipyridine (5,5′-Me2bpy), 4,5,6-trimethyl-2,2′-bipyridine (4,5,6-Me3bpy), 6-(tert-butyl)-2,2′-bipyridine (6-tBubpy) and 2-ethyl-1,10-phenanthroline (2-Etphen) and P^P is either bis(2-(diphenylphosphino)phenyl)ether (POP, PIN [oxydi(2,1-phenylene)]bis(diphenylphosphane)) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos, PIN (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane)) have been synthesized and their NMR spectroscopic, mass spectrometric, structural, electrochemical and photophysical properties were investigated. The single-crystal structures of [Cu(POP)(5,5′-Me2bpy)][PF6], [Cu(xantphos)(5,5′-Me2bpy)][PF6], [Cu(POP)(6-tBubpy)][PF6], [Cu(POP)(4,5,6-Me3bpy)][PF6]·1.5Et2O, [Cu(xantphos)(4,5,6-Me3bpy)][PF6]·2.33CH2Cl2, [Cu(POP)(2-Etphen)][PF6] and [Cu(xantphos)(2-Etphen)][PF6] are described. While alkyl substituents in general exhibit electron-donating properties, variation in the nature and substitution-position of the alkyl group in the N^N chelate leads to different effects in the photophysical properties of the [Cu(P^P)(N^N)][PF6] complexes. In the solid state, the complexes are yellow to green emitters with emission maxima between 518 and 602 nm, and photoluminescence quantum yields (PLQYs) ranging from 1.1 to 58.8%. All complexes show thermally activated delayed fluorescence (TADF). The complexes were employed in the active layer of light-emitting electrochemical cells (LECs). The device performance properties are among the best reported for copper-based LECs, with maximum luminance values of up to 462 cd m−2 and device half-lifetimes of up to 98 hours.
Introduction
In 2015, the United Nations Member States adopted the 2030 Agenda for Sustainable Development, which identified 17 sustainable development goals (SDGs).1 Many of these SDGs pose challenges that the materials science community must address with better, smarter and more sustainable materials. In particular, SDG 7 “Affordable and clean energy” places emphasis not only upon the generation of energy in clean and sustainable manners but also upon the need for new efficient technologies associated with energy usage. In 2017, the U.S. Energy Information Administration estimated that some 7% of the total U.S. energy usage was accounted for by lighting.2 The transition from energy-inefficient incandescent bulbs to light-emitting diodes (LEDs) is almost complete in the European sector; organic light-emitting diodes (OLEDs) represent an emerging technology that is currently being deployed worldwide.3OLEDs are devices in which light generation occurs in an electroluminescent organic layer and the phosphorescent organic light-emitting diodes are of particular interest.4 In OLEDs, electrons and holes are injected at the cathode and anode, respectively, and combine to form excitons in either a singlet or a triplet state in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio; exciton decay generates light through spontaneous emission. Typical organic molecules and semiconducting polymers used in OLEDs are fluorescent and the decay of the triplet exciton is spin forbidden. As a result, fluorescent OLEDs can only harvest the singlet excitons giving a limit to the internal quantum efficiency of 25%. In phosphorescent organic light-emitting diodes, the triplet excitons may also be harvested by adding a phosphorescent sensitizer, which facilitates the radiative decay of both singlet and triplet states. The commonest strategy is the addition of a heavy transition metal complex in which the large spin–orbit coupling facilitates the intersystem crossing, a process which mixes the singlet and triplet character of excited states.3
:3 ratio; exciton decay generates light through spontaneous emission. Typical organic molecules and semiconducting polymers used in OLEDs are fluorescent and the decay of the triplet exciton is spin forbidden. As a result, fluorescent OLEDs can only harvest the singlet excitons giving a limit to the internal quantum efficiency of 25%. In phosphorescent organic light-emitting diodes, the triplet excitons may also be harvested by adding a phosphorescent sensitizer, which facilitates the radiative decay of both singlet and triplet states. The commonest strategy is the addition of a heavy transition metal complex in which the large spin–orbit coupling facilitates the intersystem crossing, a process which mixes the singlet and triplet character of excited states.3
Although OLEDs can have high conversion efficiencies, they also have a number of disadvantages in terms of SDG 7. Typical sensitizers are iridium(III) complexes, which impacts on sustainability as iridium is one of the rarest elements, present in sub-ppb quantities in the Earth's crust.5 Furthermore, OLEDS are dependent on the work function of the electrodes and electron injection requires highly electropositive metals, which in turn predicates protection from air and water in fabrication and operation.3 An alternative approach is the light-emitting electrochemical cell (LEC), which uses mobile charged species as electroluminescent material. The ionic character of the light-emitting species removes the dependence on the work function of the electrode and makes the device architecture simpler. The simplicity of LECs and their processing from solution has motivated that the research activity on LECs has considerably increased in the last years.6–10 Much interest has centred upon LECs incorporating ionic transition-metal complexes (iTMCs), which are inherently more efficient than those based upon charged light-emitting polymers as the emission is based on phosphorescence rather than on fluorescence.11–13 The colour-tuning of iTMC-based LECs is well-established in the case of iridium complexes, although the use of this element makes again the sustainability questionable. We14–28 and others29–38 have investigated copper(I) complexes of the type [Cu(P^P)(N^N)]+ (P^P = chelating diphosphane, N^N = diimine or 1,10-phenanthroline) as alternative electroluminescent materials for LECs. These complexes are of particular interest as they exhibit the phenomenon of thermally activated delayed fluorescence (TADF), in which the first excited singlet (S1) and triplet (T1) states are strongly coupled, allowing intersystem crossing between the two levels without the need for a heavy metal. The energy gap between the singlet and triplet states is small, allowing reverse intersystem crossing (T1 → S1) to occur. This effectively allows the harvesting of both singlet and triplet states through repopulation of the singlet state and fluorescence to the ground state.39–43
Although a wide variety of different substituents in the N^N chelating ligand in [Cu(P^P)(N^N)]+ complexes has been studied so far, we note that relatively simple substitution, especially with alkyl groups, often gives the most promising photophysical and device properties. Encouraged by our results with [Cu(P^P)(N^N)][PF6] complexes, where P^P is either POP bis(2-(diphenylphosphano)phenyl)ether; IUPAC PIN [oxydi(2,1-phenylene)]bis(diphenylphosphane) or xantphos (4,5-bis(diphenylphosphano)-9,9-dimethylxanthene; IUPAC PIN (9,9-dimethyl-9H-xanthene-4,5-diyl)(bis(diphenylphosphane)) and N^N is a 6-methyl, 6-ethyl- or 6,6′-dimethyl-substituted bpy,24,27 we decided to investigate the effect of further alkyl substitution patterns. In this study, we focus on the four following modifications (Scheme 1): (a) a different alkyl substituent in the 6-position of the bpy ligand, in this case 6-tBubpy instead of 6-Mebpy or 6-Etbpy (because complexes of the latter led to LECs with long device lifetimes); (b) disubstitution in different positions in the bpy, here 5,5′-Me2bpy instead of 6,6′-Me2bpy (which led to LECs with high luminance values) or 4,4′-Me2bpy; (c) an unsymmetrical bpy with three electron-donating Me groups, 4,5,6-Me3bpy; and (d) the replacement of the bpy by a 1,10-phenanthroline, here with 2-Etphen instead of 6-Etbpy (which led to LECs with long device lifetimes).
 | ||
| Scheme 1 Structures of ligands with ring and atom labels for NMR spectroscopic assignments. | ||
The results of this study contribute to the rational design of emissive [Cu(P^P)(N^N)]+ complexes and reiterate the sensitivity of these systems to minor electronic and steric changes induced by ligand substitution patterns.
Results and discussion
Ligand synthesis
The ligands 6-tBubpy, 4,5,6-Me3bpy and 2-Etphen (Scheme 1) were selected on the basis of their various steric and electronic effects to compare with previous studies on 6- and 6,6′-substituted bpy ligands. Transition metal complexes bearing the 6-tBubpy ligand have been widely investigated,44 in particular because of insertions into a C–H bond of the tert-butyl group to form cyclometallated complexes.45–50 6-tBubpy has previously been prepared from the reaction of bpy with tert-butyllithium44 or from tert-butylcarbonitrile.45–50 However, we found it more convenient to use a modified Negishi procedure introduced by Hanan,51 and the reaction of 2-tert-butyl-6-chloropyridine with 2-pyridinylzinc bromide in the presence of [Pd(PPh3)4] gave 6-tBubpy in 51% yield. A single report on the synthesis of 4,5,6-Me3bpy is found in the patent literature,52 but we also prepared this compound from the Negishi reaction of 2-chloro-4,5,6-trimethylpyridine and 2-pyridinylzinc bromide in 4% yield. The low yield is most likely due to the fact that the reaction mixture was only stirred at room temperature, whereas for the synthesis of 6-tBubpy microwave conditions were employed (see ESI† for details). It can be expected that microwave conditions should lead to a similar yield for 4,5,6-Me3bpy as for 6-tBubpy. It has been reported that 2-Etphen can be prepared in low yield from the direct reaction of 2-ethyllithium with 1,10-phenanthroline53 or in 76% yield from the reaction of 8-aminoquinoline with 2-methyl-3-ethoxycyclobutanone.54 In our hands, the reaction of 2-ethyllithium with 1,10-phenanthroline gave 2-Etphen in 5% yield. Each of the ligands was fully characterized by routine spectroscopic and analytical methods (see the ESI†).Synthesis and characterization of [Cu(P^P)(N^N)][PF6] complexes
The synthesis of the [Cu(P^P)(N^N)][PF6] complexes was carried out by addition of the respective P^P and N^N ligand (Scheme 1) to a solution of [Cu(MeCN)4][PF6] in CH2Cl2. The synthetic procedure for the POP-containing complexes used the established sequential addition of the two ligands, where the bpy ligand was added after stirring POP and [Cu(MeCN)4][PF6] for two hours.28 For the preparation of the [Cu(xantphos)(N^N)][PF6] complexes, the two ligands were simultaneously added. The compounds [Cu(POP/xantphos)(N^N)][PF6] with N^N = 5,5′-Me2bpy, 4,5,6-Me3bpy, 6-tBubpy and 2-Etphen were isolated as yellow solids in yields of 69 to 96%. All compounds were characterized by elemental analysis, mass spectrometry and multinuclear NMR spectroscopies including 2D methods to assign the spectra. Detailed procedures and characterization can be found in the ESI,† together with 1H and 31P NMR spectra of the compounds (Fig. S1–S12†) as well as mass spectra (Fig. S13–S21†). In the 31P NMR spectra ((CD3)2CO or CD2Cl2), all compounds give broad signals between δ −11 and −15 ppm (Fig. S1†).Structural characterization
Layer crystallisation (diffusion of Et2O into CH2Cl2 solutions of the respective compound) yielded X-ray quality crystals of [Cu(POP)(5,5′-Me2bpy)][PF6], [Cu(xantphos)(5,5′-Me2bpy)][PF6], [Cu(POP)(6-tBubpy)][PF6], [Cu(POP)(4,5,6-Me3bpy)][PF6], [Cu(xantphos)(4,5,6-Me3bpy)][PF6], [Cu(POP)(2-Etphen)][PF6] and [Cu(xantphos)(2-Etphen)][PF6]. The geometry of the complexes is tetrahedral, with varying degrees of distortion; see Table 1 for a summary of the most important structural parameters defining the coordination sphere of copper, and Fig. 1–4 for illustrations of the complex cations. The planes defined through P–Cu–P and N–Cu–N of the P^P and N^N chelating ligand range from being almost orthogonal in [Cu(xantphos)(4,5,6-Me3bpy)]+, [Cu(POP)(4,5,6-Me3bpy)]+ and [Cu(POP)(2-Etphen)]+ (89.77, 88.44 and 88.05°, respectively) to exhibiting a greater distortion in [Cu(POP)(6-tBubpy)]+ (74.66°). The Cu–P distances are relatively similar and in the expected range, from 2.2396(4) to 2.3004(6) Å. The Cu–N distances are found between 2.0421(11) and 2.134(2) Å, with one exception: in the [Cu(POP)(t-Bubpy)]+ cation (Fig. 2a), one Cu–N bond is within the normal range with 2.0648(18) Å, however the Cu–N bond to the pyridine ring that bears the bulky CMe3 group is elongated to 2.3949(19) Å, which is around 0.26 Å longer than the second longest Cu–N distance of this complex series. Such a lengthening of the Cu–N bond has not been observed for other [Cu(POP/xantphos)(N^N)][PF6] complexes (survey of 179 structures containing chelating bisphosphane with dibenzyl ether backbone and any N^N chelating residue comprising two connected pyridyl rings as substructure in the Cambridge Structural Database (CSD),55 online search conducted 22.03.2020), not even for [Cu(POP/xantphos)(N^N)][PF6] complexes with large aromatic substituents next to the coordinating nitrogen such as for example pyrene.23 The sum of the ionic radius of Cu+ (coordination number 4) of 0.74 Å and the van der Waals radius of nitrogen (1.6 Å) is 2.34 Å.56 Although the elongated Cu–N distance of 2.3949(19) Å in the [Cu(POP)(6-tBubpy)]+ cation is found to be slightly longer than that, if we consider the van der Waals radius of 1.4 Å for Cu, the distance is still in a range where interaction can be assumed to take place. However, the coordination of this nitrogen atom to the copper could be expected to be considerably weaker, which may have an impact on the photophysical properties (see later discussion).| Complex cation | Cu–P distance/Å | Cu–N distance/Å | P–Cu–P chelating angle/deg. | N–Cu–N chelating angle/deg. | Angle between CuP2 and CuN2 planes/deg. | N–C–C–N torsion angle/deg. |
|---|---|---|---|---|---|---|
| [Cu(POP)(5,5′-Me2bpy)]+ | Cu1–P2 = 2.2535(4) | Cu1–N41 = 2.0820(11) | P2–Cu1–P8 = 110.537(15) | N41–Cu1–N44 = 80.17(5) | 76.67 | -9.1(2) |
| Cu1–P8 = 2.2396(4) | Cu1–N44 = 2.0421(11) | |||||
| [Cu(xantphos)(5,5′-Me2bpy)]+ | Cu1–P16 = 2.2506(4) | Cu1–N2 = 2.0487(11) | P16–Cu1–P45 = 114.821(14) | N2–Cu1–N15 = 80.52(4) | 77.61 | 15.7(2) |
| Cu1–P45 = 2.2465(4) | Cu1–N15 = 2.0888(11) | |||||
| [Cu(POP)(6-tBubpy)]+ | Cu1–P18 = 2.3004(6) | Cu1–N2 = 2.0648(18) | P18–Cu1–P44 = 108.71(2) | N2–Cu1–N13 = 75.36(7) | 74.66 | -28.8(3) |
| Cu1–P44 = 2.2780(6) | Cu1–N13 = 2.3949(19) | |||||
| [Cu(POP)(4,5,6-Me3bpy)]+ | Cu1–P1 = 2.2833(6) | Cu1–N1 = 2.107(2) | P2–Cu1–P1 = 113.00(2) | N1–Cu1–N2 = 80.09(8) | 88.44 | -6.53 |
| Cu1–P2 = 2.2586(7) | Cu1–N2 = 2.051(2) | |||||
| [Cu(xantphos)(4,5,6-Me3bpy)]+ | Cu1–P1 = 2.2607(18) | Cu1–N1 = 2.096(4) | P2–Cu1–P1 = 112.94(6) | N1–Cu1–N2 = 80.65(16) | 89.77 | 2.7(7) |
| Cu1–P2 = 2.2488(14) | Cu1–N2 = 2.053(4) | |||||
| [Cu(POP)(2-Etphen)]+ | Cu1–P2 = 2.2488(5) | Cu1–N2 = 2.0924(13) | P1–Cu1–P2 = 115.363(17) | N1–Cu1–N2 = 81.59(5) | 88.05 | -1.4(2) |
| Cu1–P1 = 2.2668(5) | Cu1–N1 = 2.0613(13) | |||||
| [Cu(xantphos)(2-Etphen)]+ | Cu1–P1 = 2.2915(6) | Cu1–N1 = 2.134(2) | P1–Cu1–P2 = 112.38(2) | N1–Cu1–N2 = 80.39(9) | 81.53 | -4.6(4) |
| Cu1–P2 = 2.2600(6) | Cu1–N2 = 2.068(2) |
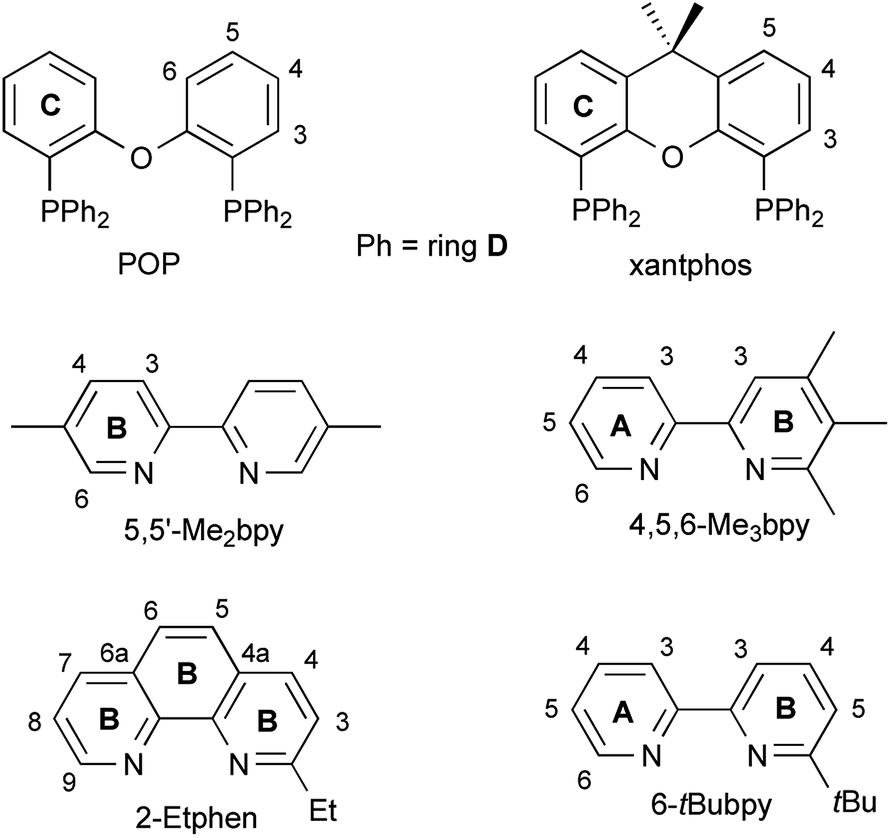
 | ||
| Fig. 1 (a) Structure of the [Cu(POP)(5,5′-Me2bpy)]+ cation with ellipsoids plotted at 50% probability, H atoms are omitted for clarity. Face to face π-stacking between the phenyl ring containing C15 and the ring of the POP backbone at P2 (angle between the ring planes 13.94°, centroid–centroid distance 3.641 Å). (b) Structure of the [Cu(xantphos)(5,5′-Me2bpy)]+ cation with ellipsoids plotted at 50% probability, H atoms are omitted for clarity. Weak π-interaction between two of the phenyl rings on each P atom (involving C23 and C46, respectively) (angle between the ring planes 21.65°, centroid–centroid distance 3.843 Å) and no intramolecular π-interaction involving the xanthene backbone. | ||
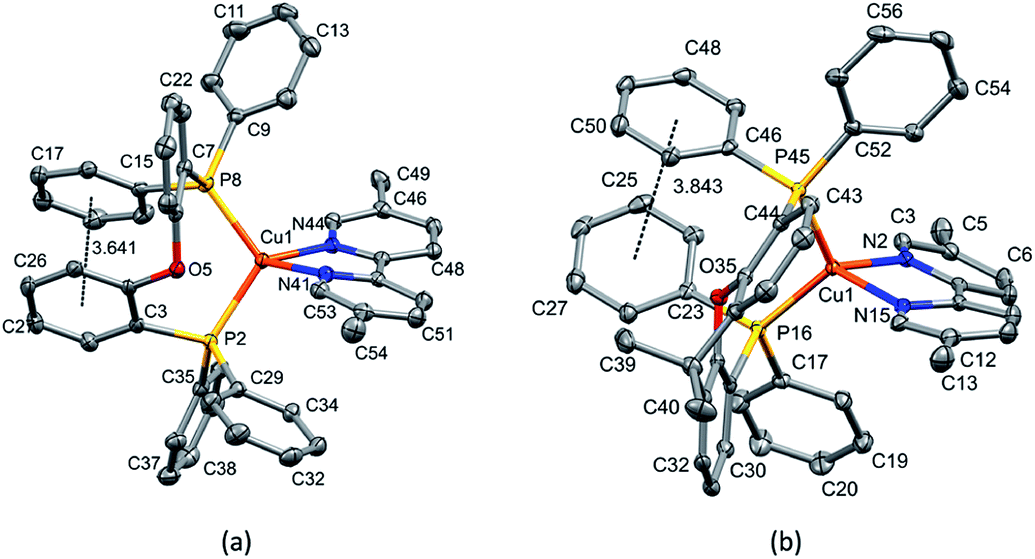
 | ||
| Fig. 2 (a) Structure of the [Cu(POP)(6-tBubpy)]+ cation with ellipsoids plotted at 50% probability, H atoms are omitted. There is a π-stacking interaction between the phenyl ring containing C45 and the ring of the POP backbone attached to P18 (angle between the ring planes 24.27°, centroid–centroid distance 3.889 Å). (b) Structure of the [Cu(POP)(4,5,6-Me3bpy)]+ cation with ellipsoids plotted at 50% probability. H atoms and solvent molecules are omitted. π-Stacking between the phenyl ring containing C35 and the ring of the POP backbone connected to P2 (angle between the ring planes 13.52°, centroid–centroid distance 3.710 Å). | ||
Another relevant parameter is the N–C–C–N torsion angle of the two pyridine rings in the bpy or 2-Etphen ligand; once again, the [Cu(POP)(6-tBubpy)]+ cation is an outlier with a torsion of −28.8(3)°, while the other complexes exhibit angles from as low as −1.4(2)° for [Cu(POP)(2-Etphen)]+ to 15.7(2)° for [Cu(xantphos)(5,5′-Me2bpy)]+. The extreme torsion angle of the bpy ligand together with the elongation of the Cu–N bond for the tBu-bearing pyridyl ring in [Cu(POP)(6-tBubpy)]+ might be the best option to accommodate the bulky tBu group while maintaining the tetracoordinated geometry at the copper centre. A possible correlation between planar coordination of the rings within the N^N chelating ligand and emissive properties of the complex will be discussed in the photophysical section of the paper.
The complexes show effective intramolecular π–π interactions between the aromatic rings,57 with relatively low offsets between the stacking rings and centroid–centroid distances between 3.641 Å and 3.924 Å. For the complexes with POP, a π–π interaction takes place between a phenyl ring at a phosphorus atom and a ring of the backbone of the bisphosphane ligand (Fig. 1a, 2a, b and 3b). In the case of the xantphos complexes, the interaction occurs between two phenyl rings on different phosphorus atoms, and does not involve the xanthene backbone (Fig. 1b, 3a and 4).
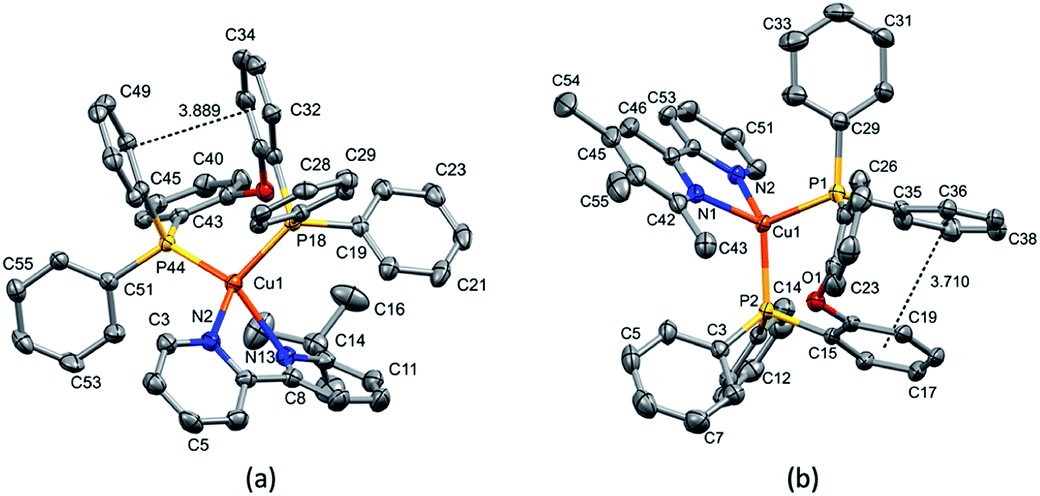
 | ||
| Fig. 3 (a) Structure of the [Cu(xantphos)(4,5,6-Me3bpy)]+ cation with ellipsoids plotted at 30% probability. H atoms and solvent molecules are omitted for clarity. π-Stacking between the phenyl ring containing C25 and the phenyl ring bonded to P2 (angle between the ring planes 7.77°, centroid–centroid distance 3.754 Å). (b) Structure of the [Cu(POP)(2-Etphen)]+ cation with ellipsoids plotted at 50% probability, H atoms are omitted for clarity. Offset π-interaction between the phenyl ring containing C21 and the ring of the POP backbone at P2 (angle between the ring planes 10.93°, centroid–centroid distance 3.733 Å). | ||
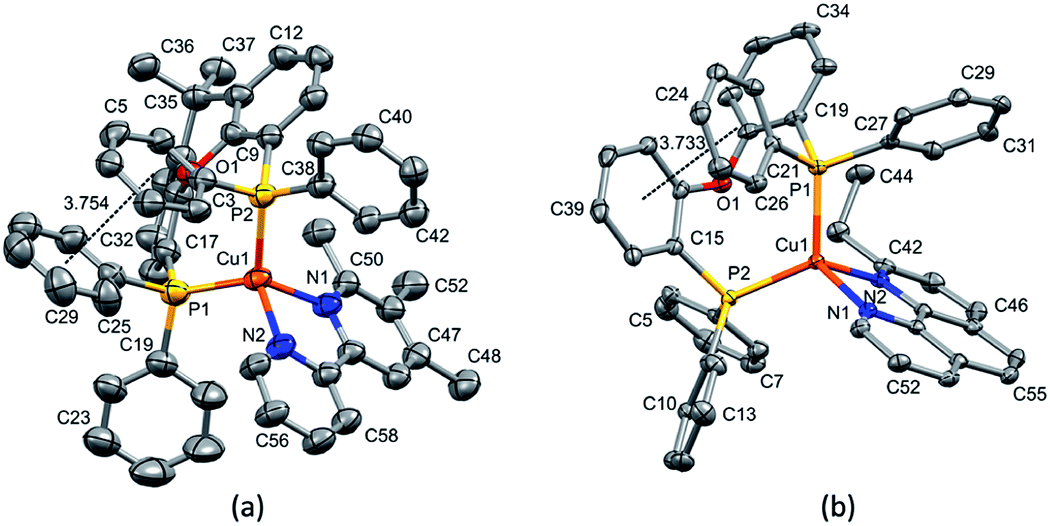
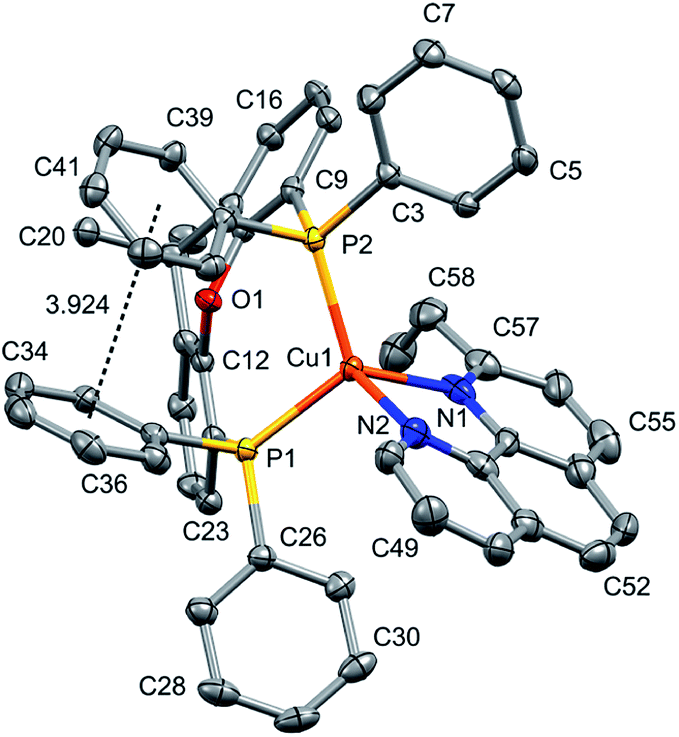 | ||
| Fig. 4 Structure of the [Cu(xantphos)(2-Etphen)]+ cation with ellipsoids plotted at 50% probability, H atoms are omitted for clarity. Negligible π-interaction between two of the phenyl rings on the different phosphorus atoms (involving C34 and C39, respectively) (angle between the ring planes 27.10°, centroid–centroid distance 3.924 Å), but none involving the xanthene backbone. | ||
Electrochemistry
The electrochemical behaviour of the eight heteroleptic [Cu(P^P)(N^N)][PF6] complexes was investigated using cyclic voltammetry (CV) and their oxidation potentials E1/2 are compared to those measured for complexes with N^N = bpy, 6-Mebpy and 6,6′-Me2bpy (Table 2 and Fig. S22–S29†). The first quasi-reversible oxidation in the positive mode is due to the oxidation of Cu+ to Cu2+, whereas the second oxidation peak is attributed to non-reversible oxidation of the phosphane ligand. The oxidation potentials for the copper(I) complexes with an alkyl group next to one coordinating nitrogen atom of the bpy ligand are shifted to slightly higher potentials (+0.77 to +0.87 V) compared to complexes with unmodified bpy (+0.72 V for [Cu(POP)(bpy)][PF6] and +0.76 V for [Cu(xantphos)(bpy)][PF6]).21 This effect of slightly impeded copper oxidation has also been observed for the analogous alkyl-substituted complexes [Cu(xantphos)(6-Mebpy)][PF6] (+0.85 V),21 [Cu(POP)(6,6′-Me2bpy)][PF6] (+0.92 V)58 and [Cu(xantphos)(6,6′-Me2bpy)][PF6] (+0.89 V),21 and for complexes with halogen substituents adjacent to one or both nitrogen atoms of the bpy ligand.59 The effect is mainly attributed to the stabilization of the tetrahedral complex geometry, independent from the nature of the substituent in 6-position, which results in a stabilization of the copper(I) state and consequently higher Cu+/Cu2+ oxidation potentials. This is further supported by the observation that the oxidation potentials for the complexes [Cu(POP)(5,5′-Me2bpy)][PF6] (+0.70 V) and [Cu(xantphos)(5,5-Me2bpy)][PF6] (+0.75 V), in which the geometric stabilization effect is much less effective, are even slightly lower than for the respective complexes with unsubstituted bpy, which means that the oxidation from Cu+ to Cu2+ is facilitated. The oxidation processes are quasi-reversible and defined reduction processes were not observed for any of the complexes, which is typical for compounds with simple alkyl substituted N^N chelating ligands, although this has been observed for complexes with CF3 substituents.21| Complex cation | E1/2/V | (Epc − Epa/mV) |
|---|---|---|
| a Data from ref. 21.b Data from ref. 27.c Data from ref. 58. | ||
| [Cu(POP)(bpy)]+a | +0.72 | 110 |
| [Cu(xantphos)(bpy)]+a | +0.76 | 110 |
| [Cu(POP)(6-Mebpy)]+b | +0.69 | 125 |
| [Cu(xantphos)(6-Mebpy)]+a | +0.85 | 100 |
| [Cu(POP)(6,6′-Me2bpy)]+c | +0.92 | 183 |
| [Cu(xantphos)(6,6′-Me2bpy)]+a | +0.89 | 145 |
| [Cu(POP)(5,5′-Me2bpy)]+ | +0.70 | 127 |
| [Cu(xantphos)(5,5′-Me2bpy)]+ | +0.75 | 131 |
| [Cu(POP)(6-tBubpy)]+ | +0.83 | 136 |
| [Cu(xantphos)(6-tBubpy)]+ | +0.87 | 99 |
| [Cu(POP)(4,5,6-Me3bpy)]+ | +0.77 | 114 |
| [Cu(xantphos)(4,5,6-Me3bpy)]+ | +0.82 | 122 |
| [Cu(POP)(2-Etphen)]+ | +0.80 | 92 |
| [Cu(xantphos)(2-Etphen)]+ | +0.86 | 91 |
DFT calculations
The geometries of the new [Cu(POP)(N^N)]+ and [Cu(xantphos)(N^N)]+ cations in their electronic ground state (S0) were optimized at the DFT B3LYP-D3/(def2-SVP + def2-TZVP) level in the presence of the solvent (CH2Cl2) using a continuum model. The values obtained for the most representative geometrical parameters defining the coordination sphere of the copper atom are summarized in Table S1.† The calculations reproduce the main features discussed above for the X-ray geometries. The atoms forming the coordination sphere of Cu(I) define a distorted tetrahedral structure with angles between the P–Cu–P and N–Cu–N planes in the range of 79.3–86.9° in good agreement with those observed experimentally (Table 1). This angle can be used as an indication of the deviation from the orthogonal disposition of the P^P and N^N ligands in the tetrahedral structure. Calculations also reproduce the π–π interaction between a phenyl ring of a phosphorus atom and a ring of the phosphane ligand in the POP complexes (centroid–centroid distances around 3.65 Å), and the less efficient π–π interactions between phenyl rings on different phosphorus atoms that take place in the xantphos complexes. The xanthene unit in the xantphos complexes adopts a “bowl-like” conformation in which the 6-alkyl substituent in the bpy complexes and the 2-ethyl group in the phen complex lie.The calculated Cu–P and Cu–N distances are in the ranges of 2.25–2.29 and 2.07–2.11 Å (Table S1†), respectively, which is in good accord with the experimental values (Table 1). In the case of the [Cu(POP)(6-tBubpy)]+ cation, calculations reproduce the unusual elongation of the Cu–N bond adjacent to the tert-butyl substituent, although the calculated bond length (2.21 Å) is not as long as the experimentally determined X-ray value (2.39 Å). This elongation is also observed in the [Cu(xantphos)(6-tBubpy)]+ complex (Table S1†), for which unfortunately no X-ray structure could be obtained. This indicates that the observed elongation is a general consequence of the presence of the bulky 6-tBubpy ligand and not a singularity of the interaction of it with POP. Therefore, a similar weakening of the Cu–N coordination should be expected for complexes bearing this ligand.
The calculations do not reproduce the higher N–C–C–N torsion observed experimentally for the bpy ligand of [Cu(POP)(6-tBubpy)]+ (28.8(3)°). The computed value of 12.0° is similar to those of the other members of the series, ranging from 12.0 to 15.8°, with the exception of the complexes with ligand 2-Etphen, where the higher rigidity of the phenanthroline moiety leads to torsion angles of 2.1° (POP complex) and 0.5° (xantphos complex). The discrepancies in the N–C–C–N torsional angle are attributed to the effects of packing and interaction with neighbouring molecules that are not considered in the calculations.
The geometry of the first triplet excited state (T1) was also optimized at the UB3LYP level for all the complexes. In the T1 state, the Cu–P bonds are lengthened to 2.30–2.40 Å and the Cu–N bonds are shortened to 1.95–2.00 Å. As a consequence, the P–Cu–P angles are as small as 103–104° in POP complexes (105–106° in xantphos complexes) and the N–Cu–N angles are widened to 83–84°. Although an elongation of the Cu–N bond in complexes bearing the 6-tBubpy ligand is not observed in the T1 state, it must be remembered that the ground-state calculations underestimate the elongation observed in the crystal geometry. Indeed, the most relevant geometrical change on going from S0 to T1 is the reduction of the angle formed by the N–Cu–N and P–Cu–P planes (Table S1†). As discussed below, upon excitation to T1 the metal atom is partially oxidized and tends to adopt the square-planar coordination sphere expected for four-coordinate d9 Cu(II) complexes, instead of the tetrahedral conformation typical of d10 Cu(I) complexes. This effect is more pronounced for the [Cu(POP)(5,5′-Me2bpy)]+ and [Cu(xantphos)(5,5′-Me2bpy)]+ complexes bearing no substituent in 6,6′-positions, for which the angle between the N–Cu–N and P–Cu–P planes decreases from 79.5 and 86.0° in S0 to 59.0 and 57.0° in T1, respectively, similarly to what happens for the unsubstituted [Cu(POP)(bpy)]+ and [Cu(xantphos)(bpy)]+ complexes (Table S1†). As discussed previously,17,21 the distortion from the tetrahedral structure is limited by the introduction of alkyl groups in the 6-positions of the bpy ligand, because substituents in these positions impede the movement of the ligands towards more planar dispositions. For instance, in the [Cu(POP)(6-tBubpy)]+ and [Cu(xantphos)(6-tBubpy)]+ complexes, the angle between the N–Cu–N and P–Cu–P planes decreases in a lower degree from 80.6 and 85.6° in S0 to 68.4 and 76.7° in T1, respectively. Thus, the presence of alkyl groups in the 6-positions affects the degree of geometrical relaxation of the T1 excited state and thereby its energy position relative to S0.
The energy and topology calculated for the frontier molecular orbitals of the new [Cu(POP)(N^N)]+ complexes are displayed in Fig. 5 (see Fig. S30† for the [Cu(xantphos)(N^N)]+ complexes). The atomic orbital composition of the molecular orbitals remains almost unchanged along both series and only the contour plots computed for the reference compound [Cu(POP)(bpy)]+ and the [Cu(POP)(2-Etphen)]+ complex are drawn in Fig. 5 as representative examples. As previously described,17,19,21,24 the highest-occupied (HOMO) and the lowest-unoccupied molecular orbital (LUMO) in these kind of complexes are mostly orthogonal. Whereas the HOMO is mainly centred on the metal with small contribution from the vicinal phosphorus atoms, the LUMO fully resides on the N^N ligand. As shown in Fig. 5, the HOMO features close energies in a small range between −5.89 eV for [Cu(POP)(5,5′-Me2bpy)]+ and −6.03 eV for [Cu(POP)(6-tBubpy)]+, which is in good correlation with the experimental values of the first oxidation potential comprised between +0.70 V for [Cu(POP)(5,5′-Me2bpy)]+ and +0.83 eV for [Cu(POP)(6-tBubpy)]+ (Table 2). The electron-donating character of the methyl groups attached to the bpy ligand significantly destabilizes the LUMO of [Cu(POP)(5,5′-Me2bpy)]+ (−2.28 eV) and [Cu(POP)(4,5,6-Me3bpy)]+ (−2.32 eV) with respect to the unsubstituted complex [Cu(POP)(bpy)]+ (−2.46 eV). In contrast, the complexes incorporating the 2-Etphen and 6-tBubpy ligands feature energies very close to that of the reference complex. For the complexes with N^N = 2-Etphen, the LUMO+1, also spreading over the phenanthroline ligand, is largely stabilized and lies close to the LUMO. In these complexes the energy difference between the HOMO and LUMO+1 is close to the HOMO–LUMO energy gap, and excited states described by the HOMO → LUMO+1 monoexcitation would appear close in energy to the expected HOMO → LUMO transition and could become competitive in the emission process.
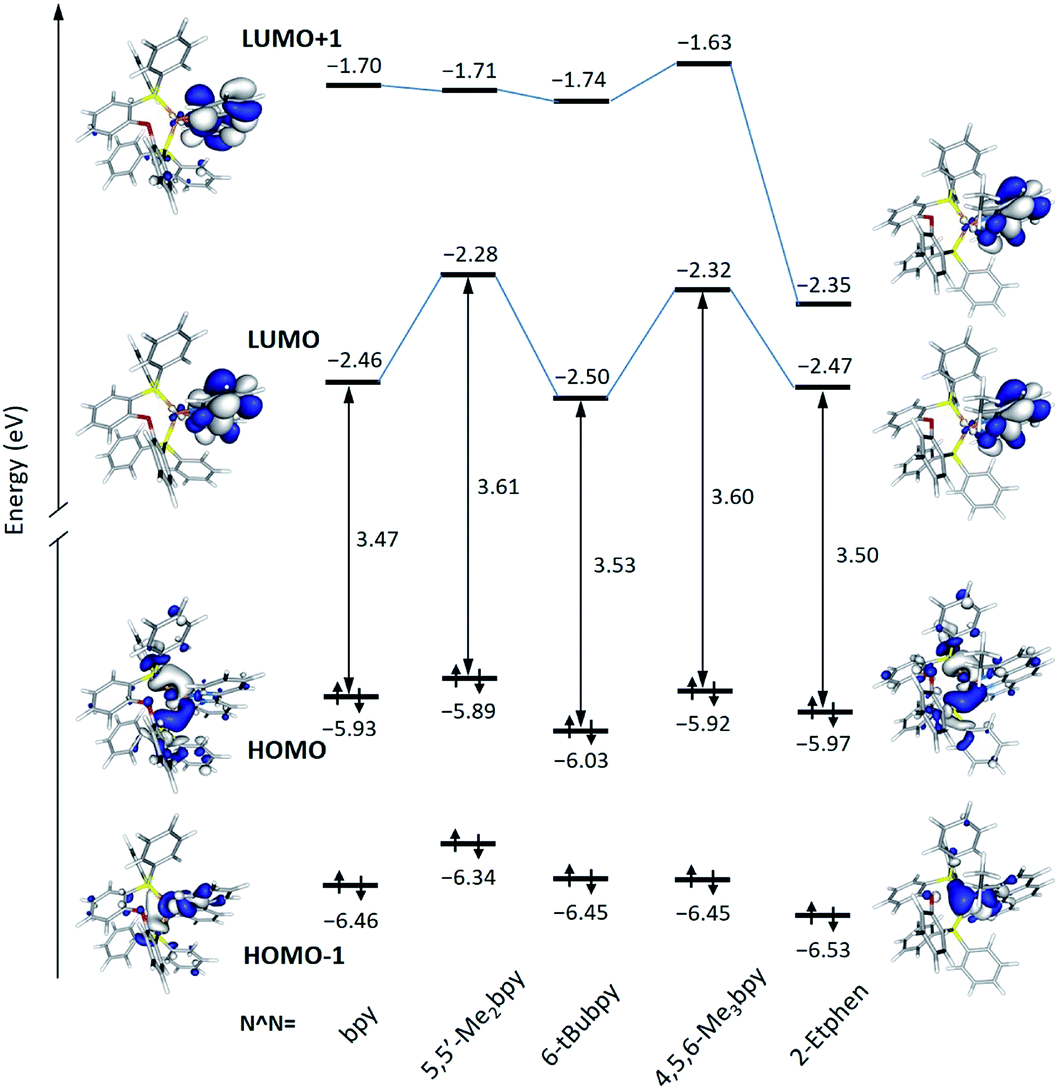 | ||
| Fig. 5 Energy diagram showing the energies calculated for the HOMO−1, HOMO, LUMO and LUMO+1 of the [Cu(POP)(N^N)]+ complexes. The HOMO–LUMO energy gap is also quoted. Isovalue contour plots (±0.03 a.u.) are shown for [Cu(POP)(bpy)]+ and [Cu(POP)(2-Etphen)]+. | ||
Photophysical properties and excited states
The complexes were subjected to detailed photophysical studies and the results compared to those of the previously studied analogous complexes with bpy, 6-Mebpy and 6,6′-Me2bpy. The absorption spectra of CH2Cl2 solutions of the xantphos complexes are presented in Fig. 6, and the spectra for the analogous POP complexes in Fig. S31.† The intense, high-energy bands at wavelengths shorter than 330 nm are typically a result of ligand-based π → π* transitions. In contrast to the complexes with bpy N^N ligands, those with 2-Etphen show a strong band at 272 nm, but lack the band at around 283 nm and the typically observed shoulders in the area between 300 and 320 nm. The broad and less intense bands between 330 and 450 nm are assigned to metal-to-ligand charge transfer (MLCT) transitions and are responsible for the yellow colour of the complexes.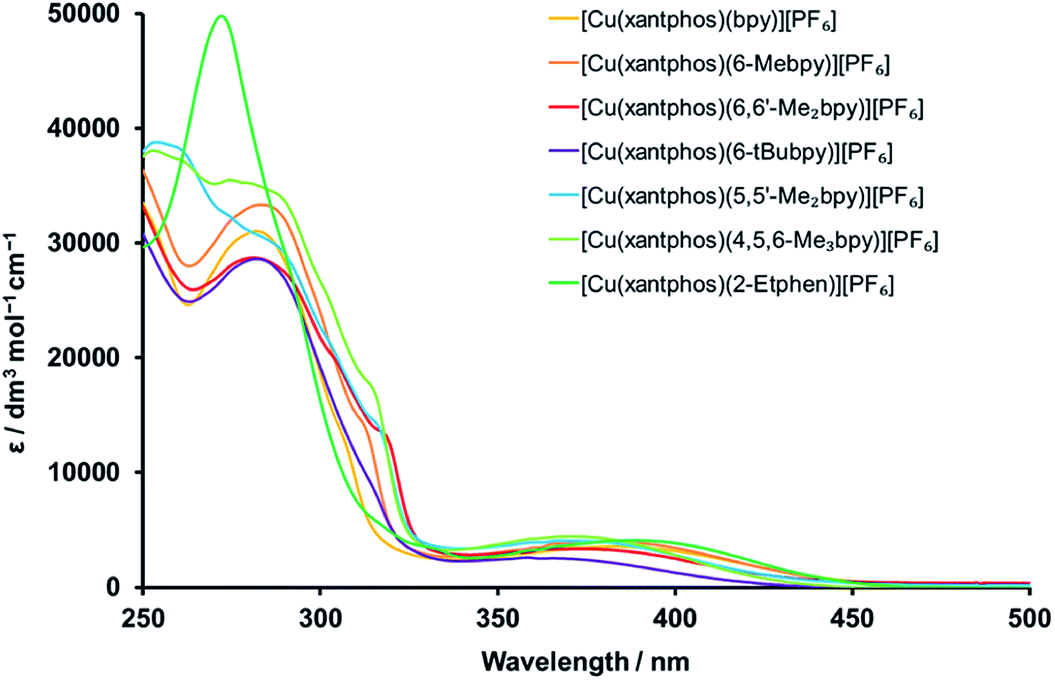 | ||
| Fig. 6 Solution absorption spectra of the [Cu(xantphos)(N^N)][PF6] complexes (CH2Cl2, 2.5 × 10−5 mol dm−3). | ||
A closer look at the absorption MLCT bands of the complexes (Fig. S32 and S33† for POP and xantphos complexes, respectively) shows that the highest-energy MLCT bands belong to the complexes with 4,5,6-Me3bpy, 6,6′-Me2bpy, 5,5′-Me2bpy and 6-tBubpy, whereas the ones with bpy and 2-Etphen are at lower energies. As alkyl groups have electron donating character and lead to a destabilization of the LUMO, which is mainly located on the N^N chelating ligand, a larger HOMO–LUMO gap is obtained for the complexes with alkyl-substituted diimine ligands. This then results in higher energies (shorter wavelengths) of the absorbed light: the more donating or larger number of alkyl groups at the bpy, the more blue-shifted the absorption. The lower energy of the MLCT bands of the complexes with 2-Etphen can be explained by the extension of the conjugated π-system. It is worth mentioning that the complex solutions with 6-tBubpy exhibit less intense MLCT bands and both the solutions as well as the solid compounds are considerably paler by eye.
The first singlet and triplet excited states were computed for all the complexes at the optimized geometry of the ground electronic state using TD-DFT calculations. The energies of the first singlet (S1) and two triplet (T1 and T2) excited states are summarized in Table 3 together with those calculated for the reference complexes [Cu(POP)(bpy)]+ and [Cu(xantphos)(bpy)]+. In all the cases, the lowest-energy singlet S1 results from the HOMO → LUMO monoexcitation, which implies an electron transfer from the Cu(P^P) environment of the complex to the N^N ligand, and therefore supports the MLCT character of S1 and thereby of the lowest-energy band around 400 nm (3.0 eV). The transition S0 → S1 is calculated at slightly higher energies compared with the reference complexes. This is in good agreement with the absorption spectra showing the MLCT maxima for all complexes displaced to bluer wavelengths with respect to those of the reference complexes. The oscillator strengths calculated for the S0 → S1 transitions are similar for all the complexes except for the complexes with the 6-tBubpy ligand, for which the significantly smaller oscillator strengths are in good agreement with the smaller intensities of the experimentally observed MLCT bands (Fig. S32 and S33†).
| Complex cation | S1 E (eV) (f) | T1 E (eV) | T2 E (eV) |
|---|---|---|---|
| [Cu(POP)(bpy)]+ | 2.79 (0.08) | 2.54 | 2.92 |
| [Cu(xantphos)(bpy)]+ | 2.82 (0.10) | 2.57 | 2.74 |
| [Cu(POP)(5,5′-Me2bpy)]+ | 2.93 (0.08) | 2.66 | 3.01 |
| [Cu(xantphos)(5,5′-Me2bpy)]+ | 2.93 (0.10) | 2.67 | 2.82 |
| [Cu(POP)(6-tBubpy)]+ | 2.81 (0.03) | 2.66 | 2.91 |
| [Cu(xantphos)(6-tBubpy)]+ | 2.84 (0.06) | 2.67 | 2.84 |
| [Cu(POP)(4,5,6-Me3bpy)]+ | 2.93 (0.08) | 2.71 | 3.00 |
| [Cu(xantphos)(4,5,6-Me3bpy)]+ | 2.94 (0.10) | 2.71 | 2.93 |
| [Cu(POP)(2-Etphen)]+ | 2.86 (0.09) | 2.61 | 2.69 |
| [Cu(xantphos)(2-Etphen)]+ | 2.86 (0.11) | 2.60 | 2.68 |
Exciting the complexes in solution (CH2Cl2, 2.5 × 10−5 mol dm−3) with wavelengths in the area of the MLCT band leads to green-yellow to orange emission, with maxima between 582 and 650 nm (Fig. 7, S34† and Table 4). All bands are structured with two emission maxima, which is a typical feature of these complexes.21,24,32,60
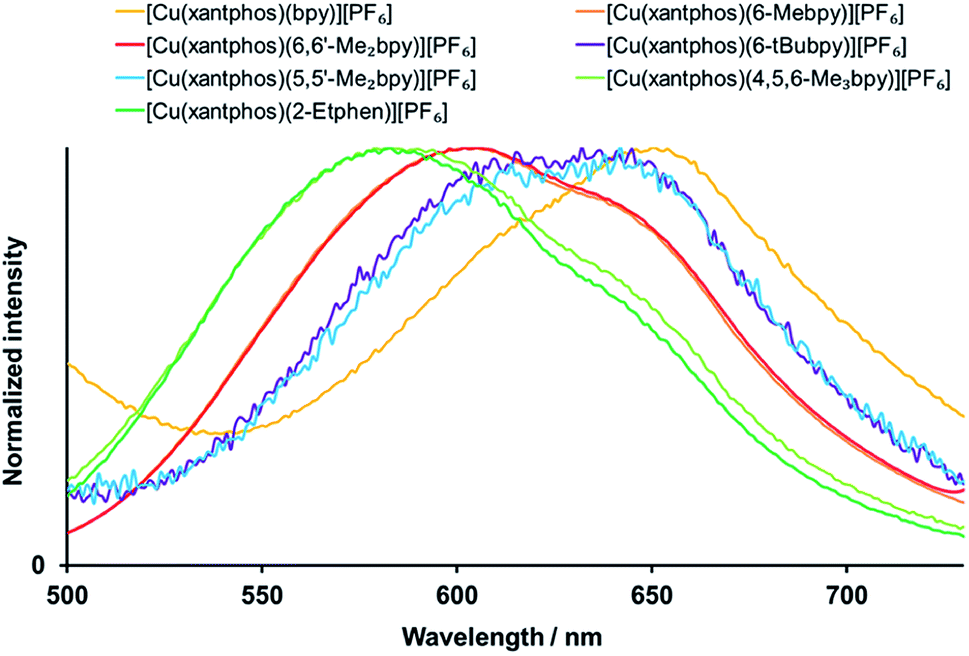 | ||
| Fig. 7 Normalized solution emission spectra of the [Cu(xantphos)(N^N)][PF6] complexes (CH2Cl2, 2.5 × 10−5 mol dm−3, for complexes with 6-tBubpy 5 × 10−5 mol dm−3). For λexc see Table 4. | ||
| Complex cation | CH2Cl2 solutiona | Powderb | Thin filmc | |||||
|---|---|---|---|---|---|---|---|---|
| λexc [nm] | λmaxem [nm] | PLQY (non-deaerated/deaerated)b [%] | τ (non-deaerated/deaerated)b [ns] | λmaxem [nm] | PLQY [%] | τ [μs] | PLQY [%] | |
| a Solution concentration = 2.5 × 10−5 mol dm−3, except where labelled with an asterisk * (5.0 × 10−5 mol dm−3).b λexc = 365 nm.c λexc = 365 nm. Thin films compose of spin-coated [Cu(P^P)(N^N)][PF6]:[Emim][PF6] in a 4:1 molar ratio, except where marked with an asterisk * (1:1 molar ratio).d Data from ref. 21.e Data from ref. 27.f Data from ref. 24. Radiative and non-radiative decay rates kr and knr are given in Table S3. |
||||||||
| [Cu(POP)(bpy)]+d | 390 | 618, 649 | 0.4/0.5 | 43/46 | 580 | 3.0 | 1.5 | 2* |
| [Cu(xantphos)(bpy)]+d | 390 | 620, 650 | 0.5/0.5 | 75/104 | 587 | 1.7 | 1.3 | — |
| [Cu(POP)(6-Mebpy)]+e | 378 | 610, 639 | 0.6/1.2 | 126/172 | 567 | 9.5 | 2.6 | 11 |
| [Cu(xantphos)(6-Mebpy)]+f | 379 | 605, 635 | 1.0/1.8 | 272/784 | 547 | 33.8 | 9.7 | 10 |
| [Cu(POP)(6,6′-Me2bpy)]+e | 372 | 564, 645 | 1.3/13.8 | 310/4032 | 535 | 43.2 | 10.5 | 38 |
| [Cu(xantphos)(6,6′-Me2bpy)]+f | 379 | 606, 635 | 1.6/10.0 | 451/3406 | 539 | 37.3 | 11.4 | 22 |
| [Cu(POP)(5,5′-Me2bpy)]+ | 390 | 622, 643 | 0.5/0.7 | 57/108 | 585 | 2.7 | 2.3 | 5 |
| [Cu(xantphos)(5,5′-Me2bpy)]+ | 390 | 616, 642 | 0.4/0.9 | 153/338 | 571 | 6.3 | 5.1 | 5 |
| [Cu(POP)(6-tBubpy)]+ | 390* | 614, 648* | 0.5/0.5 | 39/45 | 602 | 1.1 | 0.4 | <1 |
| [Cu(xantphos)(6-tBubpy)]+ | 390* | 615, 632* | 0.4/0.5 | 76/93 | 556 | 9.6 | 3.3 | <1 |
| [Cu(POP)(4,5,6-Me3bpy)]+ | 380 | 598, 630 | 1.0/1.5 | 202/730 | 518 | 42.7 | 9.3 | 16 |
| [Cu(xantphos)(4,5,6-Me3bpy)]+ | 380 | 582, 627 | 0.9/3.3 | 228/1595 | 529 | 58.8 | 9.8 | 19 |
| [Cu(POP)(2-Etphen)]+ | 390 | 597, 629 | 0.8/6.0 | 240/2401 | 558 | 27.5 | 8.7 | 26 |
| [Cu(xantphos)(2-Etphen)]+ | 390 | 583, 626 | 0.9/9.6 | 262/4987 | 550 | 9.8 | 10.2 | 10 |
For all the complexes, any alkyl substitution in the bipyridine ligand leads to a blue shift of the emission with respect to [Cu(POP/xantphos)(bpy)][PF6], and also the solution emission of [Cu(POP)(2-Etphen)][PF6] (λmaxem = 597, 629 nm) is blue shifted with respect to [Cu(POP)(phen)][PF6] (λmaxem = 648 nm).28 For the [Cu(xantphos)(N^N)][PF6] complex emission spectra in solution, the most hypsochromically shifted are the ones with 2-Etphen and 4,5,6-Me3bpy, where the wavelengths and profiles of the emission bands are very similar (Fig. 7). Slightly less blue-shifted and also very similar are the emission profiles of the complexes with 6-Mebpy and 6,6′-Me2bpy, then again very similar those with 6-tBubpy and 5,5′-Me2bpy and the most bathochromic emission is by the complex with bpy. In contrast to the absorption spectra, for the emissive properties the exact position of the alkyl group on the bpy N^N ligand appears to play a more crucial role. Substituents in position next to the coordinating nitrogen atom(s) are proven to be especially beneficial and lead to more blue-shifted emission, higher photoluminescence quantum yield (PLQY) values and longer excited-state lifetimes τ (Table 4). This is a typical observation for tetrahedral [Cu(N^N)(P^P)]+ complexes61 and can be explained by the increased rigidity of the complex and therefore reduced Jahn–Teller distortion in solution.
In the case of POP (Fig. S34† and Table 4), the situation is different in that 6,6′-Me2bpy gives the complex with considerably the most hypsochromic emission. Then, as in the xantphos analogues, the complexes with 2-Etphen and 4,5,6-Me3bpy are next and give similar emission bands, then 6-Mebpy, and afterwards 6-tBubpy and 5,5′-Me2bpy with similar maxima. Finally, the complex with bpy is again the most red-shifted one. A possible explanation is that in the case of the more flexible POP backbone, two methyl groups (or other substituents) next to the nitrogen atoms of the bpy are necessary to efficiently stabilize the tetrahedral complex geometry, whereas for the more rigid xantphos a single substituent on one side of the N^N chelating ligand is sufficient and the total number of alkyl groups (three in 4,5,6-Me3bpy versus two in 6,6′-Me2bpy) has a stronger impact on the HOMO–LUMO gap than their position on the N^N chelating ligand. The high sensitivity of these type of complexes towards number, position and type of alkyl groups on the ligands is also reflected in the quantum yield and lifetime values.
Deaeration by 20 minute argon flow through the complex solutions leads to increased PLQY and lifetime values (Table 4). By far, the best values are obtained for the complexes with 6,6′-Me2bpy, which was to be expected because this ligand has the highest steric hindrance and works best in stabilizing the tetrahedral complex geometry. Following up are the complexes with 2-Etphen. It is interesting to see that the complexes with 4,5,6-Me3bpy also give better emissive parameters than with 6-Mebpy, especially for the xantphos compounds: 1.8 vs. 3.3% PLQY and 784 vs. 1595 ns lifetime on going from 6-Mebpy to 4,5,6-Me3bpy (deaerated values). Another interesting observation is the unexpected decrease in the emissive properties on going from the complexes with 6-Mebpy to 6-tBubpy, for the xantphos complexes the PLQY is reduced from 1.8 to 0.5% and the lifetime from 784 to 93 ns (deaerated values).
Table 3 summarizes the vertical excitation energies calculated for the T1 and T2 states at the minimum-energy geometry of S0. Although those values should not be assimilated to emission energies, they follow the trend observed for the maxima of the emission spectra, being in all cases displaced to higher energies with respect to the reference complexes. Similarly to S1, the T1 state mainly originates from the HOMO → LUMO monoexcitation, which implies an electron transfer from the Cu(P^P) environment to the N^N ligand and therefore determines a partial oxidation of the Cu(I) atom upon excitation to T1. As discussed above, this partial oxidation leads to a flattening of the tetrahedral coordination that is hindered by the attachment of alkyl groups to the 6-positions of the bpy ligand. The relaxation of the T1 triplet is therefore more restricted for the complexes bearing the 4,5,6-Me3bpy and 6-tBubpy ligands (Table S1†) supporting the more blue-shifted emission energies experimentally observed in the former case for both xantphos and POP complexes (Table 4). The 6-tBubpy ligand produces a less significant effect because the bulky tert-butyl group provokes other changes in the geometry of the complex such as the increase of the Cu–N distances. In agreement with the experimental results, the emission energies calculated at the optimized geometry of T1 are significantly higher for [Cu(xantphos)(4,5,6-Me3bpy)]+ (1.62 eV) and [Cu(xantphos)(2-Etphen)]+ (1.68 eV) than for [Cu(xantphos)(6-tBubpy)]+ (1.53 eV), and a similar trend is found for the equivalent POP complexes. It should be mentioned that for the complexes with 2-Etphen the T2 state is close in energy to the T1 state, as expected from the MO study, but the HOMO → LUMO T1 state remains to be the lowest state upon relaxation and then phosphorescent emission takes place from the same MLCT triplet in all the complexes.
The solid compounds emit in the green to yellow region, with the powder emission maxima between 518 and 602 nm (Fig. 8 and S35,† Table 4). The significant blue shift with respect to the solution emission maxima is typically observed for these types of complexes. For the xantphos series (Fig. 8), alkyl substitution on the bpy ligand always leads to shorter wavelengths of the powder emission (Table 4) as well as to an increase in PLQY and lifetime, with the highest PLQY of 59% being found for [Cu(xantphos)(4,5,6-Me3bpy)][PF6], followed by 37% for [Cu(xantphos)(6,6′-Me2bpy)][PF6]. The excited state lifetimes are in the range between 1.3 and 11.4 μs, with the shortest for [Cu(xantphos)(bpy)][PF6] and the longest for [Cu(xantphos)(6,6′-Me2bpy)][PF6]. The most hypsochromic emission in powder of the complexes with xantphos is the one from [Cu(xantphos)(4,5,6-Me3bpy)][PF6] (529 nm), which is 58 nm lower (232 meV higher) than for [Cu(xantphos)(bpy)][PF6] (587 nm).
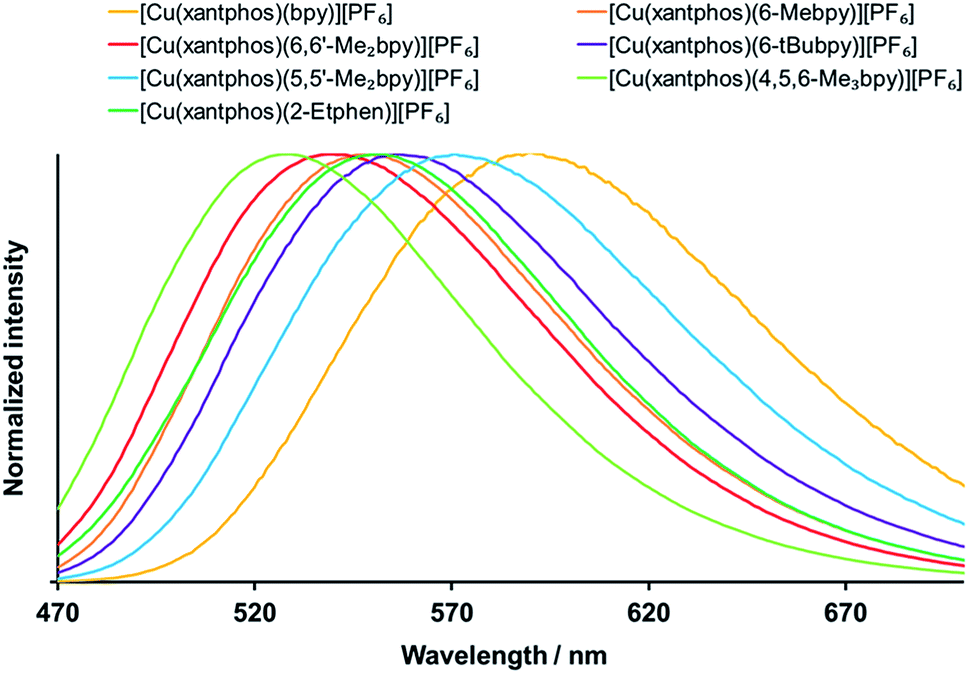 | ||
| Fig. 8 Normalized emission spectra of solid [Cu(xantphos)(bpy)][PF6] complexes, λexc = 365 nm. | ||
For the analogous complex series with POP (Fig. S35† and Table 4), also [Cu(POP)(4,5,6-Me3bpy)][PF6] is the most blue-shifted complex, 62 nm lower (256 meV higher) than [Cu(POP)(bpy)][PF6]. Similar to the observation in solution, the PLQY values increase on going from 6-Mebpy (Table 4) to 6-Etbpy (24% for [Cu(POP)(6-Etbpy)][PF6] and 37% for [Cu(POP)(6-Etbpy)][PF6]),14 but then decrease again with the larger alkyl group in 6-tBubpy (Table 4). The highest PLQY is for [Cu(POP)(6,6′-Me2bpy)][PF6] (43.2%), followed closely by [Cu(POP)(4,5,6-Me3bpy)][PF6] (42.7%). Surprisingly, it is not the [Cu(POP)(bpy)][PF6] complex that gives the lowest PLQY, but [Cu(POP)(6-tBubpy)][PF6] (1.1%), which also exhibits the most bathochromic emission and a considerably shorter excited state lifetime (0.4 μs) than the other complexes (1.5–10.5 μs for the series with POP).
Upon comparison between the POP and xantphos series, it is sometimes the POP and sometimes the xantphos complex that shows the more blue-shifted emission (Table 4). The difference of the emission energies between the respective POP and xantphos complex is in the range of 138 to 645 cm−1 (17 to 80 meV) with one outlier: the emission of [Cu(POP)(6-tBubpy)][PF6] (602 nm) is 1374 cm−1 (170 meV) lower in energy than the one from its xantphos analogue (556 nm).
A possible explanation for the considerably less efficient emissive properties of the [Cu(POP)(6-tBubpy)][PF6] complex might be the impaired coordination of the nitrogen atom in the pyridine ring bearing the bulky (C(CH3)3) group. As discussed in the crystallography section, due to the steric hindrance of the tert-butyl substituent next to the nitrogen atom, the Cu–N distance is elongated to 2.3949(19) Å, which is around 0.26 Å longer than the typical distance range for these types of complexes. In addition, the N–C–C–N torsion angle of the bpy ligand is as large as −28.8(3)°, which might also impact the overlap of the aromatic systems of the two pyridyl rings, which in return affects the photophysical properties. Unfortunately, attempts for suitable X-ray quality crystals of [Cu(xantphos)(6-tBubpy)][PF6] were not successful, so there are no structural data to support this hypothesis. We note, however, that the theoretical results predict a similar elongation of the C–N bond for both [Cu(POP)(6-tBubpy)][PF6] and [Cu(xantphos)(6-tBubpy)][PF6] complexes. In addition, it is also possible that the nine C–H bonds of the tert-butyl group in vicinity of the copper centre facilitate vibronic non-radiative deactivation of the excited states. Similar complexes where the phenyl groups at the coordinating phosphorus were replaced by tert-butyl groups also showed very weak emissive properties.62 Also, both [Cu(POP/xantphos)(6-tBubpy)][PF6] complexes have lower PLQY and shorter lifetime values than [Cu(POP/xantphos)(6-Mebpy)][PF6]. We conclude that, in addition to the negative effect of large aromatic substituents,23 large alkyl groups next to the coordinating nitrogen at the N^N chelating ligand are not beneficial for the emissive properties of these type of copper complexes. However, small substituents such as methyl, ethyl, CF3 or chloro next to the nitrogen lead to an improvement of the photophysical properties. Furthermore, an increased number of methyl groups at the N^N chelating ligand results in a positive accumulating effect, which was observed for the [Cu(POP/xantphos)(N^N)][PF6] complexes on going from bpy to 6-Mebpy to 4,5,6-Me3bpy.
[Cu(P^P)(N^N)]+complexes have been shown to be good candidates for TADF,40,41 a mechanism that allows the repopulation of S1 from T1 via the available thermal energy at room temperature and thus results in a combined singlet and triplet emission. Upon decreasing the temperature, singlet state repopulation takes place in a lower degree and the contribution of the triplet emission or phosphorescence increases at the expense of the singlet emission or fluorescence. This is experimentally observable in the red shift of the emission (because the triplet excited state is energetically lower than the singlet excited state) and a significant elongation of the excited state lifetime on going from powder at room temperature to a frozen solution of Me–THF at 77 K. As detailed in Table 5, all the complexes show significantly extended lifetimes at 77 K (11 to 81 μs) in comparison to their powders at room temperature ((0.4 to 11.4 μs, Table 4)), which is a strong indication for TADF. The emission spectra at 77 K are displayed in Fig. 9 for xantphos complexes (Fig. S36† for POP complexes). However, when it comes to the emission maxima, two complexes unexpectedly give a blue shift instead of a red shift upon cooling: for [Cu(POP)(6-tBubpy)][PF6] and [Cu(POP)(2-Etphen)][PF6] the emissions at 77 K are higher in energy by 396 cm−1 (49 meV) and 97 cm−1 (12 meV), respectively. Both complexes show significantly longer lifetimes upon cooling, consistent with a TADF mechanism, and the observed blue shift can be attributed to intermolecular matrix effects in the Me–THF glass. It is noteworthy that the complexes bearing the 6-tBubpy and 2-Etphen ligands present energy differences between T1 and S1 of around 0.16 eV, that are significantly smaller than those found for the rest of the complexes lying between 0.22 and 0.27 eV (Table 3).
| Complex cation | λmaxem [nm] powder | λmaxem [nm] Me-THFb 77 K | τb [μs] 77 K | Δ wavenumber [cm−1] | ΔE [meV] |
|---|---|---|---|---|---|
| a Data from ref. 21.b λexc = 410 nm. | |||||
| [Cu(POP)(bpy)]+a | 580 | 610 | 16 | 847 | 105 |
| [Cu(xantphos)(bpy)]+a | 587 | 613 | 11 | 722 | 89 |
| [Cu(POP)(5,5′-Me2bpy)]+ | 585 | 591 | 63 | 173 | 21 |
| [Cu(xantphos)(5,5′-Me2bpy)]+ | 571 | 594 | 44 | 678 | 84 |
| [Cu(POP)(6-tBubpy)]+ | 602 | 588 | 14 | −395 | −49 |
| [Cu(xantphos)(6-tBubpy)]+ | 556 | 578 | 25 | 684 | 84 |
| [Cu(POP)(4,5,6-Me3bpy)]+ | 518 | 566 | 81 | 1637 | 202 |
| [Cu(xantphos)(4,5,6-Me3bpy)]+ | 529 | 559 | 75 | 1014 | 125 |
| [Cu(POP)(2-Etphen)]+ | 558 | 555 | 27 | −96 | −12 |
| [Cu(xantphos)(2-Etphen)]+ | 550 | 557 | 14 | 228 | 28 |
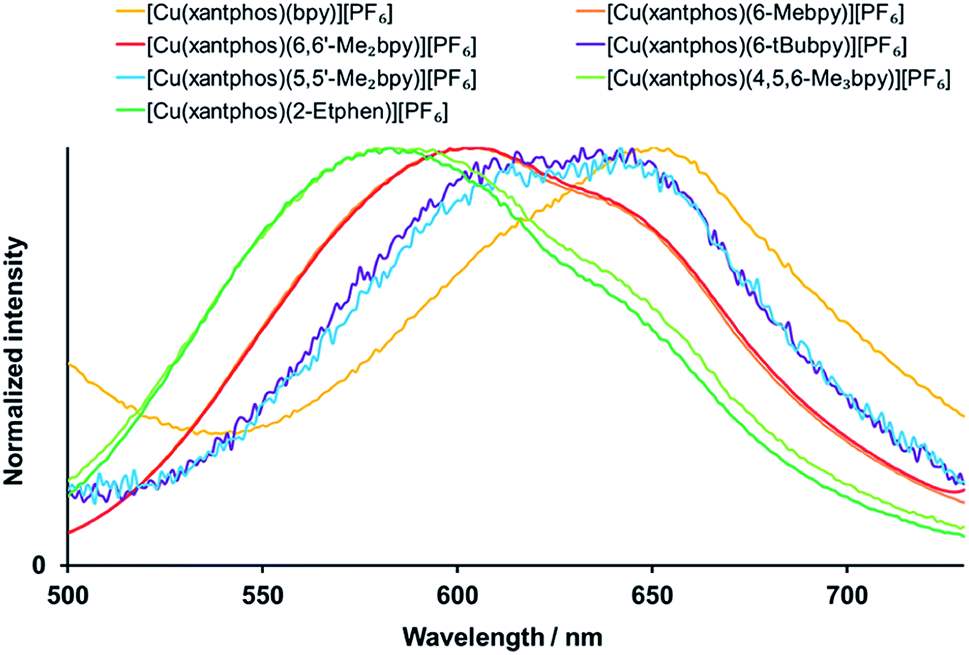 | ||
| Fig. 9 Normalized emission spectra of the [Cu(xantphos)(N^N)][PF6] complexes in Me-THF at 77 K. λexc = 410 nm. | ||
Device properties
All complexes were tested as emitters in the active layers of LECs. Details of the device fabrication are given in the ESI,† and a schematic representation of a LEC is shown in Fig. S37.† Devices were prepared on ITO-coated glass slides, using PEDOT:PSS as the hole injection layer and a thin film (about 100 nm thick) of the complex deposited by spin-coating of its dichloromethane solutions, and finished with a thermally deposited aluminium film (100 nm) as the reflective cathode. The ionic liquid [Emim][PF6] was added to the solutions of the complexes in a 4:1/[Cu(P^P)(N^N)][PF6]:[Emim][PF6] molar ratio to enhance the ionic conductivity of the resulting layers. LECs were tested by monitoring over time the average luminance and voltage upon application of a pulsed current (average current density 50 or 100 A m−2, 1 kHz, 50% duty cycle, block wave). In view of the good performance with the Cu(I) complexes with the ligands 4,5,6-Me3bpy and 2-Etphen (main performance parameters reported in Table 6 and dynamic optoelectronic behavior in Fig. 10 and S37–S40†), we mainly focus our discussion on these devices. The device performance data for the remaining complexes can be found in the ESI (Table S4†). The LECs with the four best-performing complexes show electroluminescence (EL) spectra (Fig. S38†) which are blue-shifted with respect to the PL spectra of the compounds in solution and red-shifted in comparison with the PL in the solid state (Table 4). This is expected in view of the intermediate degree of conformational freedom of the complex in the (mainly amorphous) films and in presence of ionic liquids. LECs based on [Cu(POP)(4,5,6-Me3bpy)][PF6] turned on instantaneously after biasing (initial luminance Lum0 = 64 cd m−2), and were observed to slowly increase the EL intensity up to a maximum luminance (Lummax) of 111 cd m−2 in about 18 min (time to reach the maximum luminance, ton). The slow turn-on and the limited EL intensity are likely due to a hindered charge injection/transport efficiency, as the voltage at the beginning of the test was found to exceed the limit of our setup (9 V, Fig. S39†). Upon ionic redistribution, the voltage only slightly decreased to approximately 7.5 V after 3 hours (indicating improved charge injection/transport) when, however, electroluminescence was no longer observed (the device lifetime t1/2, which is the time to decay to half of Lummax, was only 42 minutes). The analogous complex employing the xantphos ligand showed a different optoelectronic behaviour. While EL was not initially observed, again due to a saturation of the average voltage at 9 V, LECs based on [Cu(xantphos)(4,5,6-Me3bpy)][PF6] showed a sudden and fast increase of the EL signal up to a remarkable Lummax = 462 cd m−2 in only 13 minutes. Unfortunately, as is often observed in LECs using [Cu(P^P)(N^N)]+ cationic complexes, such high EL intensity was observed to decay quickly (t1/2 = 3 hours). The maximum external quantum efficiency (EQEmax) was found to be 1.7%, which is significant but still limited by substantial non-radiative recombination losses. In fact, considering the PLQY in thin film (19%), an outcoupling efficiency of 20% and neglecting charge carrier balance and exciton generation yield, the maximum achievable EQE for this compound is 3.8%. It is also reasonable to assume that [Cu(xantphos)(4,5,6-Me3bpy)][PF6] has improved charge transport/injection properties compared to the analogous POP complex, as the voltage decreased rapidly to stabilize at a lower value of about 6 V. The high efficiency and the moderate voltage resulted in a peak power conversion efficiency (PCEmax) of 2.0 lm W−1 (Fig. S40†). LECs employing Cu(I) complexes with the 2-ethyl-1,10-phenanthroline ligand showed a very different behaviour compared to those based on substituted bipyridines. Devices employing [Cu(POP)(2-Etphen)][PF6] displayed instantaneous and intense electroluminescence Lum0 = 273 cd m−2, which increased rapidly (ton = 25 min) to a maximum luminance of 451 cd m−2. However, the EL decay was also rather fast, with a lifetime t1/2 = 5.7 hours, although the device continued to emit for 24 hours, reaching a luminance of approximately 100 cd m−2. The corresponding EQEmax is 1.8%, the highest for the device series reported here but still far from the theoretical maximum (5.2%) which can be estimated from the PLQY in thin film (26%). Interestingly, LECs with [Cu(POP)(2-Etphen)][PF6] showed a fast drop of the average applied voltage to less than 6 V in about an hour, resulting in a PCEmax of 2.3 lm W−1. Distinct performance was observed for LECs based on the [Cu(xantphos)(2-Etphen)][PF6] complex, which turned on slowly (ton = 122 min) to reach a peak luminance Lummax = 153 cd m−2, corresponding to an EQEmax of 0.6%. The less intense EL compared to the analogous complex with the POP ligand correlates with the lower PLQY in thin film (10%). Nevertheless, this compound exhibited a very stable operation (t1/2 = 34 hours) with the lowest average voltage among this device series, below 5 V after 6 hours driving. This low power consumption is reflected in a moderate but stable PCEmax of 0.9 lm W−1. In general, the LEC performance observed for this series of compounds is among the highest reported for similar cationic [Cu(P^P)(N^N)]+ complexes.
:[Emim][PF6] 4:1 molar ratio/Al LECs measured using a pulsed current driving (average current density 50 or 100 A m−2, 1 kHz, 50% duty cycle, block wave). Device data for the other four complexes is summarized in Table S4. Data for previously published complexes with similar performances are added for comparison
| Complex | Javg [A m−2] | tona [min] | Lum0b [cd m−2] | Lummaxc [cd m−2] | t1/2d [hours] | EQEmaxe [%] | PCEmaxf [lm W−1] | Efficacymaxg [cd A−1] | λmaxELh [nm] |
|---|---|---|---|---|---|---|---|---|---|
| a Time to reach the maximum luminance.b Initial luminance.c Maximum luminance.d Time to reach half of the maximum luminance.e Maximum external quantum efficiency.f Maximum power conversion efficiency.g Maximum current efficiency.h Wavelength at the maximum intensity of the electroluminescence spectrum.i Data from ref. 18.j Data from ref. 24.k Data from ref. 21. | |||||||||
| [Cu(POP)(4,5,6-Me3bpy)]+ | 50 | 11 | 82 | 92 | 0.6 | 0.6 | 0.6 | 1.8 | 571 |
| 100 | 18 | 64 | 111 | 0.7 | 0.4 | 0.4 | 1.1 | ||
| [Cu(xantphos)(4,5,6-Me3bpy)]+ | 50 | 27 | 12 | 190 | 7.1 | 1.4 | 1.8 | 3.8 | 570 |
| 100 | 13 | 0 | 462 | 3.1 | 1.7 | 2.0 | 4.6 | ||
| [Cu(POP)(2-Etphen)]+ | 50 | 55 | 76 | 140 | 8.4 | 1.1 | 1.4 | 2.8 | 582 |
| 100 | 25 | 273 | 451 | 5.7 | 1.8 | 2.3 | 4.5 | ||
| [Cu(xantphos)(2-Etphen)]+ | 50 | 319 | 0 | 87 | 98.3 | 1.1 | 0.7 | 1.7 | 580 |
| 100 | 122 | 0 | 153 | 34.0 | 0.6 | 0.9 | 1.5 | ||
| [Cu(POP)(6-MeObpy)]+i | 50 | 9540 | — | 17 | 200 | — | — | 0.3 | 585 |
| [Cu(POP)(6-EtObpy)]+i | 50 | 60 | — | 63 | 102 | — | — | 1.3 | 585 |
| [Cu(POP)(6-Etbpy)]+j | 50 | 260 | 25 | 53 | 82 | 0.2 | 0.2 | 0.6 | 582 |
| [Cu(xantphos)(6-CF3bpy)]+k | 100 | 137 | 5 | 109 | 31 | 0.5 | 0.4 | 1.1 | 589 |
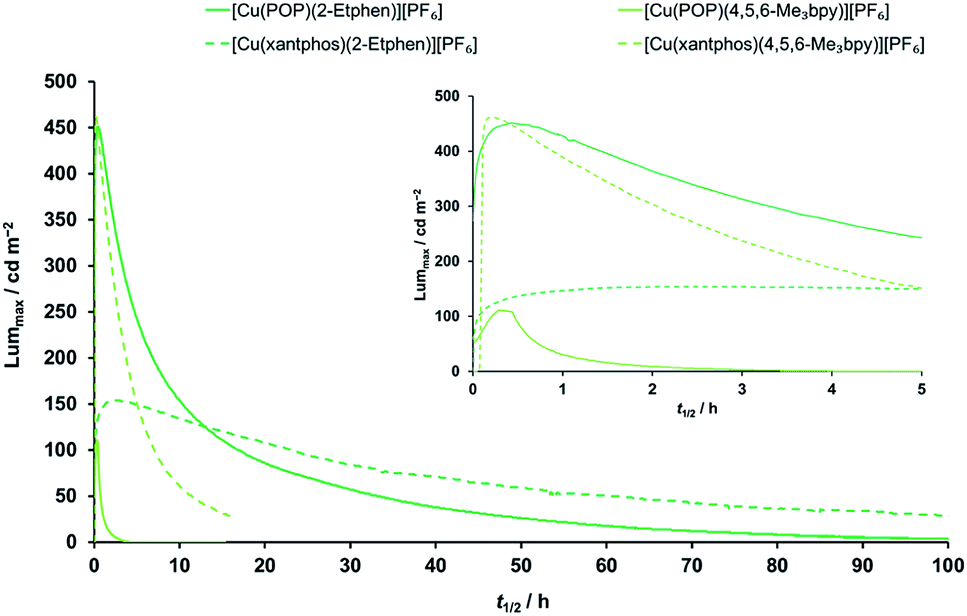 | ||
| Fig. 10 Time evolution of luminance for ITO/PEDOT:PSS/[Cu(P^P)(N^N)][PF6]:[Emim][PF6] 4:1 molar ratio/Al LECs measured using a pulsed current driving and under continuous operation (average current density 100 A m−2). Inset shows the behaviour over the first five hours. For time evolution of further parameters (average voltage, power conversion efficiency, external quantum efficiency and electroluminescence spectra) see Fig. S38–S41.† | ||
In order to better compare the device performance of this complex series with previously prepared LECs based on copper(I) complexes from our laboratories, we illustrated the two crucial device parameters that we are constantly trying to improve, namely brightness (Lummax) and device lifetime (t1/2) in two summarizing graphs which include all tested devices (Fig. 11). This overview clearly shows that three of the here- presented complexes are among the best performing copper(I) emitters that we ever tested in a LEC. Brightness records go to [Cu(xantphos)(4,5,6-Me3bpy)][PF6] and [Cu(POP)(2-Etphen)][PF6] (462 and 451 cd m−2, respectively). At an average current density of 100 A m−2, [Cu(xantphos)(2-Etphen)][PF6] is the new champion in terms of lifetime with almost 34.0 hours. And at 50 A m−2, the device with this complex is still among the most longest living ones, and by far the brightest of those with a lifetime of over three days. Compared to copper-based LECs in the literature, our devices play in the league of the best performing ones and are, to our knowledge, among the very brightest reported for copper LECs and are the devices with the longest reported lifetime.7,8,38,60,63–67
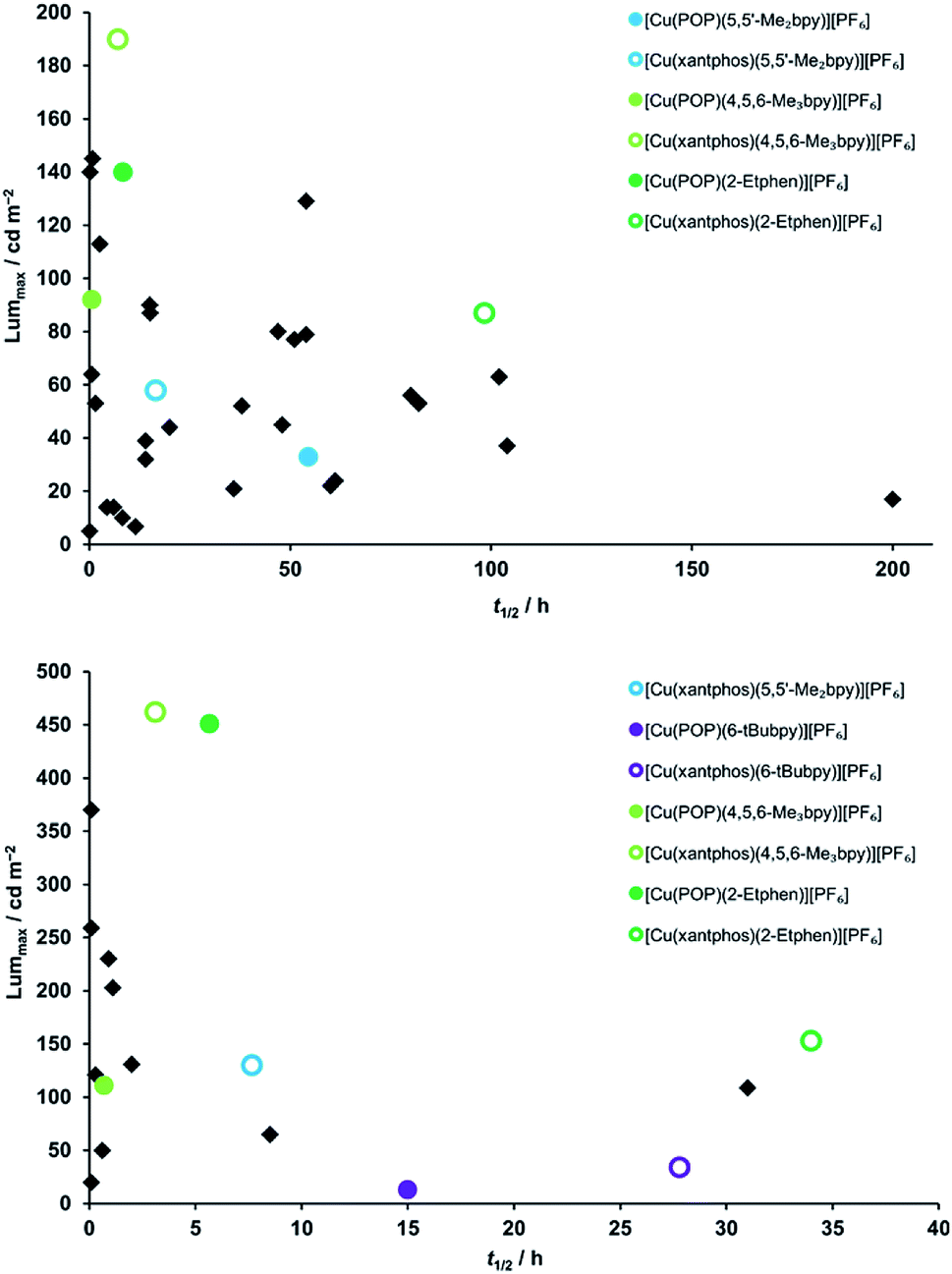 | ||
| Fig. 11 Comparison of device properties (maximum luminance and device lifetime) for the complexes described in this work (round coloured dots), compared to devices with previously tested complexes prepared in our group (black squares).15,17–19,21,24,25 Top: average current density 50 A m−2; bottom: average current density 100 A m−2. | ||
Compared to LECs, state-of-the-art OLEDs (including solution-processed OLEDs) using copper emitters are still about hundred orders of magnitudes brighter, with Lummax values in the four- and even five-digit range.68–71
Conclusions
We have described a series of eight [Cu(POP)(N^N)][PF6] and [Cu(xantphos)(N^N)][PF6] complexes with the alkyl-substituted N^N chelating ligands 5,5′-Me2bpy, 6-tBubpy, 4,5,6-Me3bpy and 2-Etphen. The study provides meaningful insights into the variety of effects that alkyl substitution has on the photophysical and device properties of the complexes. A combination of electronic and steric effects can lead to a significant improvement of PLQY values as well as the maximum luminance in the device, as was the case for the [Cu(P^P)(4,5,6-Me3bpy)][PF6] complexes. On the other hand, we have demonstrated that while an alkyl group in the 6-position of the bpy ligand is beneficial in the case of methyl or ethyl groups, a tBu substitution has a detrimental effect and leads to lower PLQY and shorter excited state lifetimes of the complexes. Another finding is that incorporation of 5,5′-Me2bpy results in less efficient emitters than 6-Mebpy, which underlines again that the stabilization of the tetrahedral complex geometry with a substitution in 6-position of the bpy is more beneficial than the electron-donating effect of two methyl groups. As the complexes with 6-Etbpy gave some of the longest living devices, we decided to replace the ligand with the analogous phenanthroline, 2-Etphen. While the resulting devices gave higher luminance values, only the complex with xantphos lead to longer device lifetimes, and the device with the analogous POP complex was considerably less stable with 2-Etphen than with 6-Etbpy. This highlights once more the sensitivity of the interplay between the P^P and N^N chelating ligands and the challenge and relative unpredictability when it comes to the complex composition of stable and efficient copper emitters. However, with a systematic approach, we have been able to improve the design of the Cu–iTMC, thereby increasing both brightness and lifetime of the devices, which are among the best reported in copper–LEC literature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swiss National Science Foundation (grant number 200020_162631), the University of Basel, the MCIU of Spain (MAT2017-88821-R, RTI2018-095362-A-I00, PGC2018-099568-B-I00, PCIN-2017-014 and Unidad de Excelencia María de Maeztu MDM-2015-0538), the Generalitat Valenciana (PROMETEO/2016/135) and European FEDER funds (PGC2018-099568-B-I00) is gratefully acknowledged. We thank Marco Meyer for recording some of the ESI-MS experiments. M. S. acknowledges the MCIU for his RyC contract.Notes and references
- Transforming our World, The 2030 Agenda for Sustainable Development, https://sustainabledevelopment.un.org/post2015/transformingourworld/publication, retrieved 09.06.2020 Search PubMed.
- https://www.eia.gov/tools/faqs/faq.php?id=99%26t=3, retrieved 22.11.2018.
- Organic Light-Emitting Diodes (OLEDs), ed. A. Buckley, Woodhead Publishing (Elsevier), Oxford, 1st edn, 2013 Search PubMed.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719 RSC.
- J. Emsley, The Elements, Oxford University Press, Oxford, 3rd edn, 1998 Search PubMed.
- S. van Reenen and M. Kemerink, in Light-Emitting Electrochemical Cells. Concepts, Advances and Challenges, ed. R. Costa, Springer, Cham, 2017, p. 3 Search PubMed.
- E. Fresta and R. D. Costa, J. Mater. Chem. C, 2017, 5, 5643 RSC.
- B. Pashaei, S. Karimi, H. Shahroosvand, P. Abbasi, M. Pilkington, A. Bartolotta, E. Fresta, J. Fernandez-Cestau, R. D. Costa and F. Bonaccorso, Chem. Soc. Rev., 2019, 48, 5033 RSC.
- S. Kanagaraj, A. Puthanveedu and Y. Choe, Adv. Funct. Mater., 2019, 1907126 CrossRef.
- E. Fresta and R. D. Costa, Adv. Funct. Mater., 2020, 1908176 CrossRef.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monit, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- C. E. Housecroft and E. C. Constable, in Light-Emitting Electrochemical Cells. Concepts, Advances and Challenges, ed. R. Costa, Springer, Cham, 2017, p. 167 Search PubMed.
- L. He, in Light-Emitting Electrochemical Cells. Concepts, Advances and Challenges, ed. R. Costa, Springer, Cham, 2017, p. 203 Search PubMed.
- S. Keller, M. Alkan-Zambada, A. Prescimone, E. C. Constable and C. E. Housecroft, Crystals, 2020, 10, 255 CrossRef CAS.
- N. Arnosti, F. Brunner, I. Susic, S. Keller, J. M. Junquera-Hernández, A. Prescimone, H. J. Bolink, M. Sessolo, E. Ortí, C. E. Housecroft and E. C. Constable, Adv. Opt. Mater., 2020, 1901689 CrossRef CAS.
- S. Keller, M. Bantle, A. Prescimone, E. C. Constable and C. E. Housecroft, Molecules, 2019, 24, 3934 CrossRef CAS PubMed.
- F. Brunner, A. Babaei, A. Pertegás, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, Dalton Trans., 2019, 48, 446 RSC.
- M. Alkan-Zambada, S. Keller, L. Martínez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 8460 RSC.
- S. Keller, A. Prescimone, H. J. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263 RSC.
- S. Keller, A. Prescimone, E. C. Constable and C. E. Housecroft, Photochem. Photobiol. Sci., 2018, 17, 375 RSC.
- S. Keller, F. Brunner, J. M. Junquera-Hernández, A. Pertegás, M.-G. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Ortí and C. E. Housecroft, ChemPlusChem, 2018, 83, 217 CrossRef CAS PubMed.
- F. Mazzeo, F. Brunner, A. Prescimone, E. C. Constable and C. E. Housecroft, Crystals, 2020, 10, 1 CrossRef CAS.
- F. Brunner, S. Graber, Y. Baumgartner, D. Häussinger, A. Prescimone, E. C. Constable and C. E. Housecroft, Dalton Trans., 2017, 46, 6379 RSC.
- S. Keller, A. Pertegás, G. Longo, L. Martínez, J. Cerdá, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, C. E. Housecroft, E. Ortí and H. J. Bolink, J. Mater. Chem. C, 2016, 4, 3857 RSC.
- F. Brunner, L. Martínez-Sarti, S. Keller, A. Pertegás, A. Prescimone, E. C. Constable, H. J. Bolink and C. E. Housecroft, Dalton Trans., 2016, 45, 15180 RSC.
- N. S. Murray, S. Keller, E. C. Constable, C. E. Housecroft, M. Neuburger and A. Prescimone, Dalton Trans., 2015, 44, 7626 RSC.
- S. Keller, E. C. Constable, C. E. Housecroft, M. Neuburger, A. Prescimone, G. Longo, A. Pertegás, M. Sessolo and H. J. Bolink, Dalton Trans., 2014, 43, 16593 RSC.
- R. D. Costa, D. Tordera, E. Orti, H. J. Bolink, J. Schönle, S. Graber, C. E. Housecroft, E. C. Constable and J. A. Zampese, J. Mater. Chem., 2011, 21, 16108 RSC.
- M. Elie, S. Gaillard and J.-L. Renaud, in Light-Emitting Electrochemical Cells. Concepts, Advances and Challenges, ed. R. Costa, Springer, Cham, 2017, p. 203 Search PubMed.
- C. M. Brown, C. Li, V. Carta, W. Li, Z. Xu, P. H. F. Stroppa, I. D. W. Samuel, E. Zysman-Colman and M. O. Wolf, Inorg. Chem., 2019, 58, 7156 CrossRef CAS PubMed.
- M. Holler, B. Delavaux-Nicot and J.-F. Nierengarten, Chem.–Eur. J., 2019, 25, 4543 CrossRef CAS PubMed.
- S. Saeedi, C. Xue, B. J. McCullough, S. E. Roe, B. J. Neyhouse and T. A. White, ACS Appl. Energy Mater., 2019, 2, 131 CrossRef CAS.
- R. Giereth, I. Reim, W. Frey, H. Junge, S. Tschierlei and M. Karnahl, Sustainable Energy Fuels, 2019, 3, 692 RSC.
- S. Yanagida, M. Yoshida, W. M. C. Sameera, A. Kobayashi and M. Kato, Bull. Chem. Soc. Jpn., 2019, 92, 1684 CrossRef CAS.
- M. Alkan-Zambada and X. Hu, Organometallics, 2018, 37, 3928 CrossRef CAS.
- Y. Shi, X. Liu, Y. Shan, X. Zhang, W. Kong, Y. Lu, Z. Tan and X.-L. Li, Dalton Trans., 2019, 48, 2430 RSC.
- X. Liu, Y. Shan, J. Xu, X. Zhang, S. Shang and X.-L. Li, Polyhedron, 2019, 164, 152 CrossRef CAS.
- E. Fresta, M. D. Weber, J. Fernandez-Cestau and R. D. Costa, Adv. Opt. Mater., 2019, 7, 1900830 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, in Light-Emitting Electrochemical Cells. Concepts, Advances and Challenges, ed. R. Costa, Springer, Cham, 2017, p. 237 Search PubMed.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508 CrossRef CAS PubMed.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2 CrossRef CAS.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127 CrossRef CAS.
- G. Lia, Z.-Q. Zhua, Q. Chenb and J. Li, Org. Electron., 2019, 69, 135 CrossRef.
- V. Hebbe-Viton, V. Desvergnes, J. J. Jodry, C. Dietrich-Buchecker, J.-P. Sauvage and J. Lacour, Dalton Trans., 2006, 17, 2058 RSC.
- A. Zucca, M. A. Cinellu, M. V. Pinna, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, Organometallics, 2000, 19, 4295 CrossRef CAS.
- S. Stoccoro, G. Chelucci, M. A. Cinellu, A. Zucca and G. Minghetti, J. Organomet. Chem., 1993, 450, C15 CrossRef CAS.
- G. Minghetti, M. A. Cinellu, S. Stoccoro, A. Zucca and M. Manassero, J. Chem. Soc., Dalton Trans., 1995, 7, 777 RSC.
- A. Zucca, S. Stoccoro, M. A. Cinellu, G. Minghetti and M. Manassero, J. Chem. Soc., Dalton Trans., 1999, 19, 3431 RSC.
- G. Minghetti, A. Doppiu, A. Zucca, S. Stoccoro, M. A. Cinellu, M. Manassero and M. Sansoni, Chem. Heterocycl. Compd., 1999, 35, 992 CrossRef CAS.
- G. Minghetti, S. Stoccoro, M. A. Cinellu, B. Soro and A. Zucca, Organometallics, 2003, 22, 4770 CrossRef CAS.
- Y.-Q. Fang and G. S. Hanan, Synlett, 2003, 6, 852 Search PubMed.
- T. Kamei, N. Kishii, T. Kobayashi, K. Kurihara, N. Matsuzawa, H. Iwamoto and H. Morita, Jpn. Pat., JP2000076640A, 20000314, Kokai Tokyo Koho, 2000.
- P. J. Pijper, H. Van der Goot, H. Timmerman and W. T. Nauta, Eur. J. Med. Chem., 1984, 19, 399 CAS.
- Y. Cheng, X. Han, H. Ouyang and Y. Rao, Chem. Commun., 2012, 48, 2906 RSC.
- The Cambridge Structural Database, C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171, DOI:10.1107/s2052520616003954.
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, 2007 Search PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- S. Keller, PhD thesis, University of Basel, 2017.
- S. Keller, A. Prescimone, H. J. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263 RSC.
- M. D. Weber, C. Garino, G. Volpi, E. Casamassa, M. Milanesio, C. Barolo and R. D. Costa, Dalton Trans., 2016, 45, 8984 RSC.
- C. M. Brown, V. Carta and M. O. Wolf, Chem. Mater., 2018, 30, 5786 CrossRef CAS.
- S. Keller, A. Prescimone, E. C. Constable and C. E. Housecroft, Photochem. Photobiol. Sci., 2018, 17, 375 RSC.
- E. Fresta, G. Volpi, M. Milanesio, C. Garino, C. Barolo and R. D. Costa, Inorg. Chem., 2018, 57, 10469 CrossRef CAS PubMed.
- M. D. Weber, E. Fresta, M. Elie, M. E. Miehlich, J.-L. Renaud, K. Meyer, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef.
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J.-F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu, J.-L. Renaud, M. Linares, M. Hamel, R. D. Costa and S. Gaillard, ACS Appl. Mater. Interfaces, 2016, 8, 14678 CrossRef CAS PubMed.
- O. Moudam, A. Kaeser, B. Delavaux-Nicot, C. Duhayon, M. Holler, G. Accorsi, N. Armaroli, I. Séguy, J. Navarro, P. Destruel and J.-F. Nierengarten, Chem. Commun., 2007, 3077 RSC.
- Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 1203 CrossRef CAS.
- G. K.-M. So, G. Cheng, J. Wang, X. Chang, C.-C. Kwok, H. Zhang and C.-M. Che, Chem.–Asian J., 2017, 12, 1490 CrossRef CAS PubMed.
- L. Lin, D.-H. Chen, R. Yu, X.-L. Chen, W.-J. Zhu, D. Liang, J.-F. Chang, Q. Zhang and C.-Z. Lu, J. Mater. Chem. C, 2017, 5, 4495 RSC.
- D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Fléchon, D. M. Zink, J. Friedrichs, H. Flügge, R. Steininger, J. Göttlicher, C. Heske, L. Weinhardt, S. Bräse, F. So and T. Baumann, Adv. Mater., 2015, 27, 2538 CrossRef CAS PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; Fig. S1–S21: NMR and ESI-MS spectra; cyclic voltammograms (Fig. S22–S29); computational results (Table S1 and Fig. S30); absorption and emission spectra and photophysical properties (Tables S2 and S3 and Fig. S31–S36); LEC device: Fig. S37; device performance parameters (Table S4 and Fig. S38–S41). CCDC 1978436–1978442. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03824e |
| This journal is © The Royal Society of Chemistry 2020 |