
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid approach to obtain high-quality BaMO3 perovskite nanocrystals†
Natalia Chamorro ab,
Jordi Martínez-Esaín‡
ab,
Teresa Puigb,
Xavier Obradorsb,
Josep Rosa,
Ramón Yáñeza and
Susagna Ricart*b
ab,
Jordi Martínez-Esaín‡
ab,
Teresa Puigb,
Xavier Obradorsb,
Josep Rosa,
Ramón Yáñeza and
Susagna Ricart*b
aDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
bInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), 08193 Bellaterra, Spain. E-mail: ricart@icmab.es
First published on 5th August 2020
Abstract
A novel hybrid solvothermal approach for perovskite nanocrystal formation via accurate control of the hydrolytic process is reported. This new synthetic methodology sets a whole general route to successfully tune the sizes of high-quality BaMO3 (M = Ti4+, Zr4+, and Hf4+) perovskite nanocrystals. Purely cubic-phase nanocrystals (stable in alcohol media) were obtained using controlled water amounts, combining the well-known aqueous sol–gel process with the classic solvothermal method. Exhaustive optimizations revealed feasibility of a fast (1 hour) and reproducible synthesis with small variations in the crystal size or agglomeration parameters. The study also reveals water content as the pivotal factor to achieve this wide range of sizes through a controlled hydrolytic step. Finally, the study of the hydrolytic process made it possible to shed some light on mechanistic insights of this synthetic route.
Introduction
Nowadays, the search for a fast, cheap, reproducible and robust approach to synthesize oxide nanocrystals (NCs) still attracts strong attention among researchers. This high interest derives from their wide range of applications in electronics,1 medicine,2,3 catalysis,4,5 energy storage6,7 and sensing.8 Among metal oxide NCs, the perovskite family has gained attention due to its stability under high-temperature treatments and its emergent applications.9–11 Although, BaTiO3 NCs have been extensively studied,12–15 the achievement of BaZrO3 and BaHfO3 NCs continues to be an entangled process, in which researchers keep finding difficulties to obtain small-size, crystalline NCs, stable in colloidal suspensions without agglomeration.16–18Although the applicability of these systems is clear and well-known, the synthesis of this family of perovskites still entails many limitations to obtain stable, homogeneous and high-quality NCs. Our main goal is to find alternative methods for these intricate perovskite NCs using the current background researches in the field. Two main strategies are highlighted: (i) non-aqueous solvothermal treatments and/or (ii) sol–gel aqueous processes. Although the hydrolytic mechanism of the aqueous sol–gel procedure is well-known,19–21 this methodology is usually avoided because the fast hydrolysis promotes formation of non-homogeneous systems; this requires a posterior calcination treatment, reducing the final NC stability in solution.12,22 On the other hand, non-aqueous approaches23–27 have risen up during last decades due to several advantages such as high crystallinity, controlled crystal growth and homogeneous NC dispersions.28,29 However, this methodology shows two main drawbacks: long reactions times and the need to use high temperatures during the process.12,17,18 We aimed to go further by trying to control the hydrolytic step with different water proportions applying same conditions as in the “non-aqueous” methodology. This approach aims to merge the advantages of both routes (i.e., sol–gel and solvothermal) of perovskite synthesis in one that combines short reaction times, high crystallinity and long stability of the NCs.
Herein, we not only report a suitable synthetic methodology for a group of perovskite NCs (BaMO3) but also a general route that allows accurate size control of high-quality NC systems. This new proposed synthetic procedure has been tested for three tetravalent cations (Ti4+, Zr4+ and Hf4+), being the first time that the same methodology can be applied to this perovskite family, obtaining comparable quality NCs. This novel approach allows to fine-tune the NC size and an excellent uniformity of NCs dispersion, with promising applications in emergent energetic devices. The present work conducted by our group showed the possibility to combine BaZrO3 and BaHfO3 NCs with YBa2Cu3O7 (YBCO) type II superconductors, to enhance the superconducting properties, unravelling a novel pathway in electronic materials science and bestowing a crucial breakthrough for the implementation of NCs to act as pinning centres.30–32 Thus, we present here a novel hybrid method (i.e., built from the classical approaches) to produce high-quality perovskite NCs and enhance the current applicability portfolio.
Experimental methods
Materials
The following chemicals were used without further purification. Zirconium(IV) n-butoxide solution 80 wt% in n-butanol, hafnium(IV) n-butoxide 99%, titanium(IV) n-butoxide, triethylene glycol (TEG 99%), barium hydroxide octahydrate ≥98% and ammonium hydroxide solution 30% v/v were purchased from Sigma Aldrich. Ethanol absolute was purchased from Scharlau.Nanocrystal synthesis
In a standard procedure, 1.24 mL (2.8 mmol) of Zr(OC4H9)4 or 2.8 mmol of the desired n-butoxide precursor (M = Ti4+, Zr4+ or Hf4+) was added to 10 mL of absolute ethanol at room temperature under N2 atmosphere and vigorous stirring for 5 min. Followed by the addition of barium hydroxide octahydrate, 1.104 g (3.5 mmol) until forming a homogeneous solution (10 min). Then, 6 mL of triethylene glycol (TEG, capping ligand) and 1 mL of NH3 30% v/v solution (or instead the corresponding mmol of water detailed later in this work) were added under continuous stirring at room temperature resulting in a milky solution (5 min). The final suspension was transferred into a steel autoclave equipped with a Teflon-line and heated up at 180 °C under constant stirring for 1 hour and slowly cooled down to room temperature. The resulting orange-coloured suspension was washed as follows. Firstly 10 mL of ethyl acetate was added, sonicated during 10 min and then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. Then, supernatant was discarded and 10 mL of ethyl acetate:ethanol (4:1) were added, the mixture was sonicated until total dispersion of the pellet and later centrifuged at 10000 rpm for 10 min (this washing procedure was repeated twice). In the final step, the pellet was dispersed into absolute ethanol. To ensure a homogeneous size distribution of the NCs an extra centrifugation of 10 min at 7000 rpm was performed, discarding the pellet and keeping the supernatant, with yield loss of 15–20% from the initial.
000 rpm for 10 min. Then, supernatant was discarded and 10 mL of ethyl acetate:ethanol (4:1) were added, the mixture was sonicated until total dispersion of the pellet and later centrifuged at 10000 rpm for 10 min (this washing procedure was repeated twice). In the final step, the pellet was dispersed into absolute ethanol. To ensure a homogeneous size distribution of the NCs an extra centrifugation of 10 min at 7000 rpm was performed, discarding the pellet and keeping the supernatant, with yield loss of 15–20% from the initial.
Microwave synthesis was performed following the same procedure as solvothermal reaction but replacing 1 mL of NH3 30% v/v solution with 0.5 mL H2O (added in the same step). After the mixing, the resulting milky solution was transferred to the MW Teflon and heated with a ramp of 36 °C min−1 until 180 °C for the BaMO3 (M = Zr4+, and Hf4+) and 100 °C for the BaTiO3, holding the corresponding final temperature for 5 min Afterwards, the same cleaning process as the solvothermal procedure was performed and finally dispersed into absolute ethanol.
Characterization
X-ray powder diffraction (XRD) samples were recorded by a Phillips XPert diffractometer equipped with a two circle diffractometers and Cu tube. Transmission electron microscopy (TEM) micrographs were obtained on a 120 kV JEOL 1210 TEM, Transmission Electron Microscopy (HRTEM) micrographs obtained on a 200 kV JEOL 2011 TEM. Dynamic (SPECS GmbH, Berlin, Germany) in ultra-high vacuum conditions (base pressure 4 × 10−10 mbar) with a monochromatic aluminium Kα X-ray source (1486.74 eV). Light scattering (DLS) was done using a Zetasizer Nano. XPS measurements were performed with a Phoibos 150 analyser. Micro-Raman measurements were performed in backscattering geometry at room temperature using the 5145 Å line of argon-ion laser with a XploRA PLUS Raman spectrometer from Horiba, attached to an Olympus microscope and equipped with CCD detector. To perform microwave reaction, a microwave advanced flexible synthesis platform (flexiwave) from milestone was used.Results and discussion
The hybrid solvothermal method
According to our previous experience in metal oxide NC synthesis, we aimed to use a modified polyol route33 and make it compatible with the BaMO3 (M = Ti4+, Zr4+ and Hf4+) perovskite oxide NCs. The optimised procedure requires mixing 10 mL of ethanol and 6 mL of TEG to prevent the agglomeration or further precipitation.The versatility of this approach is reflected by obtaining aggregation-free monodisperse BaMO3 (M = Ti4+, Zr4+ or Hf4+) NCs (Fig. 1a–c, respectively) with a uniform size and shape, as corroborated by the histogram in Fig. S1.† The transmission electron microscopy (TEM) images and powder X-ray diffraction (XRD) reveals a square-like shape of the BaZrO3 with a ∼8 nm size while the two other perovskites (Ti4+ and Hf4+) showed spherical shape with sizes of 8 and 7 nm respectively. These results are in close concordance with those calculated using the Scherrer equation (Table 1). Moreover, the resulting perovskites show high crystallinity, without the need of thermal treatments after the synthesis (Fig. 1d) contrarily to standard sol–gel procedures.
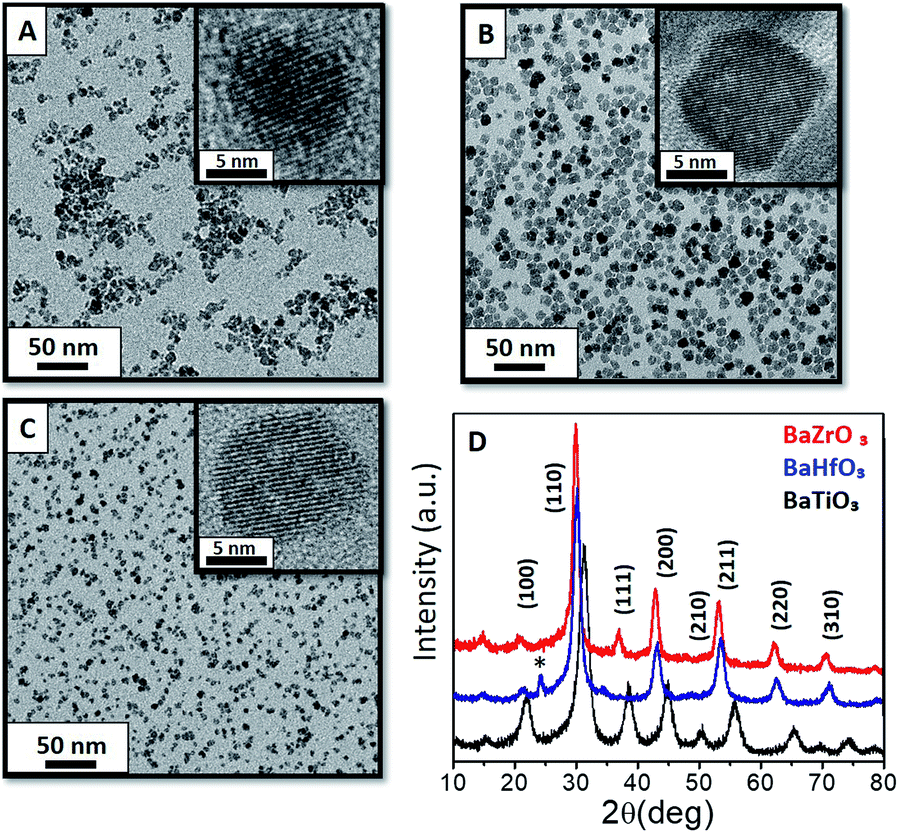 | ||
| Fig. 1 TEM images of BaTiO3 (a) BaZrO3 (b) and BaHfO3 (c) by using hybrid solvothermal reaction at 180 °C and 1 hour, showing their corresponding HRTEM. The index of Miller of the principal peaks by XRD (d) have been assigned into the diffractogram to confirm the cubic structure. On BaHfO3 appears an * BaCO3 peak suggesting a 5% of impurity on the pattern. | ||
| NCs | Scherrer size (nm) | TEM size(nm) | Morphology |
|---|---|---|---|
| BaTiO3 | 7.8 | 8.3 ± 1.4 | Spherical |
| BaZrO3 | 8.2 | 8.6 ± 1.5 | Square-like |
| BaHfO3 | 6.8 | 7.0 ± 1.2 | Spherical |
Many perovskite NCs from this family have been synthesised using stoichiometric amounts of Ba:M (1:1), even though the resulting NC systems are not homogeneous enough nor well dispersed.34–36 Nonetheless, it has been demonstrated that the addition/excess of hydroxide compounds could lead to an increment on homogeneity and NC sizes, as well as, a better defined NCs shape.37,38 We aimed to perform our hybrid solvothermal method in both conditions (i.e. stoichiometric and non-stochiometric Ba:Zr ratios) to study its effect to BaZrO3 NCs. As shown in Fig. S2a,† the use of stoichiometric Ba:Zr ratio produced a non-homogeneous NC dispersion with agglomeration observed via TEM and corroborated with DLS (i.e. NC size one magnitude order higher than non-stoichiometric ones). However, both NC dispersions show the same cubic phase in XRD (Fig. S2c†), making us to postulate a surface effect on the stabilization of the NCs allowed with an excess of Ba(OH)2.
As these NCs appear as good candidates for a wide range of material applications, assessing their stability and dispersion is mandatory. The DLS analysis revealed that the NCs remain well dispersed and stable in alcohols for 6 months, at concentration up to 80 mM for all cases. DLS analysis of the BaTiO3, BaZrO3 and BaHfO3 NCs in EtOH, show that they are stable in solution at least 6 months, exhibiting constant sizes of 7–10 nm, in close agreement with the results obtained by TEM and XRD techniques (Fig. 2).
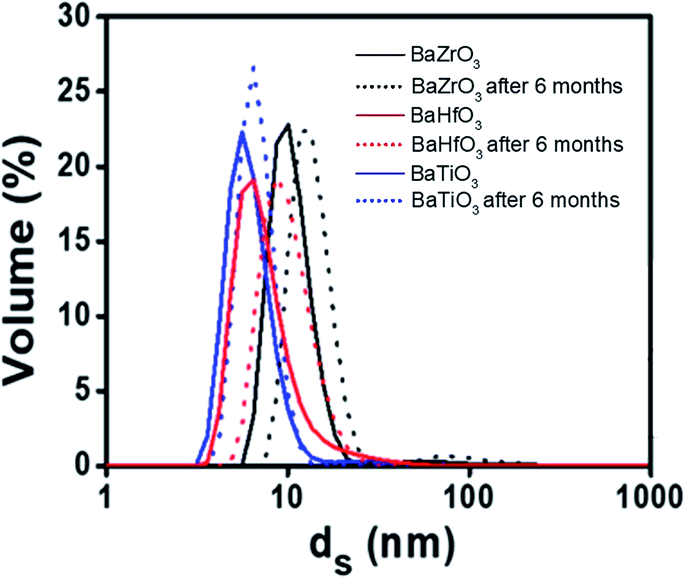 | ||
| Fig. 2 A stability study by using DLS for monitoring the stability of the nanocrystals (BaTiO3, BaZrO3 and BaHfO3 synthesised at 180 °C for 1 hour) from the moment of the synthesis to 6 month, all of them in ethanol media. | ||
To study the crystal structure stability, the Goldschmidt tolerance factor (t) equation (eqn (1))39,40 was reported as a powerful tool to predict the stability based only on the BaMO3 perovskite compounds. This equation has a direct dependence on the ionic radii of each element (BaMO3), where rB is the ionic radius of Ba, rM the ionic radius of (M = Ti4+, Zr4+ or Hf4+) and rO the ionic radius of oxygen. To corroborate the stability of a cubic perovskite by tolerance factor the range of values should be between 0.8 and 1.05. In the case of M = Zr4+ and Hf4+, a tolerance factor of t = 1 is obtained (Table 2), which is considered as the ideal cubic structure, where the ions are in a perfect cubic lattice.
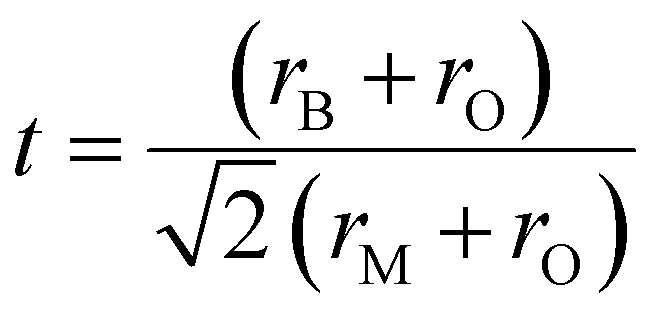 | (1) |
| Perovskites BMO3 | Ionic radius [Å] | Tolerance factor | |||
|---|---|---|---|---|---|
| Cation X2+ | Cation M4+ | ||||
| BaZrO3 | Ba2+ | 1.61 | Zr4+ | 0.72 | 1.00 |
| BaHfO3 | Ba2+ | 1.61 | Hf4+ | 0.71 | 1.00 |
| BaTiO3 | Ba2+ | 1.61 | Ti4+ | 0.61 | 1.06 |
Applying eqn (1) to the BaTiO3, a value of t > 1.05 is obtained due to a smaller size of Ti4+ cations, this might cause a distortion on the lattice structure which could entail a transition from cubic phase to tetragonal, therefore showing a ferroelectric behaviour.
To verify the possibility of tetragonal phase, XRD diffraction could show some limitation since both, cubic and tetragonal phase, may coexist together. This would be observed by a split peak over 45° in 2θ in the XRD as an evidence of tetragonality. Although Fig. 1d shows an individual broad peak, the assignation cannot be fully attributed to cubic phase as a consequence of working at such reduced sizes (8 nm), which limits the XRD analysis.
To complete the study on the phase transition a complementary analysis using Raman spectroscopy was performed (Fig. S3†). The inactive bands present on paraelectric cubic phase (176, 270 and 520 cm−1) become active on the transition to tetragonal phase13,14,42,43 at 306 cm−1 (sharp peak) and 713 cm−1 with well-defined and high intensity peaks are the footprints to determine the tetragonally. BaTiO3 NCs powder at room temperature was analysed, observing the absence of a sharp peak at 306 cm−1, and a subtle broad peak on 713 cm−1 without good definition. The information extracted from this data suggests a non-dominant tetragonal phase on the NCs which leads to cubic perovskite postulation.
Time and temperature screening
Special attention was paid to the tuning of the NC synthesis. In particular, temperature and time are claimed to have a direct effect on the size, shape and crystallinity of the final colloidal dispersions.44 By using solvothermal methodology, we are inducing a temperature and pressure (depending on the precursors and solvent) which will trigger the nucleation of the crystals and a posterior growth of these nuclei to mature crystals. For this purpose, we performed a study modifying the temperature and time to observe the correlation of these parameters with the final NCs (i.e., sizes, shape, crystallinity).Firstly, the temperature was decreased from 180 to 100 °C for 1 hour, as it is observed in Fig. S4.† The lack of crystallinity in the cases of BaZrO3 and BaHfO3 at 100 °C is shown in the XRD pattern (Fig. S5†) with a non-defined cubic shape, which entails not enough temperature to induce the final crystals. However, BaTiO3 shows a similar cubic pattern synthesised at both temperatures 100 and 180 °C (Fig. S4 and Table S1†). From this standpoint, Ti4+ precursor needs low temperatures (i.e., 100 °C) suggesting a different growth rate compared with the other two, BaZrO3 and BaHfO3 (i.e., 180 °C). This difference in growth conditions could be driven by a faster supersaturation point and different surface energy by using titanium instead of zirconium or hafnium precursors.44 Although each metal has a different chemical and physical proprieties, it should be noted the close similarities between hafnium and zirconium, showing almost identical radii compared with titanium.45,46
Then, we held the temperature and modified the reaction time from 1 to 24 hours (Fig. S6†). It is observed that 1 hour is more than enough to synthesise the NCs, reducing the current growth times for BaZrO3 and BaHfO3 on solvothermal procedure from hours to just 1 hour (Fig. S7†). XRD pattern (Fig. S7 and Table S2†) did not reveal any evidence of difference on crystallinity or size (i.e., via Scherrer equation) between the different reaction times. Consequently, it seems that the nucleation rate of the NCs is almost instantaneous once they are subjected to an adequate combination of pressure and temperature in the autoclave, without changing the morphology of the NCs. Therefore, 1 hour is enough for the NC formation, and keeping the reaction 24 hours at the same temperature did not affect the agglomeration of the NCs and even refute the possibility of the NCs to suffer grain growth caused by an Ostwald ripening.
It is well known that microwave is claimed to reduce reaction time in the synthesis of oxide NCs compared with the solvothermal ones.16,24 However, in our case we were able to achieve shorter times in the solvothermal reactions without the need of microwave procedure. Even still, the reaction was checked by microwave methodology (Fig. S8 and Table S3†) at 180 °C (BaZrO3 and BaHfO3) and 100 °C (BaTiO3) for 5 minutes, giving comparable NCs as in the autoclave. Moreover, a microwave profile of BaZrO3 has been exposed in Fig. S8b,† the graph shows the variation of pressure and temperature as functions of time, showing a pressure incrementation over 15 bars and a reduction in reaction time from 1 hour in solvothermal route to 5 minutes by applying a microwave route. In consequence, both heating procedures are compatible with the hybrid solvothermal method without significant difference in crystallinity, homogeneity or sizes. These results seem to evidence that the solvothermal reaction is only required to raise the system to a desired temperature and pressure for minutes to achieve the final crystallisation, leaving the possibility to govern size and agglomeration parameters by the previous aqueous sol–gel step, thus a kinetic control over the NCs.
Tuning the NC size
Using the standard method for the synthesis of BaZrO3, 1 mL of aqueous NH3 at 30% v/v is necessary to obtain the final NCs solution. The role of ammonia in the reaction was not clear, due to its ability to act as a base or as a cationic stabiliser on the NC surface (i.e., as ammonium cation stabilizing the system).47 In order to understand the role of the ammonia and its influence in the final nanoscale system, X-ray photoelectron spectroscopy (XPS) was performed. In Fig. S9† can be demonstrated the absence of nitrogen compounds on the NC surface, meaning that ammonia or ammonium cations are not playing a stabilization role.To evaluate the role of NH3, we compared the NCs obtained using 0.7 mL of water or 1 mL NH3 at 30% v/v (being 0.7 mL of water) as shown in Fig. S10.† Comparing TEM images and XRD results, we claim that there is not difference between the use of 1 mL ammonia 30% v/v or directly using its equivalent amount of water (0.7 mL of water). NCs form homogeneous dispersion as observed in the TEM images with square-like NCs, narrow size distributions and cubic phase (Fig. S10†). This evidence, in addition with the XPS analysis, suggest that ammonia did not play any additional role in our methodology, being just the amount of water coming from ammonia solution who govern the NC formation.
To demonstrate the specific role of the water, we used several quantities (from 0 mmol to 139 mmol) to test which is the pivotal role of the water in the system (Table 3). The most striking result emerging from this data is how an incrementation of water contain (0, 28, 55, 83, 111 and 139 mmol) produces a continuous increment in size from 4 to 31 nm (Fig. 3 and Table 3) comparing the sizes by TEM average and Scherrer equation.
| H2O amount | Scherrer size | TEM size |
|---|---|---|
| 0 mmol | — | — |
| 28 mmol | 7.4 nm | 7.0 ± 1.2 nm |
| 55 mmol | 11.8 nm | 11.9 ± 1.8 nm |
| 83 mmol | 14.1 nm | 14.7 ± 3.5 nm |
| 111 mmol | 25.0 nm | 25.8 ± 7.5 nm |
| 139 mmol | 29.0 nm | 32.6 ± 9.8 nm |
 | ||
| Fig. 3 (a) TEM images of BaZrO3 NCs using different additional water amounts during the synthetic process (from 0 to 139 mmol). (b) DLS measurements of the different BaZrO3 NCs at 28, 55, 83, 111 and 139 mmol of water (avoiding the ones at 0 mmol due to not fully formation NCs) and (c) the standard deviations of TEM histograms are showed to note the polydispersity when increasing water amounts. | ||
Firstly, a test without adding water in the media using the standard protocol as before (180 °C and 1 hour in autoclave) evidences initial seed formation of NCs (Fig. 3a). XRD pattern showed a high presence of BaCO3 as the main product (Fig. S12†), suggesting that the water amount in the Ba(OH)2·8H2O is not enough to initiate the nucleation process or the conditions are not still suitable to obtain desired final NCs.
Considering the other cases (i.e., from 28 to 139 mmol of water), DLS analysis corroborates by TEM sizes showing an increment of hydrodynamic radii while the amount of water increases, as well as, the stability of the NCs on the ethanol media (Fig. 3b). At this point, we realised that the effect of water is not only related to the increase of size but also in the polydispersity of final colloidal systems (Fig. 3c and S11†). Additionally, the standard deviation in TEM histograms increases with the size of the NCs. From a kinetic point of view, this effect could be explained by the hydrolysis step, if the nucleation process is increased without modifications on the thermodynamics, the system becomes polydisperse and with poor control on the stabilisation (i.e., we are increasing the amount of water with the same amount of capping agent, TEG). Although NC size is tuned with the addition of different amounts of water, other properties are nearly similar in terms of shape, homogeneity and crystallinity. Concerning their crystalline structure, all NCs present a cubic structure with a Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (Fig. S12†), in which Scherrer sizes match with the obtained via TEM and DLS (Table 3).
m space group (Fig. S12†), in which Scherrer sizes match with the obtained via TEM and DLS (Table 3).
Mechanistic insights in the hybrid solvothermal method
To shed more light on this growing process we need to focus on the mechanistic insights of the reaction. If the NC size relies on the amount of water, the hydrolysis step should be the key factor in the control of the NC size, and hence, the behaviour of the metal cation against hydrolysis. Our starting precursors are alkoxides, and even though their chemical reactivity has been widely studied,48,49 there is still noted a controversial aspect for knowing about the full mechanism reaction.23,29As it is well known, the strategy of sol–gel is based on the hydrolysis and polycondensation steps (eqn (3)–(5)) and it has been extensible as the main step to synthesise oxide nanoparticles.21,23,50,51 Herein by following this mechanism a sol–gel process is shown with a chemical reactivity tailored by the manipulation of the water amount. In an ethanol media, the stabilization and homogenisation of the alkoxide in the solvent is considered the first step to form the intermediate M(OBu)4−x(OEt)x (eqn (2)). Alkoxides are extremely sensitive to water, so the addition of Ba(OH)2 octahydrate plays a double role; (i) the addition of the binary cation for the perovskite structure and (ii) the source of –OH groups and water which will start the hydrolysis reaction (eqn (3)) and the formation of intermediate species based on a metal hydroxide bond M(OH)x(OBu)n−x. Polycondensations are promoted as it is described in eqn (4) and (5), forming a M–O–M framework, being a critical point for the further formation of final BaMO3. The increase of –M–OH species in our reaction media is controlled with the addition of water, allowing the formation of large –M–O–M– chains (polycondensation), producing bigger NCs.
| M(OBu)4 + xEtOH → M(OBu)4−x(OEt)x + xBuOH | (2) |
| M(OBu)n + xH2O → M(OH)x(OBu)n−x + xBuOH | (3) |
| –M–OH + H–O–M → –M–O–M– + H2O | (4) |
| –M–OH + Bu–O–M → –M–O–M– + BuOH | (5) |
Simultaneously, this experiment was carried out with the other two different metal precursors Hf4+ and Ti4+ alkoxides, revealing important insights into the growth process of the NCs. The behaviour of BaHfO3 is similar to BaZrO3, the addition of water (28 and 111 mmols) increases the NC size from 4.6 ± 0.7 nm to 14.8 ± 3.4 nm respectively (Fig. 4a and b). However, the findings with BaTiO3 do not support the previous research, showing a quite different behaviour not being the amount of water the one who governs the NCs tuning sizes. After adding 28 and 111 mmols of water, BaTiO3 NCs do not show significant differences in size from 8.7 ± 2.6 and 9.2 ± 2.1 nm, respectively (Fig. 4c and d). The experimental data taken from XRD (Fig. S13†) corroborates the TEM results.
 | ||
| Fig. 4 Image by TEM of the synthesis of BaHfO3 and BaTiO3. In the case of (a) and (b) by adding 28 mmol and 111 mmol of water for synthesized BaHfO3. On the case of (c) and (d) by adding 28 and 111 mmol of water for synthesized BaTiO3. | ||
A possible explanation for the difference of the titanium, can start from the behaviour of the alkoxide precursors on solution, as well as their tendency to polymerise. The formation of coordination polymers [M(OR)x]y (where “y” is the degree of polymerisation) determines the final crystal formation. The degree of polymerisation has a direct dependency from the physical properties of the metals (i) the radius of the metal (Ti < Zr, Hf), (ii) the coordination number and (iii) the size of group R on the alkoxide. In our case, it seems that Hf and Zr are favourable to NC growth being the hydrolysis process that triggers the formation of –M–O–M– chains. While titanium cations exposed on the surface are less favourable to the NC growth, resulting on invariable NC size after water additions. Here, we need to consider the analogy between coordination chemistry and NC surface as the well-known covalent bond classification (CBC).52,53 CBC is mainly based in the direct comparison between coordination chemistry and the surface of NCs, in which each metallic cation centre could be considered as a coordination complex. This hypothesis is supported by previous publications related to studies in coordination chemistry in which the authors found significant differences between the reactivity of titanium and zirconium alkoxides against hydrolysis and/or coordination.45,50,54,55 Consequently, by controlling the hydrolysis step, different sizes of BaZrO3 and BaHfO3 NCs are produced. However, BaTiO3 NCs show same sizes corroborating the different behaviour of these three elements from the same group of the periodic table.
Conclusions
In summary, we presented a dual-based synthetic approximation to obtain perovskite BaMO3 NCs using a hybrid methodology. This novel strategy combines both aqueous sol–gel route and solvothermal reaction in a two-step process. As far as we know, this approach allows for the first time the use of the same methodology to obtain BaTiO3, BaZrO3 and BaHfO3 NCs with high control of size, shape, reproducibility and stability.In addition, by using this approximation, we evidenced the crucial role of water in the hydrolytic step, arising as a key parameter to tune the sizes of the final NCs. This allowed us to tune BaZrO3 and BaHfO3 size from 4 to 40 nm, and 4 to 15 nm respectively without deficiency of crystallinity and shape-defined appearance. In the other case, BaTiO3 has a completely different behaviour, giving rise to constant 8–9 nm size NCs, independently of the water proportion.
Moreover, a complete study in a particular sized NCs show a stability of several months in polar solvents (i.e., ethanol), being remarkable candidates for a full range of applications without taking care of the solution storage. This new outcome will open an interesting window for tuning NC sizes by the implementation of the described methodology and open a wide range of further applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Spanish Ministry of Economy and Competitiveness through the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2015-0496), CONSOLIDER Excellence Network (MAT2015-68994-REDC), COACHSUPENERGY project (MAT2014-51778-C2-1-R, cofinanced by the European Regional Development Fund) and SUPERINKS project (RTC-2015-3640-3, cofinanced by the European Regional Development Fund). We also thank support from the European Union for EUROTAPES project (FP7-NMP-Large-2011-280432) and ULTRASUPERTAPE project (H2020 ERC-2014-ADG-669504) and from the Catalan Government with 2014-SGR-753 and Xarmae. The authors acknowledge the technical support of Servei de Microscópia, and Servei de Difracció de Raigs- X, all at the UAB, and Dr Judith Oró (Microscope Service) at the ICMAB-CSIC, Dr Guillaume Sauthier for his support in XPS analysis. NCG acknowledge the PIF predoctoral fellowship from the Universitat Autònoma de Barcelona.References
- M. Segev-Bar and H. Haick, ACS Nano, 2013, 7, 8366–8378 CrossRef CAS PubMed.
- L. E. Woodard, C. L. Dennis, J. A. Borchers, A. Attaluri, E. Velarde, C. Dawidczyk, P. C. Searson, M. G. Pomper and R. Ivkov, Sci. Rep., 2018, 8, 1–13 CrossRef CAS PubMed.
- M. Mahmoudi, M. Yu, V. Serpooshan, J. C. Wu, R. Langer, R. T. Lee, J. M. Karp and O. C. Farokhzad, Nat. Nanotechnol., 2017, 12, 845–855 CrossRef CAS PubMed.
- S. A. Bonke, A. M. Bond, L. Spiccia and A. N. Simonov, J. Am. Chem. Soc., 2016, 138, 16095–16104 CrossRef CAS PubMed.
- Q. Jia, S. Ghoshal, J. Li, W. Liang, G. Meng, H. Che, S. Zhang, Z. F. Ma and S. Mukerjee, J. Am. Chem. Soc., 2017, 139, 7893–7903 CrossRef CAS PubMed.
- A. Noori, M. F. El-Kady, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, Chem. Soc. Rev., 2019, 48, 1272–1341 RSC.
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed.
- M. E. Franke, T. J. Koplin and U. Simon, Small, 2006, 2, 36–50 CrossRef CAS PubMed.
- S. Han, X. Liu, Y. Liu, Z. Xu, Y. Li, M. Hong, J. Luo and Z. Sun, J. Am. Chem. Soc., 2019, 141, 12470–12474 CrossRef CAS PubMed.
- B.-W. Li, M. Osada, Y.-H. Kim, Y. Ebina, K. Akatsuka and T. Sasaki, J. Am. Chem. Soc., 2017, 139, 10868–10874 CrossRef CAS PubMed.
- T. Li, Y. Pan, Z. Wang, Y. Xia, Y. Chen and W. Huang, J. Mater. Chem. A, 2017, 5, 12602–12652 RSC.
- M. Niederberger, G. Garnweitner, N. Pinna and M. Antonietti, J. Am. Chem. Soc., 2004, 126, 9120–9126 CrossRef CAS PubMed.
- M. B. Smith, K. Page, T. Siegrist and A. Et, J. Am. Chem. Soc., 2008, 130, 6955–6963 CrossRef CAS PubMed.
- D. Caruntu, T. Rostamzadeh, T. Costanzo, S. Salemizadeh Parizi and G. Caruntu, Nanoscale, 2015, 7, 12955–12969 RSC.
- S. O'Brien, L. Brus and C. B. Murray, J. Am. Chem. Soc., 2001, 123, 12085–12086 CrossRef PubMed.
- K. De Keukeleere, J. Feys, M. Meire, J. De Roo, K. De Buysser, P. Lommens and I. Van Driessche, J. Nanopart. Res., 2013, 15, 2074 CrossRef.
- J. De Roo, K. De Keukeleere, J. Feys, P. Lommens, Z. Hens and I. Van Driessche, J. Nanopart. Res., 2013, 15, 1778 CrossRef.
- Z. Lu, Y. Tang, L. Chen and Y. Li, J. Cryst. Growth, 2004, 266, 539–544 CrossRef CAS.
- J. Liu and J. C. Berg, J. Mater. Chem., 2007, 17, 4430–4435 RSC.
- C. N. Chervin, B. J. Clapsaddle, H. W. Chiu, A. E. Gash, J. H. Satcher and S. M. Kauzlarich, Chem. Mater., 2006, 18, 1928–1937 CrossRef CAS.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- D. Gömpel, M. N. Tahir, M. Panthöfer, E. Mugnaioli, R. Brandscheid, U. Kolb and W. Tremel, J. Mater. Chem. A, 2014, 2, 8033–8040 RSC.
- M. Niederberger and G. Garnweitner, Chem. - Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358–1374 RSC.
- I. Bilecka, L. Luo, I. Djerdj, M. D. Rossell, M. Jagodič, Z. Jagličić, Y. Masubuchi, S. Kikkawa and M. Niederberger, J. Phys. Chem. C, 2011, 115, 1484–1495 CrossRef CAS.
- K. De Keukeleere, J. De Roo, P. Lommens, J. C. Martins, P. Van Der Voort and I. Van Driessche, Inorg. Chem., 2015, 54, 3469–3476 CrossRef CAS PubMed.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS PubMed.
- N. Pinna, J. Mater. Chem., 2007, 17, 2769–2774 RSC.
- N. Pinna, G. Garnweitner, M. Antonietti and M. Niederberger, J. Am. Chem. Soc., 2005, 127, 5608–5612 CrossRef CAS PubMed.
- Z. Li, M. Coll, B. Mundet, N. Chamorro, F. Vallès, A. Palau, J. Gazquez, S. Ricart, T. Puig and X. Obradors, Sci. Rep., 2019, 9, 1–14 CrossRef PubMed.
- X. Obradors, T. Puig, Z. Li, C. Pop, B. Mundet, N. Chamorro, F. Vallés, M. Coll, S. Ricart, B. Vallejo, F. Pino, A. Palau, J. Gázquez, J. Ros and A. Usoskin, Supercond. Sci. Technol., 2018, 31, 044001 CrossRef.
- L. Soler, J. Jareño, J. Banchewski, S. Rasi, N. Chamorro, R. Guzman, R. Yáñez, C. Mocuta, S. Ricart, J. Farjas, P. Roura-Grabulosa, X. Obradors and T. Puig, Nat. Commun., 2020, 11, 344 CrossRef CAS PubMed.
- E. Solano, L. Perez-Mirabet, F. Martinez-Julian, R. Guzmán, J. Arbiol, T. Puig, X. Obradors, R. Yañez, A. Pomar, S. Ricart and J. Ros, J. Nanopart. Res., 2012, 14, 1034 CrossRef.
- M. Han, Y. Rong, Q. Li, X. Xing and L. Kang, CrystEngComm, 2015, 17, 1944–1951 RSC.
- T. Charoonsuk, W. Vittayakorn, N. Vittayakorn, P. Seeharaj and S. Maensiri, Ceram. Int., 2015, 41, S87–S94 CrossRef CAS.
- M. L. Moreira, J. Andrés, V. R. Mastelaro, J. A. Varela and E. Longo, CrystEngComm, 2011, 13, 5818–5824 RSC.
- H. W. Lee, S. Moon, C. H. Choi and D. K. Kim, J. Am. Ceram. Soc., 2012, 95, 2429–2434 CrossRef CAS.
- K. Kanie, Y. Seino, M. Matsubara, M. Nakaya and A. Muramatsu, New J. Chem., 2014, 38, 3548–3555 RSC.
- R. Mouta, R. X. Silva and C. W. A. Paschoal, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 439–445 CrossRef CAS PubMed.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, 1–10 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. A, 1976, 32, 751–767 CrossRef.
- H. Hayashi, T. Nakamura and T. Ebina, J. Phys. Chem. Solids, 2013, 74, 957–962 CrossRef CAS.
- G. Yang, Z. Yue, T. Sun, J. Zhao, Z. Yang and L. Li, Appl. Phys. A, 2008, 91, 119–125 CrossRef CAS.
- J. Van Embden, A. S. R. Chesman and J. J. Jasieniak, Chem. Mater., 2015, 27, 2246–2285 CrossRef CAS.
- U. Schubert and N. Hüsing, Synthesis of Inorganic Materials, VCH-Wiley Verlag GmbH, Weinheim, 3rd edn, 2012 Search PubMed.
- N. Y. Turova, E. P. Turevskaya, V. G. Kessler and M. I. Yanovskaya, The chemistry of Metal Alkoxide, Kluwe Academi Publisher, 2002 Search PubMed.
- J. Martínez-Esaín, J. Faraudo, T. Puig, X. Obradors, J. Ros, S. Ricart and R. Yáñez, J. Am. Chem. Soc., 2018, 140, 2127–2134 CrossRef PubMed.
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef.
- D. C. Bradley, Chem. Rev., 2002, 89, 1317–1322 CrossRef.
- N. Y. Turova, E. P. Turevskaya, V. G. Kessler and M. I. Yanovskaya, Hydrolysis of Metal Alkoxides and Synthesis of Simple Oxides by The Sol-Gel Method, in The Chemistry of Metal Alkoxides, Springer, Boston, MA, 2002 Search PubMed.
- R. R. J. P. Corriu and D. Leclercq, Angew. Chem., Int. Ed. Engl., 1996, 35, 1420–1436 CrossRef.
- J. Owen, Science, 2015, 347, 615–616 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 364 CrossRef CAS PubMed.
- U. Schubert, E. Arpac, W. Glaubitt, A. Helmerich and C. Chau, Chem. Mater., 1992, 4, 291–295 CrossRef CAS.
- A. Kumar, S. De, A. G. Samuelson and E. D. Jemmis, Organometallics, 2008, 27, 955–960 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03861j |
| ‡ Present address: Catalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and The Barcelona Institute of Science and Technology, Campus UAB, Bellaterra, 08193 Barcelona, Spain. |
| This journal is © The Royal Society of Chemistry 2020 |