
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural properties of the chelating agent 2,6-bis(1-(3-hydroxypropyl)-1,2,3-triazol-4-yl)pyridine: a combined XRD and DFT structural study†
Greta Colombo Dugoni a,
Alberto Baggiolia,
Antonino Famularia,
Alessandro Sacchettia,
Javier Martí-Rujas*ab,
Mario Marianic,
Elena Maceratac,
Eros Mossinic and
Andrea Melea
a,
Alberto Baggiolia,
Antonino Famularia,
Alessandro Sacchettia,
Javier Martí-Rujas*ab,
Mario Marianic,
Elena Maceratac,
Eros Mossinic and
Andrea Melea
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza L. da Vinci 32, 20133 Milan, Italy. E-mail: javier.marti@polimi.it
bCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, Via Pascoli 70/3, 20133 Milano, Italy
cDepartment of Energy, Nuclear Engineering Division (CeSNEF), Politecnico di Milano, Piazza L. da Vinci 32, 20133 Milan, Italy
First published on 22nd May 2020
Abstract
The conformational isomerism of the chelating agent 2,6-bis(1-(3-hydroxypropyl)-1,2,3-triazol-4-yl)pyridine (PTD), exploited in fuel reprocessing in spent nuclear waste, has been studied by single crystal X-ray diffraction analysis in combination with an extensive DFT conformational investigation. In the solid-state, the elucidated crystal structure (i.e., not yet published) shows that by thermal treatment (DSC) no other phases are observed upon crystallization from the melt, indicating that the conformation observed by X-ray data is rather stable. Mapping of intermolecular and intramolecular noncovalent interactions has been used to elucidate the unusual arrangement of the asymmetric unit. Considerations relating to the stability of different conformational isomers in aqueous and non-aqueous solutions are also presented. The accurate structural description reported here might open various research topics such as the potential of PTD to act as an outer sphere ligand in the formation of second sphere coordination complexes and their interconversion by mechanochemical means.
Introduction
In recent decades, great effort has been devoted to the reduction of the environmental impact of spent fuel produced by nuclear power plants, both in terms of quantity and radiotoxicity.1–5 In this regard, combination of metal ions and organic ligands using crystal engineering methods and supramolecular principles is among the most promising approaches towards selective separation of metals.6–10 These rely on removal of metal ions such as lanthanides and actinides from aqueous or non-aqueous solutions via the formation of metal organic complexes by addition of chelating agents (i.e. extractants). A large number of extractants have been proposed and tested in the past decades, with the ultimate goal of selectively extracting actinide ions from the mixed actinide/lanthanide-containing aqueous nitric acid solutions produced as high level waste.11,12 Such ligands must meet several criteria in order to qualify as effective extractants for nuclear waste reprocessing, including selectivity towards actinides,13 chemical stability in strong acidic conditions, radiolytic stability,14 and fast (de)complexation kinetics.15In this regard, the flexible ligand 2,6-bis(1-(3-hydroxypropyl)-1,2,3-triazol-4-yl)pyridine (PyTri-Diol, hereafter PTD), proposed and tested by Macerata and co-workers16 (Scheme 1), is an appealing candidate. PTD also features sought-after characteristics which make it suitable for industrial application: water solubility and complete incinerability.17–19 In addition, PTD has been intensively studied as terdentate chelating agent for different applications such as catalysis or biomedical application20 both in supramolecular and coordination chemistry.21 From a fundamental point of view, the full three-dimensional characterization of PTD, despite its important role in complex formation, remains still unpublished. Moreover, studies regarding the solid-state structure of PTD are of fundamental importance to understand its chelating potential. Additionally, in view of its functional applications, not only in solution but also in the solid-state, PTD might be used in the formation of second sphere coordination adducts which can be further transformed in complexes via mechanochemical reactions (i.e., dehydrochlorination reactions).6,22–28 Thus, a full understanding of the structural aspects of PTD such as conformational behavior are necessary to fully exploit its coordination chemistry abilities.
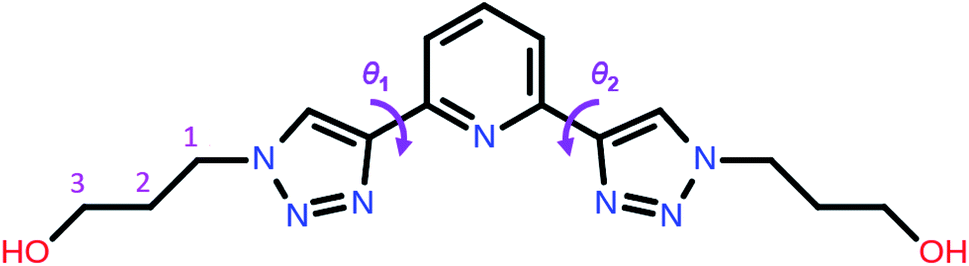 | ||
| Scheme 1 Molecular structure of 2,6-bis(1-(3-hydroxypropyl)-1,2,3-triazol-4-yl)pyridine (PTD) showing the torsion angles among the triazole and pyridine rings. | ||
Here, we report for the first time, and thoroughly discuss, the solid-state structure of PTD by means of single crystal X-ray diffraction and dispersion-corrected DFT calculations. A comprehensive analysis of weak electrostatic interactions OH⋯X, π⋯π, CH⋯π, and CH⋯X (X = O, N) among PTD molecules within the experimental crystal lattice is presented. Differential scanning calorimetry (DSC) shows that no other phases are obtained at higher temperatures, and that upon melt PTD crystallizes in the same phase, indicating the rather stable periodic solid-state conformation of PTD. Simplified structural models were also exploited to assess conformational isomerism of both the bis(triazolyl)pyridine core and the (hydroxypropyl)triazole subunit, with interesting results in the context of PTD isomerism in aqueous solution in views of its applications as chelating agent.
Results and discussion
Single crystal X-ray diffraction of PTD
PTD was synthesized following the procedure reported by Gelinas et al.29 A suitable specimen for single crystal X-ray diffraction (SC-XRD) experiments was obtained by slow evaporation of a solution of PTD in chloroform. Room-temperature SC-XRD measurements reveal that PTD crystallizes in an orthorhombic cell in the Pbca space group with lattice parameters: a = 19.985(3) Å, b = 7.6989(10) Å, c = 21.093(4) Å; V = 3245.42 Å3.The simulated powder XRD pattern is comparable to that of the bulk powder sample, suggesting the same crystal structure was observed in both cases (Fig. 1). Differential scanning calorimetry showed that PTD melts at ca. 111 °C while further heating results in the decomposition of the ligand (see ESI†). Upon cooling to room temperature and reheating, no new solid phase was observed (i.e., polymorph), as the melting point of the re-crystallized sample was the same as that measured during the first heating cycle.
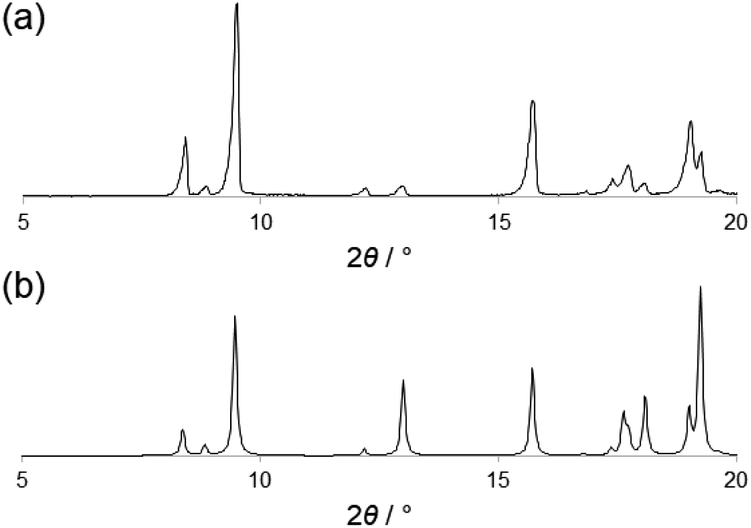 | ||
| Fig. 1 Experimental (a) and simulated (b) powder XRD pattern of PTD. | ||
PTD crystal structure description: a combined SC-XRD and DFT analysis
The asymmetric unit consists of one molecule of PTD (Fig. 2). Despite two identical side chains in 2 and 6 positions on pyridine, the PTD unit in the bulk crystal is highly asymmetric. The two triazole rings are found in opposite arrangements, one in syn and one in anti to pyridine's heteroatom. Hydroxypropyl chains also feature different conformations. As such, PTD units are not neatly aligned along crystallographic directions, and there is no segregation between aromatic and aliphatic moieties at the nanoscale.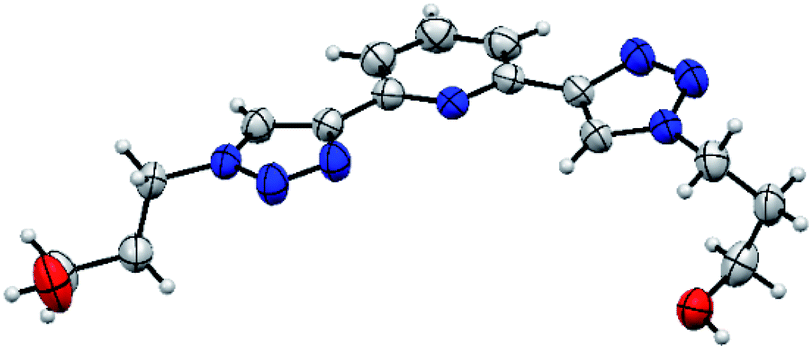 | ||
| Fig. 2 ORTEP representation of the SC-XRD structure of PTD. | ||
This combination of syn- and anti-arranged triazole rings is rather unusual, at least based on a survey of the Cambridge Structural Database. In fact, out of 30 deposited crystal structures (excluding cyclic and cationic species, see ESI† for details) comprising a 2,6-bis(1,2,3-triazol-4-yl)pyridine subunit whose N centres are not partaking in metal ion complexation, only one (refcode PEQGEA)30 features a similar configuration. It should be noted, however, that, in a few entries, an electron-rich moiety interacting with the C–H groups of both triazole rings likely drives the common anti/anti arrangement (e.g. refcodes IPEZAF,31 LITJUU,32 XUYBAV33).
In order to better understand both crystal packing and the intermolecular interactions involved into it, the noncovalent interactions (NCI) descriptor by Johnson and co-workers34 was used. This allows the visualization of noncovalent interactions as small objects separating interacting moieties and provides a semi-quantitative estimate of the strength of the corresponding interaction by highlighting specific features of these objects' surface. Pairs of neighboring PTD molecules were thus extracted from the bulk of the crystal structure, their charge density simulated by DFT means at fixed experimental geometry, and finally used to carry out NCI calculations. Overall, 54 interactions of widely different magnitude and spatial extent were detected, only half of which unique due to symmetry. Technical details on the NCI descriptor, as well as a full evaluation of all these interactions, are provided in the ESI.† Meanwhile, Fig. 3 shows a map of the interactions of a reference PTD unit with its neighbour images. Each irregular object depicted around the reference molecule represents one interaction or a group of interactions. Rather than the usual NCI colour-mapping showing interaction magnitude, crystal packing information were colour-coded instead, as to distinguish between different kinds of interactions such as strong OH⋯X hydrogen bonds (purple), π⋯π (orange), CH⋯π (cyan), or weak CH⋯X hydrogen bonds (green), with X = N, O.
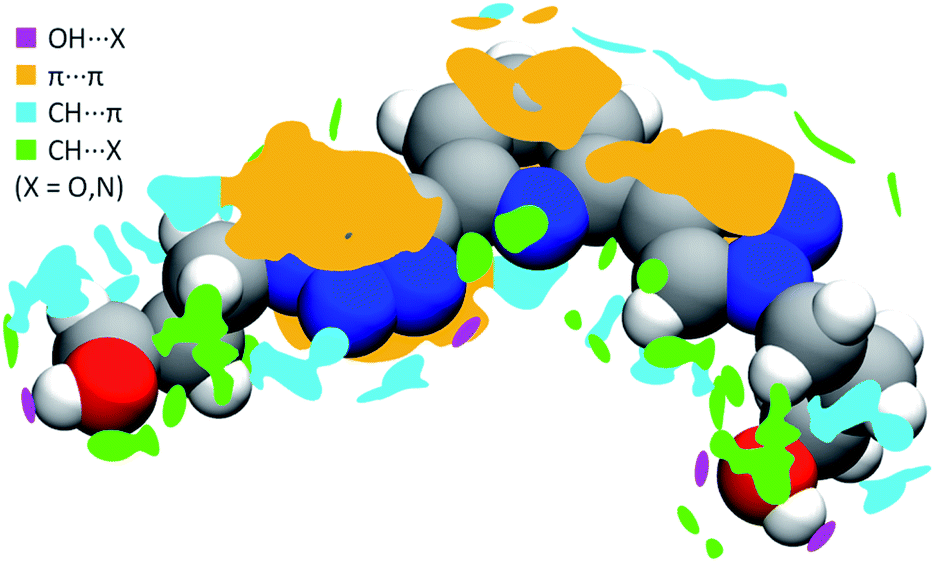 | ||
| Fig. 3 Map of the intermolecular interactions of one reference PTD molecule with its neighbours. Colour-coding was used to distinguish ‘islands’ representing strong hydrogen bonds (purple), π⋯π (orange), CH⋯π interactions (cyan), and weak CH⋯O and CH⋯N interactions (green). | ||
PTD molecules within the bulk crystal are organized in linear stacks of PTD dimers, aligned along the b direction as depicted in Fig. 4a and b. The tilt angle between units in a dimer favours CH⋯π interactions over π⋯π ones. This accounts for most of the cyan islands in Fig. 3, and for orange islands around the left-hand triazole ring. Along a stack, adjacent units are also connected via a rather strong hydrogen bond through the terminal hydroxyl groups (purple disks in Fig. 3 and 4c; dOH⋯O = 2.717 Å; 175.43°). The hydroxypropyl chain extending outwards (left-hand chain in Fig. 2 and 3) acts as a hydrogen bond donor, while the folded one (right-hand chain in Fig. 2 and 3) reaches out to the following unit in the stack to accept the hydrogen.
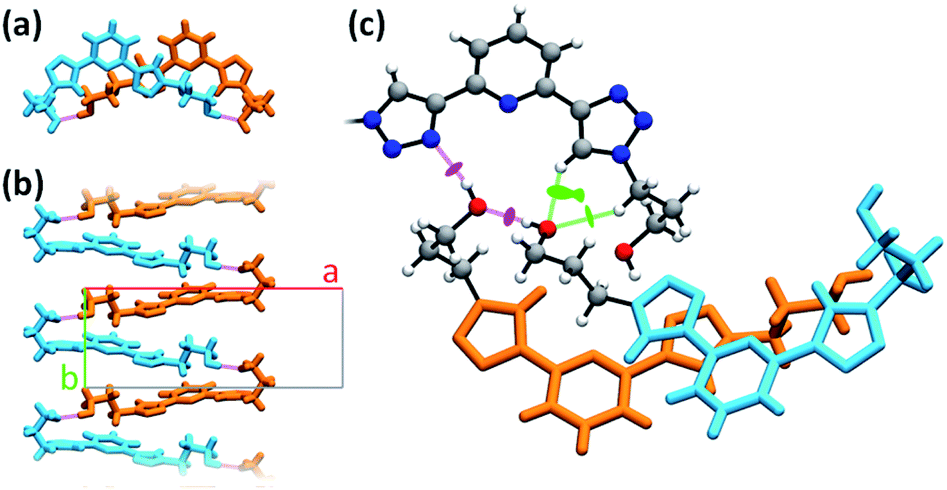 | ||
| Fig. 4 Different details of PTD crystal structure. Bright colours are used as visual aid to distinguish between superimposed molecules. Chemical detail is retained where necessary. Dashed purple lines are used to represent hydrogen bonds. (a) View of a stacked dimer along the b axis. (b) View of a stack of hydrogen-bonded PTD units along the c axis. (c) View of the linking region between neighbouring stacks (see caption of Fig. 2 for color-coding). | ||
Inter-stack interactions along the a crystallographic direction are rather poor due to aliphatic spacers. Instead, stacks are interlinked in the c direction via additional hydrogen bonds as it is depicted in Fig. 4c and 5. While already engaging in the intra-stack OH⋯O interaction described previously, both hydroxyl groups interact with a PTD unit belonging to a different stack opposite to the first in c direction, and slipped by half a unit cell along the a direction. This configuration allows one OH group to engage in an additional hydrogen bond with a triazole ring acting as hydrogen acceptor (purple disks in Fig. 3 and 4c; dOH⋯N = 2.803 Å; 166.09°), and another to engage in weaker CH⋯O interactions (green islands around hydroxyl groups in Fig. 3 and 4c).
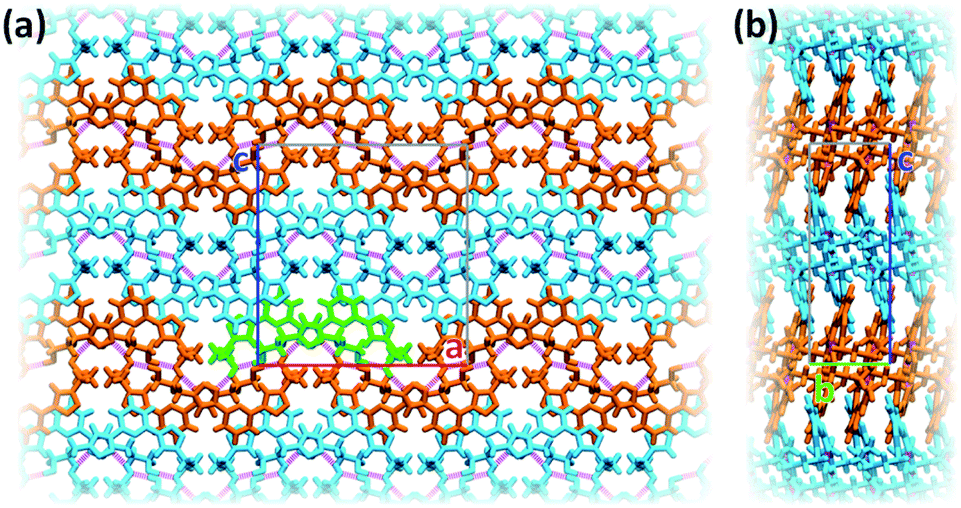 | ||
| Fig. 5 Representation of interlinked stacks within ac (a) and bc (b) crystallographic planes. Bright colours are used as visual aid to distinguish between adjacent lamellae. Dashed purple lines are used to represent strong hydrogen bonds. For clarity, one PTD dimer is highlighted in green. | ||
Fig. 4c highlights the N⋯HO⋯HO⋯HC chain of noncovalent interactions which ultimately drives the uncommon syn/anti configuration of triazole rings observed for PTD in the solid state. A similar network of hydrogen bonds, N⋯HOC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H(triazole), drives the same syn/anti arrangement in the PTD congener featured in CSD entry PEQGEA, with a carboxylic group forming a bridge between the two triazole units. These two 2,6-bis(1,2,3-triazol-4-yl)pyridine derivatives' crystal structures, however, are not the only ones containing electrophilic groups such as acidic or alcoholic protons. Among the 30 crystal structures mentioned earlier, a few include either CHCl3 solvent molecules, –NH2 or –C(O)OH groups, or iodine substituents, which qualify as potential halogen-bond donors. However, what distinguishes these electrophilic groups from those in PTD and PEQGEA derivatives is the inability to simultaneously interact with both triazole rings of another molecule as both electron donor and electron acceptor, either due to geometrical/steric constraints, or due to the availability of stronger or more convenient bonding motifs discouraging such bridged arrangements.
O⋯H(triazole), drives the same syn/anti arrangement in the PTD congener featured in CSD entry PEQGEA, with a carboxylic group forming a bridge between the two triazole units. These two 2,6-bis(1,2,3-triazol-4-yl)pyridine derivatives' crystal structures, however, are not the only ones containing electrophilic groups such as acidic or alcoholic protons. Among the 30 crystal structures mentioned earlier, a few include either CHCl3 solvent molecules, –NH2 or –C(O)OH groups, or iodine substituents, which qualify as potential halogen-bond donors. However, what distinguishes these electrophilic groups from those in PTD and PEQGEA derivatives is the inability to simultaneously interact with both triazole rings of another molecule as both electron donor and electron acceptor, either due to geometrical/steric constraints, or due to the availability of stronger or more convenient bonding motifs discouraging such bridged arrangements.
Interlinked stacks form rather rigid lamellae extending in 2D, parallel to the ac plane. Fig. 5 shows packed lamellae as seen along a and b crystallographic axes. Inter-lamellae interactions consist of slipped π⋯π interactions and extended CH⋯π networks, as PTD spacing within a stack allows for a certain degree of interdigitation, most evident in Fig. 5b (orange islands on pyridine and right-hand triazole and surrounding green/cyan islands in Fig. 3).
PTD conformational study
Now we turn to the study of PTD in its free form, either in gas phase or solvated state. PTD is in fact soluble in water, such that most of its technological applications take place in aqueous solution. Due to the inherent flexibility of its side chains, however, a full conformational study of PTD could potentially lead to a few hundreds of stable isomers. As such, a piece-wise approach was adopted instead.The extent of PTD's conformational space was assessed at first via a relaxed scan of the torsional potential of triazole rings about bridging bonds connecting to pyridine, in the gas phase. For this purpose, a simplified model congener of PTD was used, obtained by replacing side chains on triazole units with methyl groups. This is a reasonable approximation because of the modest length of the aliphatic –(CH2)3– side chain, which negates any hydroxyl/pyridine interaction and hampers hydroxyl/triazole ones significantly. Degrees of freedom θ1 and θ2 were defined as N(tr)–C(tr)–C(py)–N(py) dihedral angles of the two non-equivalent triazole units, with a 0° reference chosen to correspond to syn configuration (see Scheme 1). The resulting potential energy surface (PES) is reported in Fig. 6.
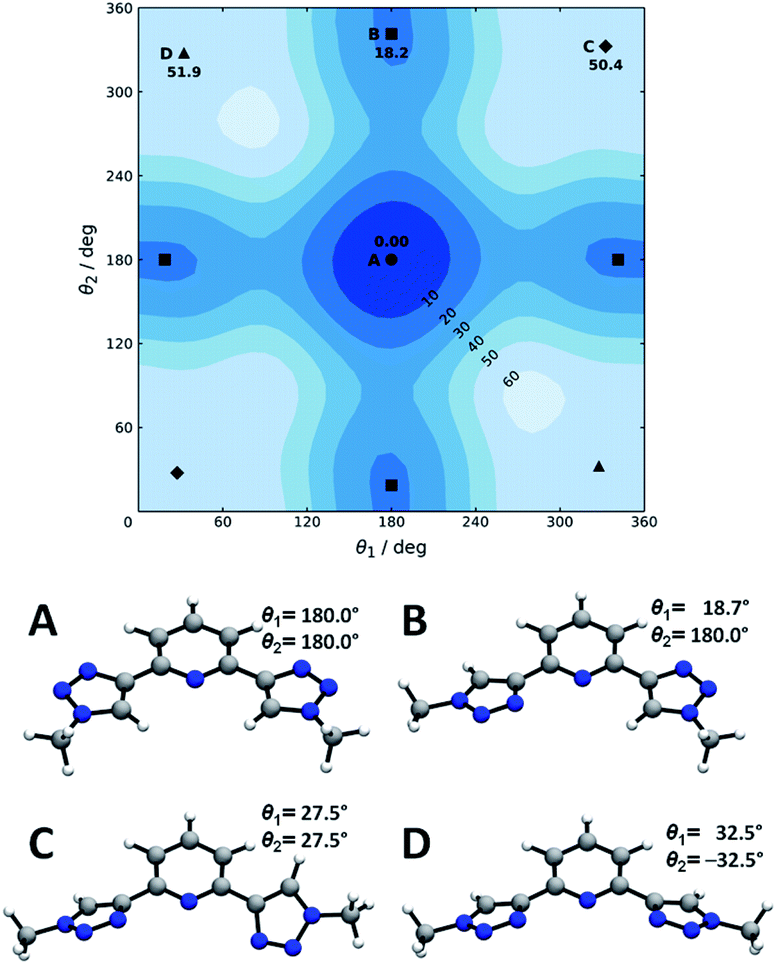 | ||
| Fig. 6 Torsional potential energy surface for a PTD congener with truncated side chains. Conformers A through D are signaled by circles, squares, diamonds, and triangles, respectively. Labels and corresponding relative electronic energies (kJ mol−1) are reported only once per symmetry-related minimum. Optimized geometries and dihedral angles are reported for quick reference. | ||
Four conformational isomers were obtained, hereafter labelled A through D from the most to the least stable, all featuring θ1 and θ2 angles in the neighborhood of either 0° or 180°. Regarding Fig. 6, conformer A is characterized by triazole units in anti arrangement to pyridine, conformer B by one triazole in anti and one in syn arrangements, and conformers C and D by both triazole units in syn position. As it is immediately evident upon comparison between Fig. 2 and 6, the geometry of PTD in the single crystal relates most closely to that of conformer B rather than to that of the more stable conformer A. This is a rather common occurrence, since higher energy conformers can oftentimes crystallize more readily than absolute minimum energy ones due to the ability to strike a better balance between intramolecular and intermolecular interactions.35 Furthermore, typical arrangements relevant to metal ion complexation, i.e. with θ1 and θ2 both in syn position (C and D), appear to be inaccessibly high in energy, at least in the gas phase. However, solvation effects in practical applications are likely to mitigate the issue (vide infra).
The soundness of the approximation introduced by neglecting hydroxypropyl side chains was assessed next by a separate conformational investigation on an isolated 1-(3-hydroxypropyl)-1,2,3-triazole unit. All permutations of anti (a, 180°), gauche+ (g+, 60°), and gauche− (g−, −60°) arrangements of the side chain were explored, starting from the same orientation of the triazole ring, and relaxing the dihedral angle between the ring and the chain. The resulting 23 unique conformational isomers are listed in Table 1. Details on dihedral angles series, along with an account of intramolecular interactions within the NCI framework, are collected in the ESI.† It should be noted that each unique conformational isomer listed in Table 1 exists alongside another representing its mirror image. The propyl spacer is only long enough to provide OH groups with limited access to π electrons on triazole. Nonetheless, aliphatic chains bound to aromatic rings are known to allow for stabilizing CH⋯π interactions.36–44 These, however weak compared to an hydrogen bond, can control the conformational space of the aliphatic chain in the absence of heteroatoms. Furthermore, bent aliphatic chains bearing an electron-rich groups such as OH are also able to produce weak CH⋯O interactions.40 However, donor–acceptor locking due to a strong interaction can sometimes generate mechanical strain in the short aliphatic spacer, so that the stabilization due to that interaction is negated. As such, despite the weak stabilizing interactions, most isomers are 6 kT or higher in energy than the absolute minimum at room temperature.
| Isomera | ΔE/kJ mol−1 | Notes |
|---|---|---|
| a Symbols represent sequences of dihedral angles in the side chain, in order: N–C(1)–C(2)–C(3), C(1)–C(2)–C(3)–O, and C(2)–C(3)–O–H (see Scheme 1), whereas a stands for anti (about 180°) and g± stands for gauche (about ±60°). | ||
| g−g−g+ | 0.0 | OH⋯π, CH⋯π, CH⋯O |
| g−g+g− | 6.9 | OH⋯π |
| g+g+a | 7.4 | CH⋯π, CH⋯O (×2) |
| g+g+g+ | 8.8 | CH⋯π, CH⋯O (×2) |
| ag−a | 13.4 | CH⋯O |
| g+g−g− | 14.7 | OH⋯π |
| ag−g− | 14.7 | CH⋯O |
| g−aa | 15.0 | CH⋯π |
| ag+g+ | 15.1 | CH⋯O |
| g+ag+ | 17.0 | CH⋯π (×2) |
| ag+a | 17.1 | CH⋯O |
| g−g−g− | 17.2 | CH⋯π, CH⋯O |
| g−ag− | 17.4 | CH⋯π |
| g−ag+ | 17.4 | CH⋯π |
| g+g−g+ | 17.7 | OH⋯π |
| g+aa | 18.4 | CH⋯π |
| g+ag− | 19.4 | CH⋯π |
| g+g−a | 19.4 | CH⋯O |
| aag− | 19.5 | — |
| aaa | 20.6 | — |
| ag+g− | 21.1 | CH⋯O |
| aag+ | 21.3 | — |
| g−g+g+ | 24.3 | OH⋯π |
It is interesting to note that hydroxypropyl chains within the asymmetric unit of the PTD single crystal show g+g+g+ and ag+a conformations, both rather far from the absolute minimum obtained for the isolated alkyltriazole unit, at g−g−g+. These conformations, evidently, allow them to engage in the two strong intermolecular OH⋯O and OH⋯N hydrogen bonds mentioned above, so that their higher energy is likely to be handsomely compensated in the process.
In hindsight, with regard to the choice of a piece-wise conformational study, we note that the results presented so far suggest that PTD's full conformational space might potentially comprise about 46 (hydroxypropyl arrangements including mirror images) squared (because of the two independent hydroxypropyl chains) times 4 (bis(triazolyl)pyridine arrangements) isomers, which justifies the approximation used in the first place. Within this framework, then, PTD isomers featuring a combination of relatively optimal bis(triazolyl)pyridine (i.e. A) and hydroxypropyl (i.e. g−g−g+, g−g+g−) conformations are likely to be more stable than isomers featuring subunits in higher energy conformations. In turn, the more stable an isomer, the larger the relative population of that isomer will be, in compliance with a Maxwell–Boltzmann distribution.
Exploring the chelating ability of PTD in typical solvents involved in the reprocessing of spent nuclear fuel
Finally, in order to investigate the ability of PTD to act as a terdentate ligand for metal ion chelation in solution, a further elaboration into the relative stability of conformers C and D of truncated PTD was attempted. In particular, the relative free energy of all four conformers was re-evaluated in the presence of an implicit solvent, modelled as a continuum dielectric medium with an aptly-shaped cavity in which the solute is lodged. While handling of explicit solvent molecules is problematic41–45 (e.g. added degrees of freedom, shallow intermolecular potential energy wells, multiplicity of spatial arrangements), implicit solvation models have been proven in numerous occasions to provide a reliable description of short-range and long-range solute–solvent interactions.46–50 Thermal corrections to free energy were computed within the rigid-rotor-harmonic-oscillator (RRHO) formalism,51,52 and manually adjusted for standard state (1 mol L−1 was adopted),41 rotational symmetry,42 and low-frequency vibrational normal modes.53,54 Typical solvents involved in the reprocessing of spent nuclear fuel include kerosene and water, the former often in combination with small amounts of fatty alcohols.16 Accordingly, n-octane, n-octanol, and water, were chosen as model solvents, the former two representing kerosene and its alcohol additives, respectively.With reference to Table 2, although A remains the most stable conformer, the relative free energy (ΔG) of other conformers decreases significantly from gas phase to octane, then to octanol, and finally to water. However, in aqueous solution, all four conformers are rather accessible, all lying roughly within a 5 kT range at room temperature. The marked difference between C8H18 and C8H17OH suggests that the latter can stabilize syn/syn arrangements of PTD (conformers C and D) beyond the capabilities of the former, hence enhancing PTD chelating activity in mixed alcohol/kerosene solvents compared to kerosene alone.
| Conf. | ΔG (298 K)/kJ mol−1 | ||
|---|---|---|---|
| C8H18 | C8H17OH | H2O | |
| A | 0.00 | 0.00 | 0.00 |
| B | 13.4 | 7.2 | 2.8 |
| C | 42.3 | 24.4 | 12.6 |
| D | 41.8 | 22.2 | 10.1 |
Conclusions
A detailed structural analysis of the chelating agent PTD has been carried out combining single crystal X-ray diffraction and dispersion-corrected DFT calculations. PTD crystallizes in linear stacks, inter-locked in loosely-interdigitated lamellae by strong hydrogen bonds. Powder X-ray diffraction and calorimetric analysis are presented as evidence of the absence of polymorphism above room temperature. An unusual asymmetric syn/anti arrangement of the two otherwise symmetric substituents on pyridine was successfully elucidated as the result of hydrogen bonds involving three separate molecules. Only one example of a similar arrangement was detected in literature, involving however multiple hydrogen bonds between just two separate molecules. Because the relative populations of different PTD isomers in solution can relate to its efficacy and efficiency in metal ion complexation, extensive exploration of the PTD conformational space has been done by a piece-wise approach.Relative Gibbs free energies comprising implicit solvation effects and free-rotor corrections suggest PTD conformational space in aqueous solution to express a rather shallow PES, implying lower reorganization energies associated with the transition from free to bound ligand, thus favouring metal ion chelation. A similar stabilizing effect, although weaker, is obtained for n-octanol solutions, hinting to a stabilization of syn/syn arrangements of PTD in kerosene/alcohol mixed solvent solutions compared to kerosene alone.
The thorough PTD structural description presented here might be useful in various research fields such polymorphism in PTD or to evaluate the potential of PTD to act as outer sphere ligand in the formation of super-complexes (i.e., second sphere coordination complexes) which is another approach to extract metals from contaminated solutions. The interconversion from second-to-first coordination spheres by mechanochemical means via dehydrohalogenation reactions in the solid-state is certainly an intriguing research topic that deserves to be explored.
Experimental details
Single-crystal XRD data were collected on a Bruker X8 Prospector APEX-II/CCD diffractometer equipped with a microfocusing mirror (Cu-Kα radiation, λ = 1.54178 Å) at room temperature. The structure was determined using direct methods (SHELXTL 97) and refined (based on F2 using all independent data) by full-matrix least-squares methods (SHELX 2014). All non-hydrogen atoms were located from different Fourier maps and refined with anisotropic displacement parameters. Hydrogen atoms were added in riding positions.Powder XRD experiments were carried out at room temperature using a Bruker D2-Phaser diffractometer equipped with Cu radiation (λ = 1.54184 Å) using Bragg–Brentano geometry.
DSC measurements were performed in air using a PerkinElmer DSC7 differential scanning calorimeter. Samples, sliced off from corks and weighing 10.79 ± 0.10 mg, were fitted in an aluminium capsule. Heat flow variations were determined over a temperature range from 20 to 200 °C at a heating rate of 10 °C min−1.
Geometry optimizations and normal modes analyses (within the rigid-rotor-harmonic-oscillator approximation, RRHO)51,52 were performed without symmetry restraints at ωB97X-D/cc-pVTZ level using Gaussian16.55–57 NCI analyses were carried out using the Multiwfn 3.6 code.58 The SMD variant of the IEFPCM formalism was used to simulate implicit solvation.59 Free energies were recomputed at 298.15 K considering a 10−3 m3 mol−1 standard state (corresponding to 1 mol L−1).41 Symmetry-related corrections to rotational entropy were considered.42 Vibrational frequencies were corrected by a factor of 0.956 based on the combination of density functional and basis set used.60 Enthalpic and entropic vibrational contributions from low-frequency modes were corrected by weighted interpolation between rigid-rotor and free-rotor terms according to Head-Gordon and co-workers53 and Grimme,54 respectively. A threshold vibrational frequency of 100 cm−1 (about 0.5 kT at room temperature) and an average molecular moment of inertia of 10−44 kg m2 were used throughout as suggested by the authors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support. A. F. thanks MIUR for the Fondo Finanziamento delle Attività Base di Ricerca (FFARB). G. C. D. thanks Politecnico di Milano for her Inter-Departments PhD scholarship. J. M.-R thanks Politecnico di Milano for funding “Fondo Chiamata Diretta Internazionalizazzione. Prg. Id. 61566”.References
- Ch. Poinssot, S. Bourg, N. Ouvrier, N. Combernoux, C. Rostaing, M. Vargas-Gonzalez and J. Bruno, Energy, 2014, 69, 199–211 CrossRef.
- C. Hill, in Ion exchange and solvent extraction. A series of advances, ed. B. A. Moyer, CRC Press, Boca Raton, FL, 2019, pp.119–193 Search PubMed.
- G. Modolo, A. Geist and M. Miguirditchian, in Reprocessing and recycling of spent nuclear fuel, ed. R. Taylor, Woodhead Publishing, Oxford, UK, 2015, pp. 245–287 Search PubMed.
- J. N. Mathur, M. S. Murali and K. L. Nash, Solvent Extr. Ion Exch., 2001, 19, 357–390 CrossRef CAS.
- S. Bourg, C. Hill, C. Caravaca, C. Rhodes, C. Ekberg, R. Taylor, A. Geist, G. Modolo, L. Cassayre, R. Malmbeck, M. Harrison, G. de Angelis, A. Espartero, S. Bouvet and N. Ouvrier, Nucl. Eng. Des., 2011, 241, 3427–3435 CrossRef CAS.
- J. Martí-Rujas and F. Guo, Dalton Trans., 2016, 45, 13648–13662 RSC.
- Z. Liu, A. Samanta, J. Lei, J. Sun, Y. Wang and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 11643–11653 CrossRef CAS PubMed.
- J. J. M. Nelson and E. J. Schelter, Inorg. Chem., 2019, 58, 979–990 CrossRef CAS.
- J. J. M. Nelson, T. Cheisson, H. J. Rugh, M. R. Gau, P. J. Carroll and E. J. Shelter, Chem. Commun., 2020, 3, 7 CrossRef CAS.
- C. Kiefer, A. T. Wagner, B. B. Beele, A. Geist, P. J. Panak and P. W. Roesky, Inorg. Chem., 2015, 54, 7301–7308 CrossRef CAS PubMed.
- J.-P. Glatz, P. Souček and R. Malmbeck, in Reprocessing and recycling of spent nuclear fuel, ed. R. Taylor, Woodhead Publishing, Oxford, UK, 2015, pp. 49–62 Search PubMed.
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367–377 RSC.
- M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2013, 52, 3414–3428 CrossRef CAS PubMed.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS.
- E. Macerata, E. Mossini, S. Scaravaggi, M. Mariani, A. Mele, W. Panzeri, N. Boubals, L. Berthon, M. C. Charbonnel, F. Sansone, A. Arduini and A. Casnati, J. Am. Chem. Soc., 2016, 138, 7232–7235 CrossRef CAS.
- E. Mossini, E. Macerata, L. Brambilla, W. Panzeri, A. Mele, C. Castiglioni and M. Mariani, J. Radioanal. Nucl. Chem., 2019, 322, 1663–1673 CrossRef CAS.
- C. Wagner, E. Mossini, E. Macerata, M. Mariani, A. Arduini, A. Casnati, A. Geist and P. J. Panak, Inorg. Chem., 2017, 56, 2135–2144 CrossRef CAS PubMed.
- E. Mossini, E. Macerata, A. Wilden, P. Kaufholz, G. Modolo, N. Iotti, A. Casnati, A. Geist and M. Mariani, Solvent Extr. Ion Exch., 2018, 36, 373–386 CrossRef CAS.
- Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692–2694 RSC.
- J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, Chem. Soc. Rev., 2014, 43, 5302–5325 RSC.
- C. J. Adams, M. H. Colquhoun, P. C. Crawford, M. Lusi and A. G. Orpen, Angew. Chem., Int. Ed., 2007, 46, 1124–1128 CrossRef CAS PubMed.
- E. Coronado, M. Gimenez-Marques, G. M. Espallargas and L. Brammer, Nat. Commun., 2012, 3, 828 CrossRef PubMed.
- F. Guo, H. D. Shao, Q. Yang, A. Famulari and J. Martí-Rujas, CrystEngComm, 2014, 16, 969–973 RSC.
- H. Y. Guan, Z. Wang, A. Famulari, X. Wang, F. Guo and J. Martí-Rujas, Inorg. Chem., 2014, 53, 7438–7445 CrossRef CAS.
- F. Guo, Z. Wang, J. J. Zhang, A. Famulari, H. Li and J. Martí-Rujas, Dalton Trans., 2017, 46, 9466–9471 RSC.
- H. Li, F. Guo, M. Kou, A. Famulari, Q. Fu and J. Martí-Rujas, Inorg. Chem., 2017, 56, 6584–6590 CrossRef CAS PubMed.
- F. Guo, H. C. Wang, A. Famulari, H. D. Lu and J. Martí-Rujas, CrystEngComm, 2018, 20, 6721–6726 RSC.
- B. S. Gelinas, J. A. Jaye, G. R. Mattos and E. H. Fort, Tetrahedron Lett., 2015, 56, 4232–4233 CrossRef CAS.
- E. P. McCarney, C. S. Hawes, J. A. Kitchen, K. Byrne, W. Schmitt and T. Gunnlaugsson, Inorg. Chem., 2018, 57, 3920–3930 CrossRef CAS PubMed.
- D. Zornik, R. M. Meudtner, T. El Malah, C. M. Thiele and S. Hecht, Chem.–Eur. J., 2011, 17, 1473–1484 CrossRef CAS.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem.–Eur. J., 2007, 13, 9834–9840 CrossRef CAS.
- M. Ostermeier, M.-A. Berlin, R. M. Meudtner, S. Demeshko, F. Meyer, C. Limberg and S. Hecht, Chem.–Eur. J., 2010, 16, 10202–10213 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- H. P. G. Thompson and G. M. Day, Chem. Sci., 2014, 5, 3173–3182 RSC.
- Y. Kodama, K. Nishihata, M. Nishio and N. Nakagawa, Tetrahedron Lett., 1977, 24, 2105–2108 CrossRef.
- A. Baggioli, S. V. Meille, G. Raos, R. Po, M. Brinkmann and A. Famulari, Int. J. Quantum Chem., 2013, 113, 2154–2162 CrossRef CAS.
- A. Baggioli and A. Famulari, Phys. Chem. Chem. Phys., 2014, 16, 3983–3994 RSC.
- A. Baggioli, C. A. Cavallotti and A. Famulari, Phys. Chem. Chem. Phys., 2016, 18, 29616–29628 RSC.
- J. R. Lane, J. Contreras-García, J.-P. Piquemal, B. J. Miller and H. G. Kjaergaard, J. Chem. Theory Comput., 2013, 9, 3263–3266 CrossRef CAS PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS.
- J. H. Jensen, Phys. Chem. Chem. Phys., 2015, 17, 12441–12451 RSC.
- J. R. Pliego, Jr. and J. M. Riveros, J. Phys. Chem. A, 2001, 105, 7241–7247 CrossRef.
- A. Baggioli, O. Crescenzi, M. J. Field, F. Castiglione and G. Raos, Phys. Chem. Chem. Phys., 2013, 15, 1130–1140 RSC.
- B. Thapa and K. Raghavachari, J. Chem. Theory Comput., 2019, 15, 6025–6035 CrossRef CAS PubMed.
- F. Egidi, V. Barone, J. Bloino and C. Cappelli, J. Chem. Theory Comput., 2012, 8, 585–597 CrossRef CAS PubMed.
- R. Cammi, C. Cappelli, S. Corni and J. Tomasi, J. Phys. Chem. A, 2000, 104, 9874–9879 CrossRef CAS.
- F. Castiglione, A. Baggioli, A. Citterio, A. Mele and G. Raos, J. Phys. Chem. A, 2012, 116, 1814–1819 CrossRef CAS PubMed.
- A. Baggioli, M. Sansotera and W. Navarrini, Chemosphere, 2018, 193, 1063–1070 CrossRef CAS PubMed.
- F. Santoro, R. Improta, A. Lami, J. Bloino and V. Barone, J. Chem. Phys., 2007, 126, 084509 CrossRef PubMed.
- J. W. Ochterski, Thermochemistry in Gaussian, Gaussian Inc., Wallingford, CT, 2000, pp 1–19 Search PubMed.
- D. A. McQuarrie and J. D. Simon, Molecular Thermodynamics, University Science Books, Sausalito, CA, 1999 Search PubMed.
- Y.-P. Li, J. Gomes, S. Mallikarjun Sharada, A. T. Bell and M. Head-Gordon, J. Phys. Chem. C, 2015, 119, 1840–1850 CrossRef CAS.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision B. 01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 20, August 2019, ed. R. D. Johnson III, http://cccbdb.nist.gov/ Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NCI details and quantitative analyses, CSD survey, model systems optimized geometries. CCDC PTD: 1984000. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra04142d |
| This journal is © The Royal Society of Chemistry 2020 |